

Abstract
Objectives
Cutaneous wound healing is one of the major medical problems worldwide. Epigenetic modifiers have been identified as important players in skin development, homeostasis and wound repair. SET domain–containing 2 (SETD2) is the only known histone H3K36 tri‐methylase; however, its role in skin wound healing remains unclear.
Materials and Methods
To elucidate the biological role of SETD2 in wound healing, conditional gene targeting was used to generate epidermis‐specific Setd2‐deficient mice. Wound‐healing experiments were performed on the backs of mice, and injured skin tissues were collected and analysed by haematoxylin and eosin (H&E) and immunohistochemical staining. In vitro, CCK8 and scratch wound‐healing assays were performed on Setd2‐knockdown and Setd2‐overexpression human immortalized keratinocyte cell line (HaCaT). In addition, RNA‐seq and H3K36me3 ChIP‐seq analyses were performed to identify the dysregulated genes modulated by SETD2. Finally, the results were validated in functional rescue experiments using AKT and mTOR inhibitors (MK2206 and rapamycin).
Results
Epidermis‐specific Setd2‐deficient mice were successfully established, and SETD2 deficiency resulted in accelerated re‐epithelialization during cutaneous wound healing by promoting keratinocyte proliferation and migration. Furthermore, the loss of SETD2 enhanced the scratch closure and proliferation of keratinocytes in vitro. Mechanistically, the deletion of Setd2 resulted in the activation of AKT/mTOR signalling pathway, while the pharmacological inhibition of AKT and mTOR with MK2206 and rapamycin, respectively, delayed wound closure.
Conclusions
Our results showed that SETD2 loss promoted cutaneous wound healing via the activation of AKT/mTOR signalling.
Keywords: AKT, cutaneous wound healing, histone modification, keratinocytes, mTOR Pathway, SETD2
The epidermis‐specific Setd2‐deficient mice showed accelerated re‐epithelialization during cutaneous wound healing by promoting keratinocytes proliferation and migration. Mechanistically, deletion of Setd2 resulted in the activation the AKT/mTOR signalling pathway and pharmacological inhibitions of AKT and mTOR with MK2206 and rapamycin delayed wound closure, respectively.
1. INTRODUCTION
Mammalian skin is composed of the inner and outer epidermis, separated by a basement membrane. 1 The skin is a natural physical and immune protective barrier that prevents not only the penetration of harmful microorganisms, but also dehydration. 2 , 3 Once the skin barrier is damaged, wound‐healing process begins immediately. Cutaneous wound healing is a complex and dynamic process that involves three phases: inflammation, re‐epithelialization and tissue remodelling. 4 , 5 Even though these three phases occur sequentially, they overlap, and the extracellular matrix (ECM), soluble growth factors and multiple cell types, such as immune cells, fibroblasts and keratinocytes (KCs), participate in these phases. 6 Re‐epithelialization is the formation of new epithelium and covering of the wound surface, which requires the migration and proliferation of KCs. 7
Epigenetic regulation controls the transcriptional activation or silencing of genes without changing the DNA sequence, governing the phenotypic plasticity of individual cells, organs or a whole organism. 8 In the skin, epigenetic regulation mechanisms play an important role in its development, homeostasis and wound repair. 9 Histone modification is a type of epigenetic regulation that affects the transcriptional activity of genes and is involved in normal physiological processes and diseases. 10 , 11 Several studies have reported that histone modification and related enzymes play an essential role in skin wound healing. For example, H3K27me3 demethylase JMJD3 interacts with NF‐κB, resulting in increased expression of inflammatory, matrix metalloproteinase and growth factor genes, while the inactivation of JMJD3 leads to delayed wound healing. 1 , 12 The hair follicle cells of mice lacking EZH1 and EZH2 histone H3K27 tri‐methylases have defective cell proliferation and wound healing, even though epidermis continues to hyperproliferate. 13 Furthermore, the hypomethylation of histone H3 K4/9/27me3 is beneficial for the differentiation and growth of hair follicles (HFs) and promotes wound healing. 14
SETD2 (SET domain‐containing protein 2) was first identified as the protein associated with Huntington's disease (HD). 15 Currently, SETD2 is the only known H3K36 tri‐methylase; it interacts with RNA polymerase II to mediate transcriptional extension, resulting in changes in gene transcription levels. 16 , 17 As a tumour suppressor, SETD2 plays an important role in gene transcription regulation, DNA damage repair and alternative splicing. 18 , 19 , 20 , 21 , 22 Recently, SETD2 has been extensively studied in various biological processes and diseases. Loss of SETD2 function has been investigated in several human tumours, including GI stromal tumours, renal cell carcinoma, pancreatic ductal adenocarcinoma (PDAC), prostate cancer, breast cancer, leukaemia and high‐grade gliomas. 23 , 24 , 25 , 26 , 27 , 28 , 29 Moreover, the role of SETD2 in hematopoietic stem cell self‐renewal, sperm development, bone marrow mesenchymal stem cell differentiation, V(D)J recombination, maternal epigenome and embryonic development has been examined. 30 , 31 , 32 , 33 , 34 In addition, four non‐histone substrates of SETD2: α‐tubulin, STAT1, EZH2 and actin were discovered. 23 , 35 , 36 , 37 However, the role of SETD2, an important histone‐modifying enzyme, in skin wound healing is still not understood.
In this study, to investigate the role of SETD2 in skin wound healing, we generated epidermis‐specific Setd2‐knockout mice and showed that SETD2 deficiency promoted cutaneous wound healing through the activation of AKT/mTOR pathway.
2. MATERIALS AND METHODS
2.1. Mice
Setd2fl/fl mice were generated by Shanghai Biomodel Organism Co. using conventional homologous recombination in embryonic stem (ES) cells as previously described. 19 , 29 Tg‐CK5CreERT2; R26R‐CAG‐lsl‐Tomato mice were purchased from the Jackson Laboratory. Setd2‐KO mice were generated by crossing Setd2‐floxed mice with K5CreERT2 mice, and tamoxifen was intraperitoneally injected at 100 mg kg−1 body weight. All mice were maintained in a specific‐pathogen‐free (SPF) facility, and all experimental procedures were approved by the Animal Ethics Committee of School of Biomedical Engineering & Med‐X Research Institute, Shanghai Jiao Tong University. Primers used for genotyping are listed in Table S1.
2.2. In vivo wound‐healing experiments
Mice (8 to 10‐week‐old littermates) were anaesthetized by intraperitoneal injection of tribromoethanol, and their backs were shaved. Four 4‐mm full‐thickness cutaneous biopsy punch wounds were made on the back of each mouse. The entry wounds were photographed on days 0, 1, 3, 5, 7 and 9. The wound areas were measured using ImageJ software. Mice were euthanized 1, 3 and 5 days after wounding by carbon dioxide inhalation, the wounds were excised, fixed overnight at 4°C with 4% paraformaldehyde and then embedded in paraffin.
2.3. Isolation of primary mouse keratinocytes
Primary mouse keratinocytes were isolated from the skin as previously described. 38 , 39 Briefly, the skin of 10‐week‐old mice was treated with dispase II (Gibco,17105‐041) overnight at 4°C. The epidermis was separated and digested for 10 min at 37°C with 0.25% trypsin/EDTA (Gibco, 25200‐072) and strained through a 70‐μm filter. The supernatants were centrifuged, and cells were collected.
