

Abstract
B-cell lymphoma 6 (BCL6) is a master regulator of germinal center formation that produce antibody-secreting plasma cells and memory B-cells for sustained immune responses. The BTB domain of BCL6 (BCL6BTB) forms a homodimer that mediates transcriptional repression by recruiting its co-repressor proteins to form biologically functional transcriptional complex. The protein-protein interaction (PPI) between the BCL6BTB and its co-repressors has emerged as a therapeutic target for the treatment of DLBCL and a number of other human cancers. This perspective provides an overview of recent advances in the development of BCL6BTB inhibitors from reversible inhibitors, irreversible inhibitors, to BCL6 degraders. Inhibitor design and medicinal chemistry strategies for the development of novel compounds will be provided. The binding mode of new inhibitors to BCL6BTB are highlighted. Also, the in vitro and in vivo assays used for the evaluation of new compounds will be discussed.
Graphical Abstract
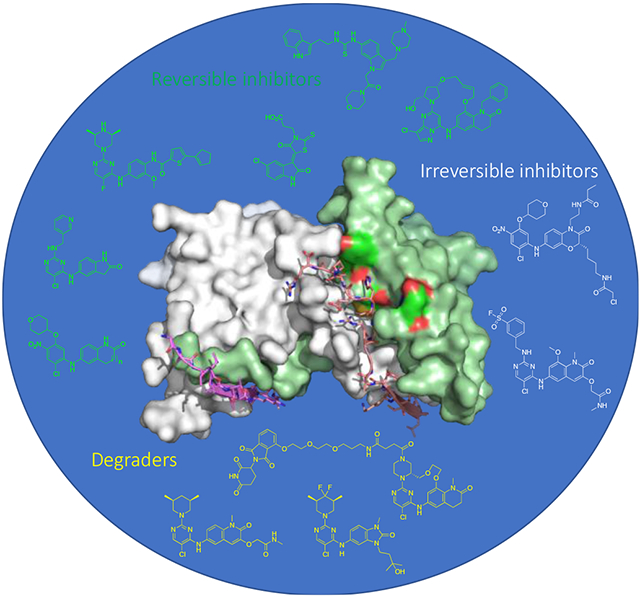
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin’s lymphomas (NHL) throughout the world with over 18,000 new cases each year. DLBCL is a fast growing cancer arising from germinal center (GC) B-cells.1 GCs are transient structures formed within lymphoid follicles responding to antigenic stimulation.2 The transcriptional repressor protein B-cell lymphoma 6 (BCL6) is needed for B-cells to form GCs.1 Normally BCL6 is upregulated when B-cells are activated to form GCs and downregulated once the GC reaction is complete.1 Failure to downregulate BCL6 leads to continuous expression of this transcriptional repressor, which is required to maintain the survival of DLBCL cell lines and primary human DLBCL tumors.3-7 BCL6 regulates lymphomagenesis by directly repressing DNA damage sensing genes as well as cell cycle and cell death and checkpoint genes.3,8-13 In addition, BCL6 also blocks differentiation of DLBCL cells by repressing the PRDM1 locus that encodes a master regulator for plasma cell differentiation.13,14 BCL6 knockdown or inhibition of its repressive activity causes expression of these many checkpoint genes, resulting in proliferation arrest and cell death.3-7,15
BCL6 has been reported as an oncoprotein in many other types of human tumors. Follicular lymphomas (FLs), the second most frequent type of NHL, are documented to be biologically dependent on the repressive function of BCL6 in GC B-cells.16 In addition, BCL6 is highly upregulated in BCR-ABL positive B-acute lymphoblastic leukemia (B-ALL) cells after treatment by tyrosine kinase inhibitors.17 Upregulation of BCL6 prevented B-ALL cells from being killed by kinase inhibitors, however, the combination of ABL kinase inhibitor plus BCL6 blockade synergistically eradicated B-ALLs in vivo.17 Moreover, BCL6 is constitutively upregulated in mixed-lineage leukemia (MLL) fusion oncoproteins in B-ALLs with MLL translocation, while suppression of BCL6 kills primary human MLL translocated B-ALL cells.18 Patients with chronic myeloid leukemia (CML) are resistant to tyrosine kinase inhibitor imatinib because CML stem cells are also dependent on BCL6,19 and blockade of BCL6 activity eliminated these critical leukemia-repopulating cells. Furthermore, BCL6 is highly expressed in acute myeloid leukemia (AML) stem cells, and blocking BCL6 kills AML cells and synergizes with chemotherapy.20 BCL6 is also required to maintain the survival of “triple-negative” breast cancer cells.21 Recently, BCL6 was characterized as a promising therapeutic target in non-small cell lung cancers, which feature frequent amplification of the BCL6 locus.22,23 Taken together, BCL6 has broad oncogenic roles in different cancers and BCL6 targeted therapy can potentially benefit many patients beyond those with lymphomas.
BCL6 has an N-terminal broad-complex, tramtrack, and bric-a-brac (BTB) domain (BCL6BTB) that controls transcriptional repression and a C-terminal C2H2 zinc finger that binds to a specific DNA consensus sequence.24 The two domains are connected by an unstructured region containing a second repression domain (RD2).25 The BCL6BTB forms a homodimer (Figure 1) that recruits corepressors silencing mediator for retinoid and thyroid receptor (SMRT), nuclear receptor corepressor (NCOR) and BCL6 corepressor (BCOR).26,27 The minimal binding domain of these corepressors contains a conserved 18 amino-acid peptide known as BCL6 binding domain (BBD)28,29 that binds to a lateral groove (LG) formed at the interface between BCL6BTB monomers (Figure 1).28,29 This LG/BBD interaction is essential for the transcriptional repression activity of BCL6BTB.28,29 Interestingly, the key BCL6BTB surface residues that contact the BBD are not conserved in any of the other BTB domains.30
Figure 1.

Surface view of the binding mode of the SMRT peptide (LVATVKEAGRSIHEIPR) to BCL6BTB LG (PDB ID: 1R2B). The monomers of BCL6BTB were shown in green and white, respectively. The SMRT peptides were shown in pink and yellow, respectively.
Besides GC B-cells, BCL6 also plays essential role in the development of follicular T-helper cells and anti-inflammatory effects in macrophages.31 Experimental evidence has shown that BCL6 knockout mice are runted, born at sub-Mendelian ratios and die within weeks of acute inflammatory diseases.31 However, in contrast to BCL6 knockout mice, those with a BCL6BTB mutant that cannot bind to the corepressors, were born at expected rates, and lived normal healthy lives and had no evidence of inflammatory syndrome. The only observed phenotype was failure to form GC B-cells in response to antigenic challenge.32 Therefore, the function of BTB LG is only essential in GC B-cells and in tumors, but is inessential for the function of BCL6 in other lineages. The effect of BCL6 in macrophages, for instance, is mediated by its DNA-binding zinc finger domain, while not affected by targeting the BTB LG.32 Transient suppression of GCs is expected to be non-toxic in humans. Thus, BCL6BTB LG inhibitors represents a safe therapeutic target.
Numerous inhibitors of BCL6BTB have been developed. In pioneering work, Polo and coworkers developed a BCL6 peptide inhibitor (BPI),4 which occupied the BTB LG, prevented recruitment of the corepressors by BCL6 and induced expression of BCL6-target genes.4 BPI also had potent anti-lymphoma activity, inducing apoptosis and growth arrest in BCL6-dependent DLBCL cell lines but not for BCL6-independent cell lines.4 Injections of BPI in mice reproduced the BCL6 null GC phenotype.4 Refinement of the BPI yielded a retro-inverso peptidomimetic RI-BPI that bind to BCL6BTB LG and potently killed DLBCL cells in vitro and in vivo and primary human DLBCL cells ex vivo.5
A fundamental lesson of these studies was that the duration of occupancy of the BCL6BTB LG was critical for killing of DLBCL cells. Whereas a single dose of short acting decoy peptides with t1/2 < 6 h could not kill DLBCL cells, the longer acting RI-BPI, which has a nuclear dwell time up to 24 h in vitro and in vivo did kill DLBCL cells similar to BCL6 siRNA or shRNA. Duration of target engagement is a key parameter for BCL6 targeted therapy.33,34 Notably, because BCL6 blockade reactivates checkpoints and cell death, remarkable synergy between RI-BPI and chemotherapy was observed.12 The lack of toxicity of LG blockade makes BCL6 inhibitors ideally suited to serve as “anchors” for developing novel combination therapy regimens. In sum, BCL6 presents a highly promising therapeutic target that can be effectively and specifically blocked by occluding the BCL6BTB LG.
Early small molecule BCL6BTB inhibitors 1 (79-6)6 and 2 (FX1)7.
The discovery and activity profiles of BCL6BTB inhibitors 1 and 2 have been reviewed previously (Figure 2A).35,36 In silico screening hit 1 could disrupt BCL6 repression complexes and kill DLBCL cells.6 Subsequent SAR efforts yielded inhibitor 2,7 which bound to BCL6 with affinity similar to endogenous corepressors, exhibited a prolonged dwell time in cells, and mirrored the efficacy and specificity of RI-BPI in vitro by killing BCL6 dependent but not BCL6 independent DLBCL cell lines. Inhibitor 2 was nontoxic and demonstrated promising efficacy in vivo in both germinal center B-cell (GCB) and activated B-cell (ABC) type DLBCL xenograft models.7 The binding modes of these inhibitors have been characterized by X-ray crystallography and NMR. The indolin-2-one rings of compounds 1 and 2 occupy an aromatic pocket (named the aromatic site) on the top region of the BCL6 LG (red, Figure 2B). The aromatic site is formed by four key residues: R’24, L’25 from one BCL6BTB monomer, and residues M51 and Y58 from the other. The same aromatic site is occupied by corepressor SMRT residues H1426 and I1428,27 or BCOR residues W509 and V511.28,29 Also, the side chains of L25’ and M51 line a hydrophobic pocket that interacts with the Br atom in inhibitor 1. The carboxylic acid group of the inhibitor fits snugly into the acid site (cyan, Figure 2B) and forms electrostatic interaction with R’28 (Figure 2C). The NH group of the indolin-2-one ring forms an H-bond (2.5 Å) with the backbone carbonyl O of residue M51. Adjacent to the aromatic site, a shallow hydrophobic pocket (named linker site, blue, Figure 2B), occupied by the side chain of a tryptophan residue in the SMRT protein, is available with the residue E115 located 7.5 Å away from the indolin-2-one ring (Figure 2C). Importantly, the backbone amide NH group, as well as the carboxylate side chain of E115 provide opportunities for interactions such as hydrogen bonds (H-bonds) and electrostatic interactions. Moreover, a large and deep pocket (H’14-D’17-C53-H116), known as the HDCH site (yellow, Figure 2B), is located below residue E115, providing an additional pocket for inhibitor design. The presence of residue C53 in the HDCH site offers a unique opportunity for the development of irreversible inhibitors for BCL6BTB. The specificity of inhibitors can be designed into irreversible inhibitors by exploiting the unique chemical characteristics and spatial orientation of the aromatic and linker sites, as well as the novel HDCH site, to impart specific binding before covalent bond formation.
Figure 2.

(A) Chemical structures of early BCL6 inhibitors 1 and 2. (B) Binding mode of compound 1 to BCL6BTB (PDB ID: 3LBZ). Compound 1 was shown in cyan and BCL6BTB monomers are shown in green and gray, respectively. The binding pocket of BCL6BTB LG includes four continuous binding sites: acid site (cyan), aromatic site (red), linker site (blue) and HDCH site (yellow). (C) Detailed binding mode of inhibitor 1 in complex with BCL6BTB. The key residues from BCL6BTB were shown as sticks. The distances of highlighted H-bond interactions were shown in angstrom (Å).
These early inhibitors 1 and 2 provided proof of principle for targeting the BCL6BTB LG; however, the PAINS classification of the rhodanine moiety37 motivated the design of novel BCL6 inhibitors with improved affinity while lacking undesirable PAINS moieties, which will be the focus of the current review.
Recent Development of BCL6BTB Inhibitors
Reversible Inhibitors
Sakamoto and coworkers disclosed their efforts using the technology of phage-displayed peptide library to identify novel BPIs (Scheme 1).38 Specifically, the authors panned T7 phage-displayed peptide libraries against the FLAG-BCL6BTB and control proteins (TWEAK and IL17),39,40 and determined the binding affinity of resultant polyclonal phage pools using an enzyme-linked immunosorbent assay (ELISA) on microplates by following the absorbance changes (λ = 450 nm). After five rounds of panning, two peptides, W-Y/F/I/V/L-T/S-D-I/V/L-R-M and W/F/Y-R/K-V/I-P, were identified. Next, the authors used two representative sequences FVRVHTRSSWRVP (3, F1323) and GVWYTDIRMRDWM (4, F1325) to design a hybrid peptide LWYTDIRMSWRVP (5, F1324).
Scheme 1.

Development and sequence structure of the hybrid BPI 5 (F1324)
The inhibitory potencies of peptides 3-5 were evaluated using cell-free ELISA assays. Initial peptides 3 and 4 indicated IC50 values of 0.26 μM and 0.032 μM, respectively. The hybrid 5 was a potent inhibitor of BCL6 with an IC50 value of 1 nM, which was ~3,000-fold more potent than that of the BCL6 corepressor BCOR peptide (IC50 = 3 μM). Note that the BCOR peptide was biotinylated and its sequence is detailed in Table 1. Next, the kinetics for the binding of peptide 5 was assessed using surface plasmon resonance (SPR). The Kd value of peptide 5 was determined to be 0.57 nM, which is 10,000-fold more potent than that of BCOR peptide (Kd = 6,000 nM). The kon and koff values were calculated to be 2.8 × 106 M−1 s−1 and 1.6 × 10−3 s−1, respectively. The binding half-life (t1/2) of peptide 5 was determined to be 441 s, significantly longer than that of the BCOR peptide (t1/2 < 1 s). These results supported that the potent inhibitory activity of peptide 5 against BCL6BTB was likely due to the small koff value.
Table 1.
Competitive Binding Assays and Conditions
| Assay | BCL6BTB a | Co-repressor | Reference |
|---|---|---|---|
| ELISA | FLAG-BCL6BTB | Biotinylated BCOR peptideb | 38, 42, 50 |
| TR-FRET | His tagged BCL6BTBc | Hilyte647 SMRT peptided | 60, 91 |
| Biotinylated BCL6BTB | TAMRA-peptide 4e | 72 | |
| Biotinylated BCL6BTB | FITC-BCOR peptidef | 83 | |
| Trx-His tagged BCL6BTBg | AF633-BCOR peptideh | 95 | |
| Avi-tag biotinylated BCL6BTBi | BCOR ULight peptidej | 69 | |
| GST-BCL6BTBk | His tagged SMRT peptidel | 70 | |
| FP | Avi-tag biotinylated BCL6BTBi | FP-labeled BCOR peptidem | 69 |
| BCL6BTB | AF633-BCOR peptideh | 95 | |
| AlphaLisa | His tagged BCL6BTB c | Biotinylated BCOR peptideb | 73 |
| IPC | Avi-tag biotinylated BCL6BTBi | 37 (BCL6-FP) | 78 |
BCL6BTB (5-129)
Biotinylated BCOR peptide = Ac-RSEIISTAPSSWVVPGP-Lys-(ε-Biotin-(AC5)2)-OH
His tagged BCL6BTB = Histidine (His) tagged BCL6BTB (5-129) triple mutant (C8Q, C67R, and C84N)
Hilyte™ Fluor 647 SMRT peptide H1426W = Hilyte™ Fluor 647-LVATVKEAGRSIWEIPR
TAMRA peptide 4 = TAMRA-Abu(4)-VWYTDIRMRDWM-OH
FITC, fluorescein isothiocyanate (Note: In the paper, BodipyFL-labeled BCOR peptide, but in SI FITC-BCOR peptide)
Trx, Thioredoxin
Alexa Fluor 633-BCOR peptide = Ac-RSEIISTAPSSWVVPGP-Cys-Alexa Fluor 633-CONH2
Avi-tag, amino acid sequence GLNDIFEAQKIEWHE
BCOR ULight peptide = Ac-RSEIISTAPSSWVVPGP-Cys-ULight
GST, Glutathione S-transferase
His tagged SMRT peptide = H6-LVATVKEAGRSIHEIPR
FP-labeled BCOR peptide = 5-TAMRA-RSEIISTAPSSWVVPGP
To identify key amino acid residues responsible for the high inhibitory potency of peptide 5, the authors synthesized and tested 19 fragment peptides. While most of the N-terminal truncated peptides showed dramatically decreased potencies, the C-terminus tetrapeptide WRVP (6, Scheme 1) displayed promising inhibition activity (IC50 = 170 μM), which is even more potent than longer sequences MSWRVP (IC50 = 620 μM) and SWRVP (IC50 = 1,500 μM). Besides, replacing the C-terminus carboxylate of WRVP, which occupies the acid site of BCL6BTB LG (Figure 4), with an amide group caused a significant decrease in inhibitory potency (IC50 = 170 μM to IC50 = 2,100 μM), highlighting the importance of this carboxylate group for the potency of WRVP.
Figure 4.

Binding mode of peptide 6 (Ac-WRVP-OH) to BCL6BTB (PDB ID: 5H7H). (A) Surface view of the binding of peptide 6 to the BCL6BTB LG. Peptide 6 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 6 in complex with BCL6BTB.
Although peptide 5 was highly potent in cell-free assays, the authors found this tetrapeptide had no significant inhibitory activity in cellular assays. While various peptide fusion strategies with cell-permeable peptides failed to improve the cellular activity, the authors pursued a cell-based mammalian two-hybrid (M2H) assay27 to deliver peptide 5 into cells. Specifically, the authors built the plasmid coding AcGFP, a nuclear localization sequence DPKKKRKV, and peptide 5. The expression level of the peptide was confirmed by following the fluorescence signal of AcGFP.41 The results confirmed that the expressed peptide AcGFP-(PKKKRKV)3-GGG-5 localized in the nucleus, causing decreased luminescence from the transcription of the Luc gene by inhibiting the PPI between BCL6BTB and BCOR. In a separate experiment, expression of BCORpep-containing sequence AcGFP-(PKKKRKV)3-GGG-BCORpep failed to decrease the luminescence signal, indicating that the level of the expressed peptide was significantly less than the Kd value of BCOR peptide.
The co-crystal structure of the complex BCL6BTB-5 (PDB 5H7G) was solved at 1.85 Å (Figure 3). Peptide 5 binds to BCL6BTB LG in a binding mode similar to those of the SMRT and BCOR peptides. The side chains of amino acids L1, R7, and R11 were completely disordered and those of W2, D5, and P13 were partly disordered. The peptide backbones of W2 and T4 made H-bonds with those of Q10 (2.7 Å) and T12 (2.9 Å) of BCL6BTB, respectively. In addition, the M8, W10, and V12 backbones formed H-bonds with the sidechains of D’17 (2.8 Å), N’21 (3.0 Å), and R’24 (2.8 Å) of BCL6BTB, respectively (Figure 3). The side-chains of amino acids W10 and P13 formed hydrophobic interactions with residue Y58 of BCL6BTB. The C-terminal -CO2H of amino acid P13 snugged into the acid site of LG and formed H-bonds with the guanidine group of R’28 (3.3 Å). The N-terminus of peptide 5 interacts with a β-strand in the bottom of the BCL6BTB LG. The sidechain of amino acid M8 is deeply buried in the HDCH site of BCL6BTB LG, with a significant conformational change of H116 in BCL6BTB. This methionine residue is substituted by serine residues in both the analog BPI 3 and the BCOR peptide, and an isoleucine (I1425) in the SMRT peptide.
Figure 3.

Binding mode of BPI 5 (F1324) to BCL6BTB (PDB ID: 5H7G). (A) Surface view of the binding of BPI 5 to the BCL6BTB LG. Peptide 5 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 5 in complex with BCL6BTB.
The authors also studied the binding mode of the tetrapeptide WRVP (6) to BCL6BTB (Figure 4). Interestingly, the truncate peptide 6 occupied the same site as peptide 5 (Figure 4A), with highly similar H-bond patterns to BCL6BTB LG. The carboxylate group of P13 interacted simultaneously to guanidine groups of R’24 and R’28 in the acid site (Figure 4B). The isopropyl group of V12 fit snugly into the aromatic site while the backbone NH group of V12 and carbonyl O atom of W10 formed two H-bonds to the sidechain of residue N’21. In addition, the guanidine group of R11 formed electrostatic interaction to residue D’17 and the indole NH of W10 formed H-bond to the backbone carbonyl O of residue M51. This unique binding mode of tetrapeptide 6 provided a potential scaffold for effective inhibition of BCL6BTB.
By screening a library of ~130,000 compounds using ELISA, Yasui and coworkers discovered diphenylamine hits 7 and 8 with IC50 values of 4.5 μM and 14 μM, respectively (Scheme 2).42 To confirm the binding of these hits to the BCL6BTB, the authors used surface plasmon resonance (SPR) and determined the Kd values of compounds 7 and 8 to be 5.0 μM and 24 μM, respectively. The co-crystal structure of BCL6BTB dimer in complex with hit 7 (Figure 5) showed the inhibitor bind to the top part of BCL6BTB LG, the same site as those of compound 1 and tetrapeptide 6. Specifically, the left ring of inhibitor 7 occupied the aromatic site formed by sidechains of Y58, N’21 as well as with R’24, and R’28 in the acid site. The Cl atom of the hit was found to fit into the small hydrophobic pocket formed by three residues M51, L’25, and A52. Large substituents (e.g., Br and CF3) were not tolerated at this position. A key H-bond (3.0 Å) was formed between the diphenylamino NH group and the backbone O atom of residue M51. Another important H-bond (3.1 Å) was detected between the benzimidazolone O atom and the backbone NH group of residue E115.
Scheme 2.

Optimization strategy of HTS hits 7 and 8 and identification of compounds 10 and 13
Figure 5.

Binding mode of hit 7 to BCL6BTB (PDB ID: 5X9O). (A) Surface view of the binding of hit 7 to the BCL6BTB LG. Compound 7 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 7 in complex with BCL6BTB.
Suggested by docking results of hit 8 in BCL6BTB, the hydroxypropylamino group of compound 8 was introduced to the left ring of hit 7, resulting in the hybridized inhibitor 9, with a 20-fold improvement in potency (IC50 = 0.24 μM) compared to that of hit 7 (Scheme 2). To improve physiochemical properties of hybrid inhibitor 9 (solubility: 0.09 μg/mL, parallel artificial membrane permeability assay (PAMPA): not detected), five additional analogs with different substituents at the 5-position of the left ring were prepared to identify the 4-tetrahydropyranyloxy analog 10 and carboxylic acid 11. Compounds 10 and 11 indicated significantly increased aqueous solubility (10: 18 μg/mL, 11: > 72 μg/mL)43 without compromising their potencies. The 4-tetrahydropyranyloxy analog 10 kept modest PAMPA permeability (45 nm/s) although the carboxylic acid analog 11 failed to show detectable PAMPA permeability, likely due to the presence of the carboxylic acid functionality. To improve membrane permeability of inhibitor 10, the authors explored the substitution effects on the right ring of the scaffold and reported that the two H-bond donors in the benzimidazolone group contributed to the low PAMPA permeability. Accordingly, oxindole analog 12 and tetrahdroquinolinone analog 13 demonstrated improved PAMPA permeabilities (288 nm/s and 206 nm/s, respectively) compared to that of 4-tetrahydropyranyloxy 10, while maintaining potency and aqueous solubilities (12, IC50 = 0.13 μM, aqueous solubility = 12 μg/mL; 13, IC50 = 0.10 μM, aqueous solubility = 12 μg/mL vs. 10, IC50 = 0.22 μM, aqueous solubility = 18 μg/mL). Besides, compounds 12 and 13 exhibited no cytotoxicity at the concentration of 30 μM in HepG2 cells.
Next, inhibitors 12 and 13 were tested in cellular M2H assays. Both inhibitors indicated good cellular potencies with IC50 values of 1.2 μM and 0.72 μM, respectively. As a control, the unoptimized parent compound 9 demonstrated no cellular activity (IC50 > 100 μM). The PK profile of the more potent inhibitor 13 was studied (0.1 mg/kg i.v.; 1 mg/kg p.o.), and the results showed that compound 13 had a good PK profile (MRT = 3.3 h, AUC = 1.27 μg·h/mL, F = 79.9%), making this compound a promising candidate for future in vitro and in vivo studies.44
The binding mode of aqueous soluble analog 11 was determined by X-ray crystallography (PDB 5X9P). As shown in Figure 6, the left ring moiety of compound 11 is positioned in the aromatic site composed of amino acid side chains of Y58, N’21, L’25, and R’28. The NO2 group of inhibitor 11 interacted with the guanidinium side chain of R’28. The Cl at the 2-position of compound 11 was found to occupy the relatively small hydrophobic pocket formed by three residues M51, L’25, and N’21. Interestingly, the carboxylic acid group of inhibitor 11 was solvent-exposing and largely disordered, while no obvious electrostatic interaction was formed between the carboxylate and the guanidine side chain of R’28 in the acid site. The linker NH group formed a clear hydrogen bond (2.8 Å) with the backbone carbonyl group of the residue M51. Finally, the bicyclic lactam occupied the linker site and the lactam carbonyl O formed a key H-bond (3.1 Å) with the backbone NH of E115.
Figure 6.

Binding mode of compound 11 to BCL6BTB (PDB ID: 5X9P). (A) Surface view of the binding of inhibitor 11 (pink) to the BCL6BTB LG. Compound 11 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 11 in complex with BCL6BTB.
Using fragment-based drug discovery (FBDD),45-49 Kamada and coauthors conducted screens of 1494 fragment-sized molecules against four BCL6BTB proteins:50 captured wt BCL6BTB, captured mt BCL6BTB, coupled wt BCL6BTB and NeutrAvidin, using SPR.51-54 From the identified hits, the authors chose 64 pan-active candidates and conducted full dose-responses. The binding of selected hits was confirmed by STD-NMR.55 Based on these efforts, a triazine hit 14 (Scheme 3) was identified with a Kd value to 1,200 μM (SPR56-58) and ligand efficiency (LE59) of 0.28 to BCL6BTB.
Scheme 3.

Identification of hit 14 and optimization to the generation of inhibitors 17
Hit 14 binds to BCL6BTB LG (PDB 5X4M) with a similar binding mode to that of inhibitor 1. The linker NH group formed a key H-bond with the backbone carbonyl O atom of M51. Next, the authors tested the Kd values of four additional pyrimidine analogs of triazine 14, and found the chloropyrimidine analog 15 with a significantly improved potency (Kd = 68 μM) and LE (0.38).
Parallelly, another hit 16 (Kd = 88 μM, LE = 0.16) was obtained from an ELISA-based HTS campaign by measuring the PPI between the BCL6BTB dimer and BCOR peptide. The co-crystals revealed that inhibitors 15 and 16 bind to BCL6BTB dimer with a similar orientation. The NH linkers bridging the two aromatic rings overlapped. The Cl atom on the pyrimidine ring in compound 15 fit into a small hydrophobic pocket formed by residues M51, L’25, and A52) (PDB 5X4N). The carbonyl O atom of the cyclic amide in hit 16 occupied the linker site and interacts with the backbone NH group of residue E115 (PDB 5X4O).
The structural similarities of compounds 15 and 16 inspired the authors to develop a hybrid compound 17, which showed significantly improved potency (Kd = 78 nM, >15,000-fold more potent than hit 14). Co-crystal structure of inhibitor 17 in complex with BCL6BTB (Figure 7) showed that its pyridine ring was solvent exposed and mostly disordered. The two key H-bonds remained between the bridging NH group and the backbone carbonyl oxygen atom of M51 (2.8 Å), and between the cyclic amide oxygen with the backbone NH of residue E115 (3.0 Å). Interestingly, one molecule of ethylene glycol was observed in the HDCH site. One of the OH groups of the ethylene glycol formed an H-bond with the sidechain of H’14, while the other OH located 3.6 Å away from the cyclic amide fragment of compound 17. These results highlighted the potential of further expansion of this inhibitor series toward the HDCH site.
Figure 7.

Binding mode of compound 17 to BCL6BTB (PDB ID: 5X4Q). (A) Surface view of the binding of inhibitor 17 (pink) to the BCL6BTB LG. Compound 17 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 17 in complex with BCL6BTB.
Based on the encouraging binding affinity and ligand efficiency of inhibitor 17, the authors further evaluated the activity of this compound in additional assays. In a competitive binding ELISA assay, compound 17 potently inhibited the PPI between BCL6BTB and BCOR peptide with the IC50 value of 0.48 μM. However, the cellular activity of this inhibitor was moderate (IC50 = 8.6 μM in M2H assay). Overall, compound 17 represented a promising BCL6BTB inhibitor for future development.
Using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay, McCoull and coauthors screened a library of ~8,000 compounds from the AstraZeneca collection, and identified a pyrazolo[1,5-a]pyrimidine-based hit 18 with the IC50 value of 61 μM (Scheme 4).60 Parallelly, the authors also used SPR to screen ~3,500 fragments and identified another hit 19 employing a same pyrazolo[1,5-a]pyrimidine core with the Kd value of 690 μM. The binding of hits 18 and 19 to BCL6BTB was confirmed using 2D 1H,15N-TROSY NMR experiments in which, competitive displacement of the SMRT peptide by testing compounds was studied.
Scheme 4.

Optimization strategy of hits 18 and 19 and generation of pyrazolo[1,5-a]pyrimidine inhibitors using TR-FRET, SPR, and cellular gene reporter assays
Based on the co-crystal structures of 18 (PDB 5N20) and 19 (PDB 5N1X) in complex with BCL6BTB, the authors found that the pyrazolo[1,5-a]pyrimidine of both hits bind to the aromatic site of BCL6BTB LG. The acid site residue R’24 formed a cation-π interaction with the pyridine ring of hit 19 while the disubstituted phenyl group of hit 18 extruded from the protein surface. Next, the authors conducted a solvent analysis (CCG, MOE) using 3D-RISM61, and identified a number of unstable H2O molecules that are present in the linker site. Specifically, one of the H2O molecules, which indicated high occupancy and low stability, interacted with the backbone NH of residue E115 through an H-bond. Then, a new analog 20 was synthesized employing a bicyclic lactam group designed to replace the E115-interacting H2O molecule. Indeed, the co-crystal structure of inhibitor 20 (PDB 5N21) in complex with BCL6BTB confirmed that the bicyclic lactam group occupied the linker site with the lactam carbonyl interacting with the E115 backbone NH via an H-bond. Interestingly, the carboxylate of inhibitor 20 did not interact directly to R’24. Instead, these two groups interacted through a structural H2O molecule. As a confirmation of this result, the hydroxy analog 21 also indicated an excellent potency (IC50 = 370 nM). Based on the structural data, a macrocyclic compound 22 was synthesized by tethering the two ring systems of compound 21 (Scheme 4). Remarkably, the new macrocyclic inhibitor 22 indicated an IC50 value of 2.9 nM, over 120-fold more potent than its parent 21. To explain the increase of affinity from acyclic inhibitor 21 to macrocycle 22, the authors conducted conformational analysis using NMR in solution.62,63 Two major conformations were observed for both compounds, with an active/inactive ratio of 79:18 for macrocycle 22 and 60:40 for acyclic 21, indicating that the flexible rings limited the inhibitor from adopting the bioactive configuration.
The authors further studied the binding kinetics of inhibitors 20-22 using SPR,64-66 and found that the Kd values correlated well with the IC50 values obtained from the TR-FRET (R2 = 0.99), such as compounds 20 (SPR Kd = 1.1 μM vs. TR-FRET IC50 = 0.35 μM), 21 (SPR Kd = 0.50 μM vs. TR-FRET IC50 = 0.37 μM), and 22 (SPR Kd = 6.5 nM vs. TR-FRET IC50 = 2.9 nM). The authors then reasoned the affinity increase from acyclic 21 to macrocycle 22 as a combination of an increased associate rate constant kon due to enhanced structural rigidity, and decreased dissociate rate constant koff achieved from additional H-bond interactions due to the favorable orientation of the (5S)-hydroxymethyl group.
A co-crystal structure of macrocycle 22 (PDB 5N1Z) in complex with BCL6BTB dimer was solved, in which, compound 22 occupied both the aromatic and linker sites similarly to that of acyclic analog 20 (Figure 8). The hydroxy group formed an H-bond to R’28 in the acid site. The linker region of the macrocycle was solvent-exposing and formed non-polar interactions with the protein. The NH group bridging the two ring systems formed a key H-bond (2.8 Å) to the backbone carbonyl O atom of M51. The bicyclic lactam of compound 22 occupied the linker site, with the lactam carbonyl O atom forming an H-bond (3.4 Å) to the side chain of E115. As the HDCH site was not occupied by the inhibitor, a chloride ion was detected in the co-crystal structure.
Figure 8.

Binding mode of compound 22 to BCL6BTB (PDB ID: 5N1Z). (A) Surface view of the binding of inhibitor 22 to the BCL6BTB LG. Compound 22 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 22 in complex with BCL6BTB.
Considering macrocyclic inhibitor 22 originated from a kinase CK2-targeting scaffold,67 a kinase screen was performed employing a panel of 126 kinases. The results indicated that across the panel only CK2 indicated > 60% inhibition at a relatively high concentration (1 μM). More detailed evaluation revealed that compound 22 inhibited CK2 with an IC50 value of 0.64 μM, which was 220-fold weaker compared to that for BCL6BTB (IC50 = 2.9 nM) and 50-fold weaker compared to that of the original hit 18 (CK2 IC50 = 12 nM). Interestingly, co-crystal structures of compound 22 bound to these two proteins (BCL6BTB, PDB 5N1Z; CK2, PDB 5N1V) showed highly similar binding modes: the diarylamino group formed a key H-bond to both proteins. While in CK2, the NH group interacted with the kinase hinge domain via residue V116, in BCL6, it formed an H-bond to the carbonyl group of M51 backbone. The binding pockets of BCL6BTB dimer and CK2 provided similar interactions and compound 22 showed similar bound conformations in both proteins, underlining the potential challenge in achieving selectivity for BCL6BTB over CK2.
To further evaluate the selectivity of the macrocyclic inhibitors, compounds were parallelly tested in cellular gene reporter assays employing either BCL6BTB or another BTB domain PLZF. All tested compounds indicated no inhibitory activity against PLZF at the concentration of 30 μM (IC50 range against BCL6BTB: 0.1-6.1 μM). One of the active macrocycle 23 was further screened (1 μM) against a panel of 398 kinases and over 30 diverse pharmacological targets.68 Inhibitor 23 indicated high selectivity68 by hitting only two targets (GABA receptor, Ki = 0.25 μM; 5-HT1B receptor, EC50 = 0.57 μM).
Finally, to examine the anticancer activity of the macrocyclic inhibitors, the authors performed cell proliferation assays using a panel of BCL6-dependent7 (e.g., OCI-Ly1, SUDHL-4, OCI-Ly3, SUDHL-2, U2932, OCI-Ly10, TMD8) and BCL6-independent7 (e.g., Karpas422, OCI-Ly19) DLBCL cell lines, along with a multiple myeloma (MM) cell line AMO-1 with undetectable levels of BCL6. Note that Karpas422 has also been reported by Kerres and coworkers as a BCL6-dependent cell line.69 Unfortunately for all macrocyclic inhibitors and the control compound FX1, the authors only observed no distinguishable features with low micromolar antiproliferative activity for all tested cell lines. While the authors didn’t expand the discussion on these results, the lack of the antiproliferative effects of these macrocycles might due to the evaluation platform used in the study. Prior to the antiproliferation studies, the cellular activity of the macrocycles was only evaluated using a luciferase reporter assay employing engineered BCL6BTB and a corepressor peptide but not the corresponding full-length proteins. Thus, one cannot exclude the possibility that the compounds could not inhibit the PPI between full length BCL6 protein and its corepressors. It is also worth noting that although good correlation between the results of TR-FRET and SPR were obtained, the authors didn’t observe reasonable correlation between cell-free assays and cellular reporter assays. Furthermore, in the antiproliferation study, only a short 3-day proliferation study was pursued, which might not provide sufficient time for tested compounds to show BCL6-mediated effects.
Guo and coauthors screened an in-house collection of 230 compounds using a TR-FRET assay at three concentrations (100, 50, and 25 μM) and identified a diaminopyrimidine compound 26 that inhibited the PPI between BCL6BTB and SMRT with an IC50 value of 19 μM (Scheme 5).70 Structural optimization of hit 26 focused on the amino-substitution of the diaminopyrimidine core and the meta- and para-substitutions of the phenyl ring. In the first stage, the isobutyl group of hit 26 was replaced by 14 different functional groups, which led to the generation of the (2S,6R)-2,6-dimethylpiperazinyl 27 with an IC50 value of 0.77 μM. Next, based on the structure of compound 27, the substitutions of the phenyl ring were investigated by 18 additional analogs to yield compound 28 with an IC50 value of 0.47 μM. Overall, the SAR study employed a total number of 32 new compounds with the IC50 range of >10 μM to 0.47 μM.
Scheme 5.

Optimization strategy of HTS hit 26 and generation of diaminopyrimidine inhibitor 28
Next, the activity of selected inhibitors in reactivating BCL6 target genes was evaluated in the BCL6-dependent SUDHL-4 cells using real-time quantitative PCR (RT-PCR). All tested compounds, at the concentration of 5 μM, reactivated ATR, CD69 and CXCR471 compared to the vehicle. Then, compound 28 was chosen for further evaluation in two BCL6-dependent DLBCL cell lines SUDHL-4 and Farage. At the concentration of 5 μM, compound 28 caused more significant reactivation effects of BCL6 target genes (p53, ATR, CD69, CXCR4, and CDKN1A)71 compared to those induced by positive controls 17 (5 μM, Takeda)50 and 2 (FX1, 50 μM).7
To explore the selectivity of inhibitor 28, the authors followed the mRNA levels of SUDHL-4 and Farage along with a BCL6-independent DLBCL cell line Toledo. The results indicated that treatment of 28 caused a dose-dependent increase in the mRNA levels of six tested BCL6 target genes (p53, ATR, CD69, CXCR4, CDKN1A, and CD80) in both BCL6 dependent lines.71 Specifically, the mRNA level of CXCR4 in SUDHL-4 cells was ~12 times greater with exposure to compound 28 compared to the control 2. Interestingly, compound 28 had no obvious effect on tested genes in the BCL6-independent Toledo cells.
The kinetics of inhibitor 28 binding to BCL6BTB was studied using bio-layer interferometry (BLI). Compound 28 binds to BCL6BTB with a Kd value of 0.37 μM, 20-fold and 3-fold greater than those of controls 2 (Kd 7.9 μM) and 17 (Kd 1.2 μM). To study its antiproliferative effects, compound 28 was tested in four BCL6 dependent DLBCL cell lines (SUDHL-4, Farage, DOHH-2, and OCI-Ly7) and a BCL6 independent Toledo cell line using the Cell Counting Kit 8 (CCK8) assay.7 Compound 28 killed all four BCL6 dependent cell lines with IC50 values ~1 μM, which was > 15-fold more potent than positive controls 2 and 17. On the other hand, compound 28 indicated an IC50 value of 3.3 μM against Toledo cells, a BCL6 independent GCB-DLBCL cell line that is less sensitive than the tested GCB-DLBCL cell lines. While the results are encouraging, the observed antiproliferative effects of compound 28 was in disagreement with other reports where significant antiproliferation could be obtained after a long study usually ranging from 14-17 days. Subsequently, selectivity of 28 over human normal cell lines (L02, HAF, NCM460, and PNT1A) were determined, and the IC50 values of compound 28 for normal cells were ~20-fold higher than that of DLBCL cells, confirming that compound 28 was selective against DLBCL cells.
The inhibitory effects of compound 28 in SUDHL-4 cell were evaluated using the EdU flow cytometry assay.7 Compound 28 significantly inhibited DLBCL cell growth. Treatment of compound 28, at the concentrations of 0.625 μM, 1.25 μM, and 2.50 μM significantly decreased the number of living cells (from 48%, to 27%, 13%, and 6.6%, respectively) in 48 h. The results were more dramatic than those of controls 2 and 17. The effect of compound 28 on DLBCL apoptosis was studied using flow cytometry, and the results revealed that compound 28 dose-dependently induced apoptosis (from 4.0%, to 6.8%, 16%, 73%), while at the same concentration, control compounds 2 and 17 indicated no obvious activity in inducing apoptosis.
To assess whether compound 28 could diminish the number of GC B cells, C57BL/6 mice were immunized with 4-hydroxy-3-nitrophenyl)acetyl-chicken gamma globulin (NP–CGG) and then treated with the compound (i.v., 10 mg/kg/3d for 12 days, vehicle: 20% (2-hydroxypropyl)-β-cyclodextrin). Flow cytometry was conducted to detect splenic GC B cells. Compared to the untreated group (frequency of GC B cells, ~2.0%), the treated group indicated a significantly decreased GC formation (frequency = 0.45%). The group treated with control 2 (50 mg/kg, i.p. daily injection) indicated a frequency of 0.73%. As BCL6 was a key regulator for the differentiation and maturation of CD4+ Tfh cell, the author further studied the development of Tfh cells upon treatment of compound 28 using flow cytometry,72 and found that the proportion of Tfh cells was significantly lower in the 28 treated group (0.99%) compared to in the control group (2.3%). Moreover, using immunofluorescent staining, the author also studied the splenic architecture, and found that both size and number of GCs were significantly decreased (~80%) upon treatment of compound 28, which was consistent with the results of the flow cytometry analysis. It was noted that control 2 indicated modest inhibitory effects (~50%) in GC B cell development and GC formation.
Next, the authors used ELISA to determine the effects of compound 28 in impairing immunoglobulin affinity maturation.32 Compared to the controls, mice treated with compound 28 indicated ~50-fold lower titers of high-affinity immunoglobulin G1 measured by NP5-BSA. A similar decrease was also observed in total NP-specific immunoglobulin G1 production determined by NP23-BSA.
To evaluate the in vivo efficacy of compound 28, an SCID DLBCL xenograft model with SUDHL-4 cells was used.7 At the dose level of 5 mg/kg/3d (i.v., vehicle: 20% (2-hydroxypropyl)-β-cyclodextrin), compound 28 suppressed the tumor weight (~70%) and volume (~60%) compared to the vehicle-control. Treatment of compound 28 caused no apparent adverse events as little difference was observed in body weight and no obvious damage was detected to major organs (heart, liver, spleen, lung and kidney) determined by H&E staining. In addition, the Ki67 staining was used to assess tumor aggressiveness, and the results indicated that the expression level of Ki67 was decreased by half in 28 treated mice, confirming the in vivo effects of compound 28 in inducing DLBCL cell growth arrest. Moreover, using RT-PCR, the authors also showed that compared to controls, the mRNA levels of the BCL6 target genes CD69, CXCR4 and ATR were increased by >150%, >60% and >200%, respectively when compound 28 was administered (5 mg/kg/3d).
Finally, the in vitro ADME profile of compound 28 was assessed. The results indicated that this lead compound had low microsomal clearance CLint (Clint(mic) < 9.6 μl/min/mg, Clint(liver) < 38.0 ml/min/kg), acceptable metabolic stabilities (t1/2 > 60 min) in human liver microsomes, and modest binding to plasma proteins (7.6% free fraction).
Cheng and coworkers reported a series of inhibitors for the BCL6BTB dimer based on a novel thiourea scaffold.73 Screening of a library of ~1,500 fragment-sized molecules by protein-observed NMR spectroscopy led to the identification of a thiourea hit 29 with a Kd value 3.2 mM (Scheme 6). The co-crystal structure of BCL6BTB-29 (PDB 6C3N) revealed that hit 29 binds in the aromatic site of the BCL6BTB LG. The S atom of this hit fit into a hydrophobic pocket formed by N’21, L’25, M51, A52, and Y58. One of the thioamide hydrogens formed an H-bond (2.6 Å) with the backbone carbonyl O atom of M51.
Scheme 6.

Identification and optimization of thiourea-based inhibitors of BCL6BTB.
Based on this result, new inhibitors were designed using a computer-aided drug design (CADD) method called site identification by ligand competitive saturation (SILCS).74-77 Optimization of hit 29 involved replacing the 3-substituted pyridine with an indole ring and expanding the chemical structure of inhibitors into the HDCH site via the indole nitrogen. Moreover, a piperazine group was introduced to the indole group to interact with the acidic sidechain of residue E115. Finally, the phenethyl group of hit 29 was replaced by a second indole ring.
The authors characterized the binding affinity of early weak inhibitors using 1H-15N HSQC spectra NMR. Specifically, the sum of chemical shift perturbations of six selected amide (T62, T48, F61, N23, R28, and V18) resonances (6PA) were used to rank the relative affinity of new compounds to BCL6BTB. Compared to hit 29, the di-indole analog 30 showed an increased 6PA value (88 Hz vs 49 Hz for hit 29). Substitution of the right-arm indole nitrogen of compound 30 using eight additional analogs yielded compound 31 with a significantly increased 6PA value of 391 Hz. Next, a methyl piperazine was introduced into the right-arm indole, followed by a further investigation of substitutions on the right-arm indole nitrogen by nine additional analogs. These efforts resulted in compound 32 with a 6PA value of 473 Hz. Overall, the SAR employed a total number of 19 new compounds with the 6PA range of 49 Hz to 473 Hz.
Using NMR-titration experiments, the Kd value of inhibitor 32 to BCL6BTB dimer was determined to be 44 μM, which was 70-fold more potent than hit 29 (Kd = 3,200 μM). Isothermal-titration calorimetry (ITC) was also performed to validate the binding of 32 to BCL6BTB dimer in which a very similar affinity (Kd = 36 μM) was reported. The crystal structure of the BCL6BTB-32 complex (Figure 9) showed 32 occupying the aromatic, linker and HDCH sites of the BCL6BTB LG. Specifically, the NH group of the thiourea functionality formed an H-bond to M51 (2.8 Å). In addition, the tertiary amino group of the piperazine ring interacted to the carboxylate sidechain of E115 via an electrostatic interaction (3.3 Å). Moreover, the morpholine group of amide tail binds into the HDCH site formed by the backbones of A52 and C53, and side chains of H’14, A’17, V’18 and N’21.
Figure 9.

Binding mode of compound 32 to BCL6BTB (PDB ID: 6C3L). (A) Surface view of the binding of inhibitor 32 (pink) to the BCL6BTB LG. Compound 32 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 32 in complex with BCL6BTB.
To test the potencies of compounds, an AlphaLisa assay was established involving labeled BCOR peptide binding to BCL6BTB dimer. As the most potent candidate, compound 32 showed an IC50 value of 27 μM. Next, differential cell-killing assays were conducted for selected inhibitors using a panel of four BCL6-dependent lymphoma cell lines (OCI-Ly1, OCI-Ly7, SUDHL-4, and SUDHL-6) and three BCL6-independent lymphoma cell lines (Karpas422, Toledo, and OCI-Ly1-B50). Note that Karpas422 has reported by Kerres and coworkers as a BCL6-dependent cell line.69 Unfortunately, the tested inhibitors only showed relatively weak cellular activity likely due to limited cell permeability.
Irreversible BCL6BTB Inhibitors
By studying the binding mode of the HTS hit 742 to the BCL6BTB protein, Sameshima and coauthors learned that compound 7 binds into the HDCH site in proximity to the nucleophilic residue C53 (Scheme 7). Based on this observation, the authors synthesized chloroacetamides 34-36 as potential C53-targeting irreversible inhibitors for BCL6BTB.78 The covalent bond formation between BCL6BTB and these chloroacetamide analogs were assessed by mass spectrometry, and the results confirmed a 1:1 covalent modification by chloroacetamide 36 and its homologated analogs 34 and 35. Interestingly, the acrylamide analogs of compound 36 failed to modify BCL6BTB.
Scheme 7.

Design of irreversible BCL6 inhibitors by targeting C53 residue from compound 7
Next, TR-FRET assays were used to evaluate the inhibitory activity of compounds 34-36 for the PPI between BCL6BTB dimer and the TAMRA peptide, a fluorescent tetramethylrhodamine-conjugated BCL6BTB binding sequence (TAMRA-Abu(4)-VWYTDIRMRDWM-OH) derived from peptide 4.38 Although none of the inhibitors gave a full dose response curve, the results confirmed that compound 36 was more potent than its homologated analog 35 at the concentration of 6.3 μM, while the other analog 34 indicated no inhibitory activity at the same concentration. To identify the specific BCL6BTB amino acid residue modified by inhibitor 36, the authors conducted MS studies in the presence of compound 5 (50 μM),38 a highly potent peptide BCL6BTB inhibitor covering the C53-containing HDCH site. The results indicated that compound 36, at concentrations as high as 20 μM, failed to modify BCL6BTB, implying that inhibitor 36 likely achieved his inhibitory activity by modifying C53 of BCL6BTB. Next, irreversible modification of BCL6BTB by inhibitor 36 was confirmed using jump dilution assays in which compound 36 (6.3 μM) or its non-covalent analog (100 μM) was incubated with BCL6BTB for 10 h, followed by a 100-fold dilution using a buffer containing excess TAMRA peptide (20-fold Kd). Different to the noncovalent analog whose inhibitory activity disappeared, the inhibitory effect of compound 36 maintained in 24 h after dilution. These results again, confirmed irreversible inhibitory mechanism of compound 36.
Next, optimization of compound 36 was pursued by following the kinact/KI value of new inhibitors in an irreversible probe competition (IPC) assay using a covalent fluorescent probe 37 (BCL6-FP) (Figure 10).79,80 The kinact/KI value of inhibitor 36 was determined to be 3.0 × 101 M−1 s−1. Introduction of a 4-tetrahydropyranyloxy group to compound 36 resulted in a new inhibitor 38 with an 18-fold increased kinact/KI value (5.4 × 102 M−1 s−1). Expansion of the five-membered benzo[d]imidazol-2-one ring to a six-membered benzoxazine ring yielded compound 39, which indicated an another 7.4-fold improvement of kinact/KI value (4.0 × 103 M−1 s−1). Finally, introduction of a 2-propanoylaminoethyl group to the chemical scaffold gave inhibitor 40 (BCL6-i) that was highlighted by an excellent kinact/KI value of 1.9 × 104 M−1 s−1, over 670-fold higher than that of the parent 36.
Figure 10.
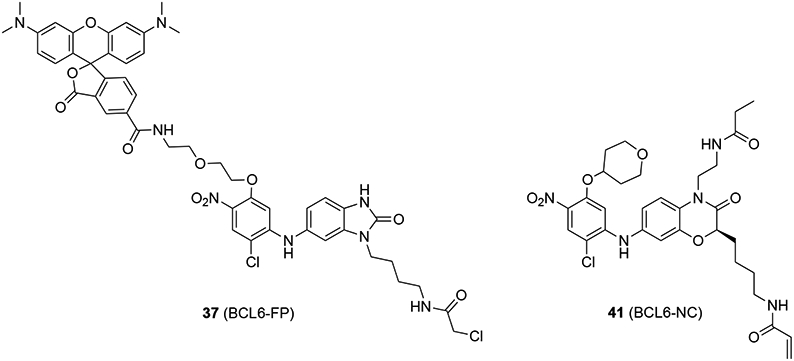
Chemical structures of 37 (BCL6-FP) and 41 (BCL6-NC).
MS analysis confirmed the covalent binding of inhibitor 40 to BCL6BTB with 1:1 stoichiometry. In addition, compound 40 indicated identical IC50 values after several hours of incubation in the IPC assay, confirming the irreversible binding of the compound to BCL6BTB. The authors further confirmed that inhibitor 40 covalently modified residue C53 in the HDCH site of BCL6BTB via time-dependent inhibition experiments. Compound 40 effectively inhibited BCL6 mutants containing C53 time-dependently, while the inhibitory activity disappeared for BCL6 mutants without C53. These results together supported that the optimized electrophilic chloroacetamide inhibitor 40 modified C53 residue in the HDCH site of BCL6BTB LG.
To address the potential toxicity of these irreversible inhibitors, the intrinsic chemical reactivity (kchem) values81,82 of compound 40 and its analogs was compared to an acrylamide-based irreversible pan-erbB kinase inhibitor CI-1033 that is under Phase II clinical trials. Compound 40 showed a 23-fold smaller kchem value (1.6 × 10−3 M−1 s−1) than that of compound CI-1033 (3.7 × 10−2 M−1 s−1), indicating an acceptable kchem value of compound 40 for further cellular or in vivo evaluations.
To assess the cellular activity of inhibitor 40, the authors used a fluorescence imaging assay to follow colocalization of BCL6 with the BCL6-FP 37. Cells expressing full length FLAG-BCL6 were treated with DMSO, 40 (1 μM), or reversible control 41 (BCL6-NC, Figure 10) (1 μM) for 1 h, then allowed to react with BCL6-FP 37 (1 μM) for another 1 h. Colocalization of BCL6 protein with BCL6-FP 37 was detected, indicating that BCL6-FP 37 covalently interacts with the BCL6 protein in cellular environment. On the other hand, pre-treatment of inhibitor 40 prohibited the fluorescence signal from BCL6-FP 37, whereas the treatment of a reversible compound 41 did not. These results together confirmed that inhibitor 40 irreversibly modifies the protein BCL6 in cell. Next, using an M2H assay, the authors confirmed that compound 40 inhibited the PPI between intracellular BCL6 and BCOR. Specifically, after a 24 h treatment, compound 40 (1 μM) inhibited the M2H signal by ~80%, whereas the negative control compound 41 (1 μM) did not (< 10% inhibition).
Finally, the authors tested the antiproliferation activity of inhibitor 40 and the negative control 41 against two BCL6-dependent GCB-DLBCL cell lines SUDHL-4 and OCI-Ly3. The results revealed that treatment of inhibitor 40, at the concentration of 3 μM after 72 h, significantly suppressed growth of SUDHL-4 (~80%) and OCI-Ly3 (~70%) compared to negative control 41 (~15% and ~25%, respectively).
Teng and coauthors reported a series of irreversible BCL6BTB inhibitors by targeting the side chain of amino acid residue Y58 in the aromatic site using a well-known sulfonyl fluoride functionality.83 The initial inhibitor 42 (TMX-1120) was designed based on the chemical scaffold of Takeda compound 1750 (Scheme 8). Based on the co-crystal structure, the authors noticed that the Y58-OH group and meta-carbon of the pyridine are located in reasonable distance (4.2 Å) for the insertion of an additional functional group. In addition, the guanidinium group of R’28 was in close proximity (2.4 Å) to Y58-OH, which could help deprotonation of the Y58-OH group. Furthermore, the solvent-exposed and disordered pyridine moiety suggested no steric hindrance to reach the Y58-OH. Therefore, the authors introduced a sulfonyl fluoride,84-87 a widely used tyrosine-targeting electrophilic warhead, to target Y58.
Scheme 8.

Development of irreversible inhibitor 43 (TMX-2164) targeting Y58 of BCL6BTB
The potency of new compounds was determined using a TR-FRET assay employing a bodipyFL-labeled BCOR peptide in 30 min. Compound 42 showed an IC50 value of 251 nM, > 10-fold more potent than parent 17 (IC50 = 2.7 μM). To verify the formation of a covalent bond of the inhibitor to Y58, the authors used protein mass spectrometry by analyzing the changes of molecular weights of the recombinant BCL6BTB after incubating with 10 equivalent compound 42 for 2 h. A mass shift was observed corresponding to 1:1 stoichiometric modification of the protein by compound 42. The site of covalent modification was further confirmed by digesting the labeled protein with trypsin followed by analyzing the resulting peptides using capillary electrophoresis-mass spectrometry (CE-MS).
Next, the authors synthesized new hybrid 43 (TMX-2164) by merging the sulfonyl fluoride warhead with the chemical scaffold of another BCL6 inhibitor 62 (BI-3812)69. Compound 43 displayed an IC50 value of 152 nM and its reversible sulfone analog 44 (TMX-2177) revealed a comparable IC50 of 368 nM. Covalent modification of BCL6 by inhibitor 43 was also confirmed by mass spectrometry. A mass shift of 572 Da, corresponding to the addition of compound 43 with the loss of an HF, was detected.
To examine the cellular activity of the compounds, the authors developed a fluorescence-activated cell sorting (FACS) assay that enabled quantification of BCL6 levels by following the ratio of eGFP/mCherry.88 Cells were treated with irreversible inhibitors 42 and 43 or reversible controls 17 and 44 at 5 μM for 30 h, followed by a BCL6 degrader 61 (BI-3802)69 at different concentrations. While treatments of covalent inhibitors 42 and 43 protected BCL6 from 61-induced degradation, treatment of reversible controls 17 and 44 did not indicate the same protection, demonstrating permanent modification of BCL6 protein by covalent inhibitors. Finally, the anti-DLBCL activity of new covalent inhibitors were evaluated using SUDHL-4 cells. After 5 days of treatment, the irreversible inhibitor 43, compared to both reversible inhibitors 17 and 44, indicated the most potent antiproliferation effect with the GI50 value in the single digit micromolar range.
BCL6 Protein Degraders
Based on the small molecule reversible inhibitors developed by the team in AstraZeneca (Scheme 4),60 McCoull and coauthors reported their efforts in the development of cell permeable BCL6 inhibitors and a subsequent PROTAC,89,90 which effectively degraded the BCL6 protein.91 A triazine hit 45 (IC50 = 830 μM, TR-FRET, Scheme 9) in this study was identified from a fragment screen and optimized by 2D-NMR-guided effective conformational analysis, to yield a macrocycle 46 with a remarkable ~93,000-fold improvement in inhibitory potency (IC50 = 8.9 nM). The co-crystal structure of compound 45 in complex with BCL6BTB homodimer (PDB 6EW6) confirmed the binding site of the ligand in the BCL6BTB LG.
Scheme 9.

Development of BCL6 PROTAC 50.
The authors assessed BCL6 target engagement in OCI-Ly1 cells using a cellular thermal shift assay (CETSA),34 in which, ligand binding could lead to increased BCL6 stability and therefore a higher protein unfolding temperature.34 The EC50 value of a testing ligand could be calculated based on the isothermal dose response curve. Using this assay, compound 46 was determined to have an EC50 value of 0.48 μM. The authors further demonstrated a good correlation between the CETSA results and an additional cellular luciferase gene reporter assay established in HEK293T/17 cells, in which, the compound 46 indicated an IC50 value of 0.71 μM. Using a similar gene reporter assay, the authors demonstrated the specificity of compound 46 to BCL6BTB over another BTB domain protein PLZF (IC50 >32 μM). Specificity of compound 46 was further evaluated via a kinase screen against a panel of 399 kinases, and the result revealed that compound 46 had no obvious inhibitory activity to the tested kinases (< 25% inhibition at 1 μM).
Analysis of the co-crystal structure of compound 47 (Figure 11), a close analog of compound 46, indicated that vectors from both the lactam and piperazine N atoms were directed toward solvent accessible space that could allow PROTAC installation. Substitution at these two N atoms were subsequently pursued yielding compounds 48 and 49, and the result indicated that compound 49 maintained higher cellular activity than compound 48 (IC50 3.1 μM vs. 7.4 μM). Accordingly, compound 49 was attached to a thalidomide warhead,92-94 through a two-unit PEG hydroxypropylamine butanediamide linker, to generate the BCL6 PROTAC 50 (Scheme 9). Compared with parent compound 49, PROTAC 50 displayed improved BCL6 inhibitory activity with purified protein (IC50 = 0.12 μM, RT-FRET), although it only indicated modest inhibitory potency in cell (IC50 = 8.8 μM, luciferase gene reporter assay). Besides, PROTAC 50 had some specificity for BCL6BTB over PLZF (IC50 > 32 μM, luciferase gene reporter assay). However, PROTAC 50 indicated decreased lipophilicity (log D = 1.6 vs 4.2 for compound 49) and Caco2 permeability (0.081×10−6 cm/s vs 31×10−6 cm/s for compound 49).
Figure 11.

Binding mode of compound 47 to BCL6BTB (PDB ID: 6EW8). (A) Surface view of the binding of inhibitor 47 (pink) to the BCL6BTB LG. Compound 47 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 47 in complex with BCL6BTB.
The degradation ability of PROTAC 50 was evaluated by following the BCL6 protein levels after treatment with the compound in OCI-Ly1 cells. PROTAC 50 dose-dependently degraded BCL6 in OCI-Ly1 cells and the effect was specific to BCL6 as it caused no degradation on another protein involved in the BCL6-corepressor complex, TBLR1. It was noted however, the degradation obtained from the treatment of PROTAC 50 was not complete at the highest concentration tested (1 μM, 82% degradation). It was also shown that significant BCL6 protein degradation was observed as early as 1 h and the effect lasted for at least 72 h. In contrast, upon treatment of reversible inhibitor 49 caused no obvious change in BCL6 levels over time, suggesting that the thalidomide group was responsible for BCL6 protein degradation.
To elucidate the mechanism of action of PROTAC 50, three control experiments were performed by including: i) proteasome inhibitor MG132, ii) BCL6 inhibitor 49, and iii) a potent thalidomide derivative pomalidomide to the tests. Reduced levels of degradation were observed for all three experiments. More complete protection from protein degradation was achieved when a combination of BCL6 inhibitor 49 and pomalidomide were added. Altogether, these results supported the mechanism of PROTAC-based BCL6 protein degradation. Furthermore, PROTAC 50 at the concentration of 1 μM, also caused significant BCL6 degradation effects (59% - 84%) for other DLBCL cells SUDHL-4, Karpas422 and ULA, and Burkitt’s lymphoma cell line Ramos.
To assess the antiproliferative activity of PROTAC 50, a three-day cell proliferation study was performed using DLBCL cell lines of both GCB (OCI-Ly1, Karpas422, SUDHL-4, WILL-2 and OCI-Ly19) and ABC (OCI-Ly10, RI-1, SUDHL-2, OCI-Ly3 and TMD8) subtypes with varying levels of BCL6, along with the MM cell line AMO-1. Unfortunately, PROTAC 50 failed to show significant selectivity with the similar low micromolar pGI50 values for all tested cell lines. Furthermore, PROTAC 50 did not indicate improved antiproliferative potency compared to the BCL6 inhibitor 49 in BCL6-dependent DLBCL cells. Finally, a long 16-day proliferation study was pursued. However, similarly modest antiproliferative effects of PROTAC 50 were obtained for OCI-Ly1, SUDHL-4, and OCI-Ly19 cell lines.
To explain the relatively weak antiproliferative effect of the compound, the authors followed the subcellular BCL6 levels after the treatment the PROTAC (10 μM, 24 h) using an immunofluorescence (IF) staining assay in OCI-Ly1 cells. Although reduced levels of diffuse and punctate staining of BCL6 were detected, significant punctate staining of BCL6 remained. Considering the incomplete degradation of BCL6 by PROTAC 50, quantitative mass spectrometry was used to estimate specific intracellular concentrations of compound 50. After treatment with PROTAC 50, the concentrations of PROTAC 50 in cytoplasm was 0.76 μM, over 10-fold below the IC50 (8.8 μM) of the compound in BCL6 cell reporter assay. Importantly, the concentration of PROTAC 50 in the nucleus, where the target BCL6 locates, was only detected as 0.05 μM. The extremely low levels of compound concentration in the nucleus might explain the relatively weak cellular potency of the PROTAC.
The authors further showed that after treatment of PROTAC 50, BCL6 levels were similar in different subcellular compartments including cytoplasmic (DMSO:50 = 7.3:2.3), soluble nuclear (DMSO:50 = 86.0:20.2), and chromatin bound (DMSO:50 = 6.7:1.9) fractions. Finally, the intracellular levels of BCL6 were measured after treatment of PROTAC 50 (1 μM, 4 h). PROTAC 50 could sufficiently access the chromatin bound (1.9 μM) and soluble nuclear (0.46 μM) fractions, to cause BCL6 degradation within cytoplasm, nucleus, and chromatin bound fractions.
Bellenie and coworkers at the Institute of Cancer Research (ICR) reported benzimidazolone-based BCL6 inhibitors.95 Interestingly a subset of compounds triggered rapid degradation of BCL6, of which, compound 58 (CCT369260) was highlighted as a probe unlike known PROTAC and non-PROTAC degraders (Scheme 10).
Scheme 10.

