

Abstract
Astrocyte reactivity is a hallmark of neuroinflammation that arises with Alzheimer’s disease (AD) and nearly every other neurodegenerative condition. While astrocytes certainly contribute to classic inflammatory processes (e.g. cytokine release, waste clearance, and tissue repair), newly emerging technologies for measuring and targeting cell specific activities in the brain have uncovered essential roles for astrocytes in synapse function, brain metabolism, neurovascular coupling, and sleep/wake patterns. In this review, we use a holistic approach to incorporate, and expand upon, classic neuroinflammatory concepts to consider how astrocyte dysfunction/reactivity modulates multiple pathological and clinical hallmarks of AD. Our ever-evolving understanding of astrocyte signaling in neurodegeneration is not only revealing new drug targets and treatments for dementia but is suggesting we reimagine AD pathophysiological mechanisms.
Keywords: Astrocytes, Reactive astrocytes, Alzheimer’s disease, Dementia, Neuroinflammation, Neurodegeneration
1. Introduction
Astrocytes are an abundant and highly ramified cell type in the brain with processes that ensheathe the cerebrovasculature as well as many, if not most, excitatory synaptic connections. Astrocytes provide essential metabolic support for neighboring neurons and other cell types, while simultaneously protecting their neighbors through the uptake of excess glutamate and K+ as well as the release of growth factors, mitogens, and other essential chemical messengers. With aging, injury, and disease, astrocytes can undergo remarkable morphologic and molecular phenotype changes, the most extensively characterized of which are cellular hypertrophy and the upregulation of the intermediate filament protein, GFAP. Astrocyte hypertrophy, in close proximity to “senile plaques”, was one of the primary pathologies identified by Alois Alzheimer in 1910 and is now recognized as a hallmark of AD and most other forms of brain injury and chronic neurodegeneration (Verkhratsky et al., 2019). Despite the long history and prominent appearance of reactive astrocytes in AD, these cells have generally taken a back seat to other major cell types in the brain, namely neurons and microglia. As a consequence, the functional impact of reactive astrocytes on AD pathophysiology has remained murky and speculative. In this review, we will discuss evolving evidence showing that several major AD pathophysiological processes including neuroinflammation, synapse dysfunction/degeneration, impaired cerebrovascular function, hypometabolism, and sleep disturbances are all fundamentally linked through astrocyte reactivity and/or dysfunction. Collectively, the evidence suggests that astrocytes provide many molecular targets that could be exploited for wide-ranging therapeutic benefits.
2. Astrocyte reactivity arises early in disease
Hallmark signs of astrocyte reactivity appear at very early stages of age-related cognitive decline (Landfield et al., 1977). In humans, most of the evidence supporting the early emergence of reactive astrocytes comes from studies on postmortem tissue showing that GFAP and/or a number of other astrocyte-related proteins and mRNA species are altered in individuals with mild cognitive impairment (MCI) or pre-clinical AD (Schipper et al., 2006; Assaraf et al., 2007; Abdul et al., 2009; Owen et al., 2009). In the last decade, postmortem evidence for the early appearance of astrocyte reactivity has been confirmed in human subjects using positron emission tomography (PET) and novel PET tracers, like 11C-deuterium-L-deprenyl (11C-DED) and (S)-(2-methylpyrid-5-yl)-6-[(3-[18F]fluoro-2-hydroxy)propoxy]quinoline (18F-SMBT-1), which bind to the reactive astrocyte marker monoamine oxidase B (MAO-B) (Carter et al., 2012; Harada et al., 2020). Nordberg and colleagues have used 11C-DED to reveal significant elevations in astrocyte reactivity throughout many cortical and subcortical regions in living humans with MCI, relative to age-matched healthy controls (Carter et al., 2012). Though 11C-DED binding was most prominent in MCI individuals with elevated 11C-PIB binding, consistent with the association of reactive astrocytes with amyloid deposits, 11C-DED was also found in MCI subjects with negligible 11C-PIB levels. Elevated 11C-DED uptake was also observed in individuals with autosomal dominant AD, long before the appearance of clear cognitive symptoms (Rodriguez-Vieitez et al., 2016). Astrocyte reactivity, detected in vivo with 11C-DED, has similarly been reported in a variety of animal models of AD-like pathology –either at the outset of, or prior to the development of significant amyloidosis and neurodegeneration (Rodriguez-Vieitez et al., 2015; Olsen et al., 2018). The early appearance of astrocyte reactivity in AD may provide a key upstream mechanism for many of the intricate, and highly interconnected processes that go awry in AD including neuroinflammation, synapse dysfunction, cerebrovascular pathology, and hypometabolism.
3. Neuroinflammation: extracellular mediators and transcription factor pathways
Though different terms, including astrocyte activation and astrogliosis have been used interchangeably with astrocyte “reactivity”, the phenotype change found in AD and other forms of neurodegeneration is probably best described as a reaction to pathological factors (Escartin et al., 2021). In AD, this change appears to encompass alterations in morphology and/or biochemical properties, rather than an increase in the number of astrocytes, per se (Serrano-Pozo et al., 2013). As astrocytes exhibit substantial heterogeneity depending on brain region and local interacting partners (e.g. different neurons and/or synapse subtypes, blood vessels, etc) (Batiuk et al., 2020), it shouldn’t be surprising that astrocyte reactivity is also a highly heterogeneous phenomenon. Reactive astrocytes may include unique morphologic features (e.g. differing levels of GFAP expression, orientation of processes toward and/or into amyloid deposits, degeneration, or clasmatodendrosis) and/or the presence of unique protein markers (Perez-Nievas and Serrano-Pozo, 2018; Sofroniew, 2020). Astrocytic tauopathies, which may be found in AD, can include up to six different astrocyte subtypes: thorn-shaped, granular/fuzzy, tufted, ramified, plaques, and globular inclusions (Kovacs, 2020). Recently, there has been much interest in the field about the binary classification of reactive astrocytes according to an “A1” neurotoxic phenotype or an “A2” neuroprotective phenotype based on distinct transcriptional profiles (Zamanian et al., 2012; Liddelow et al., 2017). While this categorization is conceptually useful, it is unlikely it effectively captures the nuances of astrocyte heterogeneity at the molecular or functional levels (Escartin et al., 2021; Sofroniew, 2020). We will therefore avoid this terminology in most cases in favor of describing discrete astrocyte-based properties/functions and how they change with AD.
3.1. Factors that modulate astrocyte reactivity
Amyloid is one of the best characterized factors for triggering astrocyte reactivity. Delivery of pathogenic Aβ peptides to primary astrocytes (Pike et al., 1994), or intracranial delivery to intact animals (Craft et al., 2004) is associated with robust changes in astrocyte morphology. And, of course, Aβ deposits in both humans and in rodent models of amyloidosis are typically surrounded by reactive astrocytes (Duffy et al., 1980; Dickson et al., 1988; Mandybur and Chuirazzi, 1990; Borchelt et al., 1997; Benzing et al., 1999; Oakley et al., 2006) (see Fig. 1A). In addition to AD specific pathology, astrocyte reactivity arises from loss of oxygen and glucose during hypoperfusion and from the entry of blood borne factors into the parenchyma following cerebral infarct/hemorrhage (Symon et al., 1975; Kowianski et al., 2003). Frank neuronal damage and the release of reactive oxygen species (ROS), nucleotides, excitatory amino acids, and myelin fragments also commonly trigger reactive astrocyte phenotypes in a variety of animals, as do numerous cytokine species arising from reactive microglia and other sources (Giovannoni and Quintana, 2020; Sofroniew, 2020). Many of these factors are found at elevated levels in AD and linked to neural dysfunction.
Fig. 1. Reactive astrocytes are a key component to neuroinflammation in AD.
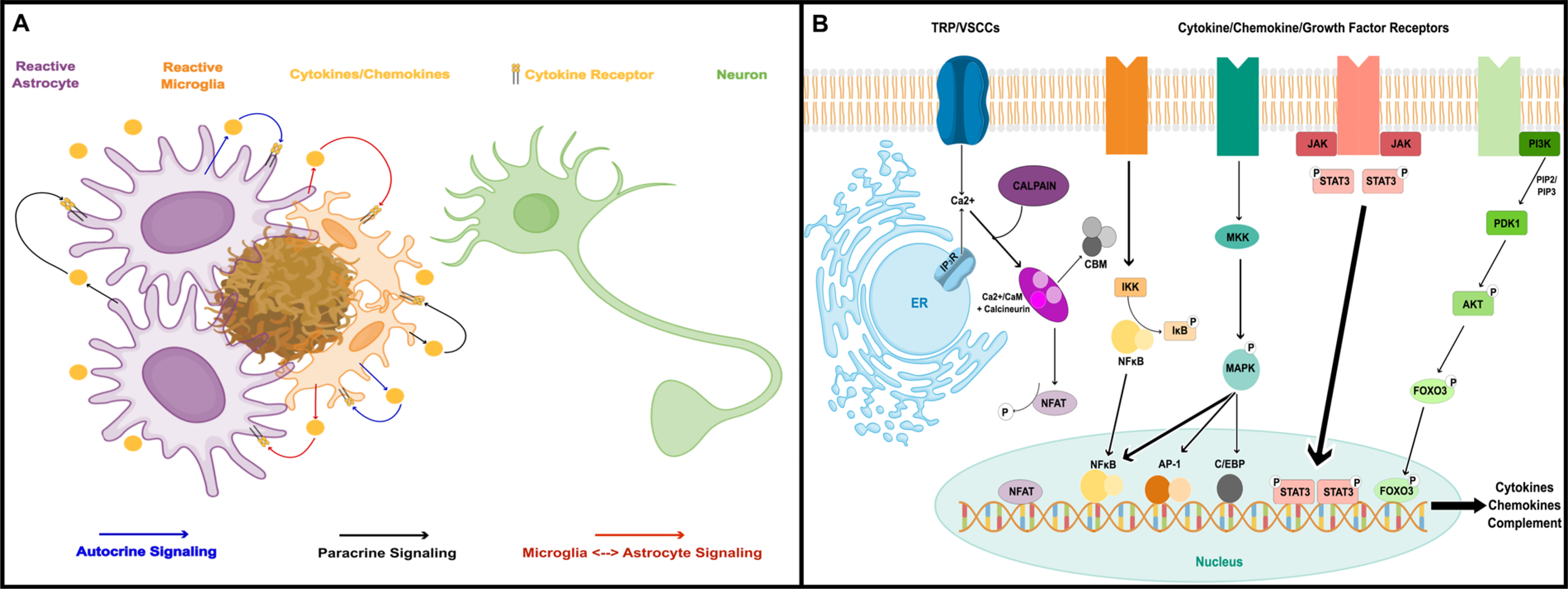
A) Cartoon showing astrocytes and microglia surrounding a parenchymal amyloid deposit. Both cell types respond to amyloid (and other extracellular factors) with morphologic and biochemical changes including the release of numerous cytokines, chemokines, and other inflammatory factors which can maintain and/or propagate glial reactivity. Neuroinflammatory processes resulting from chronic glial activation can have many deleterious (and sometimes beneficial) effects on neurons. B) Inflammatory factors trigger astrocyte reactivity through a number of transcriptional pathways involving NFκB, MAPK, Jak/Stat, and/or FOXO3. Concurrent Ca2+ signaling and/or Ca2+ dysregulation leads to the activation of the calcineurin/NFAT pathway, which further shapes astrocyte reactivity through extensive interactions with other transcription factors and second/third messenger systems. These pathways, in turn, regulate the production of numerous cytokines and chemokines involved in triggering and maintaining glial reactivity.
Similar to reactive microglia, in response to perturbations, astrocytes produce and/or release an array of inflammatory mediators, including cytokines (e.g. TNFα, TGFβ, IK-1β, IL-6, and INFγ), chemokines (e.g. MIP-1α, CXCL10, CCL5), complement factors (e.g. C3, C5-C9), and ROS (Giovannoni and Quintana, 2020). Many of these factors are upregulated in AD and have been implicated in both harmful and beneficial neuroinflammatory effects (Dansokho and Heneka, 2018). Selective knockdown of inflammatory signaling pathways in astrocytes has been shown to reduce other general markers of neuroinflammation (e.g. microglial activation, tissue cytokine levels) in disease/injury models (Brambilla et al., 2005; Furman et al., 2012; Ben Haim et al., 2015b). In addition to directly interacting with other local CNS constituents, astrocyte-based inflammatory signaling also has been shown to directly influence vascular and perivascular cells leading to alterations in blood brain barrier permeability (Daniels et al., 2017). Astrocytes may also provide chemoattractant cues to recruit peripheral macrophages, white blood cells, and lymphocytes into the brain parenchyma in response to neuronal damage and/or degeneration (Babcock et al., 2003; Moynagh, 2005). It’s clear from these observations and others, that reactive astrocytes are more than just a biomarker for neuroinflammation—they are critical effector cells.
3.2. Transcription factor pathways
Given the intimate association of reactive astrocytes with neuroinflammation, it may not be surprising that the transcriptional pathways linked to the reactive astrocyte phenotype are some of the same pathways involved in peripheral and innate immune/inflammatory responses (Fig. 1B). Numerous studies have shown that key components of JAK/STAT, FOXO3, C/EPB, AP-1 and NFκB pathways are expressed in astrocytes in vitro and in vivo where they are coupled to numerous cytokine receptors, toll-like receptors, and CD proteins, and are therefore activated by many of the same factors that trigger astrocyte reactivity (i.e. pro-inflammatory cytokines, blood-borne factors, Aβ and tau oligomers) (Cui et al., 2011; Ben Haim et al., 2015a; Brenner et al., 2019). In turn, these transcriptional pathways promote the expression of cytokines, chemokines, complement factors, prostaglandins, and extracellular matrix modifying factors that drive or maintain glial reactivity and neuroinflammation. In addition to these classic inflammatory pathways, astrocytes also use Ca2+ to regulate transcription through activation of the protein phosphatase, calcineurin, and its target transcription factor, nuclear factor of activated T cells (NFATs) (Furman and Norris, 2014; Lee et al., 2016; Sompol and Norris, 2018). Similar to the other classic inflammatory pathways, activation of calcineurin/NFATs leads to the production of numerous cytokine species and/or the modulation of other key metabolic changes linked to the reactive astrocyte phenotype. While calcineurin provides a relatively direct route between Ca2+ signals and transcriptional activity (through NFATs), many of the other classic inflammatory pathways are also affected by extensive calcineurin-mediated crosstalk (Furman and Norris, 2014). For instance, FOXO3 and NFκB are both activated in astrocytes directly (FoxO3) or indirectly (NFκB) by calcineurin (Fernandez et al., 2012). Depending on the source of cellular activation, astrocytic calcineurin can drive pro-inflammatory responses through the coordination of FOX-O3-NFκB interactions, or anti-inflammatory responses through the promotion of NFκB-PPARγ interactions. NFATs also interact closely with different transcription factors, such as NFκB, AP-1, and others, to trigger dramatic changes in cellular phenotype (Rao et al., 1997; Hogan et al., 2003).
Thus, addition of a Ca2+ signal provides a critical mechanism for shaping and/or fine-tuning reactive astrocyte responses. As a corollary, Ca2+ dysregulation in reactive astrocytes, which has been noted in multiple disease models including AD (Sompol and Norris, 2018; Verkhratsky, 2019), may be a major contributing factor to the maintenance of chronic neuroinflammation. With severe Ca2+ dysregulation resulting from excitotoxicity and/or amyloid pathology, calcineurin is proteolized into a hyperactive fragment (ΔCN) that is partially uncoupled from regulated Ca2+ signaling (Wu et al., 2004; Wu et al., 2010; Mohmmad Abdul et al., 2011). Pathologic ΔCN fragments alongside the NFAT4 isoform are found at very high levels in subsets of reactive astrocytes in humans and mouse models, usually in conjunction with amyloid deposits, vascular pathology, and upregulation of GFAP (Serrano-Perez et al., 2011; Pleiss et al., 2016; Sompol et al., 2017). Moreover, forced overexpression of ΔCN in primary astrocytes leads to the transcriptional induction of numerous immune/inflammatory genes associated with astrocyte reactivity (Norris et al., 2005, Fernandez et al., 2007) and propagates elevated CN/NFAT signaling across nearby astrocyte networks (Sama et al., 2008). In wildtype rodents, overexpression of ΔCN in astrocytes has been associated with both detrimental (Pleiss et al., 2016) and beneficial (Fernandez et al., 2012) consequences for surrounding nervous tissue, perhaps reflecting the complex role of neuroinflammation in degenerative diseases.
4. Astrocyte reactivity and synapses
Fast communication between neurons in the CNS occurs primarily through the transfer of neurotransmitter molecules across synapses. The process of synaptic transmission, primarily the vesicular release and repackaging of neurotransmitters, is energetically expensive and is overwhelmingly responsible for the disproportionate amount of oxygen and glucose consumed by the brain (Harris et al., 2012). Given their high-metabolic demand, synapses are also among the most vulnerable structures in the brain and are easily damaged by insults that occur acutely or arise with aging. Among the most fascinating and important properties of synapses is the capacity to change and adapt to new experiences. Synaptic plasticity is at the center of who we are as individuals; it’s how we learn and remember. When synapses are lost, cognitive deficits usually follow. Reduced synapse number and density, or reduced expression of synaptic proteins, is associated with the very earliest stages of cognitive decline in humans with AD (Mufson et al., 2012) and also in many animal models of AD-like pathology (Spires-Jones and Knafo, 2012; Pozueta et al., 2013). In fact, synapse loss is generally a better predictor of failing cognition than other major AD neuropathological hallmarks. Consequently, synapses are intensely studied, not only for their mechanistic role in pathophysiology, but also for their potential as a therapeutic target.
Glutamate is the most common excitatory neurotransmitter in the mammalian brain. After release from presynaptic terminals, glutamate interacts primarily with two major ionotropic glutamate receptors on the postsynaptic membrane: AMPA/kainate receptors and NMDA receptors (AMPARs and NMDARs). NMDARs provide the primary Ca2+ signal responsible for initiating receptor trafficking events (e.g. AMPAR insertion into or removal from the postsynaptic membrane) and gene expression changes necessary for long-term increases and decreases in synaptic strength implicated in new learning and memory formation: i.e. long-term potentiation (LTP) and (LTD) (Andersen et al., 2017). Though essential for mediating synaptic plasticity, high levels of Ca2+ in neurons, arising from excess glutamate receptor activation can lead to the degeneration of synapses and neurites, and ultimately cause neuronal death (Zhou et al., 2015; Carvajal et al., 2016). Glutamate-mediated excitotoxicity has been hypothesized to play a major role in the neurodegeneration that arises with AD and related disorders (Wang and Reddy, 2017; Armada-Moreira et al., 2020). In support of this hypothesis, individuals with AD are at greater risk for seizures (Vossel et al., 2017; Asadollahi et al., 2019; Gail Canter et al., 2019; Powell et al., 2019), and many rodent models of AD-like pathology exhibit signs of synaptic hyperexcitability, especially in regions of frank pathology (Siskova et al., 2014; Siwek et al., 2015; Tamagnini et al., 2015; Maeda et al., 2016; Fontana et al., 2017; Sompol et al., 2017; Hijazi et al., 2019). Alleviation of excitotoxicity is thought to underlie the modest clinical efficacy observed in AD patients treated with the weak NMDAR blocker memantine (Kabir et al., 2019).
The vast majority of research on synapse loss and dysfunction in AD has historically focused on neuron-intrinsic mechanisms, including alterations in neuronal Ca2+ regulation, oxidative stress, and gene regulation. However, it’s increasingly appreciated that synapse function and stability are also heavily regulated by other extra-neuronal cell types, including astrocytes and microglia. Astrocytes, in particular, appear to play fundamental roles in regulating synapse formation, stability, and turnover under both healthy and pathological conditions (Allen and Eroglu, 2017). Many, if not most, of the synapses in the mammalian CNS are in very close proximity to astrocyte processes. In adult rats, for instance, astrocytes appear to contact up to 90 % of synapses in the hippocampus, depending on the synapse subtype (Witcher et al., 2007). At the astrocyte/synapse interface, specialized astrocyte membranes ensheath or “cradle” pre and postsynaptic neuronal elements, extracellular matrix (ECM) components, and even microglial processes (Verkhratsky and Nedergaard, 2014). Within these cradles, astrocytes express and/or excrete numerous proteins that respond to and/or modulate the function and structure of synapses. There is presently much debate over whether astrocytes directly contribute to synaptic transmission via the release of gliotransmitters. Resolving this debate is beyond the scope of this review, but evidence for and against physiologic gliotransmission has been summarized in outstanding dual perspective articles: pro-gliotransmission (Savtchouk and Volterra, 2018) and anti-gliotransmission (Fiacco and McCarthy, 2018). In the current review, we instead focus on key astrocyte-derived proteins that both interact with synapses, and undergo changes with astrocyte reactivity and/or AD. These include (1) scaffolding proteins that modulate synapse formation and stability; (2) innate immunity factors that regulate phagocytic removal of synapses; (3) glutamate transporters that modulate the duration of chemical transmission and dampen excitability; and (4) cytokines that modulate synaptic viability and plasticity (see Fig. 2).
Fig. 2. Astrocytes regulate synaptic transmission, synapse stability, and synapse removal.

Pre- and postsynaptic neuronal compartments are cradled by specialized astrocytic processes that express numerous factors that directly shape synapse/structure and function. Astrocyte secreted factors (ASF), including thrombospondin (TSP), hevin, and sparc help anchor pre- and postsynaptic cell adhesion molecules together. This arrangement not only aligns and stabilizes presynaptic terminals with dendritic spines, but it also helps to cluster important synaptic machinery, including postsynaptic density constituents and neurotransmitter receptors, to active zones. Reactive astrocytes in AD brain tissue may release less of the pro-synaptogenetic factors thrombospondin and hevin, relative to sparc (which opposes hevin-mediated synaptogenesis), leading to a net loss of synapses. Astrocytes are also a major source for complement C3, which is released by reactive astrocytes and binds to C3 receptors (C3R) on “weakened” pre- and postsynaptic elements, leading to microglial-mediated phagocytosis. Increased C3 levels arising from reactive astrocytes have been shown to contribute to abnormal synapse loss in mouse models of amyloid pathology. Finally, astrocytes express several different types of glutamate transporters (EAATs 1 and 2, aka Glast and Glt-1) that help terminate synaptic glutamate signaling and prevent hyperactivation of extrasynaptic glutamate receptors. The downregulation of EAAT2/Glt-1 levels and/or function in reactive astrocytes is thought to be a primary mechanism for excitotoxic neuronal degeneration during AD.
4.1. Secreted scaffolding proteins
Synapses are formed and maintained in part by interactions between pre and postsynaptic adhesion molecules in the synaptic cleft. These proteins not only promote synapse stability, they help to cluster critical pre- and postsynaptic elements (e.g. synaptic vesicles and neurotransmitter receptors) in optimal locations for synaptic transmission. Very commonly, pre-synaptic adhesion molecules, like neurexins, interact with specific postsynaptic partners, like neuroligins. However, these interactions are usually indirect and require the help of scaffolding proteins to make stable connections. These scaffolding proteins are secreted and may arise from neurons and/or astrocytes (Yuzaki, 2018). The matricellular proteins thombospondin-1, hevin and sparc are some of the best characterized scaffolding proteins produced and released by astrocytes. Thrombospondin and hevin interact with cell adhesion molecules to promote synaptogenesis, whereas sparc inhibits hevin-mediated synaptogenesis (Christopherson et al., 2005; Jones et al., 2011; Kucukdereli et al., 2011). Levels of glial-derived scaffolding proteins tend to be elevated during development, as synaptic connectivity is established and fine-tuned, and then decreased somewhat during adulthood. Expression levels increase again following acute brain injury and are strongly associated with astrocyte reactivity (Jones and Bouvier, 2014). Under these conditions, reactive astrocytes and glial-derived scaffolding proteins may play a critical role in sprouting and synapse remodeling.
During AD, changes in thrombospondin 1, hevin, and sparc appear to be more complex or at least different from what is observed with acute neurodegeneration. For instance, thrombospondin-1 levels are reduced in human AD brain, in mouse models of parenchymal amyloid pathology, and in primary astrocytes treated with Aβ peptides (Son et al., 2015). Moreover, exogenous application of thrombospondin-1 prevented Aβ-mediated loss of synaptic markers, such as PSD-95. A similar reduction in thrombospondin-1 was observed in astrocytes from human subjects with Down Syndrome (Garcia et al., 2010), which shares common amyloid pathologies with AD. Notably, primary neurons co-cultured with human Down Syndrome astrocytes exhibited reduced dendritic spine density and synapse viability: a finding that was mitigated by addition of thrombospondin-1 to the cell culture medium. In addition to synaptic-modulatory properties, thrombospondin-1 is also a potent inhibitor of angiogenesis in peripheral tissues (Lawler, 2002). As discussed below, vascular inflammation and reduced cerebral perfusion lead to increased angiogenesis during AD pathology (Vagnucci and Li, 2003), which is thought to disrupt the blood-brain-barrier (BBB) and exacerbate inflammation and other brain pathologies. Thus, the loss of thrombospondin-1 in reactive astrocytes during AD may also contribute significantly to cerebrovascular dysfunction.
As mentioned, hevin and sparc are also altered in AD brain. qPCR performed on laser captured brain sections revealed mRNA for sparc is increased relative to hevin. Elevations in sparc were particularly pronounced near Aβ deposits, whereas hevin levels were generally reduced in AD relative to cognitively normal individuals (Strunz et al., 2019). The relative increase in sparc associated with astrocyte reactivity may offset hevin-induced synaptogenesis contributing to a net loss of synapses in AD. However, somewhat at odds with this study is another report that identified hevin in cerebrospinal fluid (CSF) samples from AD cases using a proteomic fingerprinting approach. In a panel of 7 peptide markers, hevin provided the greatest predictive value for discerning AD from cognitively normal controls (Vafadar-Isfahani et al., 2012). Further research is needed to clarify the extent to which hevin and sparc change with AD, and how these changes affect synaptic maintenance and/or turnover.
4.2. Complement factors
Establishing efficient neural networks not only requires the formation and maintenance of necessary synaptic contacts, it also requires the elimination of unnecessary or dysfunctional contacts. Resident microglia play the predominant role in physically removing synaptic elements, but their actions appear to be guided, in part, by the release of complement components C1q from neurons and C3 from astrocytes (Stevens et al., 2007; Stephan et al., 2012). At synapses of the developing nervous system, nearby astrocytes induce the release of C1q from neurons, leading to the cleavage of astrocyte derived C3. Activated C3 fragments (C3b), in turn, bind to or “tag” inactive or dysfunctional synaptic structures which are bound by C3 receptors (C3R) expressed on microglial cells. C3b-C3R interactions then trigger microglial-mediated phagocytosis of tagged synapses.
Several reports have shown that C3 levels are increased in reactive astrocytes in AD and mouse models of AD-like pathology (Tomimoto et al., 1997; Fonseca et al., 2011). Elevated C3 expression in astrocytes has been linked to hyperactive calcineurin signaling (Norris et al., 2005) and NFκB activation (Lian et al., 2015). In mouse models of parenchymal amyloidosis, a significantly greater proportion of synapses were associated with C1q and C3, relative to WT littermates (Hong et al., 2016). NFκB-mediated C3 expression led to deleterious changes in dendritic spines, neuronal Ca2+ dysregulation, and impaired synaptic function (Lian et al., 2015). Similarly, knockdown of C3 preserved synaptic density and improved cognition in APP/PS1 mice, despite enhanced amyloid plaque load under these conditions (Shi et al., 2017). Together, these findings suggest that astrocyte derived C3 plays a critical role in synapse loss and cognitive decline in AD.
4.3. EAATs
Excitatory amino acid transporters (EAATs), including EAAT1 (mouse homologue, GLAST1) and EAAT2 (mouse homologue, Glt1) are highly expressed in astrocyte plasmalemma near synapses, where they take up 80 % or more of the glutamate in the extracellular space (Lopez-Bayghen and Ortega, 2011) and help terminate glutamatergic synaptic transmission (Weng et al., 2007). Perhaps even more important than fine-tuning synaptic transmission, astrocytic EAATs prevent glutamate spillover at the synapse and minimize hyperactivation of extra-synaptic receptors (Shen et al., 2014). In this role, EAATs provide a fundamental protective mechanism against glutamate-related hyperexcitability and excitotoxicity. For every glutamate molecule transported, EAATs co-transport three Na+ ions and one H+, and countertransport one K+ ion (Levy et al., 1998). Once taken into the astrocyte cytosol, glutamate is converted to glutamine by glutamine synthetase and transported back to neurons where it is converted back to glutamate (i.e. the so-called glutamate-glutamine cycle) (Robinson and Jackson, 2016). The electrogenic and redox properties of EAATs stimulate glucose uptake and drive the glycolytic production of lactate, arguably the preferred energy substrate of neurons (Pellerin and Magistretti, 1994). Thus, astrocytic EAATs are also critical for ensuring that energy substrates are made available to neurons according to their demand (see Fig. 3).
Fig. 3. Astrocytic metabolic pathways are essential for meeting the energy demands of neurons.

Astrocytes are the major source of lactate in the brain, which appears to be the preferred energy substrate of neurons. Three intertwined sources drive lactate production in astrocytes: glycogen, glucose uptake through GLUT1 transporters, and glutamate uptake through EAATs. Glucose taken up from blood vessels, or derived from the breakdown of glycogen, is converted to pyruvate and subsequently lactate, which is ultimately released to nearby neurons. Glutamate uptake during neuronal activity helps create an electrochemical gradient (with nearby Na+/K+ exchangers) that facilitates both glucose uptake via GLUT1 and the conversion of glucose to lactate. Glutamate uptake via astrocytic EAATs is also recycled back to neurons in the form of glutamine. Inhibition of astrocyte glycogen metabolism at any of these steps has been shown to be detrimental to both acute neural function and for the extended processes of LTP and memory formation, which are disrupted in many rodent models of AD-like pathology.
In many brain regions, the majority of glutamate uptake is carried out by EAAT2/Glt1, which is either lost or undergoes extensive oxidative damage in a variety of neurodegenerative diseases such as amyotrophic lateral sclerosis, stroke, Alexander disease, and AD (Sheldon and Robinson, 2007). In primary astrocytes, the downregulation of EAATs are triggered by extracellular factors (e.g. pro-inflammatory cytokines, Aβ peptides) and transcriptional pathways (i.e. NFATs and NFκB) linked to astrocyte reactivity (Su et al., 2003; Prow and Irani, 2008; Sama et al., 2008; Abdul et al., 2009; Tolosa et al., 2011; Fang et al., 2012). EAAT2 protein and/or activity is lost in AD brain (Masliah et al., 1996; Abdul et al., 2009). These changes can occur at very early stages of cognitive decline (Abdul et al., 2009), and are observed in parallel with signs of astrocyte reactivity (Simpson et al., 2010), or upregulation of NFAT transcription factors (Abdul et al., 2009). A similar drop off in Glt-1 levels/function has been reported in several common rodent models of AD-like pathology (Schallier et al., 2011; Scimemi et al., 2013; Meeker et al., 2015). Downregulation of Glt-1 expression in reactive astrocytes is clearly sufficient to precipitate synaptic hyperexcitability and excitotoxic neurodegeneration in experimental models (Rothstein et al., 1996; Petr et al., 2015). Moreover, forced overexpression of EAAT2/Glt-1 in astrocytes and/or pharmacologic activation of EAAT2/Glt-1 in disease models imparts neuroprotection and enhances cognitive function (Prow and Irani, 2008; Zumkehr et al., 2015; Fontana et al., 2016; Hefendehl et al., 2016). Notably, riluzole, a polypharmacological compound that enhances EAAT2 function, is FDA approved for the treatment of ALS and is currently in Phase 2 clinical trials for AD (NCT01703117).
4.4. Alpha7 nicotinic acetylcholine receptors
Alterations in CNS cholinergic signaling in the CNS, particularly a loss of basal forebrain cholinergic neurons, has long been proposed as a mechanism of cognitive dysfunction during AD (Francis et al., 1999). The effects of acetylcholine are generally thought to occur through the activation of muscarinic and nicotinic acetylcholine receptors (AChRs) on neuronal membranes where they modulate synaptic function and plasticity. But, in addition to neurons, astrocytes and other glial cell subtypes also express AChRs and likely play a significant role in brain cholinergic signaling (Zoli, Pucci et al. 2018). Some AChR subtypes, such as the Alpha-7 nicotinic acetylcholine receptor (nAChR) have garnered much interest in AD due to its high binding affinity for, and activation by, Aβ peptides (Wang et al., 2000; Dineley et al., 2002). Interestingly, nAChR levels exhibit complex changes in AD that depend on brain region and cell type examined. For instance, though alpha7 nAChRs levels appear to be reduced in several brain regions affected by AD (Shimohama et al., 1986), the proportion of astrocytes expressing alpha-7 nAChRs appears to increase (Teaktong et al., 2003). Moreover, in multiple cell types, alpha-7 nAChRs have been proposed to either promote or inhibit the deleterious actions of amyloid pathology (Ma and Qian, 2019). In astrocytes and other non-neuronal cells, alpha-7 nAChRs can impart neuroprotection through the dampening of harmful neuroinflammatory signaling (Kalkman and Feuerbach, 2016; Foucault--Fruchard and Antier, 2017). However, in rodent hippocampal brain slices, application of Aβ was shown to increase astrocytic Ca2+ levels and trigger inward currents through extrasynaptic NMDARs in nearby CA1 pyramidal neurons, which was suggested to be a critical mechanism for excitotoxicity (Pirttimaki et al., 2013; Talantova et al., 2013). The complex role of alpha-7 nAChRs in brain circuits highlights some of the difficulties in developing therapeutic strategies for targeting cholinergic deficits in AD.
4.5. Cytokines
As discussed above, astrocytes secrete numerous cytokines as part of a coordinated (or dysregulated) neuroinflammatory response. In addition to acting on glial cells or innate immune cells, many cytokines also directly interact with receptors located on neuronal membranes, where they activate or modulate pathways involved in synaptic function and plasticity (e.g. p38 MAPK and NFκB pathways). However, the role of cytokines in shaping synaptic signaling properties is very complex. Several cytokine species, including TNFα, IL-1β, and IL-6 are elevated in local neural networks following the induction of LTP (del Rey et al., 2013), though it’s unclear whether these cytokines are produced in astrocytes or other cell types. These studies have suggested that cytokines may play an important role in the maintenance of increased synaptic strength. Cytokines may regulate basal synaptic function as well. For instance, astrocytic release of TNFα was shown to trigger the exocytosis and increased surface expression of AMPARs through a neuronal TNFR1-PI3 kinase signaling pathway (Beattie et al., 2002; Stellwagen et al., 2005). TNF-mediated elevations in postsynaptic AMPARs are especially important for “scaling-up” synaptic sensitivity in neural networks after periods of relative inactivity (Stellwagen and Malenka, 2006). Despite these beneficial actions, many other reports have shown that non-specific anti-inflammatory compounds, or compounds that inhibit specific cytokines like TNFα and IL-1, help to improve synapse function and plasticity in animal models of aging and AD-like pathology (Kotilinek et al., 2008; Bachstetter et al., 2012; Sama et al., 2012; MacPherson et al., 2017). Beneficial vs. detrimental actions could be attributable to the relative levels of cytokines in the local parenchyma (Beattie et al., 2002; Bernardino et al., 2005), to divergent signaling pathways in neurons vs. glial cells (Marchetti et al., 2004; Huang et al., 2011), or both. Indeed, TNFα, IL-1β, and other cytokines tend to impair synapse function when present at high levels in tissue (Pickering et al., 2005; Sama and Norris, 2013), which may be more likely to occur when astrocytes and microglia are highly reactive. Additionally, neurons express different receptors (relative to glia) for some cytokines (e.g. IL-1β) and, at least in some cases (e.g. the neuronal IL-1β pathway), these signaling components may preferentially impart neuroprotection (Huang et al., 2011). Some newly developed cytokine inhibitors have been designed to exploit differences in cytokine receptor pathways. For instance, XPro1595 is a dominant negative soluble TNF biologic that preferentially inhibits type 1 TNF receptors, which are linked to cytotoxic caspase pathways while preserving the activity of type 2 TNF receptors that are coupled to neuroprotective PI3K/Akt pathways (Steed et al., 2003). XPro1595 has been shown to restore LTD and LTP balance in aged rats and 5xFAD mice (Sama et al., 2012; Cavanagh et al., 2016; MacPherson et al., 2017; Cavanagh and Wong, 2018), and is in Phase 1b Clinical trials for treating AD (NCT03943264).
5. Astrocyte reactivity and the neurovascular unit
The neurovascular unit (NVU) is comprised primarily of vascular endothelial cells, pericytes, astrocytes, and neurons. More recently, the cellular anatomy of the neurovascular unit has been extended to include both microglia and perivascular macrophages (Keaney and Campbell, 2015). The multicellular NVU serves a number of functions including the tight regulation of blood flow through the vasculature, BBB permeability, neuroimmune responses, and neurovascular remodeling (Stanimirovic and Friedman, 2012; Kapasi and Schneider, 2016). The vascular endothelial cells lining the cerebral blood vessels are the core anatomical unit of the BBB. Both tight junctions and adherens junctions formed between adjacent vascular endothelial cells underlie the physical barrier responsible for limiting the paracellular diffusion of polar solutes (Keaney and Campbell, 2015). Pericytes are mural cells with elongated processes that encase the walls of pre-capillary arterioles, capillaries, and post-capillary venules (McConnell et al., 2017). Both their morphology and protein expression vary with their position along the vascular bed, reflecting the existence of subpopulations with diverse functions in regulating vessel diameter, cerebral blood flow, and extracellular matrix protein secretion (Winkler et al., 2011; Keaney and Campbell, 2015; Attwell et al., 2016). Astrocytes are centrally positioned within the brain parenchyma where they extend processes that communicate with local neurons, synapses, and blood vessels, allowing them to sense and respond to both neuronal and vascular activity. Thus, the BBB is composed of microvascular endothelial cells, astrocytes, pericytes, and neurons in close physical proximity to the endothelium, and together comprise a functional NVU. Notably, despite significant structural diversity of the NVU across the cerebrovascular network (Dahl, 1973; Roggendorf and Cervos-Navarro, 1977; Iadecola, 2017), more than 99 % of the cerebrovasculature of the brain is ensheathed in astrocytic end-feet (Nimmerjahn, 2009).
5.1. Neurovascular Astrocytes
Astrocytic end-feet are specialized processes that function to maintain the ionic and osmotic homeostasis of the brain and express a number of channels indicative of their specialized functions (Amiry-Moghaddam et al., 2003; Simard and Nedergaard, 2004). End-feet-enriched channels include the aquaporin 4 (AQP4) water channel, the inwardly rectifying K + channel Kir4.1, and the Ca2+-dependent K+ channel MaxiK (or BK) (Dunn and Nelson, 2010; Strohschein et al., 2011). The astrocytic endfoot is anchored to the vascular basement membrane via the alph a-b eta dystroglycan complex (Noell et al., 2011; Gondo et al., 2014). A common link between the Kir4.1, BK, and AQP4 channels at the astrocytic endfoot appears to be a shared anchoring protein, dystrophin 1. The brain expresses a short isoform of dystrophin 1 referred to as Dp71. Dp71 complexes with alpha-syntrophin forming the endfoot anchoring complex and is, therefore, responsible for anchoring the Kir4.1, BK, and AQP4 channels to the vascular basement membrane. Astrocytic end-feet are vital regulators of neuronal function given they modulate extracellular potassium concentrations, aid in removing neurotransmitters from synapses, and ensure metabolic needs are met via neurovascular coupling.
5.2. Potassium Buffering
The resting membrane potential (RMP) of a neuron (−70 to −80 mV), or the electrical potential difference across the plasma membrane at rest, is closer to the K+ equilibrium potential of −90 mV than the Na+ equilibrium potential of +65 mV. At rest, the neuronal plasma membrane is slightly permeable to both Na+ and K+, however, the permeability to K+ is much greater due to the presence of K+ leak channels embedded in the plasma membrane. Due to this enhanced permeability, K+ is close to electrochemical equilibrium and the neuronal membrane is close to equilibrium potential of K+. Conversely, the neuronal membrane at rest exhibits very low permeability for Na+. When an action potential is initiated, voltage-gated Na+ channels in the membrane open to allow an influx of Na+ ions (Fig. 4a). The influx of Na+ results in depolarization of the neuronal membrane, in turn opening additional voltage-gated Na+ channels via a positive feedback loop. Once the peak membrane potential (~ +35 mV) is reached, the neuronal membrane begins to repolarize by inactivating voltage-gated Na+ channels and opening voltage-gated K+ channels. The efflux of K+ ions from the neuron results in a decrease in the membrane potential towards the neuron’s resting voltage. Both voltage-gated Na+ and K+ channels begin to close once the membrane potential falls below the threshold potential. However, due to the slow kinetics of voltage-gated K+ channels they remain open longer than necessary, resulting in a brief hyperpolarization of the neuronal membrane, which ultimately prevents a second, rapid depolarization. The removal of K+ ions from the extracellular space following an action potential is critical in order for the neuronal membrane to adequately repolarize and reset channel function for the next action potential to occur. A single action potential can increase the extracellular K+ concentration by as much as 1 mM under normal conditions and ≥10–12 mM under pathologic conditions (Nwaobi et al., 2016). Even the relatively small elevations in extracellular K+ observed during physiologic neuronal activity depolarize the neuronal membrane, thereby increasing the probability of action potential propagation (Nwaobi et al., 2016). An essential function of neurovascular astrocytes is to maintain the neuronal RMP by modulating the extracellular K+ concentration, a process termed K+ siphoning (e.g. K+ buffering) (Harrower et al., 1984).
Fig. 4. Astrocytes modulate neuronal excitability through potassium spatial buffering.

Neuronal excitability relies on inward Na+ and outward K+ fluxes during action potentials. A) Schematic demonstrating the individual phases that comprise a single action potential. Once an action potential is initiated, voltage-gated Na+ channels in the membrane open to allow an influx of Na+ ions. The influx of Na+ further depolarizes the neuronal membrane, in turn opening additional voltage-gated Na+ channels. Once the peak membrane potential is reached, the neuronal membrane begins to repolarize by inactivating voltage-gated Na+ channels and opening voltage-gated K+ channels. The efflux of K+ ions from the neuron results in a decrease in the membrane potential towards the neuron’s resting voltage. B) Through Kir4.1 channels (shown in pink) and the Cx43-containing gap junctions (shown in teal), astrocytes are able to take up excess extracellular K+ and transfer it either into the circulation via the astrocytic end-feet (indicated by the black arrows) or to an area of the brain lacking K+ via their gap junctions (indicated by the red arrows). The removal of K+ ions from the extracellular space following an action potential is critical in order for the neuronal membrane to adequately repolarize and reset channel function for the next action potential to occur. A single action potential can increase the extracellular K+ concentration by as much as 1 mM under normal conditions and ≥10–12 mM under pathologic conditions. Even the relatively small elevations in extracellular K+ observed during physiologic neuronal activity depolarize the neuronal membrane, thereby increasing the probability of action potential propagation. Thus, impaired K+ buffering can lead to hyperexcitability and subsequent excitotoxicity. Murine models of AD manifest hyperexcitability, with some models also exhibiting evident epileptiform and seizure activity. Moreover, early onset hyperexcitability is a well known feature of human AD.
5.3. Connexins, Kir4.1, and K+ homeostasis
In addition to K+ channels, astrocytes abundantly express plasmalemmal hemichannels, made up of connexin proteins (primarily connexin 43 (Cx43) and connexin 30 (Cx30)) (Orellana 2016). Many hemichannels are directly apposed to hemichannels on adjacent cells where they form “gap junctions” or conduits between astrocytes to permit the rapid intercellular exchange of ions and small metabolites. Thus, astrocytes are highly interconnected via gap junctions and can form large electrically coupled syncytiums. Uptake of locally released K+ via Kir4.1 channels results in the transport of K+ down its concentration gradient through Cx43 gap junctions (Fig. 4b). Some of this K+ will be exported into the circulation via the astrocytic end-feet (Wallraff et al., 2004). Movement of K+ in this manner helps to dissipate local K+ gradients (Neusch et al., 2006) and prevent neuronal hyperexcitability. Genetic deletion of Kir4.1 channel from astrocytes has a dramatic effect in mice; mice lacking the Kir4.1 channel only live 25 days, during which they suffer from seizures, ataxia, and tremor. Electrophysiological studies in these mice reveal significant impairment of K+ uptake by astrocytes, decreased spontaneous action potential frequency and amplitude, and increased LTP (Djukic et al., 2007). Gap junction blockers have similarly disruptive and degenerative effects.
Interestingly, connexins and Kir4.1 channels are not only involved in the homeostasis of extracellular K+ ions but also in the regulation of extracellular glutamate. As discussed, astrocytes use EAATs to rapidly remove glutamate from the extracellular space and minimize excitotoxic damage to neurons. Because glutamate import across EAATs is partially coupled to K+ export, the presence of elevated extracellular K+ gradients exerts an inhibitory effect on glutamate uptake (Barbour et al., 1988). A number of studies have implicated the need for functional Kir4.1 channels in the regulation of glutamate transmission. Pharmacological inhibition of Kir4.1 led to a 33.1 % reduction in glutamate while siRNA mediated Kir4.1 knockdown in cortical astrocytes resulted in a 57 % reduction in glutamate uptake (Kucheryavykh et al., 2007). Further, threo-bet a-b enzyloxyaspartate-sensitive glutamate uptake was reduced by more than 50 % in Kir4.1 null mice when compared with wildtype littermates (Nwaobi et al., 2016). These results suggest that by allowing the astrocyte to maintain a K+ electrochemical gradient that favors K+ unbinding in the extracellular space, Kir4.1 helps facilitate the regulation of glutamate transmission.
5.4. AQP4
Water movement in the brain is critical for cellular function given it regulates cell volume and homeostasis between extracellular and intracellular compartments. Ionic movement across cell membranes is commonly coupled with the movement of water and with the maintenance of osmotic equilibrium. The net transport of water always has to be driven by osmotic forces due to solute movement considering there is no primary, active transport, ATP-driven, water pump (Kimelberg, 2004). Most water movement into and out of cells occurs via water channels in the plasma membrane known as aquaporins. AQP4 is expressed by astrocytes of the neurovascular unit and is highly polarized to the endfoot membrane where it functions to bring water into specific cells or to remove excess water to alleviate swelling (Doody et al., 2013). Under conditions of food and water deprivation, AQP4 has demonstrated an ability to alter its expression levels in order to maintain the brains normal water content and prevent cell loss (Ye et al., 2016). Astroglial water movements induced by AQP4 have also been proposed to be a driving force contributing to the paravascular clearance of interstitial solutes such as Aβ and tau.
Given the brain’s high metabolic rate and the sensitivity of neurons and glia to alterations in their extracellular environment, there exists a critical need for the rapid clearance of brain waste products. In 2012, a landmark study by Iliff et al. used in vivo two-photon imaging of small fluorescent tracers to show that CSF moves by convective (bulk) flow along the perivascular space between vessels and the astrocytic end-feet and is cleared via paravascular drainage routes (Iliff et al., 2012) (Fig. 5). Notably AQP4 null mice exhibited perturbed CSF influx through this system as well as a 70 % reduction in interstitial solute clearance, ultimately suggesting that glymphatic clearance is supported by astrocyte water transport. Furthermore, Iliff et al. demonstrated that fluorescently tagged Aβ peptides are transported through this system and deletion of AQP4 suppressed the clearance of soluble Aβ peptides, implicating a role for this pathway in removing Aβ from the brain.
Fig. 5. Waste products are cleared from the brain by a process that requires astrocytes.

Under physiological conditions, AQP4 channels (shown in navy blue) are polarized to astrocytic endfeet and support rapid water movement between the periarterial space and astroglial syncytium. This anatomic arrangement facilitates the convective bulk flow of CSF from the periarterial space across the astrocytic endfeet and into the interstitial space, where it mixes with interstitial fluid (ISF) and waste products such as Aβ (shown in brown). Waste products and excess fluids are then driven toward the perivenous space and ultimately cleared from the brain through the meningeal lymphatic vessels. Altered AQP4 localization has been described in aged brains, whereas loss of perivascular AQP4 has been demonstrated in human AD brains and is associated with increased levels of Aβ and tau pathology. It should be noted that while the astrocytic arbors appear to overlap in this figure, in reality their arbors occupy distinct fields with little to no overlap.
Recently, AQP4 has been shown to function not only as a water channel protein but also as an adhesion molecule involved in cell migration (Papadopoulos et al., 2008). During migration cells undergo rapid changes in their morphology due to the rapid formation and retraction of cell membrane protrusions. These rapid changes are accompanied by changes in cell volume attributable to water flow into and out of the cell. Granted, changes in cell volume not only facilitate morphological changes, but may also aid in propelling the cell forward. Thus, it is likely aquaporin-mediated transmembrane water movements facilitate changes in morphology and physically propel the cell forward. Evidence supporting the role of AQP4 in astrocyte migration primarily comes from acute injury models. In 2005, Saadoun et al. showed that while AQP4 is expressed strongly in astrocytes in the normal mouse brain, it is upregulated following stab wound injury, resulting in the migration of reactive astrocytes to the injury site. Notably, this same study observed enhanced polarization of AQP4 to the leading edge of migrating astrocytes as well as a greater number of cell membrane protrusions at the leading edge of migrating AQP4-expressing versus non-AQP4 expressing astrocytes (Saadoun et al., 2005). This may explain observations of robust upregulation of AQP4 in areas surrounding Aβ plaques (Yang et al., 2017).
Lastly, AQP4 plays a role in neuroexcitation given that when K+ ions are released into the extracellular space by neurons following an action potential, astrocytes on the other side of the synaptic cleft take up both excess K+ ions and water. AQP4 immunoreactivity is strongly expressed on the majority of the cerebrovasculature where it has been shown to bind alpha-syntrophin (Amiry-Moghaddam et al., 2003; Amiry-Moghaddam et al., 2004a, 2004b, Wilcock et al., 2009; Camassa et al., 2015). When alpha-syntrophin is deleted in mice, AQP4 is no longer localized to astrocytic end-feet. The mislocalization of AQP4 in the alpha syntrophin knockout mice is associated with significant functional defects including prolonged seizure durations with slowed K+ kinetics in the brain extracellular space. K+ clearance deficits are also observed in alpha-syntrophin deficient mice, where AQP4 is not properly targeted to the cell membrane.
5.5. Neurovascular Coupling
As previously discussed, the maintenance of brain homeostasis alongside cognitive processing requires substantial energy expenditures compared to the rest of the body. Though the brain only accounts for 2% of total body mass, it consumes up to 20 % of the whole-body energy budget and calculations estimate that the greatest proportion of the energy expenditure in the brain is attributable to synaptic transmission (Howarth et al., 2012). Therefore, it is likely that synaptic transmission will be heavily impacted by reductions in cerebral blood flow (CBF) that prevent sufficient energy supply to the brain. Autoregulatory mechanisms ensure that CBF is not impacted as a consequence of alterations in systemic blood pressure, thereby ensuring basal CBF is maintained and the brain continues to receive adequate blood supply at all times. In the resting brain CBF varies in proportion to the energy consumption of each brain region such that CBF is higher in regions with higher energy utilization and lower in regions consuming less energy (Iadecola, 2017). CBF is also regulated in response to brain activity such that increases in neural activity lead to increases in CBF which are highly localized to activated brain regions. This response is known as functional hyperemia or neurovascular coupling (Freygang and Sokoloff, 1958; Cox et al., 1993; Iadecola, 1993; Chaigneau et al., 2003) and is thought to reflect the need for a well-timed delivery of oxygen and glucose to activated brain regions at times of intense activity. In fact, CBF increases to such an extent that more oxygen is provided to active brain regions than is consumed (MacVicar and Newman, 2015). Granted, increases in CSF may also reflect the need to clear active regions of potentially toxic byproducts of neural activity (e.g. lactate, CO2, Aβ, tau) as well as for brain temperature regulation (Tarasoff-Conway et al., 2015). A series of studies have demonstrated that neurovascular coupling is mediated, to a significant degree, by calcium-dependent astrocytic mechanisms.
5.6. Arachidonic acid metabolite-mediated neurovascular coupling
For arterioles, glutamate released during routine neural activity plays a critical role in informing the blood vessel of the requirement for increased local CBF. Glutamate released from presynaptic neurons acts on astrocytic metabotropic glutamate receptors (mGluR) resulting in increased levels of intracellular calcium (Fig. 6a). Increased intracellular Ca2+ levels lead to the activation of phospholipase A2 (PLA2), an enzyme localized to the astrocytic endfoot responsible for liberating arachidonic acid from plasma membrane lipids. In 2004 Mulligan and MacVicar showed that Ca2+ transients in astrocytes lead to arteriolar constriction which directly contradicted a 2003 study by Zonta et al. demonstrating that Ca2+ transients induce arteriolar dilation (Zonta et al., 2003; Mulligan and MacVicar, 2004). Subsequent work by Metea and Newman (2006) showed that, in the same preparation, Ca2+ uncaging in astrocytes and retinal glial cells could trigger both arteriolar constriction and dilation (Metea and Newman, 2006).
Fig. 6. Mechanisms underlying astrocyte-mediated vascular responses.

From an anatomical standpoint, astrocytes are perfectly positioned to bi-directionally communicate information between neurons and blood vessels. In order to meet the metabolic needs of active neurons, increased neuronal activity induces a rapid vasodilatory response and consequent spatiotemporally restricted delivery of glucose and oxygen. A) Glutamate released from presynaptic neurons acts on astrocytic metabotropic glutamate receptors (mGluR5) resulting in increased intracellular Ca2+. B) PLA2 is activated in response to rises in intracellular Ca2+ concentrations, leading to the generation of arachidonic acid (AA) and its subsequent conversion to either prostaglandin E2 (PGE2) via COX enzymes or to epoxyeicosatrinoic acids (EETs) by CYP2C11 enzymes. Both PGE2 and EETs act on vascular smooth muscle cells to dilate vessels. Increases in intracellular Ca2+ also engage the Ca2+-dependent K+ channel BK (shown in yellow) on the astrocyte endfoot plasma membrane. Activation of BK results in the efflux of K+ into the extracellular space where it is taken up by vascular smooth muscle cells via Kir2.1 or Kir2.2. Like PGE2 and EETs, K+ also induces vasodilation. C) Conversely, in response to high pO2, AA is released from astrocytes and converted into 20-HETE in the vascular smooth muscle cells. The combination of low extracellular adenosine levels and 20-HETE leads to an elevation in smooth muscle cell free Ca2+ and subsequent arteriolar constriction. Thus, neurovascular coupling, which ensures that the brain has a proportionally matched cerebral blood flow in response to local neuronal activity, is largely mediated by astrocytic Ca2+ signaling. Both BBB and NVU breakdown are evident in AD and may impair neurovascular coupling by preventing astrocytes from relaying signals between the vasculature and neuronal circuitry, creating a mismatch between neuronal activity and the provision of oxygen and glucose required to meet metabolic demands.
It has since been elucidated that three mechanisms control arteriole diameter through arachidonic acid metabolism. As a consequence of mGluR activation, intracellular Ca2+ concentrations increase via IP3 signaling resulting in activation of phospholipase A2 (PLA2), which generates arachidonic acid (AA) from the plasma membrane. Arachidonic acid can itself act as a signaling molecule or be converted to several different lipid derivatives, each of which act on vascular smooth muscle cells through different mechanisms to influence vessel diameter. In order to induce vasoconstriction, AA must be converted into 20-hydroxyeicosatetraenoic acid (20-HETE) by the cytochrome P450 4A (CYP4A) enzyme (Gordon et al., 2008). 20-HETE functions to inhibit smooth muscle cell K+ conductance to depolarize and contract these cells (Lange et al., 1997) (Fig. 6c). Conversely, for vasodilation, AA must be converted to prostaglandin E2 via COX enzymes or to epoxyeicosatrinoic acids (EETs) by CYP2C11 enzymes (Fig. 6b). In 2008 work by Gordon et al. demonstrated that the vascular response to astrocyte Ca2+ transients is dictated by brain metabolic elements such as oxygen, lactate, and adenosine (all of which rapidly change during neuronal activity; both electrical and sensory stimulation triggers a fall in tissue pO2 and an increase in external lactate.
Irrespective of whether an astrocyte is inducing constriction or dilation of an arteriole, the first step involves the liberation of AA from the plasma membrane by Ca2+-sensitive PLA2. In response to high pO2 AA is converted to 20-HETE by the CYP4A enzyme. The combination of low extracellular adenosine levels and 20-HETE leads to an elevation in smooth muscle cell free Ca2+ and subsequent arteriolar constriction (Gordon et al., 2008). On the other hand, low pO2, results in AA being converted to PGE2 by COX then released via diffusion. Prostaglandin transporters normally take up PGE2 from the extracellular space; however, as external lactate level begin to rise as a consequence of enhanced glycolysis, lactate attenuates PGE2 clearance, resulting in the accumulation of this vasodilator which acts on smooth muscle cells (Gordon et al., 2008). Importantly, extracellular adenosine levels also rise as a consequence of low pO2. Extracellular adenosine reduces smooth muscle cell intracellular free Ca2+ via A2A receptor activity, thereby blocking the constrictor pathway and facilitating the switch from vasoconstriction to vasodilation (Gordon et al., 2008).
It is worth nothing that, although regulation of cerebral blood flow was traditionally thought to occur at the level of arterioles, capillaries are also enveloped by contractile pericytes and are better spatially situated to respond to neuronal activity and control blood flow at a more local level. This fact, alongside more recent studies revealing mGluR5 expression is downregulated in adult astrocytes and animals lacking the primary IP3 receptor in astrocytes display unaltered neurovascular coupling, led Mishra et al. to reinvestigate the role of astrocytes in neurovascular coupling (Mishra et al., 2016). Data now suggests that neuronal activity induces ATP release from postsynaptic neurons which acts on astrocyte ATP receptors containing P2 × 1 subunits to induce intracellular rises in Ca2+. Increased intracellular calcium in turn activates PLD2, resulting in AA synthesis via DAGL, and downstream metabolism by COX1 into vasodilatory PGE2. PGE2 then works by acting on capillary pericytes to induce dilation via the EP4 receptor. Interestingly, this study also found that, in contrast to capillary dilation, arteriole dilation does not depend on P2 × 1 receptors, PLA2, PLD2 or astrocyte calcium signaling. Rather, arteriole dilation was shown to be dependent upon NMDA receptor activation and nitric oxide synthesis. The divergence of new data may be a reflection of the different kinds of stimuli applied as well as the surveyed brain region. Thus, mGluR-driven astrocyte Ca2+ signaling likely still contributes to arteriole dilation, though this mechanism may decrease in importance with age.
5.7. Potassium-mediated neurovascular coupling
K+ is vasoactive, meaning that when K+ is infused into the arterial supply of a vascular bed, blood flow increases. K+-mediated vasodilation occurs when hyperpolarization of vascular smooth muscle cells following neuronal activity is detected by astrocytic end-feet processes adjacent to synapses. Neuronal activity results in PLC-mediated liberation of IP3 and DAG from membrane PIP2 pools, ultimately inducing the propagation of an IP3-mediated Ca2+ wave into astrocytic end-feet (Longden and Nelson, 2015). The resulting Ca2+ wave engages the large-conductance Ca2+-dependent K+ channel BK (MaxiK) on the astrocyte endfoot plasma membrane thereby initiating the efflux of K+ into the extracellular space between the astrocyte endfoot and vascular smooth muscle cell (Fig. 6b). The resulting rise in extracellular K+ levels activates strong inwardly rectifying K+ channels (Kir2.1 or Kir2.2) on vascular smooth muscle cells of intracerebral arterioles, leading to membrane hyperpolarization, closure of voltage dependent Ca2+ channels, vasodilation and subsequent increases in blood flow (Filosa et al., 2006; Longden and Nelson, 2015). In this case, increased blood flow sustains the augmented metabolic needs of the locally activated neurons. Notably, more intense neuronal activity leads to the propagation of larger Ca2+ waves into astrocytic end-feet ultimately promoting the release of higher concentrations of K+ from the astrocyte endfoot (Longden and Nelson, 2015). This, in turn, leads to the depolarization of the vascular smooth muscle cell membrane, VDCC activation, and subsequent vasoconstriction. Studies have demonstrated that blocking BK channels pharmacologically or ablating the gene encoding these channels results in a reduction of whisker stimulation-evoked blood flow increases in the cortex, ultimately garnering support for the BK channel mediated hypothesis of neurovascular coupling (Filosa et al., 2006; Girouard et al., 2010).
Of consideration is that, in response to increased metabolic demand, the dilation of arterioles in the area of activation may not increase blood flow in that region effectively unless upstream vessels also dilate. In other words, increasing blood flow into the microcirculation (i.e. capillary beds) may require a reduction in resistance upstream. The extensive coupling of endothelial cells as well as the electric coupling existing between endothelial cells and vascular smooth muscle cells allows for coordinated dilating responses along the length of the intracerebral arteriole. Once initiated via the local activation of K+ channels in endothelial cells, hyperpolarization is conducted along gap junctions and spreads into the surrounding vascular smooth muscle cells through myoendothelial gap junctions to promote their relaxation (i.e. dilation (Segal, 2015)).
6. Astrocyte endfoot disruption in AD and related disorders
6.1. Aging and AD
Several studies have demonstrated astrocytic endfoot disruption in both murine models and human AD. Astrocytic end feet surrounding vascular Aβ deposits exhibit morphological changes including retraction and swelling, as well as reduced expression of glutamate and lactate transporters (Merlini et al., 2011). These alterations were shown to occur at early stages of the disease and are consistent with neurovascular uncoupling. Further, as previously discussed, AQP4 facilitates CSF flow into the brain parenchyma allowing it to mix with ISF (Iliff et al., 2012; Kress et al., 2014). The CSF-ISF fluid mixture containing toxic proteinaceous metabolites is then driven towards the venous perivascular space where it ultimately exits into meningeal lymphatic vessels and the systemic circulation. (Xie et al., 2013) Interestingly, AQP4 gene expression has been shown to increase in the cerebral and cerebellar cortices as well as the hippocampal CA1 region of aged mice (Gupta and Kanungo, 2013; Bronzuoli et al., 2019). This increase in AQP4 gene expression may reflect a physiological need to compensate for astrocyte morphological and/or functional alterations known to occur throughout the aging process. Yet, despite this perceived physiological need, loss of perivascular localization of AQP4 has been reported in 24-month-old TgSwDI mice, which develop age-dependent accumulation of amyloid (Duncombe et al., 2017). Notably, similar results have been demonstrated in postmortem frontal cortex of cognitively normal individuals as well as individuals with histopathologically confirmed AD. In 2017, Zeppenfeld et al. demonstrated that altered AQP4 expression is associated with advancing age and that, when controlling for age, loss of perivascular AQP4 localization was associated with increased levels of Aβ and tau. Perhaps more convincing of the brain’s need to continuously remove toxic metabolic waste via glymphatic drainage is the fact that Zeppenfeld et al. also demonstrated that preservation of perivascular AQP4 localization in aged brains was predictive of preserved cognitive abilities (Zeppenfeld et al., 2017).
6.2. Cerebral amyloid angiopathy (CAA)
Cerebral amyloid angiopathy (CAA) refers to the deposition of beta amyloid in the media and adventitia of small arteries, arterioles, and (less often) the capillaries of the leptomeninges and cerebral cortex (Viswanathan and Greenberg, 2011). Although CAA and AD pathology frequently co-occur, CAA is also often present in the brains of cognitively normal individuals. Unlike AD-induced brain injury, which centers on Aβ-triggered loss of synapses and neurons, CAA-driven brain injury appears to arise from blood vessel dysfunction characterized either by the loss of vessel integrity and subsequent hemorrhage or by hypoperfusion and subsequent ischemic events (Greenberg et al., 2020). Like other vascular risk factors implicated in the development of dementia (i.e. atherosclerosis, hypertension, diabetes mellitus, hyperhomocysteinemia, and cerebrovascular small vessel disease) CAA itself, in the absence of comorbid pathologies, can cause dementia.
In 2009 Wilcock et al. crossed mouse strains expressing the Swedish APP mutation or the Swedish, Dutch, and Iowa APP mutations (APPSw or APPSwDI, respectively) with mice lacking the gene for inducible nitric oxide synthase (NOS2) to generate unique models displaying all three primary pathological features of Alzheimer’s disease (i.e. amyloid deposition, tau pathology, and neuronal loss). Interestingly, the resulting mouse models were shown to demonstrate clear differences in vascular amyloid deposition thereby allowing the investigators to compare astrocyte characteristics in mice with mild CAA (APPSw), moderate CAA with tau pathology and neuron loss (APPSw/NOS2−/−), severe CAA (APPSwDI), and severe CAA with tau pathology and neuron loss (APPSwDI/NOS2−/−). This study revealed that moderate-to-severe levels of CAA lead to decreases in the number of astrocytic processes contacting the vasculature in the cerebral cortex and hippocampus. Furthermore, this study demonstrated that mice with moderate-to-severe CAA experience significant reductions in AQP4-positive staining associated with blood vessels as well as decreased Kir4.1 and MaxiK (i.e. BK) gene and protein expression compared to mice with mild CAA (Wilcock et al., 2009). Notably, changes in Kir4.1 and MaxiK gene and protein expression were not isolated to transgenic mice but were also demonstrated in human AD cases with apparent CAA. Results of the aforementioned study are further supported by others showing that, in response to vascular amyloid deposition, astrocytes secrete inflammatory cytokines, metabolizing enzymes, and ROS thereby contributing to neuroinflammation and possibly contribute to subsequent alterations in BBB integrity and astrocytic end-feet-specific channels (Niwa et al., 2000; Yin et al., 2006; Yang et al., 2007; Miners et al., 2010; Carrano et al., 2012; Han et al., 2015).
6.3. Vascular contributions to cognitive impairment and dementia (VCID)
Vascular contributions to cognitive impairment and dementia (VCID) is an umbrella term used to define conditions arising from vascular brain injuries that lead to significant decline in memory, thinking and behavior (Price et al., 2018). It serves as the second leading cause of dementia, behind only AD, and can be attributed to a number of pathologies (Corriveau et al., 2016). Studies suggest vascular injury precedes hallmark AD pathologies, thereby highlighting a role for neurovascular dysfunction in AD progression (Canobbio et al., 2015; Janota et al., 2016). One major, yet underappreciated, modifiable risk factor for VCID is hyperhomocysteinemia (HHcy), a condition in which individuals exhibit elevated plasma homocysteine levels and are therefore more likely to suffer cardiovascular disease, stroke, VCID, and AD (Graham et al., 1997; Bostom et al., 1999; Eikelboom et al., 1999; Beydoun et al., 2014). HHcy has also been associated with hippocampal atrophy, white matter lesions, and lacunar infarcts (Vermeer et al., 2002; Firbank et al., 2010).
In 2013, Sudduth et al. described a HHcy model of VCID that emulates multiple VCID pathologies including neuroinflammation, cognitive impairment, and blood-brain barrier breakdown culminating in microhemorrhages throughout the cerebral cortex and, less frequently, the hippocampus (Sudduth et al., 2013). In this model, HHcy is induced through dietary modification that eliminates vitamins B6, B9 (folic acid), and B12 from mouse chow; all of which are essential cofactors of the enzymes responsible for converting homocysteine. In 2017, Sudduth et al. built upon this work by demonstrating that astrocytic end-feet are disrupted in mice on a HHcy-inducing diet. They found astrocytic endfoot disruption was characterized by a reduction in Dp71 labeling concurrent with reduced vascular labeling for AQP4. Their model also exhibited reduced gene and protein expression of the Kir4.1 and MaxiK potassium channels. Considering microglial activation is apparent in the HHcy model at all time points examined, Sudduth et al. concluded that microglial activation and subsequent pro-inflammatory responses precede astrocytic changes. This is important given astrocytic end-feet are anchored to the vascular basement membrane by an α-β dystroglycan complex (Noell et al., 2011; Gondo et al., 2014). There are a number of proteinases capable of degrading such protein complexes, however, matrix metalloproteinase 9 (MMP9) has been shown to be a major β-dystroglycan-degrading enzyme (See (Weekman and Wilcock, 2016) for review). As such, in 2018, Price et al. proposed that HHcy induces a pro-inflammatory response at the vasculature resulting in the activation of astrocyte-derived MMP9 which acts in two ways: 1) MMP9 cleaves the a-b dystroglycan complex leading to subsequent disruption of the astrocytic connection to the vasculature and 2) MMP9 degrades the dystrophin Dp71 anchoring complex initiating the downregulation of astrocytic endfoot channels; the end result of which is likely impaired potassium homeostasis and insufficient neurovascular coupling.
Estimates suggest at least 60 % of AD patients have co-occurring cerebrovascular pathologies (such as CAA, micro- and macro-infarcts, micro- and macro-hemorrhages, cerebral hypoperfusion, white matter hyperintensities, and stroke) hypothesized to act as a secondary “hit” to the brain that lowers the threshold for cognitive impairment in persons with existing AD pathology (Schneider and Bennett, 2010; Vemuri and Knopman, 2016). In 2019, Weekman et al. demonstrated a robust neuroinflammatory response, followed by cognitive defeats, microhemorrhages, and the redistribution of amyloid from the parenchyma to the vasculature in a VCID/AD comorbidity mouse model (Weekman et al., 2019). Given this study showed significant increases in TNFα and IL-1β, two pro-inflammatory cytokines responsible for activating MMP9, one can speculate this comorbidity model also displays astrocytic endfoot disruption. Furthermore, the pathological activation of astrocyte derived MMP9 likely has additional consequences for BBB integrity.
6.4. Additional consequences for blood-brain-Barrier (BBB) integrity
The BBB is a tightly sealed, continuous endothelial membrane enveloped by perivascular astrocytic endfeet (Sweeney et al., 2018). Tight junction proteins (occludins, claudins, and junctional adhesion molecules or JAMs) between the endothelial cells confer high transendothelial electrical resistance and low paracellular and transcellular permeability (Zlokovic, 2011). The average distance between the BBB and neurons (~8um) allows for the rapid exchange of molecules between capillaries and neurons (Pardridge, 2015). Thus, the BBB regulates the composition of the neuronal internal milieu, which is essential for proper neuronal and synaptic function (Zhao et al., 2015).
Increased BBB permeability has been reported both in normal aging and AD, among other neurodegenerative conditions (Montagne et al., 2015). Studies using advanced dynamic contrast-enhanced MRI have demonstrated BBB breakdown occurs before brain atrophy or dementia in the hippocampus (Montagne et al., 2015) and several gray and white matter regions (van de Haar et al., 2016, 2017) in both mild cognitive impairment (MCI) and early AD. BBB breakdown in AD has been further confirmed by more than 20 independent postmortem human neuropathology studies. Some studies have identified peripheral macrophages (Hultman et al., 2013) and neutrophils (Zenaro et al., 2015) suggesting BBB breakdown allows the influx of circulating leukocytes into the brain; while others have shown perivascular accumulation of blood-derived neurotoxic products (e.g. fibrinogen, thrombin, albumin, IgG, and hemosiderin) alongside pericyte and endothelial degeneration, loss of tight junction proteins, and red blood cell (RBC) extravasation (See (Nelson et al., 2016) for review). This is quite problematic given RBC-derived hemoglobin and free iron generate ROS, which subjects neurons to oxidant stress; while fibrinogen, plasminogen, thrombin and autoantibodies induce neuroinflammation, neuronal damage, and immune cell recruitment into the brain. Additionally, the presence of albumin may lead to the development of edema, followed by hypoperfusion and subsequent tissue hypoxia.
Although astrocytes are crucial for maintaining BBB characteristics in endothelial cells through the release of specific growth factors (VEGF, GDNF, bFHF, and ANG-1), astrocyte reactivity can lead to the secretion of cytokines and proteases that negatively impact endothelial tight junctions, pericyte phenotype, and BBB permeability. As previously suggested, BBB dysfunction is commonly observed alongside activation of matrix metalloproteinases (MMPs), of which astrocytes are the main source. Under physiologic conditions, secreted MMPs aid in remodeling the pericellular environment though the cleavage of extracellular matrix proteins. Conversely, MMPs also possess the ability to stimulate numerous pro-inflammatory mediators (CXCL-8, IL-1β, TNFα, etc.), and are themselves up-regulated by neuroinflammatory stimuli such as oxidative stress, cytokines, and Aβ pathology. In fact, accumulating evidence suggests MMPs are key regulators of Aβ metabolism and play a role in astrocyte-mediated Aβ degradation. The gelatinase class of MMPs, which consists of MMP2 and MMP9, can digest the endothelial basal lamina and tight junction scaffold proteins, both of which are necessary for BBB integrity (Qiu et al., 2011; Zhang et al., 2012). The gelatinase MMPs also have a high affinity for dystroglycan, which anchors the astrocytic endfoot to the vascular basement membrane. Due to its variety of substrates, the expression, translation, and activity of MMP9 are normally tightly regulated, but may become aberrant in disease. Although MMP9 is more abundant in the CSF of AD individuals compared with cognitively normal controls (Stomrud et al., 2010), astrocyte derived MMP9 is not the only contributor to BBB dysfunction.
The presence of BBB breakdown is most pronounced in individuals carrying the e4 allele of Apolipoprotein E (APOE). In fact, Montagne et al. recently demonstrated that APOE-e4 individuals are distinguished from those without APOE-e4 by breakdown of the BBB in the hippocampus and medial temporal lobe. Notably, this finding is evident in cognitively normal APOE-e4 carriers and more severe in those with cognitive impairment but is not related to CSF Aβ or tau levels as measured by PET imaging (Montagne et al., 2020). Upon analysis of human brain tissue, Montagne and colleagues noted higher activation of the cyclophilin A-MMP9 pathway in degenerating capillary pericytes of APOE-e4 carriers when compared to individuals homozygous for APOE-e3, confirming a pathogenic mechanism earlier described by Bell et al. (Bell et al., 2012). Blanchard et al. expanded upon this work by generating a reconstructed BBB model in vitro with iPSC-derived endothelial cells, pericyte-like mural cells and astrocytes (Blanchard et al., 2020). Their work shows that APOE and NFAT–calcineurin signaling are upregulated in APOE4 pericyte-like iPSC-derived mural cells (iMC) as well as in pericytes in the human brain. However, while addition of astrocytes to the iBBB decreased permeability, it did not substantially alter the majority of the functional or transcriptional pathological outcomes measured (further suggesting these APOE-mediated effects were primarily due to pericyte-like mural cells). Importantly, inhibition of calcineurin/NFAT reduced APOE expression and decreased vascular amyloid accumulation both in vitro and in vivo, highlighting the therapeutic potential of targeting this pathway (Blanchard et al., 2020).
Though the aforementioned studies draw attention to pericyte dysfunction as the major driver of BBB disruption in AD, it is important to consider the intricate crosstalk between pericytes, astrocytes, and endothelial cells. Pericytes have been shown to facilitate the attachment of astrocytic endfeet to the vascular basement membrane (Ihara and Yamamoto, 2016; Geranmayeh et al., 2019) and pericyte-deficient mice exhibit reduced AQP4 expression in astrocytes (Armulik et al., 2010). Conversely, astrocytes have been shown to control pericyte migration, differentiation, and the juxtaposition of pericytes to endothelial cells (Nakagawa et al., 2009; Brinton et al., 2015). Moreover, astrocyte-derived apolipoproteins differentially modulate cyclophilin A signaling in pericytes (Bell et al., 2012). Thus, dysfunction of either cell type influences the other, likely leading to BBB disruption and consequent cognitive impairment.
7. Astrocytes and brain metabolism
As previously discussed, the brain consumes up to 20 % of body’s oxygen (and up to 25 % of glucose). Despite this long-known outsized role in energy usage, there are still many remaining questions of how tight spatial and temporal coupling of neuronal activity to vascular supply of oxygen and nutrients is maintained. ATP is produced via two interconnected metabolic pathways, glycolysis and oxidative phosphorylation. The contribution of each of these two biochemical pathways varies depending on cell type – for example, erythrocytes lack mitochondria and thus rely exclusively on glycolysis. Generally speaking, when ample oxygen is present, most cells generate ATP via oxidative phosphorylation, whereas under anaerobic conditions, the faster yet less energetically efficient glycolytic pathway is the primary ATP supply source. Undifferentiated cells and many cancerous cell types are the prime exceptions to this rule, as they rely heavily on glycolysis even in the presence of sufficient oxygen, a feature described as “aerobic glycolysis”.
While it is widely accepted that the vast majority of ATP generated in the normal adult brain is derived from glucose, the devil remains in the details which are hotly debated— e.g. when, where and how much ATP is generated via oxidative vs non-oxidative (“anaerobic” glycolysis) glucose utilization (Schurr, 2018)? As critical as it has been in informing on global and regional rates of cerebral glucose metabolism in human health and disease, PET imaging cannot (yet) provide cell-specific resolution. Thus, much remains unknown regarding the contributions of neurons vs glia – i.e. what is the individual metabolic programming of each distinct cell type in the brain, and how do they interact? Still, in vitro studies have provided much critical information in defining the metabolic profiles of various CNS cell types, and in most cases have been confirmed ex vivo and/or in vivo. Generally speaking, these studies collectively paint a picture whereby astrocytes are primarily glycolytic, while neurons are primarily oxidative and have a high rate of glucose flux into the pentose phosphate pathway.
7.1. Cerebral metabolic changes in Alzheimer’s disease; a role for astrocytes?
Glucose hypometabolism, or decreased cerebral metabolic rate of glucose (CMRglc), is typically defined by a decrease in the uptake of 18 F-deoxyglucose (FDG) as measured by PET imaging. This cerebral glucose hypometabolism is a hallmark of AD (Small et al., 2000), and FDG-PET is able to differentiate AD from other types of dementia with a high degree of specificity due to specific regional patterns of signal (Laforce and Rabinovici, 2011). Clinical AD symptoms essentially never occur without glucose hypometabolism, and the extent of the metabolic changes are strongly correlated with the severity of clinical symptoms (Grady et al., 1986; Haxby et al., 1990; Blass, 2002). Furthermore, recent evidence suggests that these alterations in glucose metabolism occur very early in the neurodegenerative process (Small et al., 1995; Reiman et al., 1996; de Leon et al., 2001; Mosconi et al., 2008). What is the contribution of astrocytes to this signal?
In vivo studies aimed at determining the contribution of neurons vs astrocytes to glucose uptake have produced varying results, with some studies showing higher rates of uptake in neurons, while others show higher increases in NBDG accumulation in astrocytes (Chuquet et al., 2010). A major challenge for distinguishing the relative contributions of neurons and astrocytes in glucose uptake is the lack of spatial resolution inherent to modern PET imaging techniques. However, advances in cell-specific transcriptional and proteomic analyses may be able to offer some important insight. In this regard, a recent large-scale, unbiased proteomics analysis of AD brain tissue highlighted several biological pathways among the most altered across the patho-clinical progression from normal cognition, to mild cognitive impairment, to AD dementia. The pathway most affected across this “disease-span” was “glial sugar metabolism” (Johnson et al., 2020). This proteomic node was upregulated in both asymptomatic and symptomatic AD brains and consisted of proteins primarily involved in glycogen metabolism and glycolysis, including lactate dehydrogenase (LDH), the enzyme responsible for interconversion of pyruvate and lactate. Importantly, the glial sugar metabolism cluster identified was reproduced across multiple cohorts, tissues, and proteomic techniques, and thus strongly implicates astrocyte metabolism as an upstream driver of AD pathology and cognitive decline.
Despite many remaining questions, the central importance of astrocytes in cerebral metabolism has become clear. In fact, one of the most well-known neuronal support functions of astrocytes is a metabolic one – i.e. the astrocyte-neuron lactate shuttle (ANLS). The ANLS is a hypothetical framework for cerebral metabolism (backed by substantial evidence), which posits that astrocytes metabolize glucose to lactate, which is then released, taken up by nearby neurons and metabolized via oxidative phosphorylation as a fuel source. Astrocytes contain the main stores of glycogen in the brain – a store that is broken down to glucose, metabolized to lactate and shuttled to neurons. As mentioned, glucose utilization and lactate production in astrocytes is closely coupled to the activity of local neurons via EAATs (see Fig. 3), which not only take up synaptically released glutamate (for recycling via the glutamine/glutamate cycle), but also generate electrochemical gradients that stimulate glucose uptake and utilization via glycolysis. Inhibition of astrocyte glycogen metabolism at any of these steps has been shown to be detrimental to both acute neural function and for the extended processes of LTP and memory formation. Accumulating evidence over the past 25 years reveals that neurons actively utilize lactate as an energy source, and in fact, when presented with the choice, neurons preferentially utilize lactate over glucose (van Hall et al., 2009; Wyss et al., 2011). Given that lactate is an energy substrate used by the brain (Newington et al., 2013) and a competitive glucose alternative (Tabernero et al., 1996; Bouzier-Sore et al., 2006; Rasmussen et al., 2011), it is perhaps important to revisit the decreased FDG-PET signal in AD knowing that lactate itself has been shown to decrease FDG-PET signal (Smith et al., 2003). Thus, a potential increase in astrocyte-derived lactate may serve to compete with glucose as a substrate for brain metabolism and decrease CMRglc.
With the current understanding that this critical source of neuronal lactate is astrocyte derived, it becomes clear how this cell type (and its relative rate of lactate production) might play a foundational role in brain health and homeostasis. As noted above, aerobic glycolysis refers to the metabolism of glucose to lactate instead of the oxidative TCA cycle, despite the presence of abundant oxygen. This seemingly counterintuitive phenomenon actually occurs quite frequently in the young human brain, with a peak around five years of age (when up to 30 % of the brain’s glucose is processed this way), and then gradually declines with age (Goyal et al., 2017). Further, this process appears to be both regionally and cell-type specific, with astrocytes playing a major role in certain regions such as the precuneus, posterior cingulate cortex, and dorsolateral prefrontal cortex (Magistretti, 2016). Interestingly, the areas associated with high rates of aerobic glycolysis tend to overlap with areas known to accumulate amyloid β, indicating that perhaps the metabolic profile of certain brain regions may predispose them to later life amyloid burden (Vlassenko et al., 2010).
While the brain primarily relies on glucose as an energy substrate, other energy substrates – mainly fatty acids and ketone bodies – also contribute significantly to cerebral metabolism. While the traditional view has long been that the brain does not utilize a significant amount of fatty acids (FA), recent studies prove otherwise, estimating up to 20 % of cerebral ATP is generated from FA (Ebert et al., 2003; Panov et al., 2014). Although transport of FA through the BBB may be too limited to make FA a primary energy substrate in brain compared to glucose, appreciable concentrations of FA cross the BBB and are oxidized, (Allweis et al., 1966; Dhopeshwarkar and Mead, 1970; Spitzer, 1973) including FA derived from peripheral lipoproteins (Gao et al., 2017; Lee et al., 2017). Unlike neurons, astrocytes can readily β-oxidize FA (Edmond et al., 1987; Auestad et al., 1991). Despite the ability and favorable energetics of FA oxidation compared to glucose (the energy density of fat is over twice that of carbohydrate), the astrocytes (and the brain at large) still appear to largely avoid FA oxidation. Several explanations for this avoidance have been proposed (Schonfeld and Reiser, 2013). First, neuronal activation is a temporally constrained process that requires rapid responses to meet energy demands, and while FA β-oxidation is more efficient than glycolysis on a molar basis, it is a biochemically slower process. Second, an increased rate of FA oxidation would require more oxygen (1 mol of palmitate for example would require 23 mol of oxygen, versus just 6 per mole of glucose), potentially leading to cerebral hypoxia. Third, an increased rate of FA oxidation would result in increased generation of ROS, and in spite of high pentose phosphate pathway activity to manage these ROS, neurons are particularly sensitive to oxidative stress (Wang and Michaelis, 2010; Schonfeld and Reiser, 2013).
Fatty acids also serve as the precursor to ketone bodies, another important cerebral energy substrate. Ketone bodies (acetoacetate, beta-hydroxybutyrate, and acetone (the breakdown product of acetoacetate)), are small molecules produced by the liver from fatty acids. Ketones are primarily produced by the liver during periods of very low carbohydrate intake, such as prolonged fasting, carbohydrate restricted diets, intense exercise, and/or starvation. Cerebral ketone body uptake is directly correlated to circulating levels (i.e. it is “pushed” into the brain at a rate proportional to plasma concentrations), and the brain readily utilizes ketone bodies for energy production. Interestingly, ketone body utilization rates change dramatically over the course of the lifespan (Cunnane et al., 2011). Unlike the adult human brain, which appears to only utilize ketones during periods of glucose insufficiency, infants use ketones both as a primary source of ATP and as a principal substrate for lipid synthesis in the brain (Cunnane et al., 2003).
Interestingly, astrocytes appear to be a local source of ketone synthesis. In fact, astrocytes are the primary site of fatty acid oxidation and the only known source of ketone body production in the brain (Edmond et al., 1987; Edmond et al., 1998; Blazquez et al., 1999; Le Foll and Levin, 2016). In addition to using fatty acids as precursors for the synthesis of ketone bodies which are then exchanged with neurons, astrocytes can also produce ketones from amino acids (Auestad et al., 1991; Guzman and Blazquez, 2001). Importantly, cell culture studies suggest that when glucose availability is limited, astrocytes readily shift their metabolism from glycolysis to fatty acid oxidation and efficiently utilize fatty acids as their primary source for ATP generation (Weightman Potter et al., 2019). Moreover, studies show that even when sufficient glucose is available, ketone supplementation results in decreased cerebral glucose uptake, suggesting that ketones may actually be the preferred substrate for the brain (Hasselbalch et al., 1995). Additionally, 11C-acetoacetate (AcAc) PET studies have shown that, unlike glucose uptake, cerebral uptake of AcAc did not significantly differ between AD and MCI brains and those of cognitively healthy controls in all brain regions measured (Cunnane et al., 2016). These observations have led some to suggest ketones as a therapeutic energy substrate replacement for glucose, given that AD is associated with consistent and progressive decreases in glucose uptake. Further support for this idea is garnered from the fact that the menopausal transition is also characterized by reductions in brain glucose metabolism and mitochondrial respiration (Yao et al., 2010; Ding et al., 2013; Brinton et al., 2015; Yin et al., 2015; Mosconi et al., 2017a, 2017b). When ketone availability is limited myelin lipids are used to generate ketone bodies, thereby inducing myelin catabolism (Klosinski et al., 2015) and subsequent loss of white matter volume (Mosconi et al., 2017a, 2017b). Furthermore, this hypometabolic state is known to promote Aβ accumulation (Mattson and Magnus, 2006; Brinton et al., 2015); an effect that is only exacerbated in postmenopausal, APOE-e4 carriers (Mosconi et al., 2017a, 2017b). Several approaches designed to increase circulating ketone body concentrations have been proposed, including indirect pharmacological stimulation of ketone production via a variety of pathways, prolonged fasting, ketogenic diets, and dietary supplementation of medium chain triglycerides (which are metabolized to produce AcAc and BHB). However, while seemingly safe and well justified from a neuroenergetic perspective, ignoring glucose hypometabolism that accompanies aging and AD in favor of a ketogenic approach requires further long-term clinical testing to determine if it is a clinically effective treatment strategy (Cunnane et al., 2016).
7.2. Metabolic changes in reactive astrocytes
The rapidly growing field of immunometabolism has outlined a clear picture of metabolic reprogramming in myeloid cells during activation. Advances in cancer research have provided detailed descriptions of the metabolic shift from oxidative phosphorylation (resting) to glycolysis (activated) in immune cell populations – findings that have also applied to microglia as neuroinflammation and immunometabolism have pushed to the forefront in the AD field. Despite new knowledge regarding the intersection between microglial activation and metabolism, comparatively little is known about the metabolic consequences of astrocyte activation, particularly in vivo.
Unlike microglia, which at rest rely primarily on oxidative phosphorylation for ATP production, astrocytes are thought to be preferentially glycolytic. Recent transcriptomic studies examining astrocyte reactivity under a variety of injury or disease paradigms have provided invaluable datasets in which to explore metabolic changes during reactivity – some of which hint at increased glycolysis in reactive astrocytes (Zamanian et al., 2012; Liddelow et al., 2017, Boisvert et al., 2018). The relative youth of the field means that studies specifically addressing metabolic changes in reactive astrocytes are still relatively sparse. Nonetheless, a handful of studies have begun to directly examine astrocyte, as opposed to microglial, metabolic reprogramming following exposure to pro-inflammatory stimuli (Afridi et al., 2020). For example, experiments by various groups consistently revealed similar phenotypic changes characterizing the metabolic response of reactive astrocytes as increased ATP production via glycolysis and reduced oxygen consumption (Ferrick et al., 2008; Motori et al., 2013). Similarly, Allaman et al. demonstrated increases in glycolysis and lactate release in astrocytes following exposure to Aβ (Allaman et al., 2010). Additionally, a study by Jiang and Cadenas showed that rat primary astrocytes exhibit age-dependent increases in mitochondrial oxidative metabolism in concert with augmented responses to inflammatory cytokines (Jiang and Cadenas, 2014). Treatment with IL-1β and TNFα stimulate oxidative phosphorylation and mitochondrial biogenesis in these rat astrocytes, suggesting that the increased mitochondrial respiration and inflammatory response are interconnected (Jiang and Cadenas, 2014). In sum, these studies highlight an intriguing inflammation-induced shift toward glycolysis in reactive astrocytes. Granted, it remains unclear whether these changes reflect mitochondrial dysfunction or a “voluntary” reprogramming toward glycolysis.
8. Astrocytes and sleep disturbances
Although dementia disorders are not a normal part of aging, advanced age is the most profound risk factor for AD and other related dementias [62]. The risk imposed by age may be due in part to the association between normative aging and the reduced ability to initiate and maintain sleep [63]. Both sleep-wake abnormalities and circadian dysfunction are prevalent in AD (Pollak and Perlick, 1991; Vitiello and Borson, 2001; McCurry and Ancoli-Israel, 2003; Bliwise, 2004). In fact, compared to cognitively normal older adults, individuals with AD experience more fragmented sleep and insomnia, with estimates approaching 25–66 % of mild-to-moderate AD patients being affected (Bianchetti et al., 1995; Guarnieri et al., 2012). Sleep deprivation has not only been shown to impair memory consolidation but a bidirectional relationship between poor sleep quality and AD has been reported. In 2014, Ju et al. demonstrated that patients with AD experience sleep disturbances and also showed that poor sleep predisposes individuals to AD. Interestingly, emerging evidence suggests that sleep disturbances precede clinical AD diagnoses by years. In 2013 Hita-Yañez et al. reported shorter bouts of REM sleep and increased slow wave sleep fragmentation in individuals with MCI (Hita-Yanez et al., 2013). Additionally, this same study found that disruptions in REM sleep were exacerbated in APOE-e4 carriers, indicating that, even in prodromal phases, sleep disturbances exist in individuals with increased risk of developing AD.
Recent studies have shown that both neurons and glial cells are essential for sleep. In 2009, Halassa et al. provided the first evidence that the sleep/wake cycle is modulated by astrocytes [64]. It is now understood that astrocytes promote sleep drive through the release of adenosine, an endogenous sleep promoting factor in the brain (Porkka-Heiskanen et al., 1997), which acts on adenosine A1 receptors to induce presynaptic inhibition [64]. Astrocytes also promote sleep-dependent brain-waste clearance mechanisms and aid in shifting neocortical neuronal activity to synchronized slow-oscillations, a hallmark of slow-wave (i.e. deep) sleep, by altering extracellular glutamate levels (Poskanzer and Yuste, 2016). A recent study by Bojarskaite et al. quantified astrocytic Ca2+ signaling during natural sleep and found that astrocytic IP3-mediated Ca2+ signaling changes across the sleep/wake cycle, being reduced during sleep but abruptly increasing prior to behavior and neurophysiological signs of the sleep-to-wake transition (Bojarskaite et al., 2020). It should be noted that astrocytic Ca2+ signaling was found to precede awakenings from slow-wave sleep (both non-REM and IS sleep), but not from REM sleep. Interestingly, this is consistent with the temporal profile of cortical norepinephrine (NE) release from locus coeruleus (LC) neurons upon awakening, suggesting that astrocytic Ca2+ signals upon awakening are triggered by NE. Notably, LC degeneration and subsequent NE loss are among the first identifiable pathological alterations in AD, appearing around the same time sleep issues arise (German et al., 1992). Considering NE is an anti-inflammatory molecule (Heneka et al., 2002) which promotes glial-mediated degradation and phagocytosis of Aβ (Kong et al., 2010), loss of NE suppresses anti-inflammatory responses, thereby inducing a pro-inflammatory state which impairs Aβ degradation and clearance. Thus, LC degeneration and consequent impaired astrocytic Ca2+ signaling could explain the link between the origin of sleep abnormalities and the simultaneous accumulation of senile plaques (Chalermpalanupap et al., 2013).
In vivo microdialysis studies have shown that levels of Aβ rise in the ISF of the CNS during wakefulness and decline during sleep (Kang et al., 2009). A single night of sleep deprivation has been shown to increase morning Aβ42 levels in the CSF of healthy young adults (Ooms et al., 2014). Similarly, a recent study found that sleep deprivation increased overnight CSF levels of Aβ38, Aβ40, and Aβ42 by 25–30 % compared to controls who had a night of normal sleep (Lucey et al., 2018). Moreover, the clearance rate of Aβ from the CNS of individuals with AD is impaired (Mawuenyega et al., 2010). It is now understood that during wakefulness there is little exchange between the CNS and the glymphatic pathway; however, during sleep, brain waste products such as excessive Aβ and tau are cleared from the ISF by a process that requires astrocytes (Xie et al., 2013; Haydon, 2017; Shokri-Kojori et al., 2018). While it is unclear exactly how the sleep/wake cycle regulates solute exchange in this pathway, it is clear that proper perivascular AQP4 localization is crucial. A recent study investigated the association of single-nucleotide polymorphisms (SNPs) in the AQP4 gene with sleep latency, duration, and the amount of radiolabeled Aβ present on PET scans in healthy volunteers age 60 years or older (Rainey-Smith et al., 2018). Rainey-Smith et al. found one SNP associated with poor sleep quality and two associated with abbreviated sleep duration and enhanced Aβ signal on PET. These results and others suggest that Aβ and tau deposition are consequences of impaired clearance rather than of increased production and that proper perivascular AQP4 localization is necessary for glymphatic clearance (Benveniste et al., 2019).
Yet, studies show that the effect of wakefulness extends beyond control of the glymphatic pathway to regulate both structural and transcriptional aspects of astrocytes. Genes associated with the ANLS have been shown to be altered in murine cortical astrocytes following sleep deprivation (Petit et al., 2013), suggesting astrocytes link neurometabolic coupling to the sleep/wake cycle. The fact that wakefulness is associated with increases in both glutamate and lactate alongside a simultaneous reduction in glucose levels provides further support for this idea (Dash et al., 2009; Naylor et al., 2012; Dash et al., 2013). Interestingly, increased wakefulness and neural activity have been shown to regulate extracellular Aβ levels in mice (Bero et al., 2011; Roh et al., 2012). In 2012, Roh and colleagues demonstrated that in the absence of aggregation in amyloid depositing mouse models, diurnal oscillations in Aβ are closely related to the sleep/wake cycle and wake-related lactate levels (Roh et al., 2012). Given neural activity and wakefulness increase Aβ release, data collectively suggests that sleep disturbances may slow Aβ clearance while simultaneously encouraging added wakefulness-induced Aβ release. Together, these data led Vanderheyden et al. to hypothesize that slowed clearance increases the chance of Aβ oligomerization, aggregation, and subsequent plaque formation and that newly formed plaques generate a concentration gradient favoring additional plaque formation. Vanderheyden et al. further speculate that this Aβ gradient recruits both astrocyte and microglia-driven clearance mechanisms while also effectively mobilizing astrocytes away from glutamatergic uptake at synapses, thereby preventing normal ANLS coupling (Vanderheyden et al., 2018). Still, astrocytes may also respond to pathological amyloid deposition by changing their phenotype, rather than their location (Galea et al., 2015). Granted, the number of different astrocyte polarization states remains elusive and the ways in which these different astrocyte activation states impact Aβ uptake, ANLS function, and sleep require further exploration.
To further complicate matters, deficient slow-wave activity in deep sleep stages has been reported in individuals with significant tau pathology. in vivo microdialysis studies have shown that increasing excitatory neuronal activity significantly increases ISF tau levels within hours (Yamada et al., 2014), and that increased wakefulness leads to rapid elevations in extracellular monomeric tau levels, tau spreading and aggregation (Holth et al., 2019). Tau pathology is also associated with reduced NREM slow-wave activity in both cognitively normal and very mildly cognitively impaired individuals (Lucey et al., 2019). One study reported that individuals 60 years or older who present higher tau accumulation on tau PET experience decreased slow-wave sleep power, further implicating astrocyte dysfunction as a contributor to sleep disturbances in AD.
In all, sleep disturbance is now widely recognized as a highly disruptive behavioral manifestation of AD. Changes in sleep efficiency and quality precede the onset of cognitive decline in AD patients and progress in parallel with the development of AD pathology and cognitive impairment. Though the relationships between the sleep/wake cycle and the development of AD-related pathologies are just beginning to be understood, astrocytes appear to play a major role given they are essential for normal slow-wave sleep, which is itself imperative for the consolidation of new episodic memories. Not to mention, many astrocytic functions are likely to be modulated by the sleep/wake cycle considering brain metabolism, neural activity, and synaptic turnover change as a function of behavioral state.
9. Astrocyte-specific targeting approaches
While changes in astrocyte signaling molecules and pathways associated with synapses, blood vessels and metabolism reflect discrete outcomes of astrocyte reactivity, they are also extensively intertwined and may impact one another in deleterious ways, leading to a chain reaction of sorts. In fact, many of these signaling events act both upstream and downstream of one another, or impact common targets in parallel. For instance, reduced glutamate uptake and transporter expression — common biomarkers of astrocyte reactivity— are triggered by transcription factor pathways activated by amyloid pathology and neuroinflammation. In turn, reduced glutamate uptake is likely to disrupt glucose uptake and modulate the production of lactate, both of which are necessary to drive high fidelity neuronal activity. At the same time, excess extracellular glutamate can lead to excitotoxic damage of synapses, astrocyte endfeet, and the BBB, which would not only have adverse effects on neurovascular coupling and glymphatic clearance of protein aggregates but would also lead to the physical erosion of perivascular elements and synapses. The resulting damage, characterized by BBB breakdown as well as fragmentation and phagocytosis of synaptic contacts, is likely to further exacerbate and maintain elevated neuroinflammatory signaling. Fig. 7 provides an admittedly simplified overview of how the various changes in astrocyte signaling discussed in this review are interconnected. This incredible degree of integration highlights just how delicate the balance is between beneficial and abnormal astrocyte signaling en route to progressive chronic astrocyte reactivity during the development of AD. One possible benefit of this interconnectivity is that the resolution of astrocyte reactivity (and presumably the preservation of neuronal viability and function) could be achieved by targeting any number of different astrocyte functions. Testing this idea has proved to be a major challenge for pharmacologic approaches, considering most of the potential drug targets discussed here are expressed (to some degree) in other cell types of the brain and periphery where they may or may not be contributing to pathophysiology. To overcome this challenge, a growing number of studies have utilized genetic targeting approaches to selectively modulate astrocyte signaling pathways in intact animals.
Fig. 7.

Astrocyte reactivity is a primary nexus for the cerebrovascular and neuronal pathologies that arise with AD. The major functional phenotypes associated with astrocyte reactivity include neuroinflammation, impaired glutamate/potassium homeostasis, hypometabolism, and loss of endfoot integrity, all of which are extensively intertwined. Neuroinflammatory pathways directly affect nearby glial cells, neurons, and neurovascular elements (orange arrows). Glutamate and potassium dysregulation directly affect astrocyte metabolism, synapse function, and vascular endothelial cells (green arrows). Breakdown of astrocyte endfoot processes, lead to the loss of BBB integrity, Ca2+ dysregulation, and impaired glymphatic clearance. Impaired astrocyte metabolism directly erodes neuronal viability and glymphatic clearance of interstitial toxins, including Aβ (blue arrows), which, in turn, create an inhospitable environment for neurons marked by elevated neuroinflammation and excitotoxicity (purple arrow).
Below we discuss studies that have targeted transcription factor pathways, connexins, and intermediate filament proteins using astrocyte-specific recombinant viruses and/or conditional knock-out/knock-in mice. The preponderance of data at this point (summarized in Table 1) indicates that interference of astrocyte reactivity at multiple levels can reverse neural dysfunction and other biomarkers of AD. Collectively, these data have established critical proof-of-principle for astrocyte modulatory strategies in AD, while simultaneously corroborating that reactive astrocytes play a causative role in AD pathophysiology. Nonetheless, some studies have come to the opposite conclusion— that promotion (rather than reduction) of astrocyte reactivity helps to limit pathophysiology and neural dysfunction. These mixed findings reinforce the lesson that astrocytes are very heterogeneous, and that astrocyte reactivity is a highly complicated (and likely also heterogenous) process that requires further extensive investigation. From a therapeutic standpoint, the data compel more sophisticated approaches for limiting deleterious effects of reactive astrocytes while simultaneously promoting beneficial actions.
Table 1.
Astrocyte targeting strategies in intact animal models of AD-like pathology.
| Molecular target | Targeting method | Bio/behavioral Effects |
|---|---|---|
| Calcineurin/NFAT | AAV-GFa2 delivery of VIVIT to hippocampus of APP/PS1 mice | Reduced frequency of large, reactive astrocytes; reduced Iba1 immunolabeling and protein levels; reduced Aβ pathology; reduced BACE1 expression; increased synaptic strength and LTP; improved cognitive status (Furman et al., 2012) |
| AAV-Gfa2 delivery of VIVIT to hippocampus of 5xFAD mice | Improved cognitive status; reduced GFAP levels; reduced Aβ pathology; reduced frequency and duration of spontaneous glutamate transients; increased Glt-1 levels; reduced neurite atrophy; improved synaptic strength; reduced frequency of spontaneous synaptic currents; restoration of AMPA/NMDA balance (Sompol et al., 2017) | |
| Gfa-ΔCN (activated calcineurin) overexpressing mice (dox sensitive) crossed with APP/PS1 mice. | Reduced Aβ; reduced GFAP, TNFα and Cd11b1 mRNA levels; improved cognitive status (Fernandez et al., 2012) | |
| JAK/STAT | AAV-Gfa delivery of SOCS3 into the hippocampus of APP/PS1 mice | Normalized astrocytic transcriptome; reduced GFAP/Vimentin immunoreactivity; reduced Aβ pathology improved cognitive status (Ceyzeriat et al., 2018) |
| AAV-Gfa delivery of SOCS3 into the hippocampus of 3xTg mice | Improved synaptic strength and LTP (Ceyzeriat et al., 2018); reduced GFAP protein levels and immunoreactivity; reduced anxiety (Geuillemaud et al., 2020) | |
| Lentivirus delivery of SOC3 into hippocampus of 3xTg | Reduced number of GFAP + astrocytes and reduced GFAP immunoreactivity (Haim et al., 2015) | |
| Conditional knock-out of Stat3 in astrocytes of APP/PS1 mice | Increased astrocyte volume around Aβ plaques; increased microglia branching around plaques; reduced Aβ pathology and microglial-mediated internalization/degradation of Aβ; reduced levels of inflammatory cytokines; reduced neurite atrophy near Aβ plaques; reduced spontaneous Ca2+ transients in astrocytes and neurons; improved cognitive status (Reichenbach et al., 2019) | |
| EAAT2/Glt-1 | Gfa-human EAAT2 mice crossed with J20 mice. | Improved glutamate uptake; improved cognitive status; increased synapsin levels; reduced Aβ pathology; improved survival (Takahashi et al., 2015) |
| Connexins | Gfa-Cx43 knock-out mice crossed with APP/PS1 mice | Reduced unapposed hemichannel activity; reduced astrocytic Ca2+ levels; reduced release of ATP and glutamate; improved neuronal viability; reduced Aβ pathology (Yi et al., 2016); improved cognitive status; reduced number of GFAP + astrocytes; improved LTP; increased dendritic spine density |
| GFAP/vimentin | GFAP + vimentin knock-out mice crossed with | Increased Aβ pathology; increased neuritic dystrophy; reduced astrocyte coverage of Aβ plaques; increased microglia coverage of plaques (Kraft et al., 2013); increase in in neuroinflammatory genes; increased neuronal support genes; no effect on Aβ pathology (Kamphuis et al., 2015) |
9.1. Transcription factor pathways
Modulation of the calcineurin/NFAT pathway was among the very first approaches at directly modulating astrocyte specific function in a mouse model of AD-like pathology (Furman et al., 2012). Selective expression of the NFAT inhibitory peptide VIVIT in APP/PS1 mice using AAV vectors equipped with a GFAP promoter reduced the number of large hypertrophied astrocytes in the hippocampus, ameliorated microglial labeling (Iba1), stabilized synaptic function (i.e. improved synaptic strength and LTP), and protected cognitive function. The effects of VIVIT on amyloid pathology were mild (~20 % reduction), but significant. When delivered to hippocampal astrocytes of 5xFAD mice, VIVIT increased the expression of Glt-1 transporters, reduced the width and frequency of spontaneous glutamate transients, quelled synaptic hyperexcitability, and preserved neurite integrity (Sompol et al., 2017). These results are consistent with numerous other studies suggesting that reactive astrocytes negatively impact local neurons via dysregulation of glutamate transport (Prow and Irani, 2008; Sama et al., 2008; Zumkehr et al., 2015; Fontana et al., 2016; Hefendehl et al., 2016). Cell culture data suggest that calcineurin may mediate similar neuroinflammatory and degenerative effects through its interaction with NFκB (Lim et al., 2013) and FOXO3 transcription factors (Fernandez et al., 2016). In contrast to these observations, hyperactivity (rather than inhibition) of calcineurin in reactive astrocytes has been shown to mediate the neuroprotective and nootropic effects of IGF-1 in an AD mouse model through interactions with PPARγ signaling pathways, though a more recent study found that expression of a similar hyperactive calcineurin fragment disrupted hippocampal synaptic strength (Pleiss et al., 2016). Together, these results suggest that calcineurin can drive both detrimental and beneficial properties of reactive astrocytes.
Inhibition of the JAK/STAT pathway in reactive astrocytes in mouse models of AD-like pathology has produced similar results as calcineurin/NFAT inhibition. Delivery of SOCS3 (an endogenous JaK/Stat inhibitor) to astrocytes of APP/PS1 or 3xTg mice using AAV vectors reduced glial reactivity, lowered amyloid pathology, and improved synaptic function and plasticity, whereas delivery of a constitutively active JAK2 subtype exacerbated pathology and functional deficits (Ceyzeriat et al., 2018). In a separate study, conditional knockout of Stat3 in astrocytes of APP/PS1 mice led to molecular and morphologic changes in astrocytes near to amyloid plaques coincident with microglia-mediated phagocytosis of Aβ (Reichenbach et al., 2019). Astrocytic STAT3 knockout also corresponded to the reduced frequency of neuronal and astrocytic Ca2+ transients as well as improved cognition in APP/PS1 mice. In contrast to these observations, inhibition of astrocytic JAK/STAT did not reduce amyloid pathology in 3xTg mice (Guillemaud et al., 2020), even though effects on glial reactivity were similar to those reported in APP/PS1 mice.
9.2. EAATs
Loss of EAAT2/Glt-1 has been implicated in synaptic deficits, neuronal hyperexcitability, and neurodegeneration in AD. To determine if EAAT2/Glt-1 restoration ameliorates synaptic and cognitive deficits in the context of AD-like pathology, Takahashi et al., crossed APP mice with mice overexpressing EAAT2/Glt-1 selectively in astrocytes (Takahashi et al., 2015). Compared to APP control mice, APP/EAAT2 mice demonstrated improved performance on Y- and T-mazes, as well as a novel object recognition task. Restoration of EAAT2/Glt-1 also stabilized synaptophysin levels, reduced amyloid plaque pathology, and resulted in an overall increase in survival. These results are similar to other studies that have found a variety of protective effects in amyloid and tau rodent models treated with drugs like ceftriaxone and riluzole which enhance EAAT2/Glt-1 expression/function (Zumkehr et al., 2015; Hefendehl et al., 2016; Mokhtari et al., 2017; Pereira et al., 2017; Fan et al., 2018; Wu et al., 2020; Yang et al., 2020).
9.3. Connexins
As discussed, Cx43 is a critical component of gap junctions and unapposed plasmalemmal hemichannels in astrocytes. When hemichannels expressed on adjacent cells are directly apposed to one another they can form gap junction channels between astrocytes, providing a conduit for the intercellular shuttling of ions and small chemical messengers. In contrast, unapposed hemichannels provide an open route between the extracellular milieu and the astrocyte cytosol. Unapposed hemichannel permeability in primary cells and brain slices is increased by inflammatory factors and Aβ, leading to the release of cytotoxic factors (e.g. glutamate, ATP) and the damage of local neurons (Yi et al., 2016). In intact APP/PS1 mice, hemichannel activity is increased in reactive astrocytes throughout the brain but is especially pronounced near amyloid plaques. However, gap junction permeability does not appear to change in vivo, whether near to, or distant from amyloid deposits (Yi et al., 2016). Conditional knockout of Cx43 in astrocytes of APP/PS1 mice reduced astroglial reactivity, increased synapse number, improved neuronal viability, and protected cognition, but did not appreciably alter Aβ levels (Yi et al., 2016; Ren et al., 2018).
9.4. GFAP and vimentin knockouts
Astrocyte hypertrophy is perhaps the most commonly reported biomarker of astrocyte reactivity. Knockdown of key intermediate filament proteins in astrocytes, including GFAP and vimentin, is a highly effective way to suppress hypertrophy and limit the structural plasticity of astrocytes. This approach has been used to assess the impact of astrocyte hypertrophy in several disease models including traumatic brain injury, stroke, and AD (Wilhelmsson et al., 2004; Kraft et al., 2013; Laterza et al., 2018). Combined knockdown of GFAP and vimentin in APP/PS1 mice was initially found to reduce astrocyte hypertrophy and accelerate amyloid plaque pathology (Kraft et al., 2013). A subsequent study by the same group found that GFAP/vimentin knock-down altered the transcriptional profile of reactive astrocytes in APP/PS1 mice, characterized by a slight increase in neuroinflammatory programs, and the restoration of neuronal support genes (Kamphuis et al., 2015). However, while the latter study did observe some mild morphological changes in astrocytes, including reduced interaction/coverage of amyloid plaques, no effects on the size or total number of plaques were observed. The reason for this discrepancy is unclear but may be attributable to different genetic backgrounds. Unfortunately, it remains uncertain whether limitation of astrocyte hypertrophy is primarily beneficial, detrimental, or neutral in the context of AD pathophysiology.
10. Future directions for astrocyte research in AD
In order to develop and refine astrocyte-targeting strategies for AD, it is critical we better understand astrocyte heterogeneity. The recently described A1/A2 distinction for neurotoxic and neuroprotective reactive astrocyte (molecular) phenotypes (Liddelow et al., 2017) has unquestionably generated intense research around astrocytes. However, like the M1/M2 distinction for reactive microglia, it’s likely that the A1/A2 nomenclature will fall short of satisfactorily describing the complexity of astrocytes in neurodegenerative diseases. New single-cell sequencing technologies are revealing extensive differences in the molecular signatures of astrocytes depending on brain region, disease state, and proximity to neuropathological lesions (Itoh et al., 2018; Tassoni et al., 2019; Wheeler et al., 2020). Granted, this heterogeneity may only be touching the tip of the iceberg. In their efforts to identify subpopulations of astrocytes that express Glt1 glutamate transporters, Miller et al. (2019) serendipitously discovered a small group of astrocytes in cortical layer V that express a unique set of genes/proteins that respond to and maintain local synapses in a specific subpopulation of neurons (Miller et al., 2019). It was suggested that the breakdown in this highly specific astrocyte-neuron feedback loop was the underlying cause of the developmental disorder, Norrie disease. Whether unique astrocyte phenotypes and/or a breakdown in astrocyte-neuron networks contributes to the characteristic pattern of synapse loss observed at early stages of AD is not known, but it is intriguing to speculate.
Based on these findings and others, it seems reasonable to suspect that astrocytes are every bit as heterogeneous as neurons and become reactive in ways that defy a simple binary classification. Indeed, we haven’t even really factored in all the nuances of functional phenotype that are being detected with new imaging and physiologic approaches (Bindocci et al., 2017). This heterogeneity is forcing the field to reconsider what astrocyte “reactivity” really is (Escartin et al., 2019). The use of GFAP as the defining feature of the reactive astrocyte phenotype is no longer sufficient. Instead, multiple biomarkers assessed with both molecular and functional approaches (preferably with multivariate and/or clustering analyses) will be necessary to validate reactive astrocyte phenotypes. While no nomenclature or set of criteria will be perfect, new terminology will likely be needed to describe reactive astrocytes specific to unique (and perhaps overlapping) molecular and functional states, depending on brain region, disease, and/or distance from primary lesions. Reactive astrocyte phenotypes may be detrimental, beneficial, or neutral in regard to pathophysiology. Furthermore, astrocyte reactivity under some conditions may not be a static state, but instead a dynamic process that could be normalized or even reversed. Because astrocytes are inherently protective/beneficial cells, a goal for astrocyte-targeting strategies in AD and related neurodegenerative disease research should be the identification of molecular pathways that drive detrimental phenotypes and/or inhibit reversion to neutral or beneficial phenotypes.
Another pressing question in the field is whether rodent models are sufficient for understanding the impact of human astrocytes in disease. Despite all the technological advances for investigating astrocytes in animal models of AD, there is still the problem of species’ differences. Subsets of human astrocytes have morphologic features that astrocytes in rodents (and even non-human primates) simply don’t have. Using tissue excised from human brain (because of intractable epilepsy and other issues), Oberheim et al. identified two classes of GFAP + astrocytes (varicose and intralaminar) with millimeter-long processes that pass through other astrocyte territories and reach across cortical layers (Oberheim et al., 2009). These astrocytes are also bigger on average, have more processes, and transmit Ca2+ signals faster than astrocytes in corresponding rodent brain regions. The implications of these species-related differences for astrocyte reactivity, or for the role of astrocytes in AD, remain unclear but suggest that animal models may have very significant limitations. One option is to focus physiologic studies on fresh post-autopsy human AD brain tissue. While this has been accomplished in a relatively small number of studies, research on fresh autopsy material is a major challenge for investigators without access to research volunteers and/or a dedicated autopsy team. Moreover, tissue for physiologic analyses is severely complicated by long postmortem autopsy intervals (>2−3 h, at best). Biopsy tissue doesn’t suffer from postmortem delays but is usually obtained from patients with serious comorbid brain pathologies (e.g. epilepsy) leading to the issue of proper controls for comparison.
In an attempt to study experimental models that are more relevant to humans, some researchers have recently utilized human induced pluripotent stem cells to establish human astrocyte cultures and 3-D blood brain barrier models (Barbar et al., 2020; Blanchard et al., 2020). The astrocytes established in vitro exhibit morphologic diversity and express many of the same markers as human astrocytes investigated in postmortem tissue. Astrocytes in monocultures also exhibit gain of function (i.e. cytokine expression) and loss of function (i.e. glutamate uptake) changes when treated with pro-inflammatory stimuli. These models could be amenable to high throughput screening of astrocyte-targeted therapies for AD and other neurodegenerative diseases. However, it’s important to note that many of the same caveats that apply to rodent cell culture models also apply to human cultures. Specifically, astrocytes are metabolic cells that regulate and participate in the dynamic interplay between neuronal activity and blood flow. In the absence of this metabolic coupling and other factors (e.g. advanced age) astrocytes may behave very differently, and/or in unpredictable ways in response to insult and therapeutics.
11. Conclusion
Alzheimer’s disease currently afflicts 5.8 million Americans and represents a looming global health crisis that is only predicted to worsen. The current lack of disease-modifying therapies only exacerbates our need to better understand underlying disease mechanisms. Therapeutic approaches to the treatment of AD continue to focus on the major pathological hallmarks of the disease: amyloid plaques and neurofibrillary tau tangles. However, evidence presented in this review implicates astrocyte dysfunction in AD pathophysiology, urging us to move beyond the amyloid cascade hypothesis. As discussed, astrocyte signaling mechanisms are extensively intertwined and likely impact one another in deleterious ways in disease. This incredible degree of interconnectivity offers two potential benefits: 1) the resolution of astrocyte reactivity could be achieved by targeting any number of different astrocyte functions and 2) modifying pathological astrocyte responses will likely improve a number of AD clinical hallmarks (e.g. synaptic dysfunction, impaired glymphatic clearance, cerebral hypoperfusion, hypometabolism, sleep disturbances). Granted, we must first understand the breadth of astrocyte heterogeneity before we can successfully employ astrocyte-targeted treatment strategies. Thus, though much work remains to be done, it is evident astrocyte modulation could present a viable treatment strategy for AD.
Acknowledgements
This manuscript was facilitated by the Alzheimer’s Association International Society to Advance Alzheimer’s Research and Treatment (ISTAART) through the Immunity and Neurodegeneration Professional Interest Area (PIA). The views and opinions expressed in this manuscript represent those of the authors and do not necessarily reflect those of the PIA membership, ISTAART or the Alzheimer’s Association.
Funding
LAJ is supported by R01 AG060056 and R01 AG062550 from the National Institute on Aging.
CMN is supported by RF1 AG027297 and R01 AG056998 from the National Institute on Aging.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H 3rd, Kraner SD, Norris CM, 2009. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci 29 (41), 12957–12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afridi R, Kim JH, Rahman MH, Suk K, 2020. Metabolic regulation of glial phenotypes: implications in neuron-glia interactions and neurological disorders. Front. Cell. Neurosci 14, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ, 2010. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J. Neurosci 30 (9), 3326–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Eroglu C, 2017. Cell biology of astrocyte-synapse interactions. Neuron 96 (3), 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allweis C, Landau T, Abeles M, Magnes J, 1966. The oxidation of uniformly labelled albumin-bound palmitic acid to CO2 by the perfused cat brain. J. Neurochem 13 (9), 795–804. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A, 2003. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A 100 (4), 2106–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Frydenlund DS, Ottersen OP, 2004a. Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience 129 (4), 999–1010. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Xue R, Haug FM, Neely JD, Bhardwaj A, Agre P, Adams ME, Froehner SC, Mori S, Ottersen OP, 2004b. Alpha-syntrophin deletion removes the perivascular but not endothelial pool of aquaporin-4 at the blood-brain barrier and delays the development of brain edema in an experimental model of acute hyponatremia. FASEB J. 18 (3), 542–544. [DOI] [PubMed] [Google Scholar]
- Andersen N, Krauth N, Nabavi S, 2017. Hebbian plasticity in vivo: relevance and induction. Curr. Opin. Neurobiol 45, 188–192. [DOI] [PubMed] [Google Scholar]
- Armada-Moreira A, Gomes JI, Pina CC, Savchak OK, Goncalves-Ribeiro J, Rei N, Pinto S, Morais TP, Martins RS, Ribeiro FF, Sebastiao AM, Crunelli V, Vaz SH, 2020. Going the extra (Synaptic) mile: excitotoxicity as the road toward neurodegenerative diseases. Front. Cell. Neurosci 14, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C, 2010. Pericytes regulate the blood-brain barrier. Nature 468 (7323), 557–561. [DOI] [PubMed] [Google Scholar]
- Asadollahi M, Atazadeh M, Noroozian M, 2019. Seizure in alzheimer’s disease: an underestimated phenomenon. Am. J. Alzheimers Dis. Other Demen 34 (2), 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaraf MI, Diaz Z, Liberman A, Miller WH Jr., Arvanitakis Z, Li Y, Bennett DA, Schipper HM, 2007. Brain erythropoietin receptor expression in Alzheimer disease and mild cognitive impairment. J. Neuropathol. Exp. Neurol 66 (5), 389–398. [DOI] [PubMed] [Google Scholar]
- Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T, 2016. What is a pericyte? J. Cereb. Blood Flow Metab 36 (2), 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auestad N, Korsak RA, Morrow JW, Edmond J, 1991. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J. Neurochem 56 (4), 1376–1386. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T, 2003. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J. Neurosci 23 (21), 7922–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Norris CM, Sompol P, Wilcock DM, Goulding D, Neltner JH, St Clair D, Watterson DM, Van Eldik LJ, 2012. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer’s disease-related pathology. J. Neurosci 32 (30), 10201–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbar L, Jain T, Zimmer M, Kruglikov I, Sadick JS, Wang M, Kalpana K, Rose IVL, Burstein SR, Rusielewicz T, Nijsure M, Guttenplan KA, di Domenico A, Croft G, Zhang B, Nobuta H, Hebert JM, Liddelow SA, Fossati V, 2020. CD49f is a novel marker of functional and reactive human iPSC-Derived astrocytes. Neuron 107 (3), 436–453 e412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Brew H, Attwell D, 1988. Electrogenic glutamate uptake in glial cells is activated by intracellular potassium. Nature 335 (6189), 433–435. [DOI] [PubMed] [Google Scholar]
- Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, Koeppen J, Viana JF, Oliveira JF, Voet T, Ponting CP, Belgard TG, Holt MG, 2020. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun 11 (1), 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC, 2002. Control of synaptic strength by glial TNFalpha. Science 295 (5563), 2282–2285. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV, 2012. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485 (7399), 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim L, Carrillo-de Sauvage MA, Ceyzeriat K, Escartin C, 2015a. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci 9, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim L, Ceyzeriat K, Carrillo-de Sauvage MA, Aubry F, Auregan G, Guillermier M, Ruiz M, Petit F, Houitte D, Faivre E, Vandesquille M, Aron-Badin R, Dhenain M, Deglon N, Hantraye P, Brouillet E, Bonvento G, Escartin C, 2015b. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci 35 (6), 2817–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste H, Heerdt PM, Fontes M, Rothman DL, Volkow ND, 2019. Glymphatic system function in relation to anesthesia and sleep states. Anesth. Analg 128 (4), 747–758. [DOI] [PubMed] [Google Scholar]
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR, 1999. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging 20 (6), 581–589. [DOI] [PubMed] [Google Scholar]
- Bernardino L, Xapelli S, Silva AP, Jakobsen B, Poulsen FR, Oliveira CR, Vezzani A, Malva JO, Zimmer J, 2005. Modulator effects of interleukin-1beta and tumor necrosis factor-alpha on AMPA-induced excitotoxicity in mouse organotypic hippocampal slice cultures. J. Neurosci 25 (29), 6734–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM, 2011. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci 14 (6), 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beydoun MA, Beydoun HA, Gamaldo AA, Teel A, Zonderman AB, Wang Y, 2014. Epidemiologic studies of modifiable factors associated with cognition and dementia: systematic review and meta-analysis. BMC Public Health 14, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchetti A, Scuratti A, Zanetti O, Binetti G, Frisoni GB, Magni E, Trabucchi M, 1995. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia 6 (2), 108–112. [DOI] [PubMed] [Google Scholar]
- Bindocci E, Savtchouk I, Liaudet N, Becker D, Carriero G, Volterra A, 2017. Three-dimensional Ca(2+) imaging advances understanding of astrocyte biology. Science 356 (6339). [DOI] [PubMed] [Google Scholar]
- Blanchard JW, Bula M, Davila-Velderrain J, Akay LA, Zhu L, Frank A, Victor MB, Bonner JM, Mathys H, Lin YT, Ko T, Bennett DA, Cam HP, Kellis M, Tsai LH, 2020. Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med 26 (6), 952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass JP, 2002. Alzheimer’s disease and Alzheimer’s dementia: distinct but overlapping entities. Neurobiol. Aging 23 (6), 1077–1084. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Sanchez C, Daza A, Galve-Roperh I, Guzman M, 1999. The stimulation of ketogenesis by cannabinoids in cultured astrocytes defines carnitine palmitoyltransferase I as a new ceramide-activated enzyme. J. Neurochem 72 (4), 1759–1768. [DOI] [PubMed] [Google Scholar]
- Bliwise DL, 2004. Sleep disorders in Alzheimer’s disease and other dementias. Clin. Cornerstone 6 (Suppl 1A), S16–28. [DOI] [PubMed] [Google Scholar]
- Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ, 2018. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 22 (1), 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarskaite L, Bjornstad DM, Pettersen KH, Cunen C, Hermansen GH, Abjorsbraten KS, Chambers AR, Sprengel R, Vervaeke K, Tang W, Enger R, Nagelhus EA, 2020. Astrocytic Ca(2+) signaling is reduced during sleep and is involved in the regulation of slow wave sleep. Nat. Commun 11 (1), 3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS, 1997. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19 (4), 939–945. [DOI] [PubMed] [Google Scholar]
- Bostom AG, Rosenberg IH, Silbershatz H, Jacques PF, Selhub J, D’Agostino RB, Wilson PW, Wolf PA, 1999. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: the Framingham Study. Ann. Intern. Med 131 (5), 352–355. [DOI] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L, 2006. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur. J. Neurosci 24 (6), 1687–1694. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR, 2005. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med 202 (1), 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Messing A, Olsen ML, 2019. AP-1 and the injury response of the GFAP gene. J. Neurosci. Res 97 (2), 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD, Yao J, Yin F, Mack WJ, Cadenas E, 2015. Perimenopause as a neurological transition state. Nat. Rev. Endocrinol 11 (7), 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronzuoli MR, Facchinetti R, Valenza M, Cassano T, Steardo L, Scuderi C, 2019. Astrocyte function is affected by aging and not alzheimer’s disease: a preliminary investigation in hippocampi of 3xTg-AD mice. Front. Pharmacol 10, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camassa LMA, Lunde LK, Hoddevik EH, Stensland M, Boldt HB, De Souza GA, Ottersen OP, Amiry-Moghaddam M, 2015. Mechanisms underlying AQP4 accumulation in astrocyte endfeet. Glia 63 (11), 2073–2091. [DOI] [PubMed] [Google Scholar]
- Canobbio I, Abubaker AA, Visconte C, Torti M, Pula G, 2015. Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front. Cell. Neurosci 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A, Hoozemans JJ, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJ, 2012. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis 10 (1–4), 329–331. [DOI] [PubMed] [Google Scholar]
- Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, Nordberg A, 2012. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med 53 (1), 37–46. [DOI] [PubMed] [Google Scholar]
- Carvajal FJ, Mattison HA, Cerpa W, 2016. Role of NMDA receptor-mediated glutamatergic signaling in chronic and acute neuropathologies. Neural Plast. 2016, 2701526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh C, Wong TP, 2018. Preventing synaptic deficits in Alzheimer’s disease by inhibiting tumor necrosis factor alpha signaling. IBRO Rep. 4, 18–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh C, Tse YC, Nguyen HB, Krantic S, Breitner JC, Quirion R, Wong TP, 2016. Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 47, 41–49. [DOI] [PubMed] [Google Scholar]
- Ceyzeriat K, Ben Haim L, Denizot A, Pommier D, Matos M, Guillemaud O, Palomares MA, Abjean L, Petit F, Gipchtein P, Gaillard MC, Guillermier M, Bernier S, Gaudin M, Auregan G, Josephine C, Dechamps N, Veran J, Langlais V, Cambon K, Bemelmans AP, Baijer J, Bonvento G, Dhenain M, Deleuze JF, Oliet SHR, Brouillet E, Hantraye P, Carrillo-de Sauvage MA, Olaso R, Panatier A, Escartin C, 2018. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun 6 (1), 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigneau E, Oheim M, Audinat E, Charpak S, 2003. Two-photon imaging of capillary blood flow in olfactory bulb glomeruli. Proc Natl Acad Sci U S A 100 (22), 13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalermpalanupap T, Kinkead B, Hu WT, Kummer MP, Hammerschmidt T, Heneka MT, Weinshenker D, Levey AI, 2013. Targeting norepinephrine in mild cognitive impairment and Alzheimer’s disease. Alzheimers Res. Ther 5 (2), 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA, 2005. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120 (3), 421–433. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Quilichini P, Nimchinsky EA, Buzsaki G, 2010. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J. Neurosci 30 (45), 15298–15303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriveau RA, Bosetti F, Emr M, Gladman JT, Koenig JI, Moy CS, Pahigiannis K, Waddy SP, Koroshetz W, 2016. The science of vascular contributions to cognitive impairment and dementia (VCID): a framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cell. Mol. Neurobiol 36 (2), 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox SB, Woolsey TA, Rovainen CM, 1993. Localized dynamic changes in cortical blood flow with whisker stimulation corresponds to matched vascular and neuronal architecture of rat barrels. J. Cereb. Blood Flow Metab 13 (6), 899–913. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ, 2004. Aminopyridazines inhibit beta-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol. Aging 25 (10), 1283–1292. [DOI] [PubMed] [Google Scholar]
- Cui M, Huang Y, Tian C, Zhao Y, Zheng J, 2011. FOXO3a inhibits TNF-alpha- and IL-1beta-induced astrocyte proliferation:implication for reactive astrogliosis. Glia 59 (4), 641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane SC, Ryan MA, Nadeau CR, Bazinet RP, Musa-Veloso K, McCloy U, 2003. Why is carbon from some polyunsaturates extensively recycled into lipid synthesis? Lipids 38 (4), 477–484. [DOI] [PubMed] [Google Scholar]
- Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, Castellano A, Pifferi F, Bocti C, Paquet N, Begdouri H, Bentourkia M, Turcotte E, Allard M, Barberger-Gateau P, Fulop T, Rapoport SI, 2011. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27 (1), 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane SC, Courchesne-Loyer A, St-Pierre V, Vandenberghe C, Pierotti T, Fortier M, Croteau E, Castellano CA, 2016. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann. N. Y. Acad. Sci 1367 (1), 12–20. [DOI] [PubMed] [Google Scholar]
- Dahl E, 1973. The fine structure of intracerebral vessels. Z. Zellforsch. Mikrosk. Anat 145 (4), 577–586. [DOI] [PubMed] [Google Scholar]
- Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, Lazear HM, Gale M Jr., Diamond MS, Klein RS, 2017. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J. Clin. Invest 127 (3), 843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansokho C, Heneka MT, 2018. Neuroinflammatory responses in Alzheimer’s disease. J. Neural Transm. (Vienna) 125 (5), 771–779. [DOI] [PubMed] [Google Scholar]
- Dash MB, Douglas CL, Vyazovskiy VV, Cirelli C, Tononi G, 2009. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J. Neurosci 29 (3), 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash MB, Bellesi M, Tononi G, Cirelli C, 2013. Sleep/wake dependent changes in cortical glucose concentrations. J. Neurochem 124 (1), 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, Tsui W, Kandil E, Scherer AJ, Roche A, Imossi A, Thorn E, Bobinski M, Caraos C, Lesbre P, Schlyer D, Poirier J, Reisberg B, Fowler J, 2001. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc Natl Acad Sci U S A 98 (19), 10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO, 2013. A cytokine network involving brain-borne IL-1beta, IL-1ra, IL-18, IL-6, and TNFalpha operates during long-term potentiation and learning. Brain Behav. Immun 33, 15–23. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar GA, Mead JF, 1970. Fatty acid uptake by the brain. 3. Incorporation of (1–14C)oleic acid into the adult rat brain. Biochim. Biophys. Acta 210 (2), 250–256. [PubMed] [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH, 1988. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am. J. Pathol 132 (1), 86–101. [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD, 2002. Beta -Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J. Biol. Chem 277 (28), 25056–25061. [DOI] [PubMed] [Google Scholar]
- Ding F, Yao J, Rettberg JR, Chen S, Brinton RD, 2013. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8 (11), e79977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD, 2007. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci 27 (42), 11354–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Alzheimer’s C Disease Cooperative Study Steering, Siemers E, Sethuraman G, Mohs R, G. Semagacestat Study, 2013. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med 369 (4), 341–350. [DOI] [PubMed] [Google Scholar]
- Duffy PE, Rapport M, Graf L, 1980. Glial fibrillary acidic protein and Alzheimer-type senile dementia. Neurology 30 (7 Pt 1), 778–782. [DOI] [PubMed] [Google Scholar]
- Duncombe J, Lennen RJ, Jansen MA, Marshall I, Wardlaw JM, Horsburgh K, 2017. Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol. Appl. Neurobiol 43 (6), 477–491. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Nelson MT, 2010. Potassium channels and neurovascular coupling. Circ. J 74 (4), 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D, Haller RG, Walton ME, 2003. “Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci 23 (13), 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J, 1987. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res 18 (4), 551–561. [DOI] [PubMed] [Google Scholar]
- Edmond J, Higa TA, Korsak RA, Bergner EA, Lee WN, 1998. Fatty acid transport and utilization for the developing brain. J. Neurochem 70 (3), 1227–1234. [DOI] [PubMed] [Google Scholar]
- Eikelboom JW, Lonn E, Genest J Jr., Hankey G, Yusuf S, 1999. Homocyst(e)ine and cardiovascular disease: a critical review of the epidemiologic evidence. Ann. Intern. Med 131 (5), 363–375. [DOI] [PubMed] [Google Scholar]
- Escartin C, Guillemaud O, Carrillo-de Sauvage MA, 2019. Questions and (some) answers on reactive astrocytes. Glia 67 (12), 2221–2247. [DOI] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O’Callaghan J, Petzold GC, Serrano-Pozo A, Steinhauser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles D, B. G, Butt AM, Chen W-T, Cohen-Salmon M, Cunningham C, Deneen B, de Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt M, Iino M, Kastanenka KV, Kettenmann H, Khakh B, Koizumi S, Lee CJ, Liddelow SA, MacVicar B, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai K, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RR, Riquelme-Perez M, Robel S, Rose CR, Rothstein J, Rouach N, Rowitch D, S. A, S. S, Sontheimer H, Swanson RA, Vitorica J, Wanner I, Wood LB, Wu JQ, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A, 2021. Reactive astrocyte nomenclature, definitions and future directions. Nat. Neurosci 24 (3), 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S, Xian X, Li L, Yao X, Hu Y, Zhang M, Li W, 2018. Ceftriaxone improves cognitive function and upregulates GLT-1-Related glutamate-glutamine cycle in APP/PS1 mice. J. Alzheimers Dis 66 (4), 1731–1743. [DOI] [PubMed] [Google Scholar]
- Fang J, Han D, Hong J, Tan Q, Tian Y, 2012. The chemokine, macrophage inflammatory protein-2gamma, reduces the expression of glutamate transporter-1 on astrocytes and increases neuronal sensitivity to glutamate excitotoxicity. J. Neuroinflammation 9, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I, 2007. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J. Neurosci 27 (33), 8745–8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AM, Jimenez S, Mecha M, Davila D, Guaza C, Vitorica J, Torres-Aleman I, 2012. Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol. Psychiatry 17 (7), 705–718. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, Hervas R, Dominguez-Fraile M, Garrido VN, Gomez-Gutierrez P, Vega M, Vitorica J, Perez JJ, Torres Aleman I, 2016. Blockade of the interaction of Calcineurin with FOXO in astrocytes protects against amyloid-beta-Induced neuronal death. J. Alzheimers Dis 52 (4), 1471–1478. [DOI] [PubMed] [Google Scholar]
- Ferrick DA, Neilson A, Beeson C, 2008. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov. Today 13 (5–6), 268–274. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD, 2018. Multiple lines of evidence indicate that gliotransmission does not occur under physiological conditions. J. Neurosci 38 (1), 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT, 2006. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 9 (11), 1397–1403. [DOI] [PubMed] [Google Scholar]
- Firbank MJ, Narayan SK, Saxby BK, Ford GA, O’Brien JT, 2010. Homocysteine is associated with hippocampal and white matter atrophy in older subjects with mild hypertension. Int. Psychogeriatr 22 (5), 804–811. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Berci AM, Benoit ME, Peters DG, Kimura Y, Tenner AJ, 2011. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J. Neuroinflammation 8 (1), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana AC, Fox DP, Zoubroulis A, Mortensen OV, Raghupathi R, 2016. Neuroprotective effects of the glutamate transporter activator (R)-(−)-5-methyl-1-nicotinoyl-2-pyrazoline (MS-153) following traumatic brain injury in the adult rat. J. Neurotrauma 33 (11), 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana R, Agostini M, Murana E, Mahmud M, Scremin E, Rubega M, Sparacino G, Vassanelli S, Fasolato C, 2017. Early hippocampal hyperexcitability in PS2APP mice: role of mutant PS2 and APP. Neurobiol. Aging 50, 64–76. [DOI] [PubMed] [Google Scholar]
- Foucault-Fruchard L, Antier D, 2017. Therapeutic potential of alpha7 nicotinic receptor agonists to regulate neuroinflammation in neurodegenerative diseases. Neural Regen. Res 12 (9), 1418–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PT, Palmer AM, Snape M, Wilcock GK, 1999. The cholinergic hypothesis of Alzheimer’s disease: a review of progress.”. J Neurol Neurosurg Psychiatry 66 (2), 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freygang WH Jr., Sokoloff L, 1958. Quantitative measurement of regional circulation in the central nervous system by the use of radioactive inert gas.”. Adv. Biol. Med. Phys 6, 263–279. [DOI] [PubMed] [Google Scholar]
- Furman JL, Norris CM, 2014. Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 11, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM, 2012. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci 32 (46), 16129–16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gail Canter R, Huang WC, Choi H, Wang J, Ashley Watson L, Yao CG, Abdurrob F, Bousleiman SM, Young JZ, Bennett DA, Delalle I, Chung K, Tsai LH, 2019. 3D mapping reveals network-specific amyloid progression and subcortical susceptibility in mice. Commun Biol 2, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea E, Morrison W, Hudry E, Arbel-Ornath M, Bacskai BJ, Gomez-Isla T, Stanley HE, Hyman BT, 2015. Topological analyses in APP/PS1 mice reveal that astrocytes do not migrate to amyloid-beta plaques. Proc Natl Acad Sci U S A 112 (51), 15556–15561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Layritz C, Legutko B, Eichmann TO, Laperrousaz E, Moulle VS, Cruciani-Guglielmacci C, Magnan C, Luquet S, Woods SC, Eckel RH, Yi CX, Garcia-Caceres C, Tschop MH, 2017. Disruption of lipid uptake in astroglia exacerbates diet-induced obesity. Diabetes 66 (10), 2555–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia O, Torres M, Helguera P, Coskun P, Busciglio J, 2010. A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in Down’s syndrome. PLoS One 5 (12), e14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geranmayeh MH, Rahbarghazi R, Farhoudi M, 2019. Targeting pericytes for neurovascular regeneration. Cell Commun. Signal 17 (1), 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Manaye KF, White CL 3rd, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM, 1992. Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol 32 (5), 667–676. [DOI] [PubMed] [Google Scholar]
- Giovannoni F, Quintana FJ, 2020. The role of astrocytes in CNS inflammation. Trends Immunol. 41 (9), 805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT, 2010. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A 107 (8), 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondo A, Shinotsuka T, Morita A, Abe Y, Yasui M, Nuriya M, 2014. Sustained down-regulation of beta-dystroglycan and associated dysfunctions of astrocytic endfeet in epileptic cerebral cortex. J. Biol. Chem 289 (44), 30279–30288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA, 2008. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456 (7223), 745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, Bateman RJ, Benzinger TL, Morris JC, Raichle ME, 2017. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 26 (2), 353–360 e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady CL, Haxby JV, Schlageter NL, Berg G, Rapoport SI, 1986. Stability of metabolic and neuropsychological asymmetries in dementia of the Alzheimer type. Neurology 36 (10), 1390–1392. [DOI] [PubMed] [Google Scholar]
- Graham IM, Daly LE, Refsum HM, Robinson K, Brattstrom LE, Ueland PM, Palma-Reis RJ, Boers GH, Sheahan RG, Israelsson B, Uiterwaal CS, Meleady R, McMaster D, Verhoef P, Witteman J, Rubba P, Bellet H, Wautrecht JC, de Valk HW, Sales Luis AC, Parrot-Rouland FM, Tan KS, Higgins I, Garcon D, Andria G, et al. , 1997. Plasma homocysteine as a risk factor for vascular disease. The European Concerted Action Project. JAMA 277 (22), 1775–1781. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ, 2020. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat. Rev. Neurol 16 (1), 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri B, Adorni F, Musicco M, Appollonio I, Bonanni E, Caffarra P, Caltagirone C, Cerroni G, Concari L, Cosentino FI, Ferrara S, Fermi S, Ferri R, Gelosa G, Lombardi G, Mazzei D, Mearelli S, Morrone E, Murri L, Nobili FM, Passero S, Perri R, Rocchi R, Sucapane P, Tognoni G, Zabberoni S, Sorbi S, 2012. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement. Geriatr. Cogn. Disord 33 (1), 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemaud O, Ceyzeriat K, Saint-Georges T, Cambon K, Petit F, Ben Haim L, Carrillo-de Sauvage MA, Guillermier M, Bernier S, Herard AS, Josephine C, Bemelmans AP, Brouillet E, Hantraye P, Bonvento G, Escartin C, 2020. Complex roles for reactive astrocytes in the triple transgenic mouse model of Alzheimer disease. Neurobiol. Aging 90, 135–146. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Kanungo M, 2013. Glial molecular alterations with mouse brain development and aging: up-regulation of the Kir4.1 and aquaporin-4. Age Dordr. (Dordr) 35 (1), 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M, Blazquez C, 2001. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab 12 (4), 169–173. [DOI] [PubMed] [Google Scholar]
- Han BH, Zhou ML, Johnson AW, Singh I, Liao F, Vellimana AK, Nelson JW, Milner E, Cirrito JR, Basak J, Yoo M, Dietrich HH, Holtzman DM, Zipfel GJ, 2015. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc Natl Acad Sci U S A 112 (8), E881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada R, Hayakawa Y, Ezura M, Lerdsirisuk P, Du Y, Ishikawa Y, Iwata R, Shidahara M, Ishiki A, Kikuchi A, Arai H, Kudo Y, Yanai K, Furumoto S, Okamura N, 2020.). “(18)F-SMBT-1: a selective and reversible positron-emission tomography tracer for monoamine Oxidase-B imaging. J. Nucl. Med [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D, 2012. Synaptic energy use and supply. Neuron 75 (5), 762–777. [DOI] [PubMed] [Google Scholar]
- Harrower AD, Railton R, Newman P, 1984. Diabetic control and left ventricular ejection fraction during cold stimulation tests in insulin-dependent diabetes. Diabetes Res 1 (1), 35–38. [PubMed] [Google Scholar]
- Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB, 1995. Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am. J. Physiol 268 (6 Pt 1), E1161–1166. [DOI] [PubMed] [Google Scholar]
- Haxby JV, Grady CL, Koss E, Horwitz B, Heston L, Schapiro M, Friedland RP, Rapoport SI, 1990. Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch. Neurol 47 (7), 753–760. [DOI] [PubMed] [Google Scholar]
- Haydon PG, 2017. Astrocytes and the modulation of sleep. Curr. Opin. Neurobiol 44, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, LeDue J, Ko RW, Mahler J, Murphy TH, MacVicar BA, 2016. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Abeta plaques by iGluSnFR two-photon imaging. Nat. Commun 7, 13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O’Banion MK, Weinberg G, Klockgether T, Feinstein DL, 2002. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J. Neurosci 22 (7), 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi S, Heistek TS, Scheltens P, Neumann U, Shimshek DR, Mansvelder HD, Smit AB, van Kesteren RE, 2019. Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol. Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hita-Yanez E, Atienza M, Cantero JL, 2013. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep 36 (9), 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A, 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17 (18), 2205–2232. [DOI] [PubMed] [Google Scholar]
- Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM, 2019. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363 (6429), 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B, 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352 (6286), 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth C, Gleeson P, Attwell D, 2012. Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab 32 (7), 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Smith DE, Ibanez-Sandoval O, Sims JE, Friedman WJ, 2011. Neuron-specific effects of interleukin-1beta are mediated by a novel isoform of the IL-1 receptor accessory protein. J. Neurosci 31 (49), 18048–18059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman K, Strickland S, Norris EH, 2013. The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab 33 (8), 1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, 1993. Regulation of the cerebral microcirculation during neural activity: is nitric oxide the missing link? Trends Neurosci. 16 (6), 206–214. [DOI] [PubMed] [Google Scholar]
- Iadecola C, 2017. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96 (1), 17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara M, Yamamoto Y, 2016. Emerging evidence for pathogenesis of sporadic cerebral small vessel disease. Stroke 47 (2), 554–560. [DOI] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M, 2012. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med 4 (147), 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Itoh Y, Tassoni A, Ren E, Kaito M, Ohno A, Ao Y, Farkhondeh V, Johnsonbaugh H, Burda J, Sofroniew MV, Voskuhl RR, 2018. Cell-specific and region-specific transcriptomics in the multiple sclerosis model: focus on astrocytes. Proc. Natl. Acad. Sci. U. S. A 115 (2), E302–E309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janota C, Lemere CA, Brito MA, 2016. Dissecting the contribution of vascular alterations and aging to alzheimer’s disease. Mol. Neurobiol 53 (6), 3793–3811. [DOI] [PubMed] [Google Scholar]
- Jiang T, Cadenas E, 2014. Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell 13 (6), 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, Thambisetty M, Montine TJ, Lee EB, Trojanowski JQ, Beach TG, Reiman EM, Haroutunian V, Wang M, Schadt E, Zhang B, Dickson DW, Ertekin-Taner N, Golde TE, Petyuk VA, De Jager PL, Bennett DA, Wingo TS, Rangaraju S, Hajjar I, Shulman JM, Lah JJ, Levey AI, Seyfried NT, 2020. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26 (5), 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EV, Bouvier DS, 2014. Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast. 2014, 321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EV, Bernardinelli Y, Tse YC, Chierzi S, Wong TP, Murai KK, 2011. Astrocytes control glutamate receptor levels at developing synapses through SPARC-beta-integrin interactions. J. Neurosci 31 (11), 4154–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir MT, Sufian MA, Uddin MS, Begum MM, Akhter S, Islam A, Mathew B, Islam MS, Amran MS, Md Ashraf G, 2019. NMDA receptor antagonists: repositioning of memantine as a multitargeting agent for alzheimer’s therapy. Curr. Pharm. Des 25 (33), 3506–3518. [DOI] [PubMed] [Google Scholar]
- Kalkman HO, Feuerbach D, 2016. Modulatory effects of alpha7 nAChRs on the immune system and its relevance for CNS disorders. Cell. Mol. Life Sci 73 (13), 2511–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Kooijman L, Orre M, Stassen O, Pekny M, Hol EM, 2015. GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 63 (6), 1036–1056. [DOI] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM, 2009. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326 (5955), 1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapasi A, Schneider JA, 2016. Vascular contributions to cognitive impairment, clinical Alzheimer’s disease, and dementia in older persons. Biochim. Biophys. Acta 1862 (5), 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaney J, Campbell M, 2015. The dynamic blood-brain barrier. FEBS J. 282 (21), 4067–4079. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, 2004. Water homeostasis in the brain: basic concepts. Neuroscience 129 (4), 851–860. [DOI] [PubMed] [Google Scholar]
- Klosinski LP, Yao J, Yin F, Fonteh AN, Harrington MG, Christensen TA, Trushina E, Brinton RD, 2015. White matter lipids as a ketogenic fuel supply in aging female brain: implications for alzheimer’s disease. EBioMedicine 2 (12), 1888–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Ruan L, Qian L, Liu X, Le Y, 2010. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci 30 (35), 11848–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH, 2008. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain 131 (Pt 3), 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, 2020. Astroglia and tau: new perspectives. Front. Aging Neurosci 12, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowianski P, Karwacki Z, Dziewiatkowski J, Domaradzka-Pytel B, Ludkiewicz B, Wojcik S, Litwinowicz B, Narkiewicz O, Morys J, 2003. Evolution of microglial and astroglial response during experimental intracerebral haemorrhage in the rat. Folia Neuropathol. 41 (3), 123–130. [PubMed] [Google Scholar]
- Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, Cirrito JR, Holtzman DM, Kim J, Pekny M, Lee JM, 2013. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27 (1), 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, Plog BA, Ding F, Deane R, Nedergaard M, 2014. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol 76 (6), 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ, 2007. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55 (3), 274–281. [DOI] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C, 2011. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. U. S. A 108 (32), E440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforce R Jr., Rabinovici GD, 2011. Amyloid imaging in the differential diagnosis of dementia: review and potential clinical applications. Alzheimers Res. Ther 3 (6), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Rose G, Sandles L, Wohlstadter TC, Lynch G, 1977. Patterns of astroglial hypertrophy and neuronal degeneration in the hippocampus of ages, memory-deficient rats. J. Gerontol 32 (1), 3–12. [DOI] [PubMed] [Google Scholar]
- Lange A, Gebremedhin D, Narayanan J, Harder D, 1997. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J. Biol. Chem 272 (43), 27345–27352. [DOI] [PubMed] [Google Scholar]
- Laterza C, Uoshima N, Tornero D, Wilhelmsson U, Stokowska A, Ge R, Pekny M, Lindvall O, Kokaia Z, 2018. Attenuation of reactive gliosis in stroke-injured mouse brain does not affect neurogenesis from grafted human iPSC-derived neural progenitors. PLoS One 13 (2), e0192118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler J, 2002. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll C, Levin BE, 2016. Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol 310 (11), R1186–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KI, Lee HT, Lin HC, Tsay HJ, Tsai FC, Shyue SK, Lee TS, 2016. Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J. Neuroinflammation 13 (1), 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LL, Aung HH, Wilson DW, Anderson SE, Rutledge JC, Rutkowsky JM, 2017. Triglyceride-rich lipoprotein lipolysis products increase blood-brain barrier transfer coefficient and induce astrocyte lipid droplets and cell stress. Am. J. Physiol., Cell Physiol 312 (4), C500–C516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Warr O, Attwell D, 1998. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci 18 (23), 9620–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, Zheng H, 2015. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85 (1), 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541 (7638), 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E, Genazzani AA, 2013. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 61 (7), 1134–1145. [DOI] [PubMed] [Google Scholar]
- Longden TA, Nelson MT, 2015. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 22 (3), 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bayghen E, Ortega A, 2011. Glial glutamate transporters: new actors in brain signaling. IUBMB Life 63 (10), 816–823. [DOI] [PubMed] [Google Scholar]
- Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, Patterson BW, Baty J, Morris JC, Ovod V, Mawuenyega KG, Bateman RJ, 2018. Effect of sleep on overnight cerebrospinal fluid amyloid beta kinetics. Ann. Neurol 83 (1), 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TLS, Holtzman DM, 2019. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med 11 (474). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma KG, Qian YH, 2019. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 73, 96–106. [DOI] [PubMed] [Google Scholar]
- MacPherson KP, Sompol P, Kannarkat GT, Chang J, Sniffen L, Wildner ME, Norris CM, Tansey MG, 2017. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 102, 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Newman EA, 2015. Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol 7 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Djukic B, Taneja P, Yu GQ, Lo I, Davis A, Craft R, Guo W, Wang X, Kim D, Ponnusamy R, Gill TM, Masliah E, Mucke L, 2016. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 17 (4), 530–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, 2016. Imaging brain aerobic glycolysis as a marker of synaptic plasticity. Proc Natl Acad Sci U S A 113 (26), 7015–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandybur TI, Chuirazzi CC, 1990. Astrocytes and the plaques of Alzheimer’s disease. Neurology 40 (4), 635–639. [DOI] [PubMed] [Google Scholar]
- Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL, 2004. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem 279 (31), 32869–32881. [DOI] [PubMed] [Google Scholar]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L, 1996. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol 40 (5), 759–766. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Magnus T, 2006. Ageing and neuronal vulnerability. Nat. Rev. Neurosci 7 (4), 278–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ, 2010. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330 (6012), 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell HL, Kersch CN, Woltjer RL, Neuwelt EA, 2017. The translational significance of the neurovascular unit. J. Biol. Chem 292 (3), 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurry SM, Ancoli-Israel S, 2003. Sleep dysfunction in alzheimer’s disease and other dementias. Curr. Treat. Options Neurol 5 (3), 261–272. [DOI] [PubMed] [Google Scholar]
- Meeker KD, Meabon JS, Cook DG, 2015. Partial loss of the glutamate transporter GLT-1 alters brain akt and insulin signaling in a mouse model of alzheimer’s disease. J. Alzheimers Dis 45 (2), 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM, 2011. Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAbeta mice. Acta Neuropathol. 122 (3), 293–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR, Newman EA, 2006. Calcium signaling in specialized glial cells. Glia 54 (7), 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SJ, Philips T, Kim N, Dastgheyb R, Chen Z, Hsieh YC, Daigle JG, Datta M, Chew J, Vidensky S, Pham JT, Hughes EG, Robinson MB, Sattler R, Tomer R, Suk JS, Bergles DE, Haughey N, Pletnikov M, Hanes J, Rothstein JD, 2019. Molecularly defined cortical astroglia subpopulation modulates neurons via secretion of Norrin. Nat. Neurosci 22 (5), 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, van Helmond Z, Kehoe PG, Love S, 2010. Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 20 (4), 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D, 2016. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci 19 (12), 1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Baig I, Levine H 3rd, Guttmann RP, Norris CM, 2011. Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Abeta in primary cell culture. Aging Cell 10 (1), 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtari Z, Baluchnejadmojarad T, Nikbakht F, Mansouri M, Roghani M, 2017. Riluzole ameliorates learning and memory deficits in Abeta25–35-induced rat model of Alzheimer’s disease and is independent of cholinoceptor activation. Biomed. Pharmacother 87, 135–144. [DOI] [PubMed] [Google Scholar]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV, 2015. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85 (2), 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, Pachicano M, Joe E, Nelson AR, D’Orazio LM, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Reiman EM, Caselli RJ, Chui HC, Tcw J, Chen Y, Pa J, Conti PS, Law M, Toga AW, Zlokovic BV, 2020. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581 (7806), 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ, 2008. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 29 (5), 676–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Berti V, Quinn C, McHugh P, Petrongolo G, Osorio RS, Connaughty C, Pupi A, Vallabhajosula S, Isaacson RS, de Leon MJ, Swerdlow RH, Brinton RD, 2017a. Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS One 12 (10), e0185926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Berti V, Quinn C, McHugh P, Petrongolo G, Varsavsky I, Osorio RS, Pupi A, Vallabhajosula S, Isaacson RS, de Leon MJ, Brinton RD, 2017b. Sex differences in Alzheimer risk: brain imaging of endocrine vs chronologic aging. Neurology 89 (13), 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motori E, Puyal J, Toni N, Ghanem A, Angeloni C, Malaguti M, Cantelli-Forti G, Berninger B, Conzelmann KK, Gotz M, Winklhofer KF, Hrelia S, Bergami M, 2013. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 18 (6), 844–859. [DOI] [PubMed] [Google Scholar]
- Moynagh PN, 2005. The interleukin-1 signalling pathway in astrocytes: a key contributor to inflammation in the brain. J. Anat 207 (3), 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, Ikonomovic MD, Perez SE, Scheff SW, 2012. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 123 (1), 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA, 2004. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431 (7005), 195–199. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M, 2009. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int 54 (3–4), 253–263. [DOI] [PubMed] [Google Scholar]
- Naylor E, Aillon DV, Barrett BS, Wilson GS, Johnson DA, Johnson DA, Harmon HP, Gabbert S, Petillo PA, 2012. Lactate as a biomarker for sleep. Sleep 35 (9), 1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01703117. “Riluzole in Mild Alzheimer’s Disease.”.
- NCT03943264. “A Biomarker-directed Study of XPro1595 in Patients With Mild to Moderate Alzheimer’s.”.
- Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV, 2016. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 1862 (5), 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, Hulsmann S, 2006. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J. Neurophysiol 95 (3), 1843–1852. [DOI] [PubMed] [Google Scholar]
- Newington JT, Harris RA, Cumming RC, 2013. Reevaluating metabolism in alzheimer’s disease from the perspective of the astrocyte-neuron lactate shuttle model. J. Neurodegener. Dis 2013, 234572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, 2009. Astrocytes going live: advances and challenges. J. Physiol. (Paris) 587 (Pt 8), 1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, Ashe KH, Carlson GA, Iadecola C, 2000. Abeta 1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A 97 (17), 9735–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noell S, Wolburg-Buchholz K, Mack AF, Beedle AM, Satz JS, Campbell KP, Wolburg H, Fallier-Becker P, 2011. Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. Eur. J. Neurosci 33 (12), 2179–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD, 2005. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci 25 (18), 4649–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, Olsen ML, 2016. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 132 (1), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26 (40), 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, Ransom BR, Goldman SA, Nedergaard M, 2009. Uniquely hominid features of adult human astrocytes. J. Neurosci 29 (10), 3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen M, Aguilar X, Sehlin D, Fang XT, Antoni G, Erlandsson A, Syvanen S, 2018. Astroglial responses to amyloid-beta progression in a mouse model of alzheimer’s disease. Mol. Imaging Biol 20 (4), 605–614. [DOI] [PubMed] [Google Scholar]
- Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA, 2014. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71 (8), 971–977. [DOI] [PubMed] [Google Scholar]
- Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA, 2009. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer’s disease and mild cognitive impairment: implications for progression of AD. J. Proteome Res 8 (2), 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov A, Orynbayeva Z, Vavilin V, Lyakhovich V, 2014. Fatty acids in energy metabolism of the central nervous system. Biomed Res. Int 2014, 472459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Verkman AS, 2008. Aquaporins and cell migration. Pflugers Arch. 456 (4), 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, 2015. Blood-brain barrier endogenous transporters as therapeutic targets: a new model for small molecule CNS drug discovery. Expert Opin. Ther. Targets 19 (8), 1059–1072. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ, 1994. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91 (22), 10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira AC, Gray JD, Kogan JF, Davidson RL, Rubin TG, Okamoto M, Morrison JH, McEwen BS, 2017. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 22 (2), 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Nievas BG, Serrano-Pozo A, 2018. Deciphering the astrocyte reaction in alzheimer’s disease. Front. Aging Neurosci 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit JM, Gyger J, Burlet-Godinot S, Fiumelli H, Martin JL, Magistretti PJ, 2013. Genes involved in the astrocyte-neuron lactate shuttle (ANLS) are specifically regulated in cortical astrocytes following sleep deprivation in mice. Sleep 36 (10), 1445–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA, 2015. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J. Neurosci 35 (13), 5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O’Connor JJ, 2005. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol 90 (5), 663–670. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Cummings BJ, Monzavi R, Cotman CW, 1994. Beta-amyloid-induced changes in cultured astrocytes parallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuroscience 63 (2), 517–531. [DOI] [PubMed] [Google Scholar]
- Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, Dineley KT, Parri HR, 2013. alpha7 Nicotinic receptor-mediated astrocytic gliotransmitter release: abeta effects in a preclinical Alzheimer’s mouse model. PLoS One 8 (11), e81828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss MM, Sompol P, Kraner SD, Abdul HM, Furman JL, Guttmann RP, Wilcock DM, Nelson PT, Norris CM, 2016. Calcineurin proteolysis in astrocytes: implications for impaired synaptic function. Biochim. Biophys. Acta 1862 (9), 1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak CP, Perlick D, 1991. Sleep problems and institutionalization of the elderly. J. Geriatr. Psychiatry Neurol 4 (4), 204–210. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW, 1997. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276 (5316), 1265–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, Yuste R, 2016. Astrocytes regulate cortical state switching in vivo. Proc Natl Acad Sci U S A 113 (19), E2675–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell G, Ziso B, Larner AJ, 2019. The overlap between epilepsy and Alzheimer’s disease and the consequences for treatment. Expert Rev. Neurother 19 (7), 653–661. [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Shelanski ML, 2013. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 251, 51–65. [DOI] [PubMed] [Google Scholar]
- Price BR, Norris CM, Sompol P, Wilcock DM, 2018. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem 144 (5), 644–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prow NA, Irani DN, 2008. The inflammatory cytokine, interleukin-1 beta, mediates loss of astroglial glutamate transport and drives excitotoxic motor neuron injury in the spinal cord during acute viral encephalomyelitis. J. Neurochem 105 (4), 1276–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu LB, Zhou Y, Wang Q, Yang LL, Liu HQ, Xu SL, Qi YH, Ding GR, Guo GZ, 2011. Synthetic gelatinases inhibitor attenuates electromagnetic pulse-induced blood-brain barrier disruption by inhibiting gelatinases-mediated ZO-1 degradation in rats. Toxicology 285 (1–2), 31–38. [DOI] [PubMed] [Google Scholar]
- Rainey-Smith SR, Mazzucchelli GN, Villemagne VL, Brown BM, Porter T, Weinborn M, Bucks RS, Milicic L, Sohrabi HR, Taddei K, Ames D, Maruff P, Masters CL, Rowe CC, Salvado O, Martins RN, Laws SM, Group AR, 2018. Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Abeta-amyloid burden. Transl. Psychiatry 8 (1), 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG, 1997. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol 15, 707–747. [DOI] [PubMed] [Google Scholar]
- Rasmussen P, Wyss MT, Lundby C, 2011. Cerebral glucose and lactate consumption during cerebral activation by physical activity in humans. FASEB J. 25 (9), 2865–2873. [DOI] [PubMed] [Google Scholar]
- Reichenbach N, Delekate A, Plescher M, Schmitt F, Krauss S, Blank N, Halle A, Petzold GC, 2019. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med 11 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D, 1996. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med 334 (12), 752–758. [DOI] [PubMed] [Google Scholar]
- Ren R, Zhang L, Wang M, 2018. Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sci. 208, 175–191. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Jackson JG, 2016. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem. Int 98, 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vieitez E, Ni R, Gulyas B, Toth M, Haggkvist J, Halldin C, Voytenko L, Marutle A, Nordberg A, 2015. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur. J. Nucl. Med. Mol. Imaging 42 (7), 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Scholl M, Chiotis K, Thordardottir S, Graff C, Wall A, Langstrom B, Nordberg A, 2016. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 139 (Pt 3), 922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggendorf W, Cervos-Navarro J, 1977. Ultrastructure of arterioles in the cat brain. Cell Tissue Res. 178 (4), 495–515. [DOI] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM, 2012. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med 4 (150), 150ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF, 1996. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16 (3), 675–686. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS, 2005. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J. Cell. Sci . 118 (Pt 24), 5691–5698. [DOI] [PubMed] [Google Scholar]
- Sama DM, Norris CM, 2013. Calcium dysregulation and neuroinflammation: discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev 12 (4), 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM, 2008. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J. Biol. Chem 283 (32), 21953–21964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama DM, Mohmmad Abdul H, Furman JL, Artiushin IA, Szymkowski DE, Scheff SW, Norris CM, 2012. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS One 7 (5), e38170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchouk I, Volterra A, 2018. Gliotransmission: beyond black-and-White. J. Neurosci 38 (1), 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallier A, Smolders I, Van Dam D, Loyens E, De Deyn PP, Michotte A, Michotte Y, Massie A, 2011. Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer’s disease. J. Alzheimers Dis 24 (2), 287–300. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, Arvanitakis Z, 2006. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 27 (2), 252–261. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Bennett DA, 2010. Where vascular meets neurodegenerative disease. Stroke 41 (10 Suppl), S144–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfeld P, Reiser G, 2013. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab 33 (10), 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, 2018. Glycolysis paradigm shift dictates a reevaluation of glucose and oxygen metabolic rates of activated neural tissue. Front. Neurosci. 12, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, Cook DG, 2013. Amyloid-beta1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci 33 (12), 5312–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS, 2015. Integration and modulation of intercellular signaling underlying blood flow control. J. Vasc. Res 52 (2), 136–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Perez MC, Martin ED, Vaquero CF, Azcoitia I, Calvo S, Cano E, Tranque P, 2011. Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia 59 (1), 94–107. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Gomez-Isla T, Growdon JH, Frosch MP, Hyman BT, 2013. A phenotypic change but not proliferation underlies glial responses in Alzheimer disease.”. Am. J. Pathol 182 (6), 2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB, 2007. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int 51 (6–7), 333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Scofield MD, Boger H, Hensley M, Kalivas PW, 2014. Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J. Neurosci 34 (16), 5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA, 2017. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med 9 (392). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Taniguchi T, Fujiwara M, Kameyama M, 1986. Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J. Neurochem 46 (1), 288–293. [DOI] [PubMed] [Google Scholar]
- Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, Lindgren E, Ramirez V, Zehra A, Freeman C, Miller G, Manza P, Srivastava T, De Santi S, Tomasi D, Benveniste H, Volkow ND, 2018. Beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A 115 (17), 4483–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Nedergaard M, 2004. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129 (4), 877–896. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB, Function MRCC, G. Ageing Neuropathology Study, 2010. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31 (4), 578–590. [DOI] [PubMed] [Google Scholar]
- Siskova Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, Remy S, 2014. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84 (5), 1023–1033. [DOI] [PubMed] [Google Scholar]
- Siwek ME, Muller R, Henseler C, Trog A, Lundt A, Wormuth C, Broich K, Ehninger D, Weiergraber M, Papazoglou A, 2015. Altered theta oscillations and aberrant cortical excitatory activity in the 5XFAD model of Alzheimer’s disease. Neural Plast. 2015, 781731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, Kaplan A, La Rue A, Adamson CF, Chang L, et al. , 1995. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273 (12), 942–947. [PubMed] [Google Scholar]
- Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Saxena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak-Vance MA, Roses AD, Barrio JR, Phelps ME, 2000. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci U S A 97 (11), 6037–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Pernet A, Hallett WA, Bingham E, Marsden PK, Amiel SA, 2003. Lactate: a preferred fuel for human brain metabolism in vivo. J. Cereb. Blood Flow Metab 23 (6), 658–664. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, 2020. Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol. 41 (9), 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompol P, Norris CM, 2018. Ca(2+), astrocyte activation and Calcineurin/NFAT signaling in age-related neurodegenerative diseases. Front. Aging Neurosci. 10, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompol P, Furman JL, Pleiss MM, Kraner SD, Artiushin IA, Batten SR, Quintero JE, Simmerman LA, Beckett TL, Lovell MA, Murphy MP, Gerhardt GA, Norris CM, 2017. Calcineurin/NFAT signaling in activated astrocytes drives network hyperexcitability in abeta-bearing mice. J. Neurosci 37 (25), 6132–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SM, Nam DW, Cha MY, Kim KH, Byun J, Ryu H, Mook-Jung I, 2015. Thrombospondin-1 prevents amyloid beta-mediated synaptic pathology in Alzheimer’s disease. Neurobiol. Aging 36 (12), 3214–3227. [DOI] [PubMed] [Google Scholar]
- Spires-Jones T, Knafo S, 2012. Spines, plasticity, and cognition in Alzheimer’s model mice. Neural Plast. 2012, 319836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer JJ, 1973. CNS and fatty acid metabolism. Physiologist 16 (1), 55–68. [PubMed] [Google Scholar]
- Stanimirovic DB, Friedman A, 2012. Pathophysiology of the neurovascular unit: disease cause or consequence? J. Cereb. Blood Flow Metab 32 (7), 1207–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed PM, Tansey MG, Zalevsky J, Zhukovsky EA, Desjarlais JR, Szymkowski DE, Abbott C, Carmichael D, Chan C, Cherry L, Cheung P, Chirino AJ, Chung HH, Doberstein SK, Eivazi A, Filikov AV, Gao SX, Hubert RS, Hwang M, Hyun L, Kashi S, Kim A, Kim E, Kung J, Martinez SP, Muchhal US, Nguyen DH, O’Brien C, O’Keefe D, Singer K, Vafa O, Vielmetter J, Yoder SC, Dahiyat BI, 2003. Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 301 (5641), 1895–1898. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC, 2006. Synaptic scaling mediated by glial TNF-alpha. Nature 440 (7087), 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC, 2005. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci 25 (12), 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B, 2012. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci 35, 369–389. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA, 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131 (6), 1164–1178. [DOI] [PubMed] [Google Scholar]
- Stomrud E, Bjorkqvist M, Janciauskiene S, Minthon L, Hansson O, 2010. Alterations of matrix metalloproteinases in the healthy elderly with increased risk of prodromal Alzheimer’s disease. Alzheimers Res. Ther 2 (3), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohschein S, Huttmann K, Gabriel S, Binder DK, Heinemann U, Steinhauser C, 2011. Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 59 (6), 973–980. [DOI] [PubMed] [Google Scholar]
- Strunz M, Jarrell JT, Cohen DS, Rosin ER, Vanderburg CR, Huang X, 2019. Modulation of SPARC/Hevin proteins in alzheimer’s disease brain injury. J. Alzheimers Dis 68 (2), 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB, 2003. Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc Natl Acad Sci U S A 100 (4), 1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM, 2013. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J. Cereb. Blood Flow Metab 33 (5), 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, Zlokovic BV, 2018. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol 14 (3), 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symon L, Crockard HA, Dorsch NW, Branston NM, Juhasz J, 1975. Local cerebral blood flow and vascular reactivity in a chronic stable stroke in baboons. Stroke 6 (5), 482–492. [DOI] [PubMed] [Google Scholar]
- Tabernero A, Vicario C, Medina JM, 1996. Lactate spares glucose as a metabolic fuel in neurons and astrocytes from primary culture. Neurosci. Res 26 (4), 369–376. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kong Q, Lin Y, Stouffer N, Schulte DA, Lai L, Liu Q, Chang LC, Dominguez S, Xing X, Cuny GD, Hodgetts KJ, Glicksman MA, Lin CL, 2015. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J. Exp. Med 212 (3), 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, Powers ET, Kelly JW, Roberts A, Tong G, Fang-Newmeyer T, Parker J, Holland EA, Zhang D, Nakanishi N, Chen HS, Wolosker H, Wang Y, Parsons LH, Ambasudhan R, Masliah E, Heinemann SF, Pina-Crespo JC, Lipton SA, 2013. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A 110 (27), E2518–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnini F, Novelia J, Kerrigan TL, Brown JT, Tsaneva-Atanasova K, Randall AD, 2015. Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front. Cell. Neurosci 9, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Menard J, Zetterberg H, Wisniewski T, de Leon MJ, 2015. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol 11 (8), 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassoni A, Farkhondeh V, Itoh Y, Itoh N, Sofroniew MV, Voskuhl RR, 2019. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci. Rep 9 (1), 10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R, Perry E, 2003. Alzheimer’s disease is associated with a selective increase in alpha7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia 41 (2), 207–211. [DOI] [PubMed] [Google Scholar]
- Tolosa L, Caraballo-Miralles V, Olmos G, Llado J, 2011. TNF-alpha potentiates glutamate-induced spinal cord motoneuron death via NF-kappaB. Mol. Cell. Neurosci 46 (1), 176–186. [DOI] [PubMed] [Google Scholar]
- Tomimoto H, Akiguchi I, Wakita H, Suenaga T, Nakamura S, Kimura J, 1997. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol 94 (2), 146–152. [DOI] [PubMed] [Google Scholar]
- Vafadar-Isfahani B, Ball G, Coveney C, Lemetre C, Boocock D, Minthon L, Hansson O, Miles AK, Janciauskiene SM, Warden D, Smith AD, Wilcock G, Kalsheker N, Rees R, Matharoo-Ball B, Morgan K, 2012. Identification of SPARC-like 1 protein as part of a biomarker panel for Alzheimer’s disease in cerebrospinal fluid.”. J. Alzheimers Dis 28 (3), 625–636. [DOI] [PubMed] [Google Scholar]
- Vagnucci AH Jr., Li WW, 2003. Alzheimer’s disease and angiogenesis. Lancet 361 (9357), 605–608. [DOI] [PubMed] [Google Scholar]
- van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, Hofman PA, Verhey FR, Backes WH, 2016. Blood-brain barrier leakage in patients with early alzheimer disease. Radiology 281 (2), 527–535. [DOI] [PubMed] [Google Scholar]
- van de Haar HJ, Jansen JFA, Jeukens C, Burgmans S, van Buchem MA, Muller M, Hofman PAM, Verhey FRJ, van Osch MJP, Backes WH, 2017. Subtle blood-brain barrier leakage rate and spatial extent: considerations for dynamic contrast-enhanced MRI. Med. Phys 44 (8), 4112–4125. [DOI] [PubMed] [Google Scholar]
- van Hall G, Stromstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB, 2009. Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab 29 (6), 1121–1129. [DOI] [PubMed] [Google Scholar]
- Vanderheyden WM, Goodman AG, Taylor RH, Frank MG, Van Dongen HPA, Gerstner JR, 2018. Astrocyte expression of the Drosophila TNF-alpha homologue, Eiger, regulates sleep in flies. PLoS Genet. 14 (10), e1007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Knopman DS, 2016. The role of cerebrovascular disease when there is concomitant Alzheimer disease. Biochim. Biophys. Acta 1862 (5), 952–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, 2019. Astroglial calcium signaling in aging and alzheimer’s disease. Cold Spring Harb. Perspect. Biol 11 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, 2014. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond., B, Biol. Sci 369 (1654), 20130595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A, Parpura V, Rodriguez-Arellano JJ, Zorec R, 2019. Astroglia in Alzheimer’s Disease. Adv. Exp. Med. Biol 1175, 273–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeer SE, van Dijk EJ, Koudstaal PJ, Oudkerk M, Hofman A, Clarke R, Breteler MM, 2002. Homocysteine, silent brain infarcts, and white matter lesions: the Rotterdam scan Study. Ann. Neurol 51 (3), 285–289. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Greenberg SM, 2011. Cerebral amyloid angiopathy in the elderly. Ann. Neurol 70 (6), 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello MV, Borson S, 2001. Sleep disturbances in patients with Alzheimer’s disease: epidemiology, pathophysiology and treatment. CNS Drugs 15 (10), 777–796. [DOI] [PubMed] [Google Scholar]
- Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME, Mintun MA, 2010. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc Natl Acad Sci U S A 107 (41), 17763–17767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL, 2017. Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol. 16 (4), 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallraff A, Odermatt B, Willecke K, Steinhauser C, 2004. Distinct types of astroglial cells in the hippocampus differ in gap junction coupling. Glia 48 (1), 36–43. [DOI] [PubMed] [Google Scholar]
- Wang X, Michaelis EK, 2010. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Reddy PH, 2017. Role of glutamate and NMDA receptors in alzheimer’s disease. J. Alzheimers Dis 57 (4), 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB, 2000. Beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem 275 (8), 5626–5632. [DOI] [PubMed] [Google Scholar]
- Weekman EM, Wilcock DM, 2016. Matrix metalloproteinase in blood-brain barrier breakdown in dementia. J. Alzheimers Dis 49 (4), 893–903. [DOI] [PubMed] [Google Scholar]
- Weekman EM, Sudduth TL, Price BR, Woolums AE, Hawthorne D, Seaks CE, Wilcock DM, 2019. Time course of neuropathological events in hyperhomocysteinemic amyloid depositing mice reveals early neuroinflammatory changes that precede amyloid changes and cerebrovascular events. J. Neuroinflammation 16 (1), 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weightman Potter PG, Vlachaki Walker JM, Robb JL, Chilton JK, Williamson R, Randall AD, Ellacott KLJ, Beall C, 2019. Basal fatty acid oxidation increases after recurrent low glucose in human primary astrocytes. Diabetologia 62 (1), 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng HR, Chen JH, Pan ZZ, Nie H, 2007. Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience 149 (4), 898–907. [DOI] [PubMed] [Google Scholar]
- Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, Couturier CP, Watson BR, Scalisi G, Alkwai S, Rothhammer V, Rotem A, Heyman JA, Thaploo S, Sanmarco LM, Ragoussis J, Weitz DA, Petrecca K, Moffitt JