

Abstract
Understanding the ligand preferences of epigenetic reader domains enables identification of modification states of chromatin with which these domains associate and can yield insight into recruitment and catalysis of chromatin-acting complexes. However, thorough exploration of the ligand preferences of reader domains is hindered by the limitations of traditional protein–ligand binding assays. Here, we evaluate the binding preferences of the PHD1 domain of histone demethylase KDM5A using the protein interaction by SAMDI (PI-SAMDI) assay, which measures protein–ligand binding in a high-throughput and sensitive manner via binding-induced enhancement in the activity of a reporter enzyme, in combination with fluorescence polarization. The PI-SAMDI assay was validated by confirming its ability to accurately profile the relative binding affinity of a set of well-characterized histone 3 (H3) ligands of PHD1. The assay was then used to assess the affinity of PHD1 for 361 H3 mutant ligands, a select number of which were further characterized by fluorescence polarization. Together, these experiments revealed PHD1’s tolerance for H3Q5 mutations, including an unexpected tolerance for aromatic residues in this position. Motivated by this finding, we further demonstrate a high-affinity interaction between PHD1 and recently identified Q5-serotonylated H3. This work yields interesting insights into permissible PHD1-H3 interactions and demonstrates the value of interfacing PI-SAMDI and fluorescence polarization in investigations of protein–ligand binding.
Graphical Abstract

INTRODUCTION
Epigenetic reader domains recognize specific histone modifications and are often responsible for recruiting large protein complexes, such as those involved in chromatin modification and DNA repair, to their chromosomal target.1 Elucidating the ligand preferences of reader domains and their tolerance for post-translational modifications and mutations of histones will contribute to an understanding of the epigenetic environment in which they can perform their biological role. Additionally, since reader domains can regulate catalytic activities and substrate specificity of chromatin modifiers, characterizing ligand preferences of chromatin readers can help to define the mechanistic basis of cross-talk between chromatin marks.2–5 However, exploring the ligand preferences of reader domains is often challenging due to the limited throughput and sensitivity of traditional protein–ligand binding assays. Techniques such as saturation transfer difference nuclear magnetic resonance (STD NMR), heteronuclear single quantum coherence nuclear magnetic resonance (HSQC NMR), and isothermal titration calorimetry (ITC) allow for precise measurements of affinity but require significant amounts of sample and have low throughput.6–10 These techniques are therefore poorly suited for analysis of large numbers of samples, limiting the scope of their explorations into ligand preferences and their ability to uncover trends in binding affinity. Fluorescence-based assays, including Förster resonance energy transfer (FRET), fluorescence polarization (FP), and evanescent wave fluorescence biosensing, are also used to evaluate protein–ligand binding interactions.11–13 While these assays have higher throughput than ITC and NMR, they still cannot accommodate large ligand libraries and require the use of specialized fluorescently labeled ligands.14
Peptide and protein arrays are a useful tool for evaluating reader domain–ligand interactions in a high-throughput manner and can yield valuable insights into binding preferences. Hundreds of unique peptides can be immobilized to different regions of a planar surface to form a peptide array,15 where their binding to a reader domain is detected via a reporter antibody.16–22 Alternately, a protein microarray,23 in which the reader domain is patterned onto a solid substrate, can be probed with candidate peptide ligands to identify binding interactions.24,25 While current array-based methods can be used to qualitatively assess the binding affinity of proteins for hundreds of ligands, they often display poor sensitivity.25–27 Specific surface washing and blocking procedures have been developed to enhance detection of low-affinity interactions but are only compatible with certain immobilization substrates and have not been shown to be effective for interactions with Kd values beyond the tens of micromolar range.28 As a result, array-based methods display high rates of false negative identification.
Given the limitations of these methods, new high-throughput techniques with enhanced sensitivity will be important for determining the ligand preferences of epigenetic reader domains. We recently developed an assay that measures protein–ligand binding interactions in a high-throughput and sensitive manner.29 The assay is based on the self-assembled monolayers for a matrix-assisted laser desorption/ionization mass spectrometry (SAMDI-MS) platform, which uses a self-assembled monolayer of alkanethiolates on gold. A fraction of the alkanethiolates present a maleimide group for immobilization of cysteine-terminated peptides by Michael addition, and the remaining alkanethiolates present a tri(ethylene glycol) group that prevents nonspecific adsorption of proteins to the surface.30,31 An enzymatic substrate peptide is immobilized to the monolayer, where it undergoes modification upon application of the enzyme to the surface. The monolayer is then analyzed by MALDI-TOF mass spectrometry to reveal the masses of the substituted alkanethiolates, which can be integrated to determine the yield for the enzyme-mediated reaction.32–34
In the protein interaction by SAMDI (PI-SAMDI) assay, a fusion protein consisting of the reader domain of interest and a reporter enzyme is applied to a monolayer presenting two immobilized peptides: one a ligand for the reader domain and the other a substrate for the reporter enzyme (Figure 1). Binding of the reader domain to its ligand localizes the fusion protein to the surface and in turn colocalizes the reporter enzyme and its substrate, increasing the rate of the enzymatic reaction.35,36 Therefore, the extent of substrate conversion serves as a “covalent record” of the fraction of time the ligand is bound to the reader domain, and the activity of the reporter enzyme as read by mass spectrometry can be used as a measure of the affinity of the domain for the ligand.29
Figure 1.

In PI-SAMDI, a binding interaction between a reader domain (green) and an immobilized ligand brings a fused reporter enzyme (purple) to the surface, where it can more rapidly modify its immobilized substrate.
