

ABSTRACT
Background
The factors other than dietary intake that determine tissue concentrations of EPA and DHA remain obscure. Prior studies suggested that, in women, endogenous estrogen may accelerate synthesis of DHA from ɑ-linolenic acid (ALA), but the effects of exogenous estrogen on RBC n–3 (ɷ-3) PUFA concentrations are unknown.
Objective
We tested the hypothesis that menopausal hormone therapy (HT) would increase RBC n–3 PUFA concentrations.
Methods
Postmenopausal women (ages 50–79 y) were assigned to HT or placebo in the Women's Health Initiative (WHI) randomized trial. The present analyses included a subset of 1170 women (ages 65–79 y) who had RBC PUFA concentrations measured at baseline and at 1 y as participants in the WHI Memory Study. HT included conjugated equine estrogens (E) alone for women without a uterus (n = 560) and E plus medroxyprogesterone acetate (P) for those with an intact uterus (n = 610). RBC n–3 and n–6 (ɷ-6) PUFAs were quantified.
Results
Effects of E alone and E+P on PUFA profiles were similar and were thus combined in the analyses. Relative to the changes in the placebo group after 1 y of HT, docosapentaenoic acid (DPA; n–3) concentrations decreased by 10% (95% CI: 7.3%, 12.5%), whereas DHA increased by 11% (95% CI: 7.4%, 13.9%) in the HT group. Like DHA, DPA n–6 increased by 13% from baseline (95% CI: 10.0%, 20.3%), whereas linoleic acid decreased by 2.0% (95% CI: 1.0%, 4.1%; P values at least <0.01 for all). EPA and arachidonic acid concentrations were unchanged.
Conclusions
HT increased RBC concentrations of the terminal n–3 and n–6 PUFAs (DHA and DPA n–6). These findings are consistent with an estrogen-induced increase in DHA and DPA n–6 synthesis, which is consistent with an upregulation of fatty acid elongases and/or desaturases in the PUFA synthetic pathway. The clinical implications of these changes require further study. The Women's Health Initiative Memory Study is registered at clinicaltrials.gov as NCT00685009. Note that the data presented here were not planned as part of the original trial, and therefore are to be considered exploratory.
Keywords: estrogen, menopausal hormone therapy, progestin, n–3 fatty acids, n–6 fatty acids, Omega-3 Index, randomized trials
Introduction
Blood fatty acid concentrations are known to be associated with a variety of cardiometabolic traits including diabetes risk and heart disease, as well as brain function, mental health, and overall mortality. Our group has replicated these associations using RBC fatty acid concentrations in the Women's Health Initiative (WHI) Memory Study (1–4). The factors that determine RBC concentrations of the n–3 fatty acids are not completely understood. Although dietary and supplemental intake of n–3 fatty acids is obviously important (5), only ∼50% of the variability in the Omega-3 Index (6) (i.e., RBC concentrations of EPA plus DHA) could be explained by the intake of oily fish and fish-oil supplements, leaving a large proportion of variance unexplained (7). The possibility that sex hormone status could be one of these factors has been suggested from a variety of studies. For example, DHA is higher in young women than in young men (8–10), and in a novel study of male and female transsexuals undergoing sex-change procedures, males-to-females given oral conjugated equine estrogens (E) had an increase in DHA, whereas females-to-males given parenteral testosterone had a decrease in DHA (11). Burdge and colleagues (12, 13) showed that one reason young women have higher DHA concentrations than men is that the former convert the plant n–3 fatty acid, ɑ-linolenic acid (ALA; C18:3n−3) to EPA and DHA with a greater efficiency than do young men. Teleologically, young women may need to have a greater store of DHA to support childbirth. Finally, in a large (∼160,000 patients) clinical dataset, among those between 20 and 40 y of age, RBC DHA was significantly higher in women than in men, but not in those above age 40 (8). Korean women on menopausal hormone therapy (HT) also had elevated RBC DHA concentrations (14). These data are consistent with the hypothesis that E stimulates the biosynthesis of long-chain n–3 fatty acids from common dietary precursors, presumably via stimulation of the δ5 and 6 desaturases, the rate-limiting enzymes in n–3 and n–6 PUFA biosynthesis, although an effect on the elongases cannot be ruled out. Because these studies were not interventional, firm conclusions about the effects of E on the n–3 fatty acid status cannot be drawn. The WHI is an ideal setting to test this hypothesis. In this report we examine the effects of 1 y of HT or placebo on RBC n–3 and n–6 PUFA concentrations.
Methods
Sample
The WHI Memory Study examined the effects of menopausal HT on cognitive function in women aged 65–80 y (15, 16). Recruitment was from 1993 to 1998. There were 1170 women with both baseline and 1-y measurements of fatty acids who were randomly selected from the Memory Study cohort. Of these individuals, 610 had an intact uterus and 560 had undergone a hysterectomy. Among the 610 women with a uterus, 300 women were randomly assigned to receive estrogen + progestin (E+P) compared with 310 who were randomly assigned to the placebo group. Among the 560 without a uterus, 273 received E alone versus 287 who received placebo. (Throughout this article, HT refers to both E and E+P). Inclusion criteria stipulated that any women in either arm previously taking postmenopausal hormones had to undergo a washout period for 3 mo before joining the study. Supplemental Figure 1 provides a flow chart of study inclusion criteria. The Women's Health Initiative Memory Study is registered at clinicaltrials.gov as NCT00685009. Note that the data presented here were not planned as part of the original trial, and therefore are to be considered exploratory.