Optimization strategy of hits 51 and 52 and generation of benzimidazolone 58
By screening an in-house compound library using a fluorescence polarization (FP) assay, the authors identified two hits 51 and 52 with the IC50 values of 120 μM and 70 μM, respectively (Scheme 10). To improve aqueous solubility, their analogs 53 and 54 were synthesized, which indicated ~5-fold increased solubility without compromising the inhibitory potencies. Next, hybridization of compounds 53 and 54 yielded the benzimidazolone 55 (CCT365386) with an IC50 value of 11 μM. The co-crystal structure of compound 55 bound to BCL6BTB (PDB: 6TOH) was determined. Results from additional assays revealed that compound 55 had a favorable drug-like profile including low microsomal clearance, high permeability, and good ligand efficiency.
To enable the accurate measurement of inhibitors with nanomolar potency, the authors developed a TR-FRET assay with which the IC50 value of inhibitor 55 was determined to be 3.4 μM. Structural refinement of compound 55 involved the expansion of inhibitors into the HDCH site via substitution of the N3-position of the benzimidazolone, and optimization of the interactions to the aromatic site around residue Y58. The authors found that introduction of a 3-hydroxyl-n-butyl group at the N3-position of the benzimidazolone yielded an achiral tertiary alcohol 56 with a 5-fold improvement of inhibitory potency. The co-crystal structure of compound 56 bound to BCL6BTB (PDB: 6TOJ) showed that the pyridine group formed hydrophobic interactions with aromatic sidechain of Y58, and the pyridine nitrogen formed an H-bond with R’28 through a water molecule. The authors also found that a pyrimidine ring demonstrated the same effects. Based on these results, the 2-position on the pyrimidine was further explored and demonstrated that substitution was tolerated. Next, modification of the substituted pyridine was investigated by 24 additional analogs, and the (3R,5S)-3,5-dimethylpiperidine compound 57 (CCT368682) was identified as a potent BCL6 degrader with an IC50 value of 0.76 μM and DC50 value of 0.33 μM (100%) in an immunofluorescence degrader assay. To further investigate the degradation ability of compound 57, SUDHL-4 and OCI-Ly1 cells were treated with compound 57 at 37 °C for 4 h, and the protein levels of BCL6 was monitored by Western blots. Compound 57 reduced the protein levels of BCL6 dose dependently, with complete degradation at 10 μM of the compound. This result was consistent with the mechanism of compound-mediated degradation of BCL6.
Finally, to improve stability and reduce metabolic clearance of the compound, substitutions on the pyrimidine ring were investigated. These efforts yielded the difluoromethylene degrader 58 (CCT369260), which indicated an IC50 value of 0.52 μM (TR-FRET) and excellent DC50 value of 0.09 μM in SUDHL4 cells. Further evaluation indicated that compound 58 had a CLint mouse liver microsomes (MLM) value of 78 μl/min/mg.
To confirm the observed degradation was not cell line specific, the authors used meso scale discovery (MSD) assays to follow BCL6 protein degradation in different cells. Compound 58 caused >85% degradation of BCL6 in OCI-Ly1 (DC50 = 0.049 μM) and Karpas422 (DC50 = 0.062 μM) cells. Then, the antiproliferative activity of compound 58 and four analogs were examined using a 14-day proliferation assay in BCL6-dependent (SUDHL-4 and OCI-Ly1) and BCL6-independent (OCI-Ly3) cell lines. Compound 58 indicated differential cell killing effects for BCL6-dependent cells over the BCL6-independent OCI-Ly3 cells. Compound 58 was further evaluated in a panel of BCL6-independent cell lines (Toledo, OCI-AML3, PLB-985, and MM.1S) using a 17-day proliferation assay. The results indicated that compound 58 had no antiproliferative activity for these cell lines at the concentration of 1 μM.
PK studies of compound 58 were performed using female Balb/C mice (1 mg/kg, i.v.; or 5 mg/kg, p.o.). Compound 58 indicated a reasonably good profile with modest clearance (CL: 20 mL min−1 kg−1), good oral bioavailability (54%), and high plasma protein binding (0.07% free in SCID mouse plasma, n = 6). To determine if sufficient exposure could be achieved in the SCID mouse used for xenograft studies, additional PK studies were conducted at three different dose levels (5, 15, and 50 mg/kg, p.o.). At the medium dose (15 mg/kg), the mean total blood concentration of compound 58 was above the desired concentration for ~10 h.
Next, to determine if, and at what concentration BCL6 depletion could be achieved, a PK/PD study was conducted in mice using a single dose of compound 58 at 15 mg/kg (p.o.) in an OCI-Ly1 DLBCL xenograft model, with dosing of compound commencing 20 days after injection. Tumor BCL6 levels were quantified using capillary electrophoresis and normalized to a GADPH loading control. A clear decrease of BCL6 levels in the tumor was observed up to 10 h after dosing, with a maximal effect at ~4 h with free drug concentrations of > 1 nM. This result supported that small molecule-triggered BCL6 degradation is a promising therapeutic approach to develop anticancer agents.
Binding mode of BCL6 degrader 58 was studied (Figure 12, PDB 6TOM) along with a closely related non-degrader (PDB: 6TOL). Both compounds bind in the BCL6BTB LG with highly similar binding modes, suggesting that the mechanism of action of the compounds, either as an inhibitor or a degrader, depended more on the nature of the substituents than a particular binding mode. Both molecules occupied the aromatic, linker, and part of HDCH sites (Figure 12A). The Cl atom fit into the deep hydrophobic group formed by M51, L’25 and N’21. Clear H-bonds were observed between the backbone carbonyl O atom of M51 and the bridging NH group (2.7 Å) as between the backbone NH of E115 and the urea O atom (3.1 Å).
Figure 12.

Binding mode of degrader 58 to BCL6BTB (PDB ID: 6TOM). (A) Surface view of the binding of degrader 58 (pink) to the BCL6BTB LG. Degrader 58 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of degrader 58 in complex with BCL6BTB.
Kerres and coauthors screened a ~1.7 million-compound library using an FP assay and identified a 4-amino-5-chloro-pyrimidine hit 59 that inhibited the PPI between a co-repressor peptide and the BCL6BTB dimer with an IC50 value of 18 μM (Scheme 11).69 The binding affinity of the selected hit 59 to BCL6BTB of hit was further validated by SPR (Kd = 20 μM) and a biochemical ULight corepressor binding assay (IC50 > 20 μM). Optimization of hit 59 was conducted through a two-stage process focusing on the chlorophenyl ring and amino-substitution of the pyrimidine core, using a biochemical ULight corepressor binding assay. In the first stage, the chlorophenyl group of hit 59 was replaced by four different functional groups designed to interact with the backbone NH of E115. These efforts led to the identification of a new inhibitor 60 with a dramatically increased IC50 value of 12 nM. Based on compound 60, 10 additional compounds with various substitutions at the 2-position of the pyrimidine core were tested to yield two optimal inhibitors, 61 (BI-3802) and 62 (BI-3812) with IC50 values ≤ 3 nM. Overall, the SAR study included a total number of 14 new compounds with the IC50 range of 20 μM to ≤ 3 nM.
Scheme 11.

Optimization of hit 59 and generation of compounds 61 (BI-3802) and 62 (BI-3812)
During biophysical characterization of the new compounds using co-crystallization and protein NMR experiments, the authors noticed that some of the new compounds caused protein precipitation. By confirming these atypical compounds inhibited the PPI of BCL6BTB and its corepressor while not disrupting BCL6BTB dimerization using a cellular luminescence-based mammalian intractome (LUMIER) assays,96 the authors ruled out the possibility that these compounds could denature the protein. In addition, the thermal stability of BCL6BTB dimer was increased after treatment with both compounds 61 and 62 in differential scanning fluorimetry measurements, also supporting the maintenance of protein folding upon treatment of either type of compound. Moreover, the authors explored the binding regions of either compound on BCL6BTB using a hydrogen deuterium exchange mass spectrometry (HDX-MS).97 The atypical compound 61 showed more protection from HDX compared to the typical compound 62 in both close and remote regions of the inhibitor binding site. The authors attributed the extended protection to the induced dimer-dimer interaction by the atypical compounds.
To investigate whether this atypical behavior can translate into cellular effects, the authors performed Western blotting where they assessed BCL6 levels after treatment of compound 61 (1 μM, 60 min). Compound 61 showed similar effects on BCL6 levels in ten lymphoma cells. Importantly, the authors noticed that the effect of compound 61 could be reversed by proteasome inhibitor MG-132,98 indicating a mechanism of proteasome-dependent protein degradation. Moreover, treatment of compound 61 in combination with MG-132 led to the generation of a BCL6 form with decrease migration rate, in agreement with the formation of multi-ubiquitylated protein. The presence of multi-ubiquitin chains on BCL6 protein, after the treatment of degradation-inducing compound 61, was further confirmed by probing a BCL6 immune precipitate with tandem ubiquitin-binding entities (TUBEs). Together, these results revealed that compound 61 led to cellular BCL6 degradation via the mechanism of ubiquitylation followed by proteasome-dependent degradation.
Compound 61 caused degradation of BCL6 by 50% in just ~5 min, and by over 90% in 2 h. This rapid degradation was also dose dependent with a DC50 (half degradation concentration) value of 20 nM in SUDHL-4 cells. The correlation between the DC50 values and IC50 values derived from the inhibition of the PPI between BCL6BTB and co-repressor peptides are high (r2 = 0.82), supporting direct binding of the degradation-inducing compounds to BCL6BTB. In addition, the authors found no correlation between the degradation efficiency and degradation potency for more potent compounds did not cause a higher percentage of protein degradation.
Next, degrader 61 was studied for its cellular effects, along with non-degrader inhibitor 62 (Scheme 11). To assess the selectivity of degrader 61 in cell, an affinity pull-down experiment was conducted using immobilized inhibitor 61 from cell lysate of Farage. The results confirmed that BCL6 was the major target of compound 61. Importantly, no other BTB/POZ domain proteins were identified in the same pull-down experiment.
The authors found that the effective degradation of BCL6 by compound 61 required a functional ZF DNA-binding domain because corresponding construct with either deletion or mutation of the ZF domain could not get degraded when treated by the degrader 61. To study the domains required for BCL6 degradation, the authors used HEK293 cells that lack endogenous BCL6. The levels of BCL6 after treatment of compound 61 (500 nM, 90 min) were determined by Western blotting. When a group of DNA-binding domains from different transcription factors were introduced to BCL6BTB, the DNA-binding domains from ESRRA and basic helix-loop-helix domain of TCF4 could restore 61-induced BCL6 degradation. On the other hand, the isolated BTB domain lacking the DNA-binding domain wound not get degraded. These results indicated that to achieve efficient degradation, the presence of the BCL6 ZF domain is not required, however, the localization of BCL6 protein to DNA, via a DNA binding domain, is required.
The effects of compounds 61 and 62 on levels of BCL6 target genes were studied by qPCR. Upon treatment of degrader 61 and inhibitor 62 (500 nM, 20 h) in SUDHL-4 cells, the number of genes that were increased and decreased were 87 and 17, respectively. Specifically, degrader 61 significantly induced BCL6 target genes including ATM, PRDM1, PTPN6, CD69, IRF4, and DUSP5. Importantly, many induced genes also indicated proximal (95% within 5kb of the transcription start site) BCL6 binding sites confirmed by ChIP experiments. Additionally, the authors took three of the robustly induced genes (PTPN6, RAPGEF1, and CHST2) and showed that degrader 61 (50 nM and 500 nM) induced these BCL6 target genes time-dependently (4, 6, 8, 16, 24 h). In comparison, the authors found in parallel experiments that the potency of inhibitor 62 in inducing BCL6 target genes (PTPN6 and RAPGEF1) was weaker than that of degrader 61.
Finally, the antiproliferative effects of degrader 61 were evaluated in DLBCL cells using long-term proliferation assays with a concentration range of 0.01 to 3 μM. Degrader 61 indicated antiproliferative activities (GI50 in the low micromolar range) in tested BCL6-dependent DLBCL cell lines including Farage, SUDHL-4, OCI-Ly7 and OCI-Ly1, and no obvious antiproliferative activities in BCL6-independent cells such as OCI-Ly19, MV-4-11 and Toledo. The authors also found that inhibitor 62 only showed antiproliferative activity at concentrations from 0.3 to 3 μM, at least 100-fold higher than its IC50 value (≤ 3 nM). When a collection of compounds was tested, the observed antiproliferative potencies were well correlated with both the half degradation concentration (DC50) (r2 = 0.68) and co-repressor binding (IC50, LUMIER assay) with the r2 value of 0.68 and 0.89, respectively. These results highlighted that the effect of degrader 61 was through a BCL6-mediated mechanism. In summary, 61-mediated BCL6 degradation caused antiproliferative effects in DLBCL cells. However, no significant cell death was observed upon treatment of degrader 61. Subsequently, PK studies in SCID mice were performed after oral dosing of compound 61 at 10 mg/kg (mean plasma AUC 1860 nM·h and Cmax 193 nM) and 100 mg/kg (AUC, 4650 nM·h; Cmax, 599 nM). Due to the relatively poor bioavailability of the compound, the authors didn’t further explore the effect of compound 61 in animal models.
The co-crystal structure of hit 59 (PDB 5MW6) revealed the ligand binding to the BCL6BTB LG. The key interaction between hit 59 and BCL6BTB dimer was the anilinic HBD interacting with the backbone carbonyl O atom of M51, which brought the chloropyrimidine ring to fit snuggly into the aromatic site and placed the pyrazole ring in an approximate distance (3.6 Å) to R’24 for a potential hydrogen bonding interaction. The chlorophenyl moiety was found resting in the linker site, offering a handle for further optimization of binding affinity. As shown in Figure 13, the co-crystal structures of compound 61 (PDB 5MW2) revealed that the lactam carbonyl of this compound occupied the linker site and the O atom formed a key hydrogen bonding interaction (2.8 Å) to the backbone NH of E115. Furthermore, the acetamide tail of compound 61 added an H-bond interaction to the backbone NH of V117 (3.6 Å), which could also contribute to the potency improvement. Notably, the (3S,5R)-3,5-dimethylpiperidine ring on the pyrimidine ring was solvent-exposed without obvious interaction with the protein surface.
Figure 13.

Binding mode of compound 61 to BCL6BTB (PDB ID: 5MW2). (A) Surface view of the binding of inhibitor 61 (pink) to the BCL6BTB LG. Compound 61 was shown in pink and BCL6BTB monomers are shown in green and gray, respective. (B) Detailed binding mode of inhibitor 61 in complex with BCL6BTB.
Summary and Perspective
As an oncogenic transcription factor, BCL6 is a promising target for the treatment of DLBCL and other important human cancer such as FLs, various forms of leukemia, breast cancer, and lung cancer. Currently there are no clinically approved drugs available targeting BCL6. Although BCL6 knockout is lethal, experimental evidence indicated that disrupting the PPI between the BCL6BTB LG and its corepressors provides a safe therapeutic strategy.
First generation of BCL6BTB inhibitors include peptide-based BPI, RI-BPI and small molecules 1 and 2. These early inhibitors, albeit weak BCL6BTB binders, provided good supporting evidence that targeting BCL6BTB blocks the oncogenic activity of BCL6 and kills cancer cells. Also, the binding mode studies of these compounds have defined the binding pocket of the BCL6BTB LG, which includes the acid, aromatic, linker, and HDCH sites. On the other hand, these inhibitors suffered from major drawbacks. For instance, BPI and RI-BPI involved challenging synthesis and poor oral bioavailability, and rhodanine-based compounds 1 and 2 were limited with respect to their modest binding affinity and PAINS-related pharmacologic properties.
Recent discovery of novel BCL6BTB inhibitors is commonly driven by different high throughput assays (Table 1). Interestingly all the hits identified from different screen campaigns were small aromatic molecules that reversibly bind to the BCL6BTB LG into the aromatic site formed by residues R’24, L’25, M51 and Y58. This aromatic site is occupied by SMRT residues H1426 and I1428,28 or BCOR residues W509 and V511.29 Another common feature of the identified hits was the conserved HBD that interacted with the backbone carbonyl of residue M51. Note that the same backbone carbonyl O atom of M51 forms strong H-bonds with the imidazole sidechain of SMRT residue H1426 or indole sidechain of BCOR residue W509.28,29 Optimization of the chemical structure of the hits included two often parallel strategies: 1) expanding the molecule to occupy the linker and HDCH sites of the binding pocket, and 2) replacing the substitution on the aromatic core to the aid site for additional interactions to basic residues (R’28 and R’24) or favorable physiochemical properties (e.g., polarity and aqueous solubility).
Covalent inhibitors of the BCL6BTB have also been pursued by employing reactive warheads to the chemical scaffolds of different reversible inhibitors, to target either the residue C53 in the HDCH site or the phenolic sidechain of Y58 on the edge of the aromatic site. To selectively target the thiol sidechain of residue C53, chloroacetamides, which showed reasonable intrinsic chemical reactivity, were introduced to aliphatic groups that could fit into the HDCH site. On the other hand, the phenol sidechain the solvent-exposed residue Y58 was activated by its neighboring residue R’28. This unique networking around the aromatic site offered an opportunity for covalent interaction by introducing a phenol-reactive group such as sulfonyl fluoride. Interestingly, both C53- and Y58-targeting irreversible inhibitors could effectively modify intracellular BCL6. However, these compounds only showed modest antiproliferative effects against BCL6-dependent DLBCL cells.
BCL6 protein PROTACs have also been pursued by installing PROTAC warhead (e.g., thalidomide) onto solvent accessible positions of optimized inhibitors. These PROTACs were cell permeable and caused fast and dramatic degradation of the BCL6 protein in cell. Interestingly, introduction of a dimethylpiperidine (or difluoromethylene analog) group directly to BCL6BTB inhibitors without a spacer also caused sufficient degradation of the BCL6 protein in cell. The antiproliferative effects was in consistence with the observed DC50 values of these compounds. The rapid turnover of BCL6, which is maintained by continuous rapid translation through eIF4E,99 makes it challenging to sustain low levels of the protein. Moreover, the fact that BCL6 knockout is lethal also raises concerns about full elimination of BCL6.31 On the other hand, the fact that degrader 58 was well-tolerated in mouse may ameliorate these concerns.
Several cell-free high-throughput competitive binding assays have been developed to measure the inhibitory potency and enable structure-activity relationship analyses. ELISA commonly includes a FLAG-tagged BCL6BTB protein along with a biotinylated BCOR peptide immobilized onto streptavidin-coated microplates.38,42,50 Despite its success, ELISA requires wash steps to separate unbound labeled compound from bound ones in order to detect specific binding event. The involvement of the washing steps also limited this method in detecting low-affinity ligands. Compared to ELISA, the no-wash TR-FRET60,69,70,78,83,91,95 assay found their application in multiple studies, in which a long-lifetime fluorescent lanthanide (Er or Tb) labeled BCL6BTB (energy donor) and a fluorescence-tagged corepressor peptide (energy acceptor) were employed in solution, which can be used to provide highly sensitive and reproducible measurement of competitive displacement of the fluorescence-tagged corepressor peptide by ligands from the BCL6BTB-corepressor interface. In addition, FP assay represents another homogenous method to measure the inhibitory potency of compounds against the PPI between BCL6BTB dimer and fluorescence-labeled BCOR repressor peptide.69,95 The potential utility of un-labeled protein is a big advantage of FP assays. However, FP can experience significant limitation from autofluorescence of testing compounds and light scattering.100,101 Moreover, AlphaLISA assay, including a pre-coated BCOR peptide (donor bead) and a BCL6BTB-tagged acceptor bead, has also been developed to test potential inhibitors. AlphaLISA requires no wash steps, however, metal chelators can cause false positives in the assays. 73 Besides, to evaluate the potency of nonequilibrium binding of irreversible inhibitors for BCL6BTB protein, IPC assays have been developed. IPC offers a rapid determination of the specific binding constant kinact/KI for irreversible ligands,79 although a control covalent ligand (e.g. irreversible fluorescence probe 37 for BCL6BTB dimer78) must be developed before using the assay.
Direct binding assays including SPR, BLI, MST and ITC, have been utilized to characterize the binding event of small molecules to labeled or label-free BCL6BTB protein (Table 2). SPR is a widely used optical biosensor technology employing a tagged BCL6BTB protein for surface immobilization.38,42,50,60,69,91 It provides real-time measurement of the binding affinity (Kd) as well as kinetic parameters such as kon and koff with high sensitivity. By measuring the heat change during ligand binding to GST-tagged BCL6BTB protein,70 BLI represents another biosensor technology for real-time detecting and quantifying the binding affinity and kinetics. Compared to SPR, BLI has advantages including increased throughput and wider sample compatibility. Because BLI is a fluidics-free system, it requires lower instrumentation cost as well. Moreover, by measuring directed movement of molecules in a temperature gradient, MST has also been used to quantify binding event between GST-labelled BCL6BTB protein and its ligands.102 Compared to SPR and ITC, MST is a surface immobilization-free approach. Recent advances in the MST technology by following intrinsic fluorescence of tryptophan residue of a protein would make it a completely label-free method to study molecular interactions.103 Besides, MST experiment can be done in versatile environment even in plasma and cell lysate.103 ITC has been used to define the binding affinity, stoichiometry, and the enthalpy and entropy changes of the ligand binding to the BCL6BTB protein.73 In addition to binding affinity, results from ITC can provide information about structural features. However, ITC has limitations such as low throughput and relatively low resolution, it also requires relatively large amount of the protein.104 Therefore, it is usually used as a secondary confirmation assay instead of primary screening.
Table 2.
Direct Binding Assays and Conditions
Three types of cellular assays have been developed (Table 3). The luciferase reporter assay was first established based on a GAL4-DBD-BCL6BTB fusion construct. This assay was valuable not only in testing potential ligand interaction to BCL6BTB in cellular environment, but also providing a flexible system to study the specificity of small molecules for BCL6BTB over other related BTB domain proteins such as such as PLZF, Kaiso and HIC1.6,7,60,91 To follow the PPI of BCL6BTB and its corepressors in cell, an M2H assay, based on activation of the luciferase reporter upon interaction of genetically fused GAL4-DBD-BCL6BTB (“bait”) protein and GAL4-VP16-corepressor (“prey”) peptide, has also been developed.38,42,50,78 This assay platform was used by multiple groups to study the potency of reversible inhibitors in cellular environment. Besides, the bioluminescence resonance energy transfer-based assay system NanoBRET has been utilized to study the PPI between BCL6BTB and its compressors in live cells. This assay uses the NanoLuc luciferase to provide the donor signal and the HaloTag ligand (HL) as the fluorescence energy acceptor. Compared to the luciferase reporter and M2H assays, the NanoBRET assay has a significant advantage for it offers cellular evaluation of a potential inhibitor in blocking the PPI between full length BCL6 and corepressor proteins. Recently, by following the fluorescence signal of eGFP using a translational fusion eGFP-BCL6BTB, a FACS-based flow cytometry assay was developed that can be used to study the binding of irreversible inhibitors to in living cells.83
Table 3.
Cellular Assays and Conditions
| Assay | Protein | Readout | Reference |
|---|---|---|---|
| Luc reporter | GAL4-BCL6BTB | Luciferase catalysis | 60, 91 |
| M2H | aGAL4-BCL6BTB, VP16b | Luciferase catalysis | 38, 42, 50, 78 |
| NanoBRET | full length BCL6 and corepressor | HL Fluorescence | 95 |
| FACS | eGFP-BCL6BTBc | eGFP / mCherry | 83 |
GAL4-BCL6BTB = GAL4-BCL6BTB(5-129)
VP16 = VP16-BCOR(112-753)
eGFP, enhanced green fluorescent protein.
Co-crystal structures of various compounds in complex with BCL6BTB have been solved and successfully used in the design and development of new inhibitors. These compounds, although belonged to different families, bind to a common binding pocket in the BCL6BTB LG employing four continuous binding sites: acid site, aromatic site, linker site and the HDCH site. It is worth noting that numerous inhibitors included a fragment that extruded from the acid site to the solvent-exposing space. These fragments could play an important role in crystallization of the inhibitor/BCL6BTB complexes.60,69,73,91 As one example, the indole group of inhibitor 4791 was sandwiched between two BCL6BTB dimers (Figure 14A). The piperazine ring of compound 47 extended beyond the acid site and fit into a polar pocket formed by five residues E81, N84, I85, D88 and N96 (Figure 14B). Notably, this interaction mode involves the presence of crystal contacts of the ligand with the adjacent BCL6BTB dimer. Such interactions may impact details of the interaction orientation of the ligand with the primary BCL6BTB dimer to which it is bound, as previously discussed.71
Figure 14.

Binding mode of compound 47 to BCL6BTB (PDB ID: 6EW8). (A) Overall view of the packing of one BCL6BTB dimer (green and white) on top of a second BCL6BTB dimer (orange and purple) via inhibitor 47. The surface area of the neighboring BCL6BTB dimer that was interact with inhibitor 47 (yellow) was highlighted. (B) Detailed binding mode of inhibitor 47 to the neighboring BCL6BTB dimer. Key residues involved in the interactions were shown in sticks.
Using a combination of various biological and biophysical methods, numerous BCL6BTB-targeting hits have been identified, which were subsequently developed into lead compounds through medicinal chemistry efforts. These leads could disrupt BCL6 interaction with its partner corepressor-derived peptide in various binding assays with an affinity range of low-nanomolar to low-micromolar. They usually also demonstrated mid-nanomolar to micromolar activities in engineered cellular reporter system. Their binding modes to BCL6BTB LG were confirmed by X-ray co-crystal structures and 2D NMR (e.g., 1H,15N-HSQC). Of these new compounds, reversible inhibitors (e.g., compound 28) by Guo and coworkers showed strong antiproliferative effects.70 Irreversible inhibitors (e.g., compound 40) from Takeda indicated antiproliferative effects although selectivity for BCL6-dependent DLBCL cells was not determined.78 Moreover, recent BCL6 degraders from both ICR (e.g., compound 58)95 and Boehringer (e.g., compound 61)69 showed selective antiproliferative effects for BCL6-dependent over BCL6-independent DLBCL cells. It has also been shown that the antiproliferative effects of BCL6 degraders were stronger than those of the corresponding inhibitors. On the other hand, several optimized leads indicated either no effect on DLBCL proliferation,38,50,73 or non-selective antiproliferative effects for both BCL6-dependent and BCL6-independent cells.60,91 The failure of these compounds is likely due to the limited cellular permeability38,50,73 or likely the lack of adequate target engagement. Considering no detailed target engagement studies were performed in these studies, it is not known whether the compounds could actually block BCL6 mediated formation of repression complexes nor for how long the inhibitory event might occur.
The mechanism of action of potent BCL6 PROTACs and degraders was evaluated to confirm the involvement the BCL6 protein.69,91,95 For example, PROTAC 50 caused selective degradation of the BCL6 protein in cell,91 although only incomplete degradation of the protein has been observed. The degrader 58 caused rapid but incomplete degradation of the BCL6 protein,95 another degrader 61, on the other hand, caused degradation of BCL6 in a DNA-binding dependent manner.69 Although the degrader 61 caused gradual proliferation arrest of DLBCL cells without apoptosis, which was quite different than what was observed with BCL6 shRNA, siRNA, CRSPR, or BPI, similar effects by the degrader 61 was recently observed using inducible BCL6 knockout model for DLBCL.105
In summary, BCL6BTB holds excellent potential as a therapeutic target that has only recently started to be systematically studied. Numerous BCL6 inhibitors and degraders have been characterized using various in vitro and in vivo assays, with a few examples indicating promising effects in killing BCL6-dependent cancer cells. In general, the BCL6 degraders have demonstrated more favorable antiproliferative effects than those of BCL6 inhibitors. One of the major impediments to currently available studies was the challenge of transforming inhibitors with high binding potency to BCL6BTB into anticancer efficacies in cell and in vivo. This may due to the fact that none of the new compounds have been evaluated through the proper target engagement experiments such as QChIP or target occupancy over time assays,7 which are considered critical to understand the likely impact of those compounds. Specifically, because BCL6 must recruit its repression complex in order to perform its transcriptional repression function,28,29 it is critical to test the compounds’ effects in disrupting the repression complex by compounds. Without these results, one cannot know if the compounds had on-target efficacy. For example, the new compounds from Takeda, AstraZeneca, and Boehringer have not been tested for the effects of disrupting the BCL6 repression complex on chromatin of BCL6, as occurred with the early BPI,4 small molecule inhibitor 2,7 or by CRISPR knockdown.106 BCL6 CRISPR is lethal to lymphoma cells.106 Up to now, there have been no compounds able to sufficiently down-regulate BCL6 so as to mimic the effect of genetic ablation by CRISPR. Therefore, none of the new compounds (except for inhibitor 28) to date has been shown to kill lymphoma cells in short period of time (48-96 h) and has indicated anticancer effects in DLBCL animal models. Continuous efforts are needed in the development of potent, selective, and cellularly active BCL6BTB inhibitors to facilitate studies on the therapeutic potential of the oncogenic transcription repressor BCL6 in drug discovery. We expect that the fundamental insights gained both published and from future studies will ultimately enhance the ability of medicinal chemists to design inhibitors for BCL6BTB as well as a wide range of disease-relevant proteins that contain BTB domains. Such novel compounds, in addition to their potential therapeutic utility, are expected to prove useful in investigating the pharmacology of other protein members of the BTB/POZ family such as Kaiso and LRF.
ACKNOWLEDGMENTS
We thank the National Science Foundation 1904514 (UTA19-000870) to FX; the Samuel Waxman Cancer Research Foundation to AM, ADM and FX; University of Maryland Baltimore CADD Center, National Institute of General Medical Sciences GM131710 to ADM.
ABBREVIATIONS USED
- ABL
abelson proto-oncogene
- AML
acute myeloid leukemia
- B-ALL
B-acute lymphoblastic leukemia
- BBD
BCL6 binding domain
- BCL6
B-cell Lymphoma 6
- BCOR
BCL6 corepressor
- BPI
BCL6 peptide inhibitor
- BTB
Broad-Complex, tramtrack and bric a brac
- GCB
germinal center B-cell
- ABC
activated B-cell
- CADD
computer-aided drug design
- CETSA
cellular thermal shift assay
- CML
chronic myeloid leukemia
- CCK8
Cell Counting Kit 8
- DC50
the concentration at which the target is degraded by fifty percent (50%)
- DLBCL
diffuse large B-cell lymphomas
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- ELISA
enzyme-linked immunosorbent assay
- FBDD
fragment-based drug discovery
- FLs
follicular lymphomas
- FP
fluorescence polarization
- GC
germinal center
- HDX-MS
hydrogen deuterium exchange mass spectrometry
- HTS
high-throughput screening
- IC50
the concentration of drug at which 50% of the target is inhibited
- IPC
irreversible probe competition
- ITC
isothermal-titration calorimetry
- LE
ligand efficiency
- LG
lateral groove
- LUMIER
luminescence-based mammalian interactome
- M2H
mammalian two-hybrid
- MLL
mixed-lineage leukemia
- MSD
Meso Scale Discovery
- MST
microscale thermophoresis
- NCOR
nuclear receptor corepressor
- NHL
non-Hodgkin’s lymphomas
- NMR
nuclear magnetic resonance
- PAMPA
parallel artificial membrane permeability assay
- PDB
RSCB Protein Data Bank
- PPI
protein-protein interaction
- PRDM1
positive regulatory domain zinc finger protein 1
- PROTAC
proteolysis targeting chimera
- RD2
second repression domain
- RTHS
reverse two-hybrid system
- SILCS
site identification by ligand competitive saturation
- SMRT
silencing mediator for retinoid or thyroid hormone receptors
- SPR
surface plasmon resonance
- STD-NMR
saturation transfer difference-nuclear magnetic resonance
- TR-FRET
time-resolved fluorescence resonance energy transfer
- NP-CGG
4-hydroxy-3-nitrophenyl)acetyl-chicken gamma globulin
- MLM
mouse liver microsomes
- BLI
biolayer interferometry
- ICR
Institute of Cancer Research.
Biography
Yong Ai earned his Ph.D. in Medicinal Chemistry from China Pharmaceutical University under the supervision of Professors Yihua Zhang and Sixun Peng. He is currently a Postdoctoral Fellow of the Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore. His research interests mainly focus on inhibitors of the Wnt signaling pathway and BCL6 BTB domain.
Lucia Hwang is a second-year PharmD candidate at the University of Maryland School of Pharmacy. Her research focuses on developing small molecules to inhibit the BCL6BTB and heme uptake in Pseudomonas aeruginosa.
Alexander D. MacKerell Jr. received a Ph.D. in Biochemistry in 1985 from Rutgers University, which was followed by postdoctoral fellowships in the Department of Medical Biophysics, Karolinska Institutet, Stockholm, Sweden and the Department of Chemistry, Harvard University. In 1992 he assumed his faculty position in the School of Pharmacy, University of Maryland where he is currently the Grollman-Glick Professor of Pharmaceutical Sciences and the Director of University of Maryland Computer-Aided Drug Design Center. MacKerell is also Co-founder and CSO of SilcsBio LLC, a biotech company in the area of computer-aided drug design and development.
Ari Melnick received his doctorate in Medicine from the University of Buenos Aires in Argentina. He completed a residency in internal medicine and a fellowship in hematology/oncology at the Mount Sinai School of Medicine in New York City. Then, he did a post-doctoral fellowship with Dr. Jonathan Licht at Mount Sinai on the topic of transcriptional regulation in leukemia. Dr. Melnick started his independent laboratory at the Albert Einstein College of Medicine, and then moved to the Weill Cornell Medical College in New York City, where he is currently the Gebroe Family Professor of Hematology/Oncology and Professor of Pharmacology and Immunology.
Fengtian Xue received his BS in chemistry from the University of Science and Technology of China in 2001. He completed his PhD in chemistry under the direction of Professor Christopher Seto at Brown University in 2007, and later pursued a post-doctoral fellowship in Medicinal Chemistry under the supervision of Professor Richard Silverman at Northwestern University from 2007 to 2009. Dr. Xue is currently an Associate Professor in the Department of Pharmaceutical Sciences at University of Maryland Baltimore. His research focuses in using experimental methods from Organic Chemistry and Medicinal Chemistry to develop small molecule therapeutic agents for various diseases including various cancers, bacterial/parasitic infections, and metabolic diseases.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Mlynarczyk C; Fontan L; Melnick A Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev 2019, 288, 214–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mesin L; Ersching J; Victora GD Germinal center B cell dynamics. Immunity 2016, 45, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ranuncolo SM; Polo JM; Dierov J; Singer M; Kuo T; Greally J; Green R; Carroll M; Melnick A Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat. Immunol 2007, 8, 705–714. [DOI] [PubMed] [Google Scholar]
- (4).Polo JM; Dell'Oso T; Ranuncolo SM; Cerchietti L; Beck D; Da Silva GF; Prive GG; Licht JD; Melnick A Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat. Med 2004, 10, 1329–1335. [DOI] [PubMed] [Google Scholar]
- (5).Cerchietti LC; Yang SN; Shaknovich R; Hatzi K; Polo JM; Chadburn A; Dowdy SF; Melnick A A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 2009, 113, 3397–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cerchietti LC; Ghetu AF; Zhu X; Da Silva GF; Zhong S; Matthews M; Bunting KL; Polo JM; Fares C; Arrowsmith CH; Yang SN; Garcia M; Coop A; Mackerell AD Jr.; Prive GG; Melnick A A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 2010, 17, 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cardenas MG; Yu W; Beguelin W; Teater MR; Geng H; Goldstein RL; Oswald E; Hatzi K; Yang SN; Cohen J; Shaknovich R; Vanommeslaeghe K; Cheng H; Liang D; Cho HJ; Abbott J; Tam W; Du W; Leonard JP; Elemento O; Cerchietti L; Cierpicki T; Xue F; MacKerell AD Jr.; Melnick AM Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J. Clin. Invest 2016, 126, 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ranuncolo SM; Polo JM; Melnick A BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol. Dis 2008, 41, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ci W; Polo JM; Cerchietti L; Shaknovich R; Wang L; Yang SN; Ye K; Farinha P; Horsman DE; Gascoyne RD; Elemento O; Melnick A The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood 2009, 113, 5536–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Phan RT; Saito M; Basso K; Niu H; Dalla-Favera R BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol 2005, 6, 1054–1060. [DOI] [PubMed] [Google Scholar]
- (11).Phan RT; Dalla-Favera R The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004, 432, 635–639. [DOI] [PubMed] [Google Scholar]
- (12).Cerchietti LC; Hatzi K; Caldas-Lopes E; Yang SN; Figueroa ME; Morin RD; Hirst M; Mendez L; Shaknovich R; Cole PA; Bhalla K; Gascoyne RD; Marra M; Chiosis G; Melnick A BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. J. Clin. Invest 2010, 120, 4569–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Parekh S; Polo JM; Shaknovich R; Juszczynski P; Lev P; Ranuncolo SM; Yin Y; Klein U; Cattoretti G; Dalla Favera R; Shipp MA; Melnick A BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood 2007, 110, 2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shaffer AL; Yu X; He YS; Boldrick J; Chan EP; Staudt LM BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity 2000, 13, 199–212. [DOI] [PubMed] [Google Scholar]
- (15).Cerchietti LC; Polo JM; Da Silva GF; Farinha P; Shaknovich R; Gascoyne RD; Dowdy SF; Melnick A Sequential transcription factor targeting for diffuse large B-cell lymphomas. Cancer Res. 2008, 68, 3361–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Valls E; Lobry C; Geng H; Wang L; Cardenas M; Rivas M; Cerchietti L; Oh P; Yang SN; Oswald E; Graham CW; Jiang Y; Hatzi K; Agirre X; Perkey E; Li Z; Tam W; Bhatt K; Leonard JP; Zweidler-McKay PA; Maillard I; Elemento O; Ci W; Aifantis I; Melnick A BCL6 antagonizes NOTCH2 to maintain survival of human follicular lymphoma cells. Cancer Discov. 2017, 7, 506–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Duy C; Hurtz C; Shojaee S; Cerchietti L; Geng H; Swaminathan S; Klemm L; Kweon SM; Nahar R; Braig M; Park E; Kim YM; Hofmann WK; Herzog S; Jumaa H; Koeffler HP; Yu JJ; Heisterkamp N; Graeber TG; Wu H; Ye BH; Melnick A; Muschen M BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature 2011, 473, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Geng H; Brennan S; Milne TA; Chen WY; Li Y; Hurtz C; Kweon SM; Zickl L; Shojaee S; Neuberg D; Huang C; Biswas D; Xin Y; Racevskis J; Ketterling RP; Luger SM; Lazarus H; Tallman MS; Rowe JM; Litzow MR; Guzman ML; Allis CD; Roeder RG; Muschen M; Paietta E; Elemento O; Melnick AM Integrative epigenomic analysis identifies biomarkers and therapeutic targets in adult B-acute lymphoblastic leukemia. Cancer Discov. 2012, 2, 1004–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hurtz C; Hatzi K; Cerchietti L; Braig M; Park E; Kim YM; Herzog S; Ramezani-Rad P; Jumaa H; Muller MC; Hofmann WK; Hochhaus A; Ye BH; Agarwal A; Druker BJ; Shah NP; Melnick AM; Muschen M BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J. Exp. Med 2011, 208, 2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kawabata KC; Zong H; Meydan C; Wyman S; Wouters BJ; Sugita M; Goswami S; Albert M; Yip W; Roboz GJ; Chen Z; Delwel R; Carroll MP; Mason C; Melnick A; Guzman ML BCL6 maintains survival and self-renewal of primary human acute myeloid leukemia cells. Blood 2021,137, 812–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Walker SR; Liu S; Xiang M; Nicolais M; Hatzi K; Giannopoulou E; Elemento O; Cerchietti L; Melnick A; Frank DA The transcriptional modulator BCL6 as a molecular target for breast cancer therapy. Oncogene 2015, 34, 1073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Deb D; Rajaram S; Larsen JE; Dospoy PD; Marullo R; Li LS; Avila K; Xue F; Cerchietti L; Minna JD; Altschuler SJ; Wu LF Combination therapy targeting BCL6 and phospho-STAT3 defeats intratumor heterogeneity in a subset of non-small cell lung cancers. Cancer Res. 2017, 77, 3070–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fernando TM; Marullo R; Pera Gresely B; Phillip JM; Yang SN; Lundell-Smith G; Torregroza I; Ahn H; Evans T; Gyorffy B; Prive GG; Hirano M; Melnick AM; Cerchietti L BCL6 evolved to enable stress tolerance in vertebrates and is broadly required by cancer cells to adapt to stress. Cancer Discov. 2019, 9, 662–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ye BH BCL-6 in the pathogenesis of non-Hodgkin's lymphoma. Cancer Invest. 2000, 18, 356–365. [DOI] [PubMed] [Google Scholar]
- (25).Zhang H; Okada S; Hatano M; Okabe S; Tokuhisa T A new functional domain of Bcl6 family that recruits histone deacetylases. Biochim. Biophys. Acta 2001, 1540, 188–200. [DOI] [PubMed] [Google Scholar]
- (26).Huynh KD; Bardwell VJ The BCL-6 POZ domain and other POZ domains interact with the co-repressors N-CoR and SMRT. Oncogene 1998, 17, 2473–2484. [DOI] [PubMed] [Google Scholar]
- (27).Huynh KD; Fischle W; Verdin E; Bardwell VJ BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000, 14, 1810–1823. [PMC free article] [PubMed] [Google Scholar]
- (28).Ahmad KF; Melnick A; Lax S; Bouchard D; Liu J; Kiang CL; Mayer S; Takahashi S; Licht JD; Prive GG Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol. Cell 2003, 12, 1551–1564. [DOI] [PubMed] [Google Scholar]
- (29).Ghetu AF; Corcoran CM; Cerchietti L; Bardwell VJ; Melnick A; Prive GG Structure of a BCOR corepressor peptide in complex with the BCL6 BTB domain dimer. Mol. Cell 2008, 29, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Stogios PJ; Chen L; Prive GG Crystal structure of the BTB domain from the LRF/ZBTB7 transcriptional regulator. Protein Sci. 2007, 16, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bunting KL; Melnick AM New effector functions and regulatory mechanisms of BCL6 in normal and malignant lymphocytes. Curr. Opin. Immunol 2013, 25, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huang C; Hatzi K; Melnick A Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms. Nat. Immunol 2013, 14, 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dubach JM; Kim E; Yang K; Cuccarese M; Giedt RJ; Meimetis LG; Vinegoni C; Weissleder R Quantitating drug-target engagement in single cells in vitro and in vivo. Nat. Chem. Biol 2017, 13, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Martinez Molina D; Jafari R; Ignatushchenko M; Seki T; Larsson EA; Dan C; Sreekumar L; Cao Y; Nordlund P Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [DOI] [PubMed] [Google Scholar]
- (35).Cardenas MG; Oswald E; Yu W; Xue F; MacKerell AD Jr.; Melnick AM The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin. Cancer Res 2017, 23, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Leeman-Neill RJ; Bhagat G BCL6 as a therapeutic target for lymphoma. Expert Opin. Ther. Targets 2018, 22, 143–152. [DOI] [PubMed] [Google Scholar]
- (37).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (38).Sakamoto K; Sogabe S; Kamada Y; Sakai N; Asano K; Yoshimatsu M; Ida K; Imaeda Y; Sakamoto JI Discovery of high-affinity BCL6-binding peptide and its structure-activity relationship. Biochem. Biophys. Res. Commun 2017, 482, 310–316. [DOI] [PubMed] [Google Scholar]
- (39).Hatanaka T; Ohzono S; Park M; Sakamoto K; Tsukamoto S; Sugita R; Ishitobi H; Mori T; Ito O; Sorajo K; Sugimura K; Ham S; Ito Y Human IgA-binding peptides selected from random peptide libraries: affinity maturation and application in IgA purification. J. Biol. Chem 2012, 287, 43126–43136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sakamoto K; Ito Y; Hatanaka T; Soni PB; Mori T; Sugimura K Discovery and characterization of a peptide motif that specifically recognizes a non-native conformation of human IgG induced by acidic pH conditions. J. Biol. Chem 2009, 284, 9986–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bechara C; Sagan S Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [DOI] [PubMed] [Google Scholar]
- (42).Yasui T; Yamamoto T; Sakai N; Asano K; Takai T; Yoshitomi Y; Davis M; Takagi T; Sakamoto K; Sogabe S; Kamada Y; Lane W; Snell G; Iwata M; Goto M; Inooka H; Sakamoto JI; Nakada Y; Imaeda Y Discovery of a novel B-cell lymphoma 6 (BCL6)-corepressor interaction inhibitor by utilizing structure-based drug design. Bioorg. Med. Chem 2017, 25, 4876–4886. [DOI] [PubMed] [Google Scholar]
- (43).Ishikawa M; Hashimoto Y Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem 2011, 54, 1539–1554. [DOI] [PubMed] [Google Scholar]
- (44).Buchwald P Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life 2010, 62, 724–731. [DOI] [PubMed] [Google Scholar]
- (45).Erlanson DA; Fesik SW; Hubbard RE; Jahnke W; Jhoti H Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov 2016, 15, 605–619. [DOI] [PubMed] [Google Scholar]
- (46).Ludlow RF; Verdonk ML; Saini HK; Tickle IJ; Jhoti H Detection of secondary binding sites in proteins using fragment screening. Proc. Natl. Acad. Sci. U S A 2015, 112, 15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jordan JB; Whittington DA; Bartberger MD; Sickmier EA; Chen K; Cheng Y; Judd T Fragment-linking approach using (19)F NMR spectroscopy to obtain highly potent and selective inhibitors of beta-secretase. J. Med. Chem 2016, 59, 3732–3749. [DOI] [PubMed] [Google Scholar]
- (48).Kohlmann A; Zech SG; Li F; Zhou T; Squillace RM; Commodore L; Greenfield MT; Lu X; Miller DP; Huang WS; Qi J; Thomas RM; Wang Y; Zhang S; Dodd R; Liu S; Xu R; Xu Y; Miret JJ; Rivera V; Clackson T; Shakespeare WC; Zhu X; Dalgarno DC Fragment growing and linking lead to novel nanomolar lactate dehydrogenase inhibitors. J. Med. Chem 2013, 56, 1023–1040. [DOI] [PubMed] [Google Scholar]
- (49).Varnes JG; Geschwindner S; Holmquist CR; Forst J; Wang X; Dekker N; Scott CW; Tian G; Wood MW; Albert JS Fragment-assisted hit investigation involving integrated HTS and fragment screening: Application to the identification of phosphodiesterase 10A (PDE10A) inhibitors. Bioorg. Med. Chem. Lett 2016, 26, 197–202. [DOI] [PubMed] [Google Scholar]
- (50).Kamada Y; Sakai N; Sogabe S; Ida K; Oki H; Sakamoto K; Lane W; Snell G; Iida M; Imaeda Y; Sakamoto J; Matsui J Discovery of a B-cell lymphoma 6 protein-protein interaction inhibitor by a biophysics-driven fragment-based approach. J. Med. Chem 2017, 60, 4358–4368. [DOI] [PubMed] [Google Scholar]
- (51).Giannetti AM; Koch BD; Browner MF Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J. Med. Chem 2008, 51, 574–580. [DOI] [PubMed] [Google Scholar]
- (52).Giannetti AM; Zheng X; Skelton NJ; Wang W; Bravo BJ; Bair KW; Baumeister T; Cheng E; Crocker L; Feng Y; Gunzner-Toste J; Ho YC; Hua R; Liederer BM; Liu Y; Ma X; O'Brien T; Oeh J; Sampath D; Shen Y; Wang C; Wang L; Wu H; Xiao Y; Yuen PW; Zak M; Zhao G; Zhao Q; Dragovich PS Fragment-based identification of amides derived from trans-2-(pyridin-3-yl)cyclopropanecarboxylic acid as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). J. Med. Chem 2014, 57, 770–792. [DOI] [PubMed] [Google Scholar]
- (53).Hendrick AG; Muller I; Willems H; Leonard PM; Irving S; Davenport R; Ito T; Reeves J; Wright S; Allen V; Wilkinson S; Heffron H; Bazin R; Turney J; Mitchell PJ Identification and investigation of novel binding fragments in the fatty acid binding protein 6 (FABP6). J. Med. Chem 2016, 59, 8094–8102. [DOI] [PubMed] [Google Scholar]
- (54).Lund BA; Christopeit T; Guttormsen Y; Bayer A; Leiros HK Screening and design of inhibitor scaffolds for the antibiotic resistance oxacillinase-48 (OXA-48) through surface plasmon resonance screening. J. Med. Chem 2016, 59, 5542–5554. [DOI] [PubMed] [Google Scholar]
- (55).Viegas A; Manso J; Nobrega FL; Cabrita EJ Saturation-transfer difference (STD) NMR: a simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educ 2011, 88, 990–994. [Google Scholar]
- (56).Elinder M; Geitmann M; Gossas T; Kallblad P; Winquist J; Nordstrom H; Hamalainen M; Danielson UH Experimental validation of a fragment library for lead discovery using SPR biosensor technology. J. Biomol. Screen 2011, 16, 15–25. [DOI] [PubMed] [Google Scholar]
- (57).Navratilova I; Macdonald G; Robinson C; Hughes S; Mathias J; Phillips C; Cook A Biosensor-based approach to the identification of protein kinase ligands with dual-site modes of action. J. Biomol. Screen 2012, 17, 183–193. [DOI] [PubMed] [Google Scholar]
- (58).Perspicace S; Banner D; Benz J; Muller F; Schlatter D; Huber W Fragment-based screening using surface plasmon resonance technology. J. Biomol. Screen 2009, 14, 337–349. [DOI] [PubMed] [Google Scholar]
- (59).Hopkins AL; Groom CR; Alex A Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- (60).McCoull W; Abrams RD; Anderson E; Blades K; Barton P; Box M; Burgess J; Byth K; Cao Q; Chuaqui C; Carbajo RJ; Cheung T; Code E; Ferguson AD; Fillery S; Fuller NO; Gangl E; Gao N; Grist M; Hargreaves D; Howard MR; Hu J; Kemmitt PD; Nelson JE; O'Connell N; Prince DB; Raubo P; Rawlins PB; Robb GR; Shi J; Waring MJ; Whittaker D; Wylot M; Zhu X Discovery of pyrazolo[1,5-a]pyrimidine B-cell lymphoma 6 (BCL6) binders and optimization to high affinity macrocyclic inhibitors. J. Med. Chem 2017, 60, 4386–4402. [DOI] [PubMed] [Google Scholar]
- (61).Kovalenko A; Hirata F Potentials of mean force of simple ions in ambient aqueous solution. I. Three-dimensional reference interaction site model approach. J. Chem. Phys 2000, 112, 10391–10402. [Google Scholar]
- (62).Butts CP; Jones CR; Towers EC; Flynn JL; Appleby L; Barron NJ Interproton distance determinations by NOE--surprising accuracy and precision in a rigid organic molecule. Org. Biomol. Chem 2011, 9, 177–184. [DOI] [PubMed] [Google Scholar]
- (63).Troche-Pesqueira E; Anklin C; Gil RR; Navarro-Vazquez A Computer-assisted 3D structure elucidation of natural products using residual dipolar couplings. Angew. Chem. Int. Ed. Engl 2017, 56, 3660–3664. [DOI] [PubMed] [Google Scholar]
- (64).Copeland RA; Pompliano DL; Meek TD Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov 2006, 5, 730–739. [DOI] [PubMed] [Google Scholar]
- (65).IJzerman AP; Guo D Drug-target association kinetics in drug discovery. Trends Biochem. Sci 2019, 44, 861–871. [DOI] [PubMed] [Google Scholar]
- (66).Vauquelin G Link between a high kon for drug binding and a fast clinical action: to be or not to be? Medchemcomm 2018, 9, 1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Dowling JE; Alimzhanov M; Bao L; Chuaqui C; Denz CR; Jenkins E; Larsen NA; Lyne PD; Pontz T; Ye Q; Holdgate GA; Snow L; O'Connell N; Ferguson AD Potent and selective CK2 kinase inhibitors with effects on Wnt pathway signaling in vivo. ACS Med. Chem. Lett 2016, 7, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Bowes J; Brown AJ; Hamon J; Jarolimek W; Sridhar A; Waldron G; Whitebread S Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discov 2012, 11, 909–922. [DOI] [PubMed] [Google Scholar]
- (69).Kerres N; Steurer S; Schlager S; Bader G; Berger H; Caligiuri M; Dank C; Engen JR; Ettmayer P; Fischerauer B; Flotzinger G; Gerlach D; Gerstberger T; Gmaschitz T; Greb P; Han B; Heyes E; Iacob RE; Kessler D; Kolle H; Lamarre L; Lancia DR; Lucas S; Mayer M; Mayr K; Mischerikow N; Muck K; Peinsipp C; Petermann O; Reiser U; Rudolph D; Rumpel K; Salomon C; Scharn D; Schnitzer R; Schrenk A; Schweifer N; Thompson D; Traxler E; Varecka R; Voss T; Weiss-Puxbaum A; Winkler S; Zheng X; Zoephel A; Kraut N; McConnell D; Pearson M; Koegl M Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Rep. 2017, 20, 2860–2875. [DOI] [PubMed] [Google Scholar]
- (70).Guo W; Xing Y; Zhang Q; Xie J; Huang D; Gu H; He P; Zhou M; Xu S; Pang X; Liu M; Yi Z; Chen Y Synthesis and biological evaluation of B-cell lymphoma 6 inhibitors of N-phenyl-4-pyrimidinamine derivatives bearing potent activities against tumor growth. J. Med. Chem 2020, 63, 676–695. [DOI] [PubMed] [Google Scholar]
- (71).Hatzi K; Melnick A Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med 2014, 20, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Nurieva RI; Chung Y; Martinez GJ; Yang XO; Tanaka S; Matskevitch TD; Wang YH; Dong C Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Cheng H; Linhares BM; Yu W; Cardenas MG; Ai Y; Jiang W; Winkler A; Cohen S; Melnick A; MacKerell A Jr.; Cierpicki T; Xue F Identification of thiourea-based inhibitors of the B-cell lymphoma 6 BTB domain via NMR-based fragment screening and computer-aided drug design. J. Med. Chem 2018, 61, 7573–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Guvench O; MacKerell AD Jr. Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput. Biol 2009, 5, e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Lakkaraju SK; Yu W; Raman EP; Hershfeld AV; Fang L; Deshpande DA; MacKerell AD Jr. Mapping functional group free energy patterns at protein occluded sites: nuclear receptors and G-protein coupled receptors. J. Chem. Inf. Model 2015, 55, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Raman EP; Yu W; Lakkaraju SK; MacKerell AD Jr. Inclusion of multiple fragment types in the site identification by ligand competitive saturation (SILCS) approach. J. Chem. Inf. Model 2013, 53, 3384–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Oashi T; Ringer AL; Raman EP; Mackerell AD Automated selection of compounds with physicochemical properties to maximize bioavailability and druglikeness. J. Chem. Inf. Model 2011, 51, 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Sameshima T; Yamamoto T; Sano O; Sogabe S; Igaki S; Sakamoto K; Ida K; Gotou M; Imaeda Y; Sakamoto J; Miyahisa I Discovery of an irreversible and cell-active BCL6 inhibitor selectively targeting Cys53 located at the protein-protein interaction interface. Biochemistry 2018, 57, 1369–1379. [DOI] [PubMed] [Google Scholar]
- (79).Miyahisa I; Sameshima T; Hixon MS Rapid determination of the specificity constant of irreversible inhibitors (kinact/KI) by means of an endpoint competition assay. Angew. Chem. Int. Ed. Engl 2015, 54, 14099–14102. [DOI] [PubMed] [Google Scholar]
- (80).Sameshima T; Tanaka Y; Miyahisa I Universal and quantitative method to evaluate inhibitor potency for cysteinome proteins using a nonspecific activity-based protein profiling probe. Biochemistry 2017, 56, 2921–2927. [DOI] [PubMed] [Google Scholar]
- (81).Potashman MH; Duggan ME Covalent modifiers: an orthogonal approach to drug design. J. Med. Chem 2009, 52, 1231–1246. [DOI] [PubMed] [Google Scholar]
- (82).Johnson DS; Weerapana E; Cravatt BF Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem 2010, 2, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Teng M; Ficarro SB; Yoon H; Che J; Zhou J; Fischer ES; Marto JA; Zhang T; Gray NS Rationally designed covalent BCL6 inhibitor that targets a tyrosine residue in the homodimer interface. ACS Med. Chem. Lett 2020, 11, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Hett EC; Xu H; Geoghegan KF; Gopalsamy A; Kyne RE Jr.; Menard CA; Narayanan A; Parikh MD; Liu S; Roberts L; Robinson RP; Tones MA; Jones LH Rational targeting of active-site tyrosine residues using sulfonyl fluoride probes. ACS Chem. Biol 2015, 10, 1094–1098. [DOI] [PubMed] [Google Scholar]
- (85).Dong J; Krasnova L; Finn MG; Sharpless KB Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew. Chem. Int. Ed. Engl 2014, 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- (86).Hatcher JM; Wu G; Zeng C; Zhu J; Meng F; Patel S; Wang W; Ficarro SB; Leggett AL; Powell CE; Marto JA; Zhang K; Ki Ngo JC; Fu XD; Zhang T; Gray NS SRPKIN-1: a covalent SRPK1/2 inhibitor that potently converts VEGF from pro-angiogenic to anti-angiogenic isoform. Cell Chem. Biol 2018, 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Mukherjee H; Su N; Belmonte MA; Hargreaves D; Patel J; Tentarelli S; Aquila B; Grimster NP Discovery and optimization of covalent Bcl-xL antagonists. Bioorg. Med. Chem. Lett 2019, 29, 126682. [DOI] [PubMed] [Google Scholar]
- (88).Sievers QL; Petzold G; Bunker RD; Renneville A; Slabicki M; Liddicoat BJ; Abdulrahman W; Mikkelsen T; Ebert BL; Thoma NH Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Toure M; Crews CM Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem. Int. Ed. Engl 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- (90).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- (91).McCoull W; Cheung T; Anderson E; Barton P; Burgess J; Byth K; Cao Q; Castaldi MP; Chen H; Chiarparin E; Carbajo RJ; Code E; Cowan S; Davey PR; Ferguson AD; Fillery S; Fuller NO; Gao N; Hargreaves D; Howard MR; Hu J; Kawatkar A; Kemmitt PD; Leo E; Molina DM; O'Connell N; Petteruti P; Rasmusson T; Raubo P; Rawlins PB; Ricchiuto P; Robb GR; Schenone M; Waring MJ; Zinda M; Fawell S; Wilson DM Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem. Biol 2018, 13, 3131–3141. [DOI] [PubMed] [Google Scholar]
- (92).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Collins I; Wang H; Caldwell JJ; Chopra R Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem. J 2017, 474, 1127–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Bellenie BR; Cheung KJ; Varela A; Pierrat OA; Collie GW; Box GM; Bright MD; Gowan S; Hayes A; Rodrigues MJ; Shetty KN; Carter M; Davis OA; Henley AT; Innocenti P; Johnson LD; Liu M; de Klerk S; Le Bihan YV; Lloyd MG; McAndrew PC; Shehu E; Talbot R; Woodward HL; Burke R; Kirkin V; van Montfort RLM; Raynaud FI; Rossanese OW; Hoelder S Achieving in vivo target depletion through the discovery and optimization of benzimidazolone BCL6 degraders. J. Med. Chem 2020, 63, 4047–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Blasche S; Koegl M Analysis of protein-protein interactions using LUMIER assays. Methods Mol. Biol 2013, 1064, 17–27. [DOI] [PubMed] [Google Scholar]
- (97).Engen JR Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem 2009, 81, 7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Lee DH; Goldberg AL Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [DOI] [PubMed] [Google Scholar]
- (99).Culjkovic-Kraljacic B; Fernando TM; Marullo R; Calvo-Vidal N; Verma A; Yang S; Tabbo F; Gaudiano M; Zahreddine H; Goldstein RL; Patel J; Taldone T; Chiosis G; Ladetto M; Ghione P; Machiorlatti R; Elemento O; Inghirami G; Melnick A; Borden KL; Cerchietti L Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood 2016, 127, 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Owicki JC Fluorescence polarization and anisotropy in high throughput screening: perspectives and primer. J. Biomol. Screen 2000, 5, 297–306. [DOI] [PubMed] [Google Scholar]
- (101).Pope AJ; Haupts UM; Moore KJ Homogeneous fluorescence readouts for miniaturized high-throughput screening: theory and practice. Drug Discov. Today 1999, 4, 350–362. [DOI] [PubMed] [Google Scholar]
- (102).Osher EL; Castillo F; Elumalai N; Waring MJ; Pairaudeau G; Tavassoli A A genetically selected cyclic peptide inhibitor of BCL6 homodimerization. Bioorg. Med. Chem 2018, 26, 3034–3038. [DOI] [PubMed] [Google Scholar]
- (103).Jerabek-Willemsen M; André T; Wanner R; Roth HM; Duhr S; Baaske P; Breitsprecher D MicroScale thermophoresis: interaction analysis and beyond. J. Mol. Struct 2014, 1077, 101–113. [Google Scholar]
- (104).Duff MR Jr.; Grubbs J; Howell EE Isothermal titration calorimetry for measuring macromolecule-ligand affinity. J. Vis. Exp 2011, 55, e2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Schlager S; Salomon C; Olt S; Albrecht C; Ebert A; Bergner O; Wachter J; Trapani F; Gerlach D; Voss T; Traunbauer A; Jude J; Hinterndorfer M; Minnich M; Schweifer N; Blake SM; Zinzalla V; Drobits B; McConnell DB; Kraut N; Pearson M; Zuber J; Koegl M Inducible knock-out of BCL6 in lymphoma cells results in tumor stasis. Oncotarget 2020, 11, 875–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Hatzi K; Geng H; Doane AS; Meydan C; LaRiviere R; Cardenas M; Duy C; Shen H; Vidal MNC; Baslan T; Mohammad HP; Kruger RG; Shaknovich R; Haberman AM; Inghirami G; Lowe SW; Melnick AM Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat. Immunol 2019, 20, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]