In our first report of PI-SAMDI, we characterized the relative affinity of chromodomain proteins for histone 3 (H3) ligands displaying various post-translational modifications.29 This study revealed that the PI-SAMDI assay provided an accurate profile of the relative affinity of the individual chromodomains for the ligands and demonstrated the advantages of PI-SAMDI over traditional protein–ligand binding assays.29 It requires only microliter volumes of protein, is compatible with standard array format liquid handling instrumentation on 384- and 1536-spot plates, and uses mass spectrometry as a fast readout method, which collectively enable a throughput of tens of thousands of samples per day.29,37 Furthermore, by coupling transient protein–ligand binding to a covalent enzymatic modification, this assay can detect even low-affinity interactions that could not survive a rinsing step and therefore displays superior sensitivity relative to array-based methods.29
In the present report, we use PI-SAMDI in combination with FP to explore the binding preferences of the plant homeodomain 1 (PHD1) reader domain of histone demethylase KDM5A. PHD domains are a large family of reader domains that associate with chromatin as a function of the site and extent of lysine methylation in histone proteins.38 While the binding affinity of many PHD domains for H3 ligands containing post-translational modifications—particularly of the target lysine—has been explored, the tolerance of PHD domains for modifications of adjacent residues is poorly understood.39 Furthermore, a number of PHD domains encoded by the human genome, including the PHD2 domains of the KDM5 family of histone demethylases, the tandem PHD domains of the KDM4 family,5 the PHD domains of the KDM2 family,40 and the PHD domains of the KMT2/MLL family,41 among others, have no known ligands.
PHD1 preferentially binds the N-terminal region of H3 tails and is known to be involved in the allosteric regulation of the demethylation activity of KDM5A.2,3 PHD1 has been investigated for its binding to H3-derived ligands featuring lysine methylation and a limited number of additional modifications and mutations.2,3 Here, we rely on known ligands of PHD1 to first validate the application of the PI-SAMDI method to this new reader domain system and then use PI-SAMDI and FP to evaluate the affinity of PHD1 for uncharacterized H3 mutant ligands. Our findings revealed PHD1’s tolerance for mutations of the Q5 residue and enabled identification of a previously unknown Q5-serotonylated H3 ligand of PHD1. In this way, this work helps to better define the epigenetic context of KDM5A activity, contributes to the knowledge gap in the binding preferences of PHD domains, and demonstrates the utility of combining PI-SAMDI and FP in explorations of reader domain-ligand binding.
METHODS
Preparation of Monolayer Arrays.
Array plates with 384 gold spots on steel plates were soaked in a solution of disulfides for 48 h at 4 °C to allow formation of a self-assembled monolayer on the gold surface. The solution consisted of a mixture of EG3-alkane disulfide and a mixed disulfide of EG3-alkanethiol and a maleimide-terminated EG3-alkanethiol. The solution of disulfides contained an overall concentration of 1 mM of the two monolayer compounds in an appropriate stoichiometric ratio to yield a 10% maleimide surface density.
Substrate and Ligand Synthesis.
The peptide substrates for KDAC8 were synthesized using standard FMOC solid-phase peptide synthesis on Rink-amide resin, N-terminally acetylated, and purified by reversed-phase HPLC. Peptide ligands used in all PI-SAMDI experiments except the 361-ligand array were synthesized using standard FMOC solid-phase peptide synthesis on Rink-amide resin and purified by reversed-phase HPLC. The peptide ligands in the 361-ligand array were synthesized using standard FMOC solid-phase peptide synthesis on Rink-amide lanterns in 96-well filter plates. When synthesizing the array peptides, all amino acids were coupled twice to maximize purity. The array was characterized by MALDI-TOF mass spectrometry. Spectra of 20 (~5%) of the peptides were acquired and showed >90% purity (see Figure S1 for a representative spectrum). The peptide ligands selected from the array for further characterization by FP were purchased from Genscript. The H3Q5ser peptide ligand was purchased from CPC Scientific, and the T3D and unmodified H3 peptide ligands evaluated in the same experiment were synthesized using standard FMOC solid-phase peptide synthesis on Rink-amide resin and purified by reversed-phase HPLC. All H3 ligands for PI-SAMDI experiments were synthesized with an additional terminal GC (for 12-mer peptides) or C (for truncated peptides) moiety to enable immobilization to the maleimide-presenting monolayer.
Surface Preparation.
Peptide solutions were prepared at a total peptide concentration of 50 μM in PBS buffer at pH 7.3. For coimmobilization of a PHD1 ligand and the KDAC8 substrate, the two peptides were premixed in a stoichiometric ratio of PHD1 ligand to KDAC8 substrate of 1:4 when 12-mer ligands were used and 3:7 when truncated ligands were used. The peptide solutions were applied to the gold spots at 1 μL each using a liquid handler and incubated on the surface presenting 10% maleimide for 1 h. This procedure yields a surface of 10% total peptide density, with 2% PHD1 ligand and 8% KDAC8 substrate when 12-mer ligands are used and 3% ligand and 7% substrate when truncated ligands are used.
Expression and Purification of PHD1-KDAC8.
A PHD1287–347-KDAC8–6xHis fusion construct was generated in a pET-303 CT-His vector containing an AAAGGSS linker between PHD1 and KDAC8. BL21 (DE3) cells were used to express the fusion construct. Protein expression was induced with 200 μM IPTG at an OD600 of ~0.6, and 50 μM ZnCl2 was added to the media before incubation at 18 °C overnight before harvesting. Cell pellets were resuspended in lysis buffer (50 mM Tris, 200 mM NaCl, 1 mM MgCl2, 5 mM β- mercaptoethanol, 1 mM PMSF, and 5% glycerol pH 7.5) before being lysed by sonication followed by recovery of the supernatant after centrifugation at 35 000 rpm. The supernatant was then incubated with Ni-NTA resin for 1 h at 4 °C before being washed with lysis buffer. The resin was then washed with wash buffer (50 mM Tris, 200 mM NaCl, 1 mM MgCl2, 5 mM β-mercaptoethanol, 1 mM PMSF, 20 mM imidazole, and 5% glycerol pH 7.5) before the protein was eluted with elution buffer (50 mM Tris, 200 mM NaCl, 1 mM MgCl2, 5 mM β-mercaptoethanol, 1 mM PMSF, 250 mM imidazole, and 5% glycerol pH 7.5). The sample was then dialyzed into running buffer (50 mM Tris, 150 mM NaCl, 1 mM MgCl2, 5 mM β-mercatoethanol, and 10% glycerol pH7.5) before being concentrated and loaded onto a Superdex S75 size exclusion column, pre-equilibrated in running buffer. Following size exclusion, the eluted protein was concentrated before being flash frozen.
PI-SAMDI Assays.
All assays were performed on 384-spot array surfaces, in a standard 16 × 24 formatting, with each spot having a diameter of 2.5 mm. The fusion protein was diluted in PBS to the working concentration. The working concentration of protein and reaction time ranged between 1.5 and 5 μM and 5–30 min depending on protein purity. The enzyme was applied to array plates presenting substrate and ligand peptides at 1 μL per spot. At the end of the reaction period, 0.5 μL of 15% formic acid was applied to each spot to quench the reaction. A Multidrop Combi benchtop robot was used to simultaneously initiate and quench the reaction across all spots to ensure uniform reaction times. The surface was then rinsed with water and ethanol and dried. A matrix of 15 mg mL−1 of 2,4,6-trihydroxyacetophenone in acetonitrile was applied directly to the surface, and after drying, each spot was analyzed by MALDI-TOF mass spectrometry using an AB Sciex 5800 series instrument. The sum of the area under the peaks (∑AUP) corresponding to the acetylated and deacetylated peptide in the mass spectra was determined using custom software and was used to calculate the extent of enzymatic conversion of the substrate for each reaction according to the equation:
| (1) |
By quantifying the amount of deacetylated peptide relative to the amount of total (acetylated and deacetylated) peptide, no internal standard is required. In order to quantify the increase in substrate modification observed upon domain-ligand binding and enable a comparison of binding affinity between multiple ligands, we developed a numerical parameter termed “fold enhancement.”29 Fold enhancement is calculated by dividing the average yield in substrate modification in the presence of the ligand by the average yield in substrate modification in the absence of the ligand and represents the enhancement in substrate modification due to ligand binding. A fold enhancement of 1 represents the degree of substrate modification with no binding-induced enhancement and indicates the level of background in PI-SAMDI experiments. We used the reaction between the fusion construct and spots presenting only the KDAC8 substrate rather than one between an isolated KDAC8 enzyme and spots presenting the coimmobilized H3 ligand and KDAC8 substrate as a baseline measure of KDAC8 activity because simultaneous application of one enzyme to all spots ensures consistent reaction times and reliable activity comparisons. As the substrate modification yield measured in the presence of each ligand is normalized to the same substrate modification yield measured in the absence of ligand, the difference in the composition of the monolayer between the two conditions does not affect the affinity profile obtained.
Fluorescence Polarization Assays.
For the FP assays, a GST-PHD1 (S287–K347) construct, which has been described previously,2 was used. The binding of GST-PHD1 to H3 peptides was measured by competition-based fluorescence polarization (FP). The 1 μM GST-PHD1 was incubated with 10 nM of H3 peptide (1–10) carrying a C-terminal fluorescein label, and different concentrations of unlabeled peptides were used as competitors. Data from competition-based FP assays were fitted to the equation:
| (2) |
where FPobs is the observed FP, FPmax is the maximum FP value, FPmin is the minimum FP value, [PHD1] is the concentration of PHD1, Kd is the dissociation constant, Ki is the inhibition constant referring to the competing peptide, and [I] is the competing peptide concentration.
RESULTS AND DISCUSSION
Validation of PI-SAMDI Assay.
In order to apply the PI-SAMDI assay to evaluation of PHD1’s binding preferences, we first generated a construct of PHD1 fused to lysine deacetylase 8 (KDAC8), the reporter enzyme used in our previous PI-SAMDI study29 (Figure 2A). We then determined that the GSK(Ac)FGC peptide is an appropriate substrate for the KDAC8 reporter enzyme in the assay. GSK(Ac)FGC is a poor KDAC8 substrate that displays minimal activity toward the soluble enzyme in the absence of a PHD1-binding ligand but significant modification in the presence of the ligand upon binding-induced colocalization of the enzyme and substrate. This substrate therefore effectively enables detection of binding interactions upon coimmobilization with prospective PHD1-binding ligands (Figure 2B, Figure S2).
Figure 2.
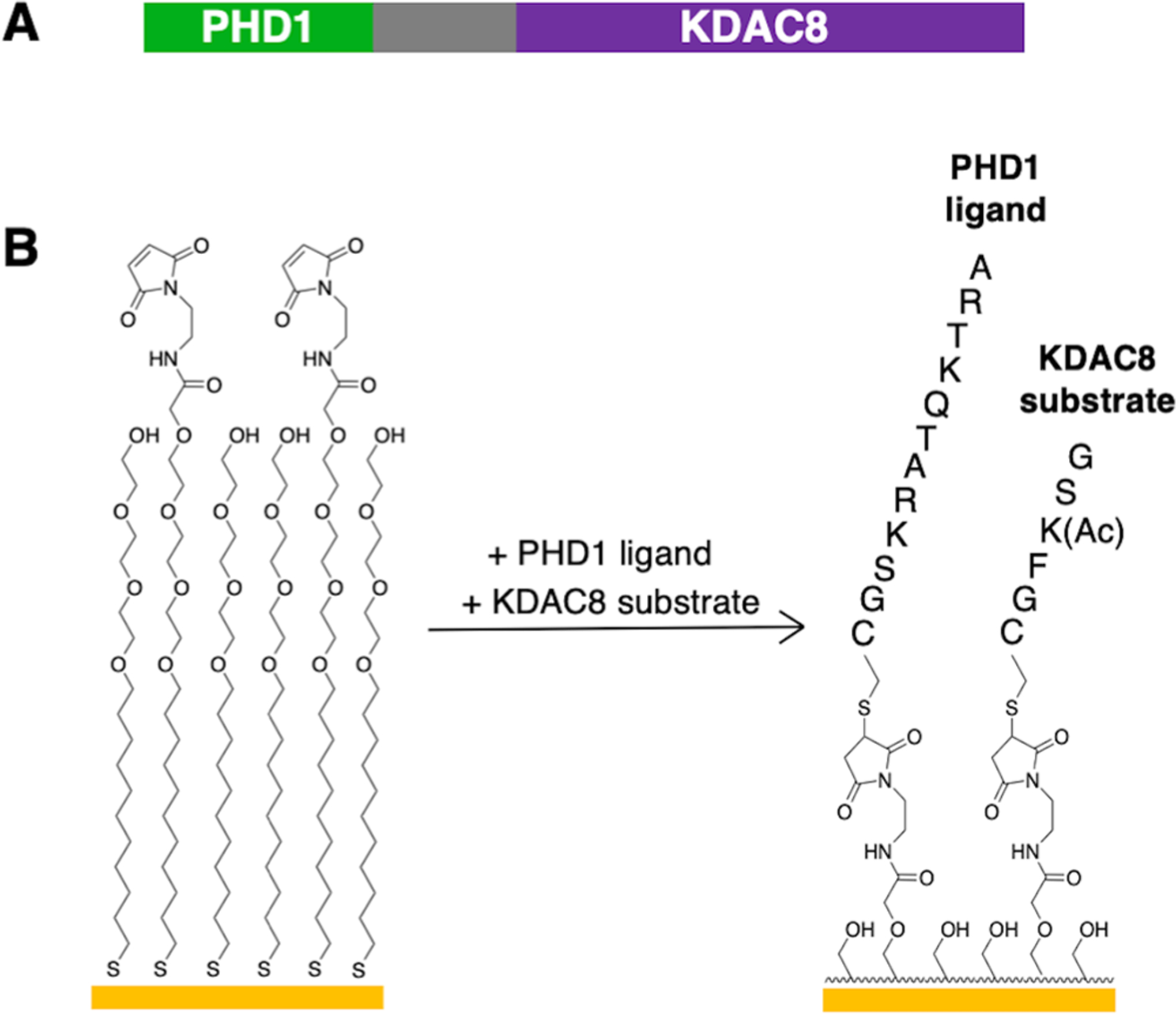
Design rationale of the PI-SAMDI assay for PHD1. (A) Domain map of the PHD1-KDAC8 fusion construct used. (B) A candidate PHD1 ligand peptide and the KDAC8 substrate peptide are coimmobilized to the maleimide-presenting monolayer via their terminal cysteine residues.
To evaluate if the function of PHD1 is maintained in the context of its KDAC8 fusion, we used the PI-SAMDI assay to measure the relative binding affinity of the PHD1 reader domain for a set of well-characterized H3 ligands.2,3 We measured the extent of substrate deacetylation by the fusion construct with and without coimmobilization of each H3 ligand and calculated the fold enhancement in substrate deacetylation produced by each ligand as described in the Methods section (a larger fold enhancement corresponds to a higher affinity of the domain-ligand binding interaction; Figure 3). We observed the highest fold enhancement in the presence of unmodified H3 as well as H3K9me3, a mark that does not impact PHD1-H3 association.2,3 Fold enhancement was observed to decrease with an increase in the extent of methylation of the H3K4 residue, which is consistent with a corresponding decrease in the affinity of PHD1 for H3K4 peptides upon sequential lysine methylation (Kd H3K4me3 > me2 > me1 > me0).2,3 A further reduction in fold enhancement was observed for the H3 R2A ligand, a mutation that strongly reduces binding of PHD1 to the H3 peptide.2,3 The lowest fold enhancement was measured for the N-terminally acetylated H3 peptide, which has been shown to not bind PHD1.2,3 The characterization of relative affinity of ligands according to their fold enhancement factors parallels the binding preferences of PHD1 established by prior FP measurements of the dissociation constants of the protein–ligand interactions.2,3 We note that as the density of the bound fusion protein increases with increasing affinity of the domain for the ligand, there will be an overlap in the regions of the monolayer that the reporter enzyme can access, leading to a decrease in fold enhancement.29 For this reason, not all differences in fold enhancement between ligands with consecutive affinity rankings are statistically significant, and we do not expect fold enhancement to correlate with Kd in a linear fashion. However, the observed agreement between the affinity profile provided by PI-SAMDI and FP indicates that the fused KDAC8 enzyme does not interfere with PHD1 binding and validates the application of PI-SAMDI as a platform for characterization of the ligand preferences of PHD1.
Figure 3.

Fold enhancement in deacetylation of the KDAC8 substrate produced by well-characterized PHD1 ligands, as measured by PI-SAMDI. Ligands are 12-mer peptides comprised of the first 10 N-terminal residues of H3 for PHD1 binding and a GC moiety for immobilization to the monolayer. Ligands (featuring various post-translational modifications or mutations) are presented in order of increasing Kd from left to right. Error bars represent the standard deviation of four replicates.
Exploration of Ligand Preferences of PHD1.
In order to probe the ligand preferences of PHD1, we used the PI-SAMDI assay to evaluate PHD1’s affinity for an array of mutant H3 peptides. While previous evaluation of mutated and post-translationally modified ligands supports the importance of the A1, R2, and K4 residues in PHD1 recognition,42 the roles of T3 and Q5 have not been addressed. Therefore, we systematically varied the T3 and Q5 residues of H3 to alternate amino acids and measured the fold enhancements produced by each of the ligands. As synthesis yields decrease with the length of the peptide, we used truncated, 6-mer ligands (comprised of the first five residues of H3 and a terminal cysteine residue for immobilization) to obtain peptides that were sufficiently pure without subsequent chromatographic purification for this experiment.43 Although PHD1 has a preference for longer H3 ligands in solution and binding to H3 6-mer ligands is not observed by FP, sufficiently high fold enhancements were produced by the truncated ligands, demonstrating the unique ability of PI-SAMDI to detect low-affinity interactions (Figure S3). We synthesized a library of ligands with the sequence ARXKZC in which X and Z represent all amino acids excluding cysteine, producing 361 unique peptides. We combined each ligand with the GSK(Ac)FGC substrate at a fixed ratio in a microtiter plate and immobilized each peptide mixture to the monolayer presented on individual gold spots of a 384-spot plate. To enable calculation of fold enhancement in substrate modification upon ligand binding, we immobilized the KDAC8 substrate alone to the remaining spots. We then applied the PHD1-KDAC8 fusion protein to the immobilized peptides and analyzed the surface by mass spectrometry after the reaction period (Figure 4A).
Figure 4.

PI-SAMDI characterization of PHD1 relative affinity for an array of H3 mutant ligands. (A) Experimental scheme for PI-SAMDI analysis of PHD1 binding to the H3 ligand array. (B) Heat map of fold enhancements produced by H3 mutant ligands of the sequence ARXKZC in which X and Z represent variable residues. The wild-type H3 sequence is indicated by a black box.
The fold enhancements produced by each ligand are presented in Figure 4B and represent an average of two replicate experiments using two arrays that were prepared separately from the same peptide stock solutions (see Table S1 for numerical fold enhancement values for each ligand). The average standard deviation in relative fold enhancement (normalized to the highest fold enhancement measured in each replicate) across all ligands was 10% between the two replicates, indicating good reproducibility in the assay. We observed a clear trend wherein PHD1 displayed low affinity toward ligands containing an acidic residue (aspartate or glutamate) in either the X or Z position. Also of note was a high tolerance for substitution in the Z (Q5) position, with the exception of Q5D and Q5E (as consistent with the observed trend of low affinity for ligands containing an acidic residue) as well as Q5H. The ligands containing an aromatic residue in the Q5 position (Q5F, Q5Y, and Q5W) showed particularly high affinity.
Guided by the results we obtained with the H3 ligand array, we selected several ligands that showed a range of fold enhancements and measured the binding affinity of their nontruncated counterparts using an FP assay. We found that binding of PHD1 toward T3 single mutant ligands is drastically diminished with the exception of T3V and T3S, which showed higher affinity compared to other T3 mutants, but still diminished binding relative to wild-type (Table 1, entries 2–7; Figure S4). These results indicate that both the methyl and hydroxyl groups of the threonine side chain are important in stabilizing interactions with PHD1. This finding is consistent with the observation of hydrogen bonding between the hydroxyl group of H3T3 and the amide of H3T6, which contributes to the H3 peptide’s adoption of a stabilized helical conformation when bound to PHD1, and hydrophobic interactions between the methyl group of H3T3 and the L309 and V330 residues of PHD1.42 The FP results for the T3 single mutant ligands largely agree with the PI-SAMDI array data, which showed relatively low affinity for each of these ligands except T3Y, which displayed a higher fold enhancement (Figure 4B). Additionally, we observed that PHD1 displays an affinity similar to the wild-type ligand for every Q5 single mutant ligand tested in the FP assay, suggesting that Q5 is dispensable for binding (Table 1, entries 8–14; Figure S4). The FP results for the Q5 single mutant ligands are mostly consistent with the PI-SAMDI array data, where each of these ligands displayed high affinity with the exception of Q5E, which showed a very low fold enhancement (Figure 4B). Several double mutants that displayed strong fold enhancements in the PI-SAMDI array were also evaluated by FP. Contrary to the PI-SAMDI data, each of these ligands showed weaker binding relative to the wild-type ligand, and binding affinity was abolished for the T3Y Q5Y ligand (Table 1, entries 15–18; Figure S4). The diminished affinity of the double mutant ligands likely stems from the loss of critical T3 side chain interactions. Loss of binding of the double tyrosine mutant suggests that PHD1 is unable to accommodate bulky aromatic residues when present in both positions 3 and 5. Interestingly, we observed stronger binding for the double mutants containing Q5R relative to their T3 single mutant counterparts (e.g., T3Y Q5R compared to T3Y). This may be due to charge–charge interactions between the positively charged Q5R residue and the negatively charged E305 and D306 residues in PHD1.42
Table 1.
Inhibition Constants Measured by Fluorescence Polarization for H3 Mutant Ligands (10-mer Peptides Comprised of the First 10 N-Terminal Residues of H3 Containing T3 and/or Q5 Mutations As Selected from the Array); Data Presented As Average ± Standard Deviation of Three Replicates
| Ligand | Ki (μM) | Ligand | Ki (μM) | ||
|---|---|---|---|---|---|
| 1. | wild-type | 0.82 ± 0.09 | 10. | Q5A | 0.75 ± 0.16 |
| 2. | T3V | 7.17 ± 1.00 | 11. | Q5F | 0.98 ± 0.18 |
| 3. | T3S | 18.8 ± 3.00 | 12. | Q5W | 1.05 ± 0.21 |
| 4. | T3A | >100 | 13. | Q5S | 1.85 ± 0.13 |
| 5. | T3Y | >100 | 14. | Q5E | 2.53 ± 0.91 |
| 6. | T3F | >100 | 15. | T3V Q5R | 2.54 ± 0.59 |
| 7. | T3D | >100 | 16. | T3Y Q5R | 12.8 ± 1.0 |
| 8. | Q5R | 0.62 ± 0.04 | 17. | T3V Q5W | 15.6 ± 2.5 |
| 9. | Q5Y | 0.65 ±0.15 | 18. | T3Y Q5Y | >100 |
We did observe several inconsistencies between the PI-SAMDI and FP results and therefore sought to better understand the sources of this discrepancy. We selected three ligands with which to compare the two assay formats: wild-type, a well-characterized, high-affinity ligand that showed only moderate fold enhancement in the PI-SAMDI array; Q5E, which displayed low fold enhancement in the PI-SAMDI array but showed high affinity in the FP assay; and Q5Y, which showed high affinity in both assays. As ligand length could play a role in the discrepancy, we synthesized the three ligands as both 6-mer and 12-mer peptides on a preparative scale and measured their fold enhancement with PI-SAMDI. Unlike our initial result, we observed that the wild-type and Q5Y ligands displayed very similar fold enhancements to one another when present as both 12-mers and 6-mers, suggesting that the fold enhancement produced by the wild-type ligand in the array was artificially low (Figure 5). As the increase in the rate of substrate modification by the reporter enzyme is influenced by the ratio of immobilized ligand to the substrate, the relative density of ligand peptide on the monolayer can affect the fold enhancement.29,36 We believe that the low and inconsistent yields that are common in small scale peptide synthesis and the number of transfer steps performed lead to errors in ligand concentration that affected the fold enhancement measured for the wild-type ligand and likely other ligands in the array. We also observed that the Q5E 12-mer ligand showed a very similar fold enhancement to the wild-type and Q5Y 12-mers but that the Q5E 6-mer ligand showed a significantly lower fold enhancement relative to the wild-type and Q5Y 6-mers (Figure 5). This disproportionate decrease in fold enhancement upon truncation of the Q5E ligand indicates that PHD1 binding to the 6-mer Q5E ligand is not representative of its binding to the 12-mer ligand. This finding explains why the Q5E ligand was characterized as low-affinity in the PI-SAMDI array but as high-affinity by FP and shows that ligand length can be an important consideration when interpreting the PI-SAMDI array. This experiment suggests that the disagreement between the PI-SAMDI array and FP data for the exceptional T3 and Q5 single mutant ligands and the double mutant ligands can be attributed to imprecision in ligand concentration and/or ligand length truncation in the PI-SAMDI array. In light of these considerations, we recommend that the array results presented here be treated not as an “answer key” but as a discovery tool to guide further exploration of PHD1’s binding preferences.
Figure 5.

Exploration of sources of discrepancy between PI-SAMDI and FP characterization of binding affinity for ligands in the 361-ligand array. Fold enhancements measured by PI-SAMDI for the resynthesized wild-type, Q5Y, and Q5E 12-mer ligands (left) and 6-mer ligands (right) are shown in the bar graph. Error bars represent the standard deviation of five replicates. Fold enhancements initially produced by the wild-type, Q5Y, and Q5E ligands in the PI-SAMDI array are shown in the heat map square below the corresponding label.
PHD1 Tolerates H3 Serotonylation at Q5.
We observed that PHD1 maintained high affinity for all selected H3 Q5 single mutants, even those featuring amino acids with bulky aromatic side chains. The Q5W ligand was a particularly notable high-affinity ligand identified by both the PI-SAMDI and FP assays. While this H3 mutation has not been observed among histone mutants, the indole side chain of tryptophan closely resembles the recently discovered serotonylated H3Q5 (H3Q5ser) mark.44 This modification has been found to colocalize with H3K4me3 marks, and several H3K4me3-specific reader domains have been shown to tolerate serotonylation at the Q5 position.44 Due to the structural similarity of tryptophan and serotonin and the newfound relevance of serotonylation of H3, we evaluated the affinity of PHD1 for the H3Q5ser ligand relative to the unmodified H3 ligand using both PI-SAMDI and FP. The wild-type and H3Q5ser ligands yielded highly similar fold enhancements and Ki values, indicating that serotonylation is well-tolerated by PHD1 (Figure 6). As expected, PHD1 displayed low affinity for an H3 T3D peptide, a negative control ligand of the same length, when analyzed by both PI-SAMDI and FP.
Figure 6.

PHD1 binds the Q5-serotonylated H3 ligand. FP binding curves (top) and inhibition constants and fold enhancements (bottom) for wild-type, Q5ser, and T3D H3 ligands (both FP and PI-SAMDI experiments used 12-mer peptides comprised of the first 10 N-terminal residues of H3 and a GC immobilization moiety). Inhibition constants are presented as average ± standard deviation of three replicates. Fold enhancements are presented as average ± standard deviation of eight replicates.
CONCLUSION
Here, we describe the combined use of the PI-SAMDI assay and FP to characterize the binding preferences of the PHD1 domain of KDM5A. In PI-SAMDI, binding between PHD1 and an immobilized ligand creates a proximity-dependent enhancement in the activity of a fused KDAC8 reporter enzyme on its coimmobilized substrate, enabling evaluation of relative affinities of the PHD1 domain for H3-derived ligands. We note that the fold enhancements for high-affinity ligands (Kd values in the range of 1–5 μM) were on average ~2.5 fold higher than those observed in our previous PI-SAMDI study of chromodomain binding.29 This difference is attributed to a more sensitive detection of the GSK(Ac)FGC substrate selected for this study relative to the GMK(Ac)FGC substrate previously used, which had a lower ionization efficiency and produced additional mass peaks due to oxidized adducts of the methionine residue.29,45 Superior detection of the new substrate further enhances the assay’s unique capacity to detect very low-affinity interactions, such as PHD1 binding to H3R2A and N-terminally acetylated H3.2,3 We note that due to potential overlap in the regions of the monolayer the reporter enzyme can access, the difference in binding affinity that can be discriminated by the PI-SAMDI assay depends on the affinity range of the ligands probed.29
Our use of the PI-SAMDI assay to efficiently characterize a 361-ligand array illuminates its role among the various protein–ligand binding assays. Analysis of such a large number of ligands by NMR, ITC, and fluorescence-based assays is simply not feasible due to the low throughput and larger sample needs of these methods. In contrast, the small material requirements of surface-phase reactions, the ability to integrate liquid handling robotics into sample preparation, the compatibility with common microtiter plates, and the fast readout provided by mass spectrometry make PI-SAMDI well-suited for efficient characterization of large numbers of ligands, particularly when they are organized in an array format. While assays of modified histone peptide and reader domain arrays have the throughput necessary to evaluate ligand libraries of comparable size, their poor sensitivity to low-affinity interactions often yields false negative results. By detecting a permanent enzymatic modification rather than directly observing a short-lived protein–ligand complex, PI-SAMDI is more sensitive to low-affinity binding interactions compared to affinity-based methods that require rinsing steps. Additionally, the use of self-assembled monolayers enables control over the relative density of the two immobilized peptides—at least across spots within an array—which allows the sensitivity of the assay to be tuned depending on the precise application.29,36 For example, the relative density of substrate and ligand was optimized to enhance the sensitivity of the assay toward the lower-affinity truncated ligands and facilitated detection of their binding to PHD1 by PI-SAMDI. Additionally, because PI-SAMDI does not require the development of fluorescently labeled ligands or an antibody that selectively recognizes the array-bound protein domain of interest, we expect it can be applied to a broader range of candidate receptors. Furthermore, as the assay detects modification of the enzymatic substrate as a measure of binding affinity, there are no restrictions in molecular weight or mass spectrometry compatibility of the ligand that can be probed. We also note that a variety of reporter activities can be used, as SAMDI-MS has been used to measure the activity of a wide range of enzymes—including phosphatases, proteases and kinases—that could serve as the reporter enzyme.46–48 For these reasons, the assay is versatile and was easily adapted from the previous study of chromodomain binding to the present investigation of PHD1 ligand preferences.
The results reported here reveal novel insights into the tolerance of the PHD1 domain toward the residues adjacent to the target lysine residue in H3-derived ligands. The observation that PHD1 is sensitive to modifications to the T3 position is consistent with the previous finding that PHD1 binding is abolished upon phosphorylation of H3T3 and highlights the importance of specific T3 side chain interactions in reader domain binding.2 The high affinity observed for ligands in which Q5 was mutated to hydrophilic and hydrophobic residues alike indicates that Q5 side chain interactions are not necessary for PHD1 binding. The discovery that the H3Q5ser mark is tolerated by PHD1 suggests that association of PHD1 with chromatin may be maintained in the context of H3Q5-serotonylated chromatin. This finding is consistent with the observation that several H3K4me3-specific reader domains are unaffected by Q5ser.44 While the precise biological implications of PHD1’s tolerance for H3Q5ser and the impact of this modification on the catalytic domain of KDM5A are yet to be elucidated, our investigation into substrate recognition combined with the analysis of the available structural data suggest that serotonylation could inhibit demethylation.49 Additionally, the ability of PHD1 to bind H3Q5ser marks suggests permissible interactions with the recently identified dopaminy-lated H3Q5 mark and other structurally similar H3Q5 modifications that may be discovered.50
Beyond contributing to the current understanding of PHD1-H3 binding, this work establishes PI-SAMDI as a useful preliminary screening tool for the high-throughput evaluation of interactions of epigenetic reader domains and their ligands. Using PI-SAMDI to evaluate a library of ligands for the first time revealed several challenges that are unique to its application to high-throughput studies and demonstrated that, as with many screening tools, the assay straddles a trade-off between throughput and accuracy. However, PI-SAMDI is valuable in systematic investigations of protein–ligand binding in that it can generate informed hypotheses to be tested by traditional methods. For example, the number of Q5 mutant ligands that showed high affinity in the array allowed us to confidently surmise that Q5 is dispensable for binding—an insight that would have been difficult to independently arrive at using lower-throughout techniques. Furthermore, the observed trend of promiscuity of Q5 substitution (including tolerance for tryptophan and other aromatic residues in this position) in the array led us to identify the serotonylated H3 as a well-tolerated ligand of PHD1. Collectively, our findings suggest that PI-SAMDI is best suited for profiling large numbers of prospective binding ligands and nominating interactions of interest for further characterization by more rigorous, though lower throughput, techniques. In this way, the combination of PI-SAMDI and FP represents a valuable platform in the exploration of reader domain-ligand binding.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institutes of Health under award number R01GM114044 (to D.G.F.). This material is based on research sponsored by the Air Force Research Laboratory under agreement number FA8650-15-2-5518 (to M.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Air Force Research Laboratory.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00891.
Representative MALDI-TOF spectrum of ligand array peptides (Figure S1), PI-SAMDI spectra showing degree of deacetylation of different KDAC8 substrates upon modification by the PHD1-KDAC8 fusion with and without coimmobilization of an H3 ligand (Figure S2), PI-SAMDI fold enhancements produced by H3 ligands of different lengths (Figure S3), FP binding curves and inhibition constants for PHD1 binding to H3 mutant ligands (Figure S4), and table of numerical fold enhancement values produced by ligands in the 361-ligand array (Table S1) (PDF)
The authors declare the following competing financial interest(s): M.M. is Founder of SAMDI Tech Inc., which uses SAMDI-MS to perform high-throughput screening and assay chemistry to clients in the pharmaceutical industry.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00891
Contributor Information
Sarah E. Anderson, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
James E. Longbotham, Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, California 94158, United States
Patrick T. O’Kane, Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Fatima S. Ugur, Chemistry and Chemical Biology Graduate Program, University of California San Francisco, San Francisco, California 94158, United States
Danica Galonić Fujimori, Department of Cellular and Molecular Pharmacology, Department of Pharmaceutical Chemistry, and Quantitative Biosciences Institute, University of California San Francisco, San Francisco, California 94158, United States.
Milan Mrksich, Department of Chemistry, Department of Biomedical Engineering, and Department of Cell and Developmental Biology, Northwestern University, Evanston, Illinois 60208, United States.
REFERENCES
- (1).Musselman CA, Lalonde M-E, Côte J, and Kutateladze TG (2012) Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Longbotham JE, Chio CM, Dharmarajan V, Trnka MJ, Torres IO, Goswami D, Ruiz K, Burlingame AL, Griffin PR, and Fujimori DG (2019) Histone H3 binding to the PHD1 domain of histone demethylase KDM5A enables active site remodeling. Nat. Commun 10, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Torres IO, et al. (2015) Histone demethylase KDM5A is regulated by its reader domain through a positive-feedback mechanism. Nat. Commun 6, 6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Horton JR, et al. (2010) Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol 17, 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pack LR, Yamamoto KR, and Fujimori DG (2016) Opposing chromatin signals direct and regulate the activity of lysine demethylase 4C (KDM4C). J. Biol. Chem 291, 6060–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li Y, and Kang C (2017) Solution NMR spectroscopy in target-based drug discovery. Molecules 22, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Meyer B, and Peters T (2003) NMR Spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem., Int. Ed 42, 864–890. [DOI] [PubMed] [Google Scholar]
- (8).Bhunia A, Bhattacharjya S, and Chatterjee S (2012) Applications of saturation transfer difference NMR in biological systems. Drug Discovery Today 17, 505–513. [DOI] [PubMed] [Google Scholar]
- (9).Ghai R, Falconer RJ, and Collins BM (2012) Applications of isothermal titration calorimetry in pure and applied research-survey of the literature from 2010. J. Mol. Recognit 25, 32–52. [DOI] [PubMed] [Google Scholar]
- (10).Maity S, Gundampati R, and Suresh Kumar T (2019) NMR methods to characterize protein-ligand interactions. Nat. Prod. Commun 14, 1934578–1984929. [Google Scholar]
- (11).Margineanu A, Chan JJ, Kelly DJ, Warren SC, Flatters D, Kumar S, Katan M, Dunsby CW, and French PMW (2016) Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Sci. Rep 6, 28186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rossi AM, and Taylor CW (2011) Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc 6, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Taitt CR, Anderson GP, and Ligler FS (2016) Evanescent wave fluorescence biosensors: Advances of the last decade. Biosens. Bioelectron 76, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mocz G, and Ross JA (2013) Fluorescence techniques in analysis of protein–ligand interactions, in Protein-Ligand Interactions: Methods and Applications (Williams MA, and Daviter T, Eds.), pp 169–210, Springer. [DOI] [PubMed] [Google Scholar]