RBC fatty acid protocol
RBCs were isolated from blood drawn after a 10–12-h fast and frozen at −80°C after collection. RBC fatty acid composition was analyzed by GC with flame ionization detection as previously described in Pottala et al. (17). As reported therein, prior to arrival at our laboratory, all of the RBC samples were stored at −20°C for a period of ∼2 wk at the central laboratory during the aliquoting phase. This caused varying degrees of oxidative degradation to the PUFAs. Experiments were undertaken to quantify the degree of degradation, and models were generated and applied to the original dataset to estimate true values. In doing so, a dataset containing 10 imputations of each fatty acid for each individual was generated. To properly account for the imputation process, all statistical analyses included multiple imputation of all fatty acid values, which consists of estimating the average effect across all imputations, as well as the variability within and between imputations (17).
The PUFAs of interest in this study were 4 n–6 and 4 n–3 species plus the Omega-3 Index, which is EPA + DHA (see Table 1). All PUFA levels are expressed as a percent of total erythrocyte fatty acids. The CVs (i.e., the reproducibility) of each RBC PUFA determined by our laboratory method are given in Supplemental Table 1. In general, for PUFAs present at >3% of total fatty acids, the CVs were <5%; for those present at 1% to 3%, <8%; and for the 2 PUFAs present at <0.5% (ALA) and docosapentaenoic acid n-6 (DPA n–6), the CVs were 31% and 13%, respectively.
TABLE 1.
RBC fatty acid concentrations at baseline1
| Overall differences between groups (estrogen + progestin minus estrogen alone) | ||||
|---|---|---|---|---|
| Fatty acid | Estrogen + progestin (n = 610)2 | Estrogen alone (n = 560)2 | Unadjusted (95% CI; P value)3 | Adjusted (95% CI; P value)3,4 |
| Alpha linolenic acid (ALA, C18:3n–3) | 0.16 ± 0.08 | 0.16 ± 0.07 | 0.0015 (−0.0012, 0.0042; 0.27) | −0.0006 (−0.004, 0.0028; 0.73) |
| EPA (C20:5n–3) | 0.72 ± 0.45 | 0.68 ± 0.39 | 0.043 (0.028, 0.058; <0.0001) | 0.032 (0.015, 0.05; 0.0004) |
| Docosapentaenoic acid n–3 (DPA n-3, C22:5n–3) | 2.55 ± 0.42 | 2.5 ± 0.41 | 0.044 (0.029, 0.06; <0.0001) | 0.025 (0.0062, 0.044; 0.009) |
| DHA (C22:6n–3) | 4.58 ± 1.46 | 4.42 ± 1.39 | 0.16 (0.11, 0.21; <0.0001) | 0.13 (0.071, 0.18; <0.0001) |
| Omega-3 Index (EPA + DHA) | 5.3 ± 1.75 | 5.1 ± 1.61 | 0.2 (0.14, 0.26; <0.0001) | 0.16 (0.094, 0.22; <0.0001) |
| Linoleic acid (LA, C18:2n–6) | 11.67 ± 1.68 | 11.7 ± 1.7 | −0.029 (−0.09, 0.033; 0.36) | −0.079 (−0.16, −0.0018; 0.045) |
| Arachidonic acid (ARA, C20:4n–6) | 16.93 ± 1.71 | 17.02 ± 1.73 | −0.09 (−0.15, −0.028; 0.0045) | −0.089 (−0.17, −0.011; 0.026) |
| Adrenic acid (ADA, C22:4n–6) | 3.99 ± 0.86 | 4.09 ± 0.82 | −0.1 (−0.13, −0.073; <0.0001) | −0.055 (−0.091, −0.02; 0.0024) |
| DPA n–6 (C22:5n–6) | 0.78 ± 0.25 | 0.8 ± 0.25 | −0.023 (−0.032, −0.014; <0.0001) | −0.0036 (−0.014, 0.0072; 0.52) |
Values are mean ± SD percentage points unless otherwise indicated.
Combined samples give the pooled mean and SD for all women.
Predicting fatty acid concentration at baseline by group using a linear model accounting for multiple imputation.
Predicting fatty acid concentration at baseline by group and adjusting for age, race, BMI, alcohol consumption, smoking status, ovarian status, dietary EPA + DHA, and dietary n–6 using a linear model accounting for multiple imputation (denoted as model 1 in the Statistical Analysis section).
Statistical analyses
This was a randomized, placebo-controlled trial in which the effects of 1 y of HT on RBC PUFA composition was the primary endpoint in this analysis. Two-sample t tests and chi-square tests were used to evaluate demographic differences between the 4 treatment arms, depending on whether variables were continuous or categorical. When comparing women in the 2 groups (E+P vs. E alone) at baseline, we used linear models predicting fatty acid concentrations by group plus demographic variables—with a separate model for each fatty acid and the Omega-3 Index [model 1: fatty acid level at baseline (y) = group (0/1) + demographic covariates]. Demographic covariates included in the model were age (y), race (Caucasian), BMI (kg/m2), alcohol (at least 1 drink/wk), smoking (ever), ovaries (still present), dietary EPA + DHA (g/d), and dietary n–6 (g/d). When evaluating change in fatty acid concentrations over time, we used paired t tests within each of the 4 treatment arms, for each of the 8 fatty acids plus the Omega-3 Index. Linear models predicting change in fatty acid concentrations with group status, HT (yes/no), and their interaction were used to evaluate the potential effects of treatment arm on 1-y change in fatty acid status, and the potential modifying effect of group status on HT. We first fit a model with the interaction term between group status and HT [model 2a: change in fatty acid concentration from baseline (y) = group status (0/1) + HT (0/1) + group status × HT + demographic covariates + baseline fatty acid concentration], where the list of demographic covariates was the same as in model 1 with the exception of dietary intake. We subsequently fit models separately within women in the E+P group [model 2b: change in fatty acid concentration from baseline (y) = HT (0/1) + demographic covariates + baseline fatty acid concentration] and women in the E-alone group [model 2c: change in fatty acid concentration from baseline (y) = HT (0/1) + demographic covariates + baseline fatty acid concentration].
Two sensitivity analyses were performed. First, to determine if ovarian status confounded the analysis of women with no uterus, we compared the change in PUFAs between HT and placebo among women with bilateral oophorectomy compared with women with conserved ovaries [model 3a: change in fatty acid concentration (y) = ovarian status (0/1) + HT (0/1) + ovarian status (0/1) × HT (0/1) + demographic covariates + baseline fatty acid concentration]. We also estimated the effects of HT separately on women with ovaries [model 3b: change in fatty acid concentration (y) = HT (0/1) + demographic covariates + baseline fatty acid concentration] and women without ovaries [model 3c: change in fatty acid concentration (y) = HT (0/1) + demographic covariates + baseline fatty acid concentration]. In the second sensitivity analysis, we evaluated whether pre-study use of E or E+P affected the results. To do this, we compared the change in PUFAs with HT versus placebo among women with or without prior (pre-baseline) use of HT [model 4a: change in fatty acid concentration (y) = history of HT (0/1) + HT (0/1) + history of HT (0/1) × HT (0/1) + demographic covariates + baseline fatty acid concentration]. We also estimated the effects of HT separately on women with a history of HT [model 4b: change in fatty acid concentration (y) = HT (0/1) + demographic covariates + baseline fatty acid concentration] and women without a history of HT [model 4c: change in fatty acid concentration (y) = HT (0/1) + demographic covariates + baseline fatty acid concentration]. Due to the nature of the fatty acid data, all analyses were adjusted for multiple imputations (18). All statistical analyses were conducted using R (19). Case-wise deletion was used when missing data were present [missing data rates were low (<3% on all variables) and typically <1%]. In all cases we used 2-sided tests and a significance level of 0.05.
Results
Participant demographic profile by group status and treatment arm
There were no statistically significant differences (P < 0.05) at baseline between the women randomly assigned to HT and the women randomly assigned to placebo in either HT trial across a variety of demographic characteristics including age, race, BMI, alcohol consumption, smoking status, ovarian status, EPA + DHA intake, and n–6 intake (Table 2). However, there were some significant demographic differences between women in the 2 groups. Notably, women in the E+P group were more likely to be white, have a lower BMI, more likely to consume at least 1 alcoholic beverage/wk, and were more than twice as likely to still have their ovaries (99% vs. 46%).
TABLE 2.
Demographic characteristics of the sample1
| Estrogen + progestin (n = 610) | Estrogen alone (n = 560) | Combined sample (n = 1170) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Characteristic | Active (n = 300) | Placebo (n = 310) | P 2 | Active (n = 273) | Placebo (n = 287) | P 2 | Estrogen + progestin (n = 610) | Estrogen alone (n = 560) | P 2 |
| Age, y | 71.94 ± 4.10 | 72.20 ± 3.97 | 0.44 | 71.46 ± 3.95 | 71.93 ± 4.14 | 0.17 | 72.07 ± 4.04 | 71.70 ± 4.06 | 0.12 |
| Race, Caucasian | 91.3 (273/299) | 91.3 (282/309) | 1 | 87.5 (239/272) | 85.3 (243/285) | 0.44 | 91.3 (555/608) | 86.5 (482/557) | 0.01 |
| BMI, kg/m2 | 27.54 ± 5.23 | 27.25 ± 5.24 | 0.49 | 28.65 ± 5.65 | 28.26 ± 5.11 | 0.40 | 27.40 ± 5.23 | 28.45 ± 5.38 | <0.001 |
| Alcohol, ≥1 drink/wk | 35.2 (105/298) | 37 (113/305) | 0.7 | 28.6 (78/272) | 31.3 (89/284) | 0.55 | 36.2 (218/603) | 30 (167/556) | 0.03 |
| Smoking, ever | 47.5 (141/297) | 42.7 (132/309) | 0.27 | 38.5 (105/272) | 41 (116/283) | 0.63 | 45 (273/606) | 39.8 (221/555) | 0.08 |
| Ovaries, still present | 99 (295/298) | 100 (307/307) | 0.24 | 45.8 (125/239) | 51.8 (132/255) | 0.98 | 99.5 (602/605) | 52 (257/494) | <0.001 |
| Dietary EPA + DHA,3 g/d | 0.11 ± 0.12 | 0.11 ± 0.10 | 0.87 | 0.10 ± 0.10 | 0.10 ± 0.10 | 0.86 | 0.11 ± 0.11 | 0.10 ± 0.10 | 0.15 |
| Dietary n–6,3 g/d | 9.89 ± 5.40 | 10.50 ± 6.22 | 0.20 | 11.27 ± 6.67 | 10.45 ± 6.58 | 0.20 | 10.20 ± 5.83 | 10.80 ± 6.63 | 0.10 |
Values are means ± SDs or % (n/total n). Continuous variable means ± SDs ignore missing data (BMI missing n = 4; dietary EPA + DHA and dietary n-6 missing n = 1). Categorical variable percentages also ignore missing data with denominator sample sizes reflective of missing data.
P values are from 2-sample t tests or chi-square tests.
Derived from FFQs.
Differences in fatty acid concentrations at baseline
As shown in Table 1, EPA, DHA, the Omega-3 Index, and docosapentaenoic acid n-3 (DPA n–3, 22:5n−3) concentrations at baseline were higher in women in the E+P group compared with those in the E-alone group; differences remained statistically significant after adjusting for age, race, BMI, alcohol consumption, smoking status, ovarian status, dietary EPA + DHA, and dietary n–6. ALA concentrations were not significantly different in either analysis. A different pattern was seen for the n–6 fatty acids, with all 3 of the long-chain metabolites of linoleic acid (LA; 18:2n–6) being lower in the women in the E+P group versus the E-alone group in unadjusted analyses. After adjustment, differences between groups remained significant for arachidonic acid (ARA, 20:4n−6) and adrenic acid (ADA, 22:4n-6), the difference for DPA n–6 (22:5n-6) became nonsignificant, and the difference for LA became statistically significant.
Changes in fatty acid concentrations after 1 y of HT
The observed changes in long-chain n–6 and n–3 fatty acid concentrations across the 4 treatment arms after 1 y are detailed in Table 3. The pooled effects (i.e., the adjusted effect of HT compared with placebo for all participants regardless of group status; linear model predicting change in fatty acid by HT) for 4 PUFAs in each family are summarized in Figure 1 as both absolute percentage point changes and percentage changes from baseline. For the pooled analysis there was a decrease of ∼10% relative to placebo for DPA n–3, and an 11% increase for DHA (P < 0.001 for both). There were no significant effects on ALA or EPA. HT had a qualitatively similar effect on concentrations of the n–6 metabolites in the pooled analysis. That is, DPA n–6 increased with HT treatment by ∼13%, whereas its metabolic precursors, ARA and ADA, were unaffected. LA was, however, slightly but significantly lowered (−2.6%) by HT (Figure 1). Effects of group status on 1-y change (linear model predicting 1-y change in fatty acid by group status) were not significant for any RBC PUFA before and after adjusting for other variables, and group status did not modify the effect of HT on any PUFA (linear model predicting 1-y change in fatty acid by an interaction between group status and HT; Supplemental Table 2). Two sensitivity analyses were undertaken examining the effects of HT on RBC PUFAs. The first included women in the E+P group stratified by ovarian status (yes/no). No differences in RBC PUFA response were observed by ovarian status (linear model predicting 1-y change in fatty acid concentrations by HT, separately for women with and without ovaries; Supplemental Table 3). The second sensitivity analysis examined whether pre-study hormone use in all participants affected the response, and again, no differences were seen (linear model predicting 1-y change in fatty acid concentrations by HT, separately for women with and without a history of HT; Supplemental Table 4). Finally, we calculated the fraction of the variability in the change in RBC PUFA concentrations that was explained by HT at year 1. The highest fraction explained was for DPA n–3 (9%) and the lowest for ADA and ALA (<0.1%). For EPA, DHA, and the Omega-3 Index, the explained proportions were ∼ 0.3%, 5%, and 3.2%, respectively (Supplemental Table 5).
TABLE 3.
Change in n–3 and n–6 PUFAs after 1 y: effects of randomized assignment to menopausal hormone therapy1
| Estrogen + progestin (n = 610) | Estrogen alone (n = 560) | |||||
|---|---|---|---|---|---|---|
| Fatty acid | Active2 (n = 300) | Placebo2 (n = 310) | Adjusted treatment effect (active − placebo)3 | Active2 (n = 273) | Placebo2 (n = 287) | Adjusted treatment effect (active − placebo)3 |
| ALA (C18:3n–3) | 0.0046 (−0.0073, 0.016; 0.45) | −0.0028 (−0.018, 0.012; 0.72) | 0.0054 (−0.0079, 0.019; 0.43) | 0.0091 (−0.0078, 0.026; 0.29) | 0.0072 (−0.0056, 0.02; 0.27) | −0.00097 (−0.016, 0.014; 0.9) |
| EPA (C20:5n–3) | −0.063 (−0.14, 0.014; 0.11) | 0.015 (−0.051, 0.082; 0.65) | −0.067 (−0.14, 0.011; 0.092) | −0.049 (−0.11, 0.012; 0.11) | −0.015 (−0.13, 0.1; 0.8) | −0.033 (−0.13, 0.069; 0.53) |
| Docosapentaenoic acid n-3 (DPA n–3, C22:5n–3) | −0.25 (−0.31, −0.18; <0.0001) | 0.014 (−0.078, 0.11; 0.77) | −0.26 (−0.34, −0.19; <0.0001) | −0.25 (−0.33, −0.17; <0.0001) | −0.0055 (−0.063, 0.052; 0.85) | −0.25 (−0.33, −0.17; <0.0001) |
| DHA (C22:6n–3) | 0.52 (0.37, 0.66; <0.0001) | −0.0042 (−0.16, 0.15; 0.96) | 0.49 (0.31, 0.68; <0.0001) | 0.47 (0.28, 0.65; <0.0001) | 0.017 (−0.16, 0.19; 0.85) | 0.48 (0.29, 0.67; <0.0001) |
| Omega-3 Index (EPA + DHA) | 0.45 (0.28, 0.62; <0.0001) | 0.011 (−0.16, 0.19; 0.9) | 0.43 (0.21, 0.64; <0.0001) | 0.42 (0.23, 0.61; <0.0001) | 0.0016 (−0.22, 0.22; 0.99) | 0.44 (0.21, 0.66; <0.0002) |
| Linoleic acid (LA, C18:2n–6) | −0.45 (−0.63, −0.27; <0.0001) | −0.17 (−0.34, 0.0088; 0.063) | −0.22 (−0.44, −0.014; 0.037) | −0.55 (−0.75, −0.35; <0.0001) | −0.051 (−0.25, 0.15; 0.62) | −0.45 (−0.71, −0.18; 0.0011) |
| Arachidonic acid (ARA, C20:4n–6) | −0.16 (−0.36, 0.052; 0.14) | 0.034 (−0.17, 0.24; 0.74) | −0.19 (−0.45, 0.078; 0.17) | 0.026 (−0.23, 0.28; 0.84) | −0.021 (−0.27, 0.22; 0.86) | 0.087 (−0.19, 0.36; 0.53) |
| Adrenic acid (ADA, C22:4n–6) | 0.077 (−0.027, 0.18; 0.15) | −0.024 (−0.11, 0.062; 0.58) | 0.059 (−0.059, 0.18; 0.32) | −0.0073 (−0.11, 0.1; 0.89) | −0.04 (−0.13, 0.054; 0.4) | 0.0057 (−0.11, 0.13; 0.93) |
| DPA n–6 (C22:5n–6) | 0.1 (0.053, 0.16; <0.0001) | −0.0022 (−0.035, 0.03; 0.89) | 0.1 (0.057, 0.15; <0.0001) | 0.13 (0.077, 0.18; <0.0001) | −0.012 (−0.063, 0.04; 0.66) | 0.14 (0.069, 0.2; <0.0001) |
Values are mean percentage point changes (95% CIs; P value).
Post minus pre, so that a positive number means that the fatty acid concentration increased over time. P values represent paired t-test P values testing for evidence of significant change over time from baseline to follow-up within each treatment group (evidence that change is nonzero) and accounting for multiple imputations.
Adjusted effects estimates are adjusted for age, race, BMI, alcohol consumption, smoking status, ovarian status, and baseline fatty acid concentration and show the difference in the changes observed in the placebo vs. the hormone therapy groups from a linear model accounting for multiple imputation. Separate models are run for women by group (denoted as models 2b and 2c in the Statistical Analysis section).
FIGURE 1.

Summary of changes in RBC long-chain n–6 and n–3 fatty acids after 1 y of hormone therapy in all women regardless of group. Values are means ± SDs. (A) Absolute change in RBC n–3 and n–6 PUFA concentrations versus placebo. For example, the placebo-adjusted change from baseline for RBC DHA was ∼0.49%. This came from Table 3 where the placebo and hormone effects are presented by group. There, the net adjusted change in RBC DHA was 0.49% for those women in the estrogen + progestin group and 0.48% for those in the estrogen-alone group. When the data are pooled, the overall net change was 0.49%, as indicated above the DHA bar. (B) Relative (or %) change in RBC n–3 and n–6 PUFA concentrations from baseline versus placebo. For example, from Table 1, the baseline RBC DHA for all women combined was ∼4.5% (i.e., the average of 4.42% and 4.58%). The average change in DHA for the 2 groups was 0.49% as described above. Thus, the overall percentage change from baseline in DHA was 0.49%/4.5%, or 11.0%. *P < 0.01, ***P < 0.001. ARA, arachidonic acid; ADA, adrenic acid; DPA, docosapentaenoic acid; LA, linoleic acid.
Discussion
In this study we found that postmenopausal women with a mean age of ∼72 y who were in the E+P group had higher concentrations of the long-chain n–3 fatty acids in their RBC membranes than women in the E-alone group. Second, we found that, largely regardless of group status, HT (whether E or E+P) for 1 y reduced concentrations of DPA n–3 and increased concentrations of DHA and DPA n–6. The latter finding supports our hypothesis that estrogen enhances the synthesis of long-chain n–6 and n–3 PUFAs in vivo.
Focusing on women randomly assigned to HT versus placebo, we found that HT increased DHA and reduced concentrations of its immediate precursor, DPA n–3, strongly suggesting accelerated production of the former from the latter. The increase in DPA n–6 further supports this hypothesis. With regard to the extent to which E may contribute to the interindividual variability in the Omega-3 Index, in this study HT explained <2%. In this study, the Omega-3 Index increased from a mean of ∼5.2% to 5.6% in the HT group. Although the clinical implications of this relatively small increase are not clear, the adjusted decrease in risk of death from any cause associated with a 1-SD higher Omega-3 Index in the WHI Memory Study cohort was 8% (1). An HT-induced increase in the Omega-3 Index of 0.4% (which was approximately one-fourth of 1 SD) would be associated with a reduction in risk of only ∼2%. This is unlikely to have major clinical significance.
The metabolic route from DPA n–3 to DHA has been a source of some controversy. The pathway proposed first by Voss et al. (20) in 1991 included the peroxisomal B-oxidation of C24:6 n–3 to DHA (Figure 2). Park et al. (21) have more recently proposed a more direct conversion of DPA to DHA via δ4 desaturation via a gene product of fatty acid desaturase 2 (FADS2). Metherel and Bazinet (22) have suggested that both pathways may exist, but definitive support for 1 pathway versus the other (or contributions from both) is presently lacking. Nevertheless, estrogen has been shown to upregulate Fads2 (23, 24), and although this could explain the increase in DHA, an upregulation of FADS2—without concomitant changes in turnover—would be expected to also increase EPA and DPA n–3, and therefore cannot explain the lack of change in EPA concentrations or the significant lowering of DPA n–3 seen here with HT.
FIGURE 2.
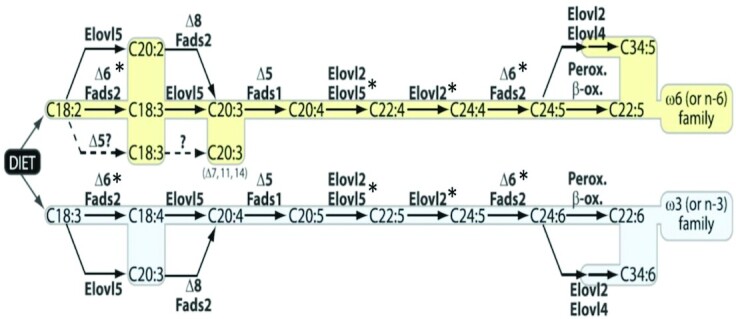
Biosynthetic pathways for the n–6 (top) and n–3 (bottom) PUFAs. *Enzymes upregulated by estrogen in various model systems. Elovl, elongation of very-long-chain fatty acid; Fads, fatty acid desaturase; Perox., peroxidation; B-ox., B-oxidation. Reproduced from reference (25) with permission.
Alternatively, the product of the ELOVL2 gene (elongation of very-long-chain fatty acids 2) shares, along with that of ELOVL5, the elongation of EPA to DPA n–3 (elongase 2 and 5, respectively). The former is the primary enzyme responsible for the elongation of DPA n–3 to C24:5n–3 (tetracosapentaenoic acid). Two genome-wide association studies (26, 27) revealed that ELOVL2 variants were associated with shifts in EPA or DPA n–3 to DHA ratios mirroring the effect of HT shown in our study. In further support, a breast cancer cell line treated with estradiol shows increased ELOVL2 gene expression compared with nontreated cells, an effect that was blocked by the estrogen receptor ɑ agonist tamoxifen (28). Furthermore, ovariectomized rats treated with estradiol had higher DHA concentrations compared with controls that corresponded to a higher expression of Elovl2 [and Elovl5 and Fads2 (23)]. DPA n–6, the n–6 analog to DHA, also increases in response to HT and further supports an upregulation of the pathway. The lack of decrease (i.e., no change) in the n–6 precursors analogous to EPA (i.e., ARA) and in particular, analogous to DPA n–3 (i.e., ADA) does not necessarily refute the role for elongase 2, but might actually be expected due to the significantly higher affinity of elongase 2 for n–3 PUFAs (29) compared with the n–6 PUFAs. The lower elongase 2 affinity for ARA and ADA suggests that any upregulation of elongation may continue to be matched by synthesis from n–6 precursors, such as LA, whereas on the n-3 side, synthesis rates of DPA n–3 cannot keep up with the rapid DPA n–3 elongation to C24:5n-3. Moreover, the affinity of elongase 2 is also much higher for 22-carbon fatty acids like DPA n–3 compared with 20-carbon fatty acids like EPA (29) and could thus explain the significant lowering of DPA n–3 relative to EPA. Although not definitive, our study combined with the aforementioned studies, suggests a mechanistic role for estrogen in the modulation of ELOVL2 expression and or elongase 2 activity which may ultimately affect blood PUFA concentrations.
Finally, the serum concentrations of estradiol and estrone achieved with the HT doses used in the WHI study are similar to natural concentrations seen in young, premenopausal women (30). Accordingly, our findings would suggest that the higher DHA concentrations seen in young women (vs. young men) (8) are likely due to an enhancement of DHA synthesis driven by higher E concentrations.
Limitations
Although the overall WHI HT study was randomized, the cohort making up this study was not truly “randomized” due to selection processes and missing data. However, there were no observed statistically significant differences in the distributions at baseline, which suggests that confounding by these variables is unlikely. This study included women aged ≥65 y. Whether the results would apply to younger women taking HT in early menopause is unknown. This study lasted only 1 y; potential adjustments to homeostasis could have occurred with prolonged treatment. As noted under Methods, the need to use multiple imputations for fatty acid concentrations in the WHI Memory Study, although not affecting point estimates, did widen CIs.
Conclusions
Randomization to HT in this cohort of postmenopausal women increased RBC concentrations of both the n–6 and n–3 22-carbon PUFAs. Together with previous studies, these findings suggest that HT activates the ELOVL2 enzyme, resulting in altered PUFA profiles. The clinical implications of such changes warrant further study.
Supplementary Material
Acknowledgments
For a list of all the investigators who have contributed to WHI science, please visit: https://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Long%20List.pdf.
The authors’ responsibilities were as follows––WSH and JGR: designed the research project; WSH: analyzed the RBC samples; NLT: collected data and performed the statistical analysis; WSH, NLT, AHM, and JEM: wrote the manuscript; WSH: had primary responsibility for final content; and all authors: read and approved the final manuscript.
WSH holds stock in OmegaQuant Analytics, LLC, a commercial laboratory that offers RBC fatty analysis. Biostatistical work was performed by NLT and was funded by OmegaQuant Analytics. All the other authors report no conflicts of interest.
Notes
This study was supported in part under a contract with the National Heart, Lung, and Blood Institute (BAA 19).
Supplemental Tables 1–5 and Supplemental Figure 1 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/ajcn/.
WSH is an AJCN Editorial Board Member.
Abbreviations used: ALA, ɑ-linolenic acid; DPA, docosapentaenoic acid; E, conjugated equine estrogens; ELOVL, elongation of very-long-chain fatty acid; FADS2, fatty acid desaturase 2; HT, hormone therapy; LA, linoleic acid; P, medroxyprogesterone acetate; WHI, Women's Health Initiative.
Contributor Information
William S Harris, Department of Internal Medicine, Sanford School of Medicine, University of South Dakota, Sioux Falls, SD, USA; Fatty Acid Research Institute, Sioux Falls, SD, USA.
Nathan L Tintle, Fatty Acid Research Institute, Sioux Falls, SD, USA; Department of Mathematics and Statistics, Dordt College, Sioux Center, IA, USA.
JoAnn E Manson, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA, USA; Department of Epidemiology, Harvard TH Chan School of Public Health, Boston, MA, USA.
Adam H Metherel, Department of Nutritional Sciences, Faculty of Medicine, University of Toronto, Toronto, Canada.
Jennifer G Robinson, Department of Epidemiology, College of Public Health, Iowa City, IA, USA; Department of Internal Medicine, College of Medicine, University of Iowa, Iowa City, IA, USA.
Data Availability
Data described in the manuscript, code book, and analytic code will be made available upon request pending application and approval.
References
- 1. Harris WS, Luo J, Pottala JV, Espeland MA, Margolis KL, Manson JE, Wang L, Brasky TM, Robinson JG. Red blood cell polyunsaturated fatty acids and mortality in the Women's Health Initiative Memory Study. J Clin Lipidol. 2017;11(1):250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harris WS, Luo J, Pottala JV, Margolis KL, Espeland MA, Robinson JG. Red blood cell fatty acids and incident diabetes mellitus in the women's health initiative memory study. PLoS One. 2016;11(2):e0147894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ammann EM, Pottala JV, Harris WS, Espeland MA, Wallace R, Denburg NL, Carnahan RM, Robinson JG. Omega-3 fatty acids and domain-specific cognitive aging: secondary analyses of data from WHISCA. Neurology. 2013;81(17):1484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Persons JE, Robinson JG, Ammann EM, Coryell WH, Espeland MA, Harris WS, Manson JE, Fiedorowicz JG. Omega-3 fatty acid biomarkers and subsequent depressive symptoms. Int J Geriatr Psychiatry. 2014;29(7):747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jackson KH, Polreis JM, Tintle NL, Kris-Etherton PM, Harris WS. Association of reported fish intake and supplementation status with the omega-3 index. Prostaglandins Leukotrienes Essent Fatty Acids. 2019;142:4–10. [DOI] [PubMed] [Google Scholar]
- 6. Harris W, von Schacky C. The Omega-3 Index: a new risk factor for death from coronary heart disease. Prev Med. 2004;39:212–20. [DOI] [PubMed] [Google Scholar]
- 7. Harris WS, Pottala JV, Lacey SM, Vasan RS, Larson MG, Robins SJ. Clinical correlates and heritability of erythrocyte eicosapentaenoic and docosahexaenoic acid content in the Framingham Heart Study. Atherosclerosis. 2012;225:425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harris WS, Pottala JV, Varvel SA, Borowski JJ, Ward JN, McConnell JP. Erythrocyte omega-3 fatty acids increase and linoleic acid decreases with age: observations from 160,000 patients. Prostaglandins Leukotrienes Essent Fatty Acids. 2013;88(4):257–63. [DOI] [PubMed] [Google Scholar]
- 9. de Groot RHM, Emmett R, Meyer BJ. Non-dietary factors associated with n-3 long-chain PUFA levels in humans—a systematic literature review. Br J Nutr. 2019;121(7):793–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lohner S, Fekete K, Marosvolgyi T, Decsi T. Gender differences in the long-chain polyunsaturated fatty acid status: systematic review of 51 publications. Ann Nutr Metab. 2013;62(2):98–112. [DOI] [PubMed] [Google Scholar]
- 11. Giltay EJ, Gooren LJG, Toorians A, Katan MB, Zock PL. Docosahexaenoic acid concentrations are higher in women than in men because of estrogenic effects. Am J Clin Nutr. 2004;80(5):1167–74. [DOI] [PubMed] [Google Scholar]
- 12. Burdge G, Wootton S. Conversion of alpha-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br J Nutr. 2002;88:411–20. [DOI] [PubMed] [Google Scholar]
- 13. Burdge G, Jones A, Wootton S. Eicosapentaenoic and docosapentaenoic acids are the principal products of alpha-linolenic acid metabolism in young men. Br J Nutr. 2002;88:355–63. [DOI] [PubMed] [Google Scholar]
- 14. Jin Y, Kim TH, Park Y. Association between erythrocyte levels of n-3 polyunsaturated fatty acids and depression in postmenopausal women using or not using hormone therapy. Menopause. 2016;23(9):1012–18. [DOI] [PubMed] [Google Scholar]
- 15. Shumaker SA, Reboussin BA, Espeland MA, Rapp SR, McBee WL, Dailey M, Bowen D, Terrell T, Jones BN. The Women's Health Initiative Memory Study (WHIMS): a trial of the effect of estrogen therapy in preventing and slowing the progression of dementia. Control Clin Trials. 1998;19(6):604–21. [DOI] [PubMed] [Google Scholar]
- 16. Manson JE, Chlebowski RT, Stefanick ML, Aragaki AK, Rossouw JE, Prentice RL, Anderson G, Howard BV, Thomson CA, LaCroix AZet al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women's Health Initiative randomized trials. JAMA. 2013;310(13):1353–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pottala JV, Espeland MA, Polreis J, Robinson J, Harris WS. Correcting the effects of -20 degrees C storage and aliquot size on erythrocyte fatty acid content in the Women's Health Initiative. Lipids. 2012;47:835–46. [DOI] [PubMed] [Google Scholar]
- 18. Rubin DB. Multiple imputation after 18+ years. J Am Statist Assoc. 1996;91:473–89. [Google Scholar]
- 19. R Core Team . Available from: https://www.R-project.org/ (accessed 27 May 2020).
- 20. Voss A, Reinhart M, Sankarappa S, Sprecher H. The metabolism of 7,10,13,16,19-docosapentaenoic acid to 4,7,10,13,16,19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J Biol Chem. 1991;266(30):19995–20000. [PubMed] [Google Scholar]
- 21. Park HG, Park WJ, Kothapalli KS, Brenna JT. The fatty acid desaturase 2 (FADS2) gene product catalyzes delta4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J. 2015;29(9):3911–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Metherel AH, Bazinet RP. Updates to the n-3 polyunsaturated fatty acid biosynthesis pathway: DHA synthesis rates, tetracosahexaenoic acid and (minimal) retroconversion. Prog Lipid Res. 2019;76:101008. [DOI] [PubMed] [Google Scholar]
- 23. Kim D, Choi JE, Park Y. Low-linoleic acid diet and oestrogen enhance the conversion of alpha-linolenic acid into DHA through modification of conversion enzymes and transcription factors. Br J Nutr. 2019;121(2):137–45. [DOI] [PubMed] [Google Scholar]
- 24. Kitson AP, Marks KA, Shaw B, Mutch DM, Stark KD. Treatment of ovariectomized rats with 17beta-estradiol increases hepatic delta-6 desaturase enzyme expression and docosahexaenoic acid levels in hepatic and plasma phospholipids. Prostaglandins Leukotrienes Essent Fatty Acids. 2013;89(2-3):81–8. [DOI] [PubMed] [Google Scholar]
- 25. Guillou H, Zadravec D, Martin PG, Jacobsson A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: insights from transgenic mice. Prog Lipid Res. 2010;49(2):186–99. [DOI] [PubMed] [Google Scholar]
- 26. Tanaka T, Shen J, Abecasis GR, Kisialiou A, Ordovas JM, Guralnik JM, Singleton A, Bandinelli S, Cherubini A, Arnett Det al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI study. PLos Genet. 2009;5(1):e1000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalsbeek A, Veenstra J, Westra J, Disselkoen C, Koch K, McKenzie KA, O'Bott J, Vander Woude J, Fischer K, Shearer GCet al. A genome-wide association study of red-blood cell fatty acids and ratios incorporating dietary covariates: Framingham Heart Study Offspring Cohort. PLoS One. 2018;13(4):e0194882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez-Bengtsson A, Asadi A, Gao H, Dahlman-Wright K, Jacobsson A. Estrogen enhances the expression of the polyunsaturated fatty acid elongase Elovl2 via ERalpha in breast cancer cells. PLoS One. 2016;11(10):e0164241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gregory MK, Gibson RA, Cook-Johnson RJ, Cleland LG, James MJ. Elongase reactions as control points in long-chain polyunsaturated fatty acid synthesis. PLoS One. 2011;6(12):e29662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harman SM, Black DM, Naftolin F, Brinton EA, Budoff MJ, Cedars MI, Hopkins PN, Lobo RA, Manson JE, Merriam GRet al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women: a randomized trial. Ann Intern Med. 2014;161(4):249–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data described in the manuscript, code book, and analytic code will be made available upon request pending application and approval.