2.4. Cell culture
Human immortalized HaCaT keratinocyte cell line 40 was cultured in DMEM supplemented with 10% foetal bovine serum (FBS) and 1% penicillin‐streptomycin at 37°C in a humidified 5% CO2 atmosphere.
2.5. Histology, H&E staining and immunohistochemistry (IHC)
Tissues were fixed in 4% paraformaldehyde overnight at 4°C, dehydrated and embedded in paraffin. Sections (5 μm) were cut and stained with haematoxylin and eosin (H&E). For IHC staining, sections were deparaffinized, rehydrated, subjected to antigen retrieval in citrate buffer, and endogenous peroxidases were quenched with 3% H2O2. Blocking was performed with 5% BSA for 1 hour at room temperature. Next, the samples were incubated with primary antibodies for 12‐16 hours at 4°C. The primary antibodies used were anti‐Ki67 (Abcam, ab15580) and anti‐K5 (Abcam, ab52635). After three washes in PBS, the sections were incubated with an HRP‐conjugated secondary antibody for 1 hour at room temperature and then counterstained with haematoxylin. Images were acquired using a Leica microscope, and staining intensities were calculated using ImageJ software. The antibodies used for staining are listed in Table S2.
2.6. Immunofluorescence
Skin samples were fixed in 4% paraformaldehyde for 30 minutes at 4°C, transferred to 30% sucrose overnight and then embedded in OCT. Sections (7 μm) were cut, permeabilized with Triton X‐100 and blocked with 5% BSA. Next, the sections were incubated with primary antibodies (anti‐SETD2 (LS‐C332416), anti‐p‐AKT (CST, #4060) and anti‐p‐mTOR (CST, #5536)) at 4°C for 12‐16 hours, followed by the incubation with the secondary antibodies at 37°C for 1 hour. Nuclei were counterstained with DAPI. All antibodies used for immunofluorescence are listed in Table S2.
2.7. Scratch wound‐healing assay
HaCaT cells were plated at 1 × 105 cells/well in triplicate in 6‐well plates. Scratch assays were performed using completely confluent HaCaT cells. Scratches were generated using a 200 μL plastic pipette tip. Suspended cells were washed off, and the remaining cells were cultured in the medium without FBS to inhibit cell proliferation. Images were acquired immediately after scratches were generated, as well as 12 hours and 24 hours after scratching. The plates were washed with PBS before imaging to reduce the number of dead cells in the field of view.
2.8. RNA isolation and quantitative RT‐PCR
Total RNA was extracted from cultured cells or tissues using an RNA extraction kit (BioTeke) following the manufacturer's protocol. RNA was reverse transcribed using an RT reagent kit (Takara). The cDNA was subsequently subjected to TB Green‐based real‐time PCR analysis. GAPDH was used to normalize the results, and the data were presented as the mean ± SD. The P‐value was calculated using Student's t test. The primers used for the qPCR analysis are listed in Table S1.
2.9. Western blot analysis and antibodies
Cell and tissue samples were lysed in RIPA buffer (Beyotime, P0013B) supplemented with protease and phosphatase inhibitors (MCE). Protein concentrations were measured using the BCA Protein Assay (Thermo Fisher Scientific). Proteins were separated using 6% and 10% SDS‐PAGE gels and then transferred to polyvinylidene fluoride (PVDF) membranes or nitrocellulose membranes (Millipore). The membranes were blocked with 5% skim milk in TBST for 1.5 hours at room temperature and subsequently incubated with primary antibodies overnight at 4°C, followed by incubation with secondary antibodies for 1 hours. The primary antibodies used in this study were as follows: anti‐SETD2 (LS‐C332416), anti‐H3K36me3 (Abcam, ab9050), anti‐H3 (Abcam, ab10799), anti‐AKT (CST, #2920), anti‐mTOR (CST, #2983), anti‐p‐AKT (CST, #4060), anti‐p‐mTOR (CST, #5536) and anti‐GAPDH (CST, #5174). The antibodies used for Western blotting are listed in Table S2.
2.10. Plasmids, transfections and lentiviruses
Human Setd2 cDNA was generated by polymerase chain reaction and cloned into the pCMV6‐Entry vector with an HA‐tag. HaCaT cells were transfected with EZ transfection reagent according to the manufacturer's instructions. Cell lines transiently expressing exogenous Setd2 were obtained 24‐48 hours after transfection. shRNA sequences for Setd2 (sh‐Setd2) were cloned into the lentiviral vector pLV‐H1‐SGIPZ. Lentiviral packaging plasmids PSPAX2 and pMD2.G were co‐transfected with the sh‐Setd2 plasmid into 293T cells for virus production. Virus‐infected cells were selected using 1.5 µg/mL puromycin for 2 weeks to generate stable transfections. sh‐Setd2 sequences were as follows: 5'‐AAGCAAAGAAGTATTCAGAAATAGTGAAGCCACAGA.
TGTATTTCTGAATACTTCTTTGCTT‐3'.
2.11. AKT/mTOR inhibition experiments
Setd2‐knockdown HaCaT cells were treated with MK2206 (MCE, HY10358) and rapamycin (MCE, HY‐10219) at concentrations of 10 μmol/L and 50 nmol/L, respectively. In vivo inhibition of AKT and mTOR signalling by MK2206 and rapamycin, respectively, was performed as previously described. 41 , 42 , 43 Rapamycin was injected intraperitoneally (i.p.) at 1 mg/kg every other day. MK2206 was administered via oral gavage at 100 mg/kg per day, three times a week. Rapamycin or MK2206 treatments were initiated 7 days before the wounding experiment.
2.12. RNA‐seq and analyses
Skin tissue mRNA was obtained from 12‐week‐old Setd2‐KO and Setd2fl/fl mice. Differential gene expression was analysed using DESeq2 package. The list of significance was determined by setting a false discovery rate (FDR) threshold at a <0.05, and |log2FC| > 0.585. All differentially expressed genes were subsequently analysed using GO and pathway analyses.
2.13. ChIP‐Seq and analyses
Mouse primary keratinocytes (>1 × 107) were crosslinked with 1% formaldehyde for 5 minutes at room temperature and quenched with 0.125 mol/L glycine. The fragmented chromatin fragments were pre‐cleared and then immunoprecipitated with protein A + G magnetic beads coupled to the anti‐H3K36me3 (ab9050) antibody. After reverse crosslinking, ChIP and input DNA fragments were end‐repaired and A‐tailed using the NEBNext End Repair/dA‐Tailing Module (E7442, NEB) followed by adaptor ligation with the NEBNext Ultra Ligation Module (E7445, NEB). The DNA libraries were amplified for 15 cycles and sequenced using Illumina NextSeq 500 with single‐end 1 × 75 as the sequencing mode. Raw reads were filtered to obtain high‐quality clean reads by removing sequencing adapters, short reads (length <50 bp) and low‐quality reads using Cutadapt (v1.9.1) and Trimmomatic23 (v0.35). Next, FastQC was used to ensure high reads quality. The clean reads were mapped to the mouse genome (assembly GRCm38) using the Bowtie2 (v2.2.6) software. Peak detection was performed using the MACS (v2.1.1) peak finding algorithm with .01 set as P‐value cut‐off. Annotation of peak sites to gene features was performed using the ChIPseeker R package.
2.14. Statistical analysis
All experiments were repeated at least three times. Unless otherwise indicated, data were presented as the mean ± SD and analysed for statistical significance by two‐way ANOVA or Student's t test using GraphPad Prism 8.0.2 software. Statistical significance was set at P < .05. *P < .05, **P < .01, ***P < .001 and ****P < .0001.
3. RESULTS
3.1. Generation of epidermis‐specific Setd2‐deficient mice
To determine whether SETD2 plays a role in re‐epithelialization after injury, we analysed microarray data obtained from the NCBI GEO Datasets GSE30355. 44 The results showed a decrease in Setd2 in injured KCs compared to normal KCs (Figure 1A). RT‐qPCR analysis also showed decreased expression of Setd2 in the full‐thickness wound tissues of wild‐type (WT) mice after injury (Figure 1B). These results suggested that SETD2 played a role in skin wound repair. To further elucidate the role of SETD2 in skin wound healing, we crossed Setd2‐flox (Setd2fl/fl) mice with K5CreERT2 mice to obtain the epidermis‐specific Setd2 knockout (Setd2‐KO) mice (Figure 1C). 40 , 44 In this system, Cre expression is driven by the K5 promoter, which directs gene expression in the basal cells of the skin, cells believed to be the stem cells of the skin. The immunofluorescence results showed that keratin 5 was mainly expressed in the epidermis and hair follicles; SETD2‐positive cells were also observed in the epidermis and hair follicles (Figure 1D). Furthermore, there was colocalization between SETD2 and keratin 5. Therefore, in our model system, Setd2 was deleted in K5‐positive cells. We confirmed that the expression levels of SETD2 and H3K36me3 were significantly decreased in Setd2‐KO mice by RT‐qPCR, Western blotting and immunofluorescent (IF) staining (Figure 1E‐G). Setd2‐KO mice had no visible skin abnormalities during the growth process until 10 months of age compared to control (Setd2fl/fl) mice (data not shown).
FIGURE 1.
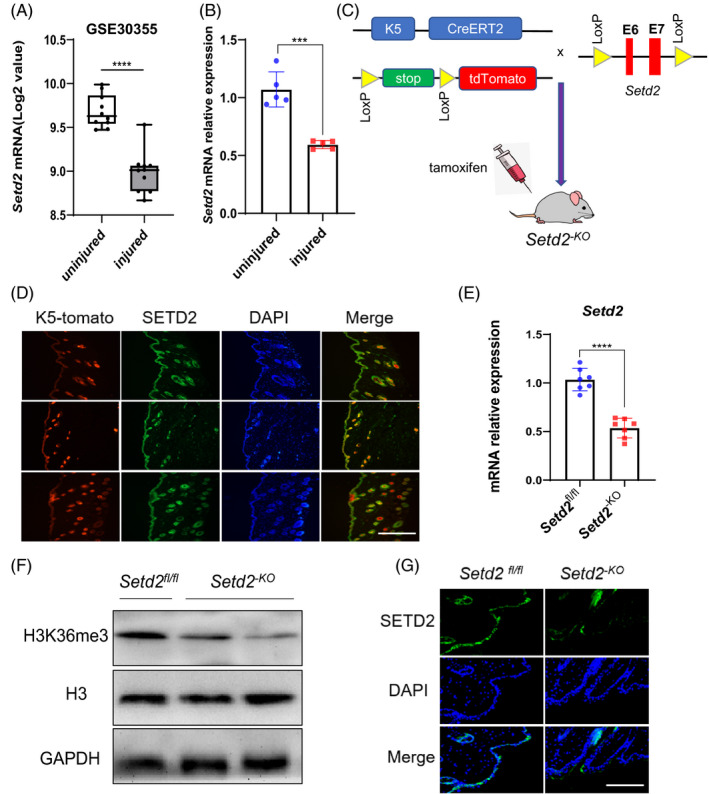
The expression of Setd2 in KCs and the generation of epidermis‐specific Setd2‐deficient mice. A, Box plot of Setd2 expression levels in normal and injured KCs (using dataset GSE30355; n = 10). B, RT‐qPCR analysis of the Setd2 mRNA expression in injured and uninjured skin tissues of wild‐type mice (n = 5). C, Schematic representation of generating the Setd2‐KO mouse model. D, Colocalization of SETD2 (green) and CK5‐Tomato (red) was visualized on frozen skin sections from CK5CreERT2; Tomato adult mice induced by tamoxifen. The nuclei were stained with DAPI (blue). Scale bars: 200 μm. E, RT‐qPCR analysis of Setd2 mRNA in control and Setd2‐KO mice (n = 7). F, Western blot analysis of H3K36me3 protein levels. Representative blots are shown. G, Representative immunofluorescence images showing protein expression of SETD2 in Setd2‐KO and Setd2fl/fl mice. Scale bars: 200 μm. Data are presented as the mean ± SD; statistical significance was determined using a two‐tailed Student's t test, ****P < .0001
3.2. Setd2 deficiency promotes cutaneous wound healing and thickens the wound epithelium in mice
Next, we investigated the effect of SETD2 deletion on cutaneous wound healing. Skin punch biopsies were obtained from Setd2‐KO and age‐matched Setd2fl/fl (control) mice to assess the wound‐healing process every 2 days for up to 9 days. Three days after wounding, the wound areas in Setd2‐KO mice were significantly reduced compared to Setd2fl/fl mice as measured by image analysis, suggesting accelerated wound healing in Setd2‐KO mice (Figure 2A). Furthermore, the differences in the wound area between Setd2‐KO and Setd2fl/fl mice were statistically significant on days 3 and 5 after wounding (Figure 2B). Increased thickness of the wound epithelium (WE) contributed to the re‐epithelialization process. The H&E staining of injured tissues showed that the thickness of WE was significantly increased in Setd2‐KO mice compared to control mice (Figure 2C,D). These results indicate that the absence of Setd2 accelerates skin wound healing in mice.
FIGURE 2.
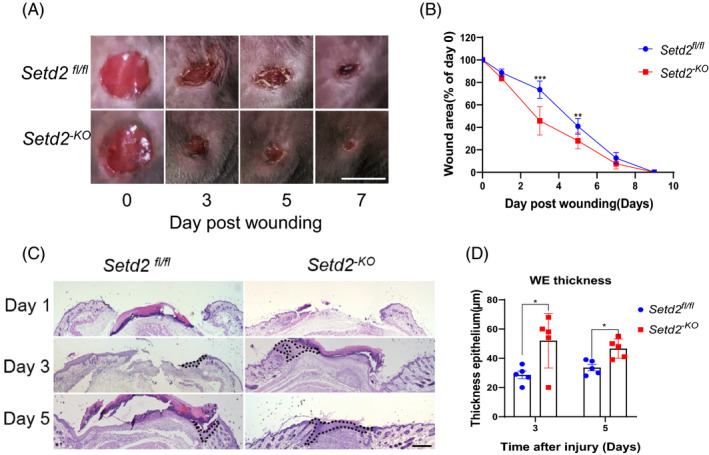
Wound closure is accelerated in the skin of Setd2‐KO mice. A, Appearance of wound areas of Setd2‐KO and Setd2fl/fl mice on days 0, 3, 5 and 7 post‐wounding. Scale bars: 4 mm. B, Quantification of the wound areas. Data are presented as percent wound area at each time point relative to the original wound area in Setd2‐KO and Setd2fl/fl mice, n = 8 wounds/group. The analysis was performed using ImageJ software. C, H&E staining of wounds in Setd2fl/fl and Setd2‐KO mice at the specified time points after injury. The wound epithelium is depicted by a dotted line. Scale bars: 500 μm. D, Thickness of the wound epidermis in Setd2fl/fl and Setd2‐KO mice, n = 5 wounds/groups. Quantification of the thickness was performed using ImageJ software. Data are presented as the mean ± SD; statistical significance was determined using a two‐tailed Student's t test, *P < .05, **P < .01 and ***P < .001
3.3. Accelerated wound closure in Setd2‐KO mice is associated with enhanced keratinocytes proliferation and migration
Since KC proliferation and migration are involved in the re‐epithelialization process, the process essential for wound healing, 45 , 46 , 47 and SETD2 is primarily expressed in skin KCs, we further hypothesized that accelerated wound healing is caused by the increased proliferation and migration of these cells. To test this hypothesis, we evaluated the differences in the migration rate of KCs from Setd2‐KO and Setd2fl/fl mice by K5 immunostaining. KCs migrated from the edge to the centre of the wound in both Setd2fl/fl and Setd2‐KO mice. However, re‐epithelialization occurred significantly faster in Setd2‐KO mice compared to control mice (Figure 3A). As shown in Figure 3B, the distance between the migration edge in Setd2‐KO KCs was significantly shorter than in controls. Next, to determine whether accelerated wound closure was due to increased cell proliferation, we used Ki67 (a marker of proliferation) immunohistochemical staining and counted the number of Ki67‐positive KCs in wound areas on different days. The results demonstrated that cell proliferation was significantly enhanced on days 3 and 5 after injury (Figure 3C,D). These results confirmed that Setd2 deletion accelerated the proliferation and migration of KCs in vivo.
FIGURE 3.
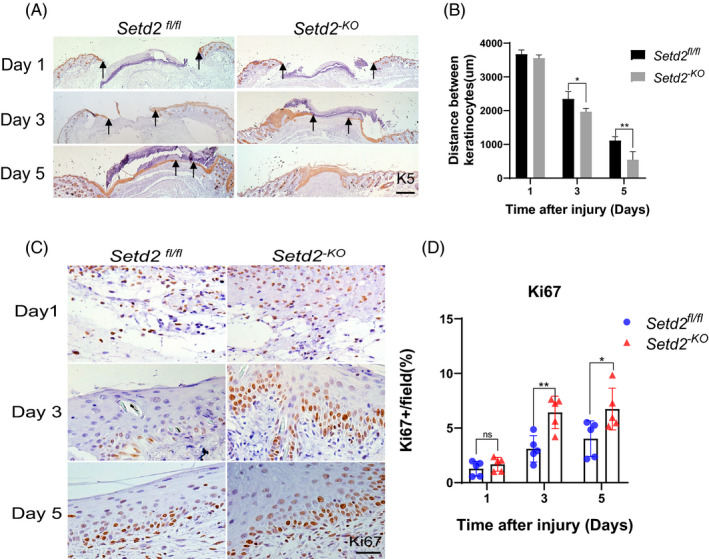
Setd2 deficiency leads to accelerated KC proliferation and migration during wound healing A, K5 staining (brown) of wounds in Setd2fl/fl and Setd2‐KO mice at the specified time points after injury. Arrows indicate the edges of migrated KCs. Scale bars: 500 μm. B, Quantification of the distance between the KCs in the wounds of Setd2fl/fl and Setd2‐KO mice; n = 5 wounds/group. C, Immunohistochemical staining using anti‐Ki67 antibody performed on Setd2fl/fl and Setd2‐KO wound sections collected on days 1, 3 and 5. Representative images. Scale bars: 50 μm. D, Quantification of Ki67‐positive KCs; data are presented as the percentage of total KC number; n = 5 wounds/group. Data are presented as the mean ± SD; and statistical significance was determined using a two‐tailed Student's t test; n.s, no significance, *P < .05, **P < .01 and ***P < .001
3.4. Loss of Setd2 promotes scratch closure and proliferation of KCs in vitro
To further analyse the effects of SETD2 loss on KC proliferation and migration in vitro, we performed CCK8 and scratch wound‐healing assays on control and Setd2‐knockdown HaCaT cells. First, we confirmed that the expression level of SETD2 was significantly decreased after transfecting HaCaT cells with the sh‐Setd2 plasmid (Figure 4A). As shown by CCK8 assays, Setd2‐knockdown promoted cell proliferation (Figure 4B). Scratch assays demonstrated that Setd2‐knockdown in HaCaT cells resulted in increased cell migration (Figure 4C,D). Closure of the wound gaps was almost completed by 12 hours and fully achieved by 24 hours in Setd2‐knockdown HaCaT cells. This process was significantly delayed in control cells. As expected, Setd2 overexpression inhibited the proliferation and migration of HaCaT cells (Figure 4E‐H). Therefore, our results further confirmed that Setd2‐knockdown accelerated KC proliferation and migration in vitro.
FIGURE 4.
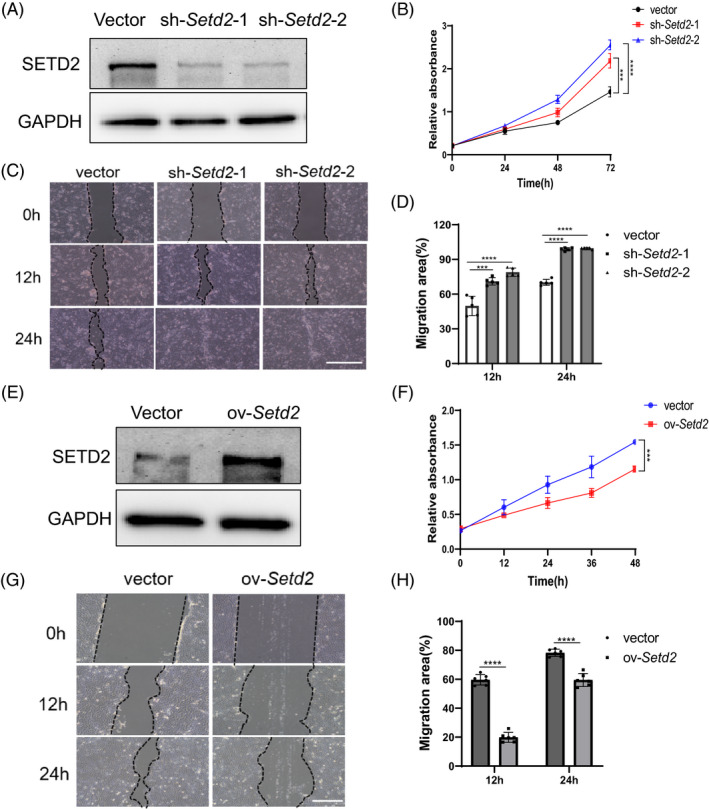
Setd2 expression levels affect the proliferation and migration of HaCaT cells in vitro. A, Western blot analysis of SETD2 expression in the Setd2‐knockdown HaCaT cells transfected either with empty vector or sh‐Setd2 plasmids. Representative blots are shown. B, CCK8 assay of HaCaT cells transfected either with empty vector or sh‐Setd2 plasmids. C, Scratch assay of HaCaT cells transfected either with empty vector or sh‐Setd2 plasmids. The gap closure was significantly accelerated at 12 h and completely closed at 24 h in Setd2‐knockdown KCs. Scale bars: 200 μm. D, Quantification of migration area at 12 h and 24 h; n = 5/group. E, Western blot analysis of SETD2 expression in the Setd2‐overexpressing HaCaT cells. Representative blots are shown. F, CCK8 assay of HaCaT cells transfected either with empty vector or HA‐Setd2 plasmids. G, Cell migration ability of Setd2‐overexpressing cells compared to the control cells. Scale bars: 100 μm. H, Quantification of migration area; n = 6/group. Data are presented as the mean ± SD; statistical significance was determined using a two‐tailed Student's t test and two‐way ANOVA; n.s, no significance, ***P < .001 and ****P < .0001.
3.5. Setd2‐deficient skin displays hyperactive AKT/mTOR Signalling
To understand how SETD2 loss enhances wound healing, we carried out a transcriptomic analysis using skin tissues derived from Setd2‐KO and control mice at the unwound (day 0) time point to identify early driving events. Clustering analysis showed that the global transcriptome changed dramatically in Setd2‐KO mice compared to control mice (Figure 5A). Gene ontology (GO) term analysis indicated that SETD2 loss significantly enriched the genes associated with wound healing, including cell differentiation, cell adhesion, inflammatory response, plasma membrane repair and angiogenesis (Figure 5B,C). The GO data were consistent with our phenotypic observations. To elucidate the SETD2‐mediated signalling pathway, we performed gene set enrichment analysis (GSEA). Our results showed that genes related to the PI3K/AKT pathway were significantly enriched in Setd2‐KO mice (Figure 5D). The mRNA expression levels of these genes were validated using RT‐qPCR (Figure 5E). Furthermore, IF staining showed that the protein expression levels of p‐AKT and p‐mTOR were enhanced in WE areas on days 1 and 3 after injury in Setd2‐KO mice (Figure 5F). These data suggested that SETD2 deficiency resulted in the activation of AKT/mTOR signalling.
FIGURE 5.
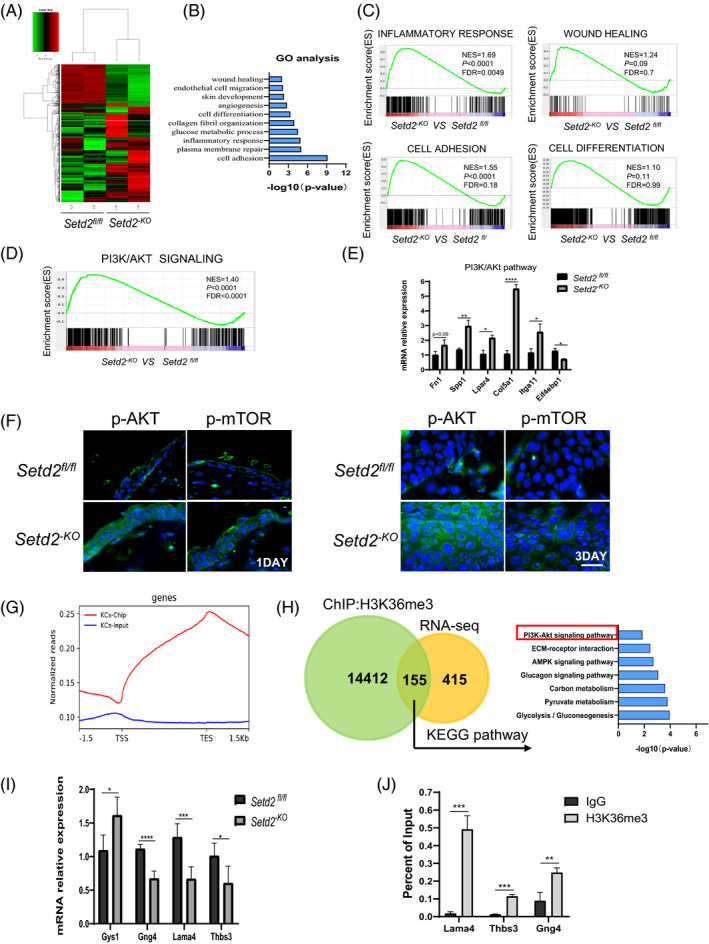
Setd2‐deficient skin displays hyperactive AKT/mTOR Signalling A, Heat map of differentially expressed genes in age‐matched Setd2fl/fl and Setd2‐KO mice. B, GO term analysis demonstrating upregulated biological processes in age‐matched Setd2fl/fl and Setd2‐KO mice. C, GSEA enrichment plots of differentially expressed genes associated with SETD2 deletion. D, GSEA enrichment plots of differentially expressed genes that belong to PI3K/AKT signalling pathway and associated with SETD2 deletion. E, RT‐qPCR analysis of genes related to PI3K/AKT/mTOR signalling. F, Immunofluorescent staining of Setd2fl/fl and Setd2‐KO wound sections using anti‐p‐AKT and anti‐p‐mTOR (green) antibodies on days 1 and 3 post‐wounding. Representative images are showing the wound epithelium areas. Nuclei are stained with DAPI (blue). Scale bars: 50 μm. G, Normalized read density of H3K36me3 ChIP‐seq and input signals of primary KCs from Setd2fl/fl mice, from 1.5 kb upstream of the TSS to 1.5 kb downstream of the TES. H, Venn diagram illustrating H3K36me3 peaks in Setd2fl/fl primary KCs and the overlap with total differential expression genes determined by RNA sequencing. Right panel shows the KEGG analysis of the overlapping genes. I, RT‐qPCR analysis of PI3K/AKT related gene expression in Setd2fl/fl and Setd2‐KO mice as indicated. J, ChIP‐qPCR analysis of H3K36me3 binding for Gng4, Lama4 and Thbs3 loci in primary KCs from Setd2fl/fl mice; IgG was used as the control. Data are presented as the mean ± SD; statistical significance was determined using a two‐tailed Student's t test; *P < .05, **P < .01, ***P < .001 and ****P < .0001
Since SETD2 mainly regulates the expression of downstream genes through H3K36me3, we performed chromatin immunoprecipitation experiments followed by next‐generation sequencing (ChIP‐seq) assays using a H3K36me3‐specific antibody in primary KCs isolated from Setd2fl/fl mice. H3K36me3 peaks were enriched around the transcription area (Figure 5G). A total of 45 584 H3K36me3 peaks (on 14 567 genes) were identified. To correlate chromatin binding with transcriptional regulation, we integrated ChIP‐seq data with RNA‐Seq data. The results indicated that 155 misregulated genes showed direct H3K36me3 occupancy. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that these overlapping 155 genes were functionally associated with ECM‐receptor interaction, AMPK signalling pathway and glucagon signalling pathway, among others. Furthermore, these genes were still closely related to the PI3K/AKT signalling pathway (Figure 5H). RT‐qPCR analysis demonstrated that genes related to the PI3K/AKT signalling pathway, including Gng4, Thbs3 and Lama4, were downregulated in Setd2‐KO compared to Setd2fl/fl mice (Figure 5I). We further validated the existence of H3K36me3 binding at the gene bodies of Gng4, Lama4 and Thbs3 by ChIP‐qPCR assay (Figure 5J), and found that the intensity of H3K36me3 binding within these gene loci significantly increased in the H3K36me3 group compared to the IgG group. These results suggested that SETD2 deletion might activate the AKT/mTOR signalling pathway via the regulation of these H3K36me3‐directly occupied genes.
3.6. Activation of AKT/mTOR in the epithelial compartment accelerates wound healing
To further elucidate the causal link between SETD2 and the AKT/mTOR pathway in wound healing, we transfected HaCaT cells with sh‐Setd2 plasmids and then treated these cells with an AKT inhibitor (MK‐2206) or an mTOR inhibitor (rapamycin). Western blotting confirmed that the protein levels of p‐AKT and p‐mTOR were decreased in Setd2‐knockdown HaCaT cells treated with MK2206 or rapamycin (Figure 6A). As shown by CCK8 assays, Setd2‐knockdown promoted cell proliferation, while the treatment with either MK‐2206 or rapamycin significantly inhibited cell proliferation. (Figure 6B,C). Next, scratch assays showed delayed wound closure after the treatment either with MK2206 or rapamycin (Figure 6D). Importantly, Setd2‐KO mice treated with rapamycin or MK2206 also demonstrated delayed wound closure compared to the vehicle‐treated group. These results indicated that the inhibition of AKT or mTOR in vivo abrogated the accelerated wound healing caused by SETD2 deletion in the epithelial compartment (Figure 6E‐G). Collectively, these results suggest that the accelerated wound healing due to SETD2 deletion is mediated by the activation of AKT/mTOR signalling.
FIGURE 6.
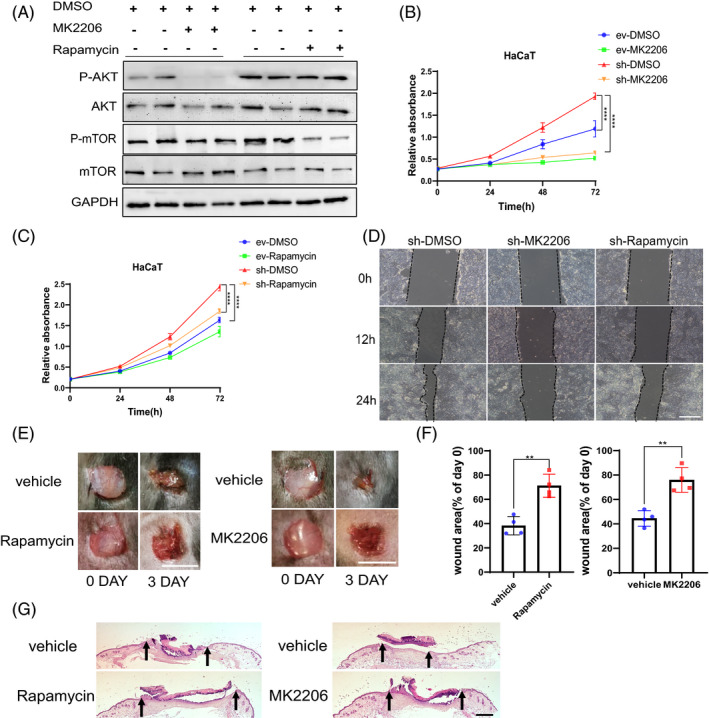
Inhibition of AKT/mTOR signalling pathway suppresses cell proliferation and migration in Setd2‐deficient HaCaT cells. A, Western blot analysis of p‐AKT and p‐mTOR expression in Setd2‐deficient HaCaT cells treated with either AKT inhibitor MK2206 or mTOR inhibitor rapamycin. B, CCK8 assay of HaCaT cells transfected either with empty vector (ev) or sh‐Setd2 plasmids (sh) and treated with the AKT inhibitor MK‐2206 or DMSO (vehicle control). C, CCK8 assay of HaCaT cells transfected either with empty vector (ev) or sh‐Setd2 plasmids (sh) and treated with the mTOR inhibitor rapamycin or DMSO (vehicle control). D, Confluent Setd2‐knockdown HaCaT cells were wounded by scraping and treated with MK2206, rapamycin or DMSO (vehicle control). Cell migration towards the wound surface was evaluated at 12 h and 24 h. Scale bars: 100 μm. E, Appearance of wound areas in Setd2‐KO mice treated either with the vehicle control or inhibitors (rapamycin or MK2206) on days 0 and 3 post‐wounding. Scale bars: 4 mm. F, Quantification of the wound areas. Results are presented as percent wound area at day 3 relative to the original wound area in Setd2‐KO mice treated with the vehicle control or inhibitors (rapamycin or MK2206); n = 4 wounds/group. The analysis was performed using ImageJ software. G, H&E staining of wound tissues of Setd2‐KO mice treated either with the vehicle control or inhibitors (rapamycin and MK2206) on day 3 after injury. The arrows indicate the boundary of the wound. Scale bars: 500 μm. Data are presented as the mean ± SD; statistical significance was determined using a two‐tailed Student's t test and two‐way ANOVA; **P < .01 and ****P < .0001
4. DISCUSSION
This study investigated the mechanistic role of SETD2 in epidermal KCs during cutaneous wound healing. We identified two novel findings: (a) the deletion of Setd2 accelerated re‐epithelialization during wound healing via increased KC proliferation and migration; and (b) SETD2 deficiency activated AKT/mTOR signalling. Furthermore, the inhibition of AKT/mTOR signalling with MK2206 or rapamycin, significantly reduced KC proliferation and migration. Therefore, we showed, for the first time, that SETD2 plays an essential role in the re‐epithelialization process during cutaneous wound healing.
Histone modification is important for cutaneous wound healing; however, little is known about the role of SETD2, a key histone modifier, in skin wound healing. In this study, we showed that mice lacking Setd2 had accelerated wound healing. Skin wound healing is a physiological (or a pathological) process in which multiple cells and signalling molecules work together. Previous studies have shown that several signalling pathways, including Wnt, Notch, TGFβ and PI3K/AKT/mTOR, are involved in skin wound healing. 38 , 48 , 49 , 50 The PI3K/AKT/mTOR signalling pathway plays an important role in numerous diseases, including cancer. This pathway controls several key cellular processes responsible for cellular homeostasis, while aberrant activation of the pathway has been observed in human cancers. 51 , 52 In the skin, this pathway is crucial for skin development and homeostasis. 53 It has been well established that activation of the PI3K/AKT/mTOR pathway can promote cell migration and proliferation, and enhance cutaneous wound healing. 49 , 54 , 55 Therefore, transient pharmacologic activation of the PI3K/AKT/mTOR signalling axis may represent a novel clinical intervention strategy to accelerate the healing of debilitating and life‐threatening wounds. 50 In our study, we demonstrated that the loss of Setd2 led to the activation of the AKT/mTOR signalling pathway during wound healing.
RNA‐seq and ChIP‐seq data indicated that seven misregulated genes associated with the PI3K/AKT/mTOR pathway showed direct H3K36me3 occupancies. We verified the mRNA expression levels of these genes including Fn1, Gys1, Itga7, Pik3r3, Gng4, Lama4 and Thbs3 by RT‐qPCR. ChIP‐qPCR validated the existence of H3K36me3 binding at these genes. These results suggested that SETD2 might regulate these downstream genes through H3K36me3, leading to the activation of the AKT/mTOR pathway, and the specific regulatory mechanism needs to be further elucidated.
As a tumour suppressor, SETD2 has been extensively studied in cancers of different tissues. Here, we utilized an animal model of a conditional epidermis‐specific knockout of Setd2. However, Setd2‐KO mice showed no tumour‐associated phenotype in their skin for the first 10 months. Therefore, the biological function of SETD2 in skin cancer could be further studied either by crossing Setd2‐KO mice with other mutant mice, or by stimulating Setd2‐KO mice with different environmental factors. In summary, our study contributes to a better understanding of the wound‐healing process and suggests that SETD2 may be considered as a therapeutic target to improve skin wound healing.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
XL mainly performed the experiments, analysed the data and wrote the paper. CL and YZ contributed to the animal breeding and identification. HR, ML, LG, WF, HT and JX helped with the experiments. LL and WQG conceived the concept. LL designed the experiments, drafted and revised manuscript. All authors edited and approved the final manuscript.
Supporting information
Table S1‐S2
ACKNOWLEDGEMENTS
This study was supported by funds from National Natural Science Foundation of China (81772938 and 82073104 to LL), State Key Laboratory of Oncogenes and Related Genes (KF01801 to LL), Science and Technology Commission of Shanghai Municipality (18140902700 and 19140905500 to LL), Innovation Research Plan from Shanghai Municipal Education Commission (ZXGF082101 to LL), the 111 Project (B21024), the Shenzhen Basic Research Foundation (JCYJ20170818090044949), and the Guangdong Province Basic Research Foundation (2018A030310012). The study is also supported by Bio‐ID Center, School of Biomedical Engineering, Shanghai Jiao Tong University.
Li X, Liu C, Zhu Y, et al. SETD2 epidermal deficiency promotes cutaneous wound healing via activation of AKT/mTOR Signalling. Cell Prolif. 2021;54:e13045. 10.1111/cpr.13045
DATA AVAILABILITY STATEMENT
All data are available from the authors upon request. RNA‐Seq and ChIP‐Seq raw data were deposited in the Gene Expression Omnibus (GEO) under accession number GEO: GSE163861.
REFERENCES
- 1. Na J, Lee K, Na W, et al. Histone H3K27 demethylase JMJD3 in cooperation with NF‐kappaB regulates keratinocyte wound healing. J Invest Dermatol. 2016;136(4):847‐858. [DOI] [PubMed] [Google Scholar]
- 2. Fuchs E. Skin stem cells: rising to the surface. J Cell Biol. 2008;180(2):273‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol. 2008;17(12):1063‐1072. [DOI] [PubMed] [Google Scholar]
- 4. Yang X, Teng Y, Hou N, et al. Delayed re‐epithelialization in Ppm1a gene‐deficient mice is mediated by enhanced activation of Smad2. J Biol Chem. 2011;286(49):42267‐42273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314‐321. [DOI] [PubMed] [Google Scholar]
- 6. Yang L, Li W, Wang S, et al. Smad4 disruption accelerates keratinocyte reepithelialization in murine cutaneous wound repair. Histochem Cell Biol. 2012;138(4):573‐582. [DOI] [PubMed] [Google Scholar]
- 7. Du H, Zhou Y, Suo Y, et al. CCN1 accelerates re‐epithelialization by promoting keratinocyte migration and proliferation during cutaneous wound healing. Biochem Biophys Res Commun. 2018;505(4):966‐972. [DOI] [PubMed] [Google Scholar]
- 8. Lewis CJ, Stevenson A, Fear MW, Wood FM. A review of epigenetic regulation in wound healing: implications for the future of wound care. Wound Repair Regen. 2020;28(6):710‐718. [DOI] [PubMed] [Google Scholar]
- 9. Kang S, Chovatiya G, Tumbar T. Epigenetic control in skin development, homeostasis and injury repair. Exp Dermatol. 2019;28(4):453‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Botchkarev VA, Gdula MR, Mardaryev AN, Sharov AA, Fessing MY. Epigenetic regulation of gene expression in keratinocytes. J Invest Dermatol. 2012;132(11):2505‐2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewis CJ, Mardaryev AN, Sharov AA, Fessing MY, Botchkarev VA. The epigenetic regulation of wound healing. Adv Wound Care (New Rochelle). 2014;3(7):468‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Na J, Shin JY, Jeong H, et al. JMJD3 and NF‐kappaB‐dependent activation of Notch1 gene is required for keratinocyte migration during skin wound healing. Sci Rep. 2017;7(1):6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011;25(5):485‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang S, Long K, Wang S, Sada A, Tumbar T. Histone H3 K4/9/27 trimethylation levels affect wound healing and stem cell dynamics in adult skin. Stem Cell Reports. 2020;14(1):34‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhattacharya S, Workman JL. Regulation of SETD2 stability is important for the fidelity of H3K36me3 deposition. Epigenetics Chromatin. 2020;13(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun XJ, Wei J, Wu XY, et al. Identification and characterization of a novel human histone H3 lysine 36‐specific methyltransferase. J Biol Chem. 2005;280(42):35261‐35271. [DOI] [PubMed] [Google Scholar]
- 17. Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem. 2002;277(51):49383‐49388. [DOI] [PubMed] [Google Scholar]
- 18. Skucha A, Ebner J, Grebien F. Roles of SETD2 in leukemia‐transcription, DNA‐damage, and beyond. Int J Mol Sci. 2019;20(5):1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yuan H, Li N, Fu D, et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J Clin Invest. 2017;127(9):3375‐3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li F, Mao G, Tong D, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153(3):590‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kanu N, Gronroos E, Martinez P, et al. SETD2 loss‐of‐function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. 2015;34(46):5699‐5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carvalho S, Vitor AC, Sridhara SC, et al. SETD2 is required for DNA double‐strand break repair and activation of the p53‐mediated checkpoint. eLife. 2014;3:e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan H, Han Y, Wang X, et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell. 2020;38(3):350‐365 e357. [DOI] [PubMed] [Google Scholar]
- 24. Zhu X, He F, Zeng H, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46(3):287‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Al Sarakbi W, Sasi W, Jiang WG, Roberts T, Newbold RF, Mokbel K. The mRNA expression of SETD2 in human breast cancer: correlation with clinico‐pathological parameters. BMC Cancer. 2009;9(1):290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fontebasso AM, Schwartzentruber J, Khuong‐Quang DA, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high‐grade gliomas. Acta Neuropathol. 2013;125(5):659‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang KK, McPherson JR, Tay ST, et al. SETD2 histone modifier loss in aggressive GI stromal tumours. Gut. 2016;65(12):1960‐1972. [DOI] [PubMed] [Google Scholar]
- 28. Duns G, van den Berg E, van Duivenbode I, et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70(11):4287‐4291. [DOI] [PubMed] [Google Scholar]
- 29. Niu N, Lu P, Yang Y, et al. Loss of Setd2 promotes Kras‐induced acinar‐to‐ductal metaplasia and epithelia‐mesenchymal transition during pancreatic carcinogenesis. Gut. 2020;69(4):715‐726. [DOI] [PubMed] [Google Scholar]
- 30. Xu Q, Xiang Y, Wang Q, et al. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet. 2019;51(5):844‐856. [DOI] [PubMed] [Google Scholar]
- 31. Ji Z, Sheng Y, Miao J, et al. The histone methyltransferase Setd2 is indispensable for V(D)J recombination. Nat Commun. 2019;10(1):3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang YL, Sun JW, Xie YY, et al. Setd2 deficiency impairs hematopoietic stem cell self‐renewal and causes malignant transformation. Cell Res. 2018;28(4):476‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L, Niu N, Li L, Shao R, Ouyang H, Zou W. H3K36 trimethylation mediated by SETD2 regulates the fate of bone marrow mesenchymal stem cells. PLoS Biol. 2018;16(11):e2006522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zuo X, Rong B, Li L, Lv R, Lan F, Tong MH. The histone methyltransferase SETD2 is required for expression of acrosin‐binding protein 1 and protamines and essential for spermiogenesis in mice. J Biol Chem. 2018;293(24):9188‐9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen K, Liu J, Liu S, et al. Methyltransferase SETD2‐mediated methylation of STAT1 Is critical for interferon antiviral activity. Cell. 2017;170(3):492‐506 e414. [DOI] [PubMed] [Google Scholar]
- 36. Park IY, Powell RT, Tripathi DN, et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell. 2016;166(4):950‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seervai RNH, Jangid RK, Karki M, et al. The Huntingtin‐interacting protein SETD2HYPB is an actin lysine methyltransferase. Sci Adv. 2020;6(40):eabb7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meng F, Cheng X, Yang L, Hou N, Yang X, Meng A. Accelerated re‐epithelialization in Dpr2‐deficient mice is associated with enhanced response to TGFbeta signaling. J Cell Sci. 2008;121(Pt 17):2904‐2912. [DOI] [PubMed] [Google Scholar]
- 39. Stepp MA, Liu Y, Pal‐Ghosh S, et al. Reduced migration, altered matrix and enhanced TGFbeta1 signaling are signatures of mouse keratinocytes lacking Sdc1. J Cell Sci. 2007;120(Pt 16):2851‐2863. [DOI] [PubMed] [Google Scholar]
- 40. Linke A, Goren I, Bosl MR, Pfeilschifter J, Frank S. The suppressor of cytokine signaling (SOCS)‐3 determines keratinocyte proliferative and migratory potential during skin repair. J Invest Dermatol. 2010;130(3):876‐885. [DOI] [PubMed] [Google Scholar]
- 41. Xing W, Guo W, Zou CH, et al. Acemannan accelerates cell proliferation and skin wound healing through AKT/mTOR signaling pathway. J Dermatol Sci. 2015;79(2):101‐109. [DOI] [PubMed] [Google Scholar]
- 42. Petit V, Raymond J, Alberti C, et al. C57BL/6 congenic mouse NRAS(Q61K) melanoma cell lines are highly sensitive to the combination of Mek and Akt inhibitors in vitro and in vivo. Pigment Cell Melanoma Res. 2019;32(6):829‐841. [DOI] [PubMed] [Google Scholar]
- 43. Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden's disease by mTOR inhibition with rapamycin. Cancer Res. 2008;68(17):7066‐7072. [DOI] [PubMed] [Google Scholar]
- 44. Dahlhoff M, Muzumdar S, Schafer M, Schneider MR. ERBB2 is essential for the growth of chemically induced skin tumors in mice. J Invest Dermatol. 2017;137(4):921‐930. [DOI] [PubMed] [Google Scholar]
- 45. Uchiyama A, Nayak S, Graf R, et al. SOX2 epidermal overexpression promotes cutaneous wound healing via activation of EGFR/MEK/ERK signaling mediated by EGFR ligands. J Invest Dermatol. 2019;139(8):1809‐1820 e1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miao Q, Ku AT, Nishino Y, et al. Tcf3 promotes cell migration and wound repair through regulation of lipocalin 2. Nat Commun. 2014;5(1):4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J, Li L, Zhang Q, et al. Microtubule‐associated protein 4 phosphorylation regulates epidermal keratinocyte migration and proliferation. Int J Biol Sci. 2019;15(9):1962‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi Y, Shu B, Yang R, et al. Wnt and Notch signaling pathway involved in wound healing by targeting c‐Myc and Hes1 separately. Stem Cell Res Ther. 2015;6(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xiao W, Tang H, Wu M, et al. Ozone oil promotes wound healing by increasing the migration of fibroblasts via PI3K/Akt/mTOR signaling pathway. Biosci Rep. 2017;37(6):BSR20170658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cristiane H, Squarize RMC, Bugge TH, Silvio Gutkind J. Accelerated wound healing by mTOR activation in genetically defined mouse models. PLoS One. 2010;5(5):e10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fattahi S, Amjadi‐Moheb F, Tabaripour R, Ashrafi GH, Akhavan‐Niaki H. PI3K/AKT/mTOR signaling in gastric cancer: epigenetics and beyond. Life Sci. 2020;262:118513. [DOI] [PubMed] [Google Scholar]
- 52. Jafari M, Ghadami E, Dadkhah T, Akhavan‐Niaki H. PI3k/AKT signaling pathway: erythropoiesis and beyond. J Cell Physiol. 2019;234(3):2373‐2385. [DOI] [PubMed] [Google Scholar]
- 53. Castilho RM, Squarize CH, Gutkind JS. Exploiting PI3K/mTOR signaling to accelerate epithelial wound healing. Oral Dis. 2013;19(6):551‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jere SW, Houreld NN, Abrahamse H. Role of the PI3K/AKT (mTOR and GSK3beta) signalling pathway and photobiomodulation in diabetic wound healing. Cytokine Growth Factor Rev. 2019;50:52‐59. [DOI] [PubMed] [Google Scholar]
- 55. Yang HL, Tsai YC, Korivi M, Chang CT, Hseu YC. Lucidone promotes the cutaneous wound healing process via activation of the PI3K/AKT, Wnt/beta‐catenin and NF‐kappaB signaling pathways. Biochim Biophys Acta Mol Cell Res. 2017;1864(1):151‐168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
All data are available from the authors upon request. RNA‐Seq and ChIP‐Seq raw data were deposited in the Gene Expression Omnibus (GEO) under accession number GEO: GSE163861.