- (15).Szymczak LC, Kuo H-Y, and Mrksich M (2018) Peptide arrays: development and application. Anal. Chem 90, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shanle EK, Shinsky SA, Bridgers JB, Bae N, Sagum C, Krajewski K, Rothbart SB, Bedford MT, and Strahl BD (2017) Histone peptide microarray screen of chromo and Tudor domains defines new histone lysine methylation interactions. Epigenet. Chromatin 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nady N, Min J, Kareta MS, Chédin F, and Arrowsmith CH (2008) A SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem. Sci 33, 305–313. [DOI] [PubMed] [Google Scholar]
- (18).Bock I, et al. (2011) Application of Celluspots peptide arrays for the analysis of the binding specificity of epigenetic reading domains to modified histone tails. BMC Biochem. 12, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bua DJ, et al. (2009) Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS One 4, e6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rothbart SB, Krajewski K, Strahl BD, and Fuchs SM (2012) Peptide microarrays to interrogate the “histone code. Methods Enzymol 512, 107–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cornett EM, et al. (2016) Substrate specificity profiling of histone-modifying enzymes by peptide microarray, in Methods in Enzymology (Marmorstein R, Ed.), pp 32–50, Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Price JV, et al. (2012) On silico peptide microarrays for high-resolution mapping of antibody epitopes and diverse protein-protein interactions. Nat. Med 18, 1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sutandy FXR, Qian J, Chen C-S, and Zhu H (2013) Overview of protein microarrays. Curr. Protoc. Protein Sci 72, 27.1.1–27.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kim J, et al. (2006) Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 7, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen J, Sagum C, and Bedford MT (2020) Protein domain microarrays as a platform to decipher signaling pathways and the histone code. Methods 184, 4. [DOI] [PubMed] [Google Scholar]
- (26).Kaustov L, et al. (2011) Recognition and specificity determinants of the human Cbx chromodomains. J. Biol. Chem 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Volkmer R, Tapia V, and Landgraf C (2012) Synthetic peptide arrays for investigating protein interaction domains. FEBS Lett. 586, 2780–2786. [DOI] [PubMed] [Google Scholar]
- (28).Petell CJ, Pham AT, Skela J, and Strahl BD (2019) Improved methods for the detection of histone interactions with peptide microarrays. Sci. Rep 9, 6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).O’Kane PT, and Mrksich M (2017) An assay based on SAMDI mass spectrometry for profiling protein interaction domains. J. Am. Chem. Soc 139, 10320–10327. [DOI] [PubMed] [Google Scholar]
- (30).Gurard-Levin ZA, and Mrksich M (2008) Combining self-assembled monolayers and mass spectrometry for applications in biochips. Annu. Rev. Anal. Chem 1, 767–800. [DOI] [PubMed] [Google Scholar]
- (31).Prime K, and Whitesides G (1991) Self-assembled organic monolayers: model systems for studying adsorption of proteins at surfaces. Science 252, 1164–1167. [DOI] [PubMed] [Google Scholar]
- (32).Szymczak LC, and Mrksich M (2019) Using peptide arrays to discover the sequence-specific acetylation of the histidine-tyrosine dyad. Biochemistry 58, 1810–1817. [DOI] [PubMed] [Google Scholar]
- (33).Szymczak LC, Huang C-F, Berns EJ, and Mrksich M (2018) Combining SAMDI mass spectrometry and peptide arrays to profile phosphatase activities, in Methods in Enzymology (Allen KN, Ed.), pp 389–403, Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kim J, and Mrksich M (2010) Profiling the selectivity of DNA ligases in an array format with mass spectrometry. Nucleic Acids Res. 38, e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liao X, Su J, and Mrksich M (2009) An adaptor domain-mediated autocatalytic interfacial kinase reaction. Chem. - Eur. J 15, 12303–12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li J, Nayak S, and Mrksich M (2010) Rate enhancement of an interfacial biochemical reaction through localization of substrate and enzyme by an adaptor domain. J. Phys. Chem. B 114, 15113–15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gurard-Levin ZA, Scholle MD, Eisenberg AH, and Mrksich M (2011) High-throughput screening of small molecule libraries using SAMDI mass spectrometry. ACS Comb. Sci 13, 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sanchez R, and Zhou M-M (2011) The PHD finger: a versatile epigenome reader. Trends Biochem. Sci 36, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Jain K, Fraser CS, Marunde MR, Parker MM, Sagum C, Burg JM, Hall N, Popova IK, Rodriguez KL, Vaidya A, Krajewski K, Keogh M-C, Bedford MT, and Strahl BD (2020) Characterization of the plant homeodomain (PHD) reader family for their histone tail interactions. Epigenet. Chromatin 13, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Horton JR, Gale M, Yan Q, and Cheng X (2017) The molecular basis of histone demethylation, in DNA and Histone Methylation as Cancer Targets (Horton JR, Gale M, Yan Q, and Cheng X, Eds.), pp 151–219, Springer. [Google Scholar]
- (41).Ali M, Hom RA, Blakeslee W, Ikenouye L, and Kutateladze TG (2014) Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochim. Biophys. Acta, Mol. Cell Res 1843, 366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Longbotham JE, Kelly MSJ, and Fujimori DG (2019) Recognition of histone H3 methylation states by the PHD1 domain of histone demethylase KDM5A, BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Young JD, et al. (1990) Coupling efficiencies of amino acids in the solid phase synthesis of peptides. Pept. Res 3, 194–200. [PubMed] [Google Scholar]
- (44).Farrelly LA, et al. (2019) Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hollemeyer K, Heinzle E, and Tholey A (2002) Identification of oxidized methionine residues in peptides containing two methionine residues by derivatization and matrix-assisted laser desorption/ionization mass spectrometry. Proteomics 2, 1524–1531. [DOI] [PubMed] [Google Scholar]
- (46).Moully EH, Berns EJ, and Mrksich M (2019) Label-free assay of protein tyrosine phosphatase activity in single cells. Anal. Chem 91, 13206–13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Dai R, Ten AS, and Mrksich M (2019) Profiling protease activity in laundry detergents with peptide arrays and SAMDI mass spectrometry. Ind. Eng. Chem. Res 58, 10692–10697. [Google Scholar]
- (48).Min D-H, Su J, and Mrksich M (2004) Profiling kinase activities by using a peptide chip and mass spectrometry. Angew. Chem., Int. Ed 43, 5973–5977. [DOI] [PubMed] [Google Scholar]
- (49).Petronikolou N, Longbotham JE, and Fujimori DG (2020) Extended recognition of the histone H3 tail by histone demethylase KDM5A. Biochemistry 59, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lepack AE, et al. (2020) Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 368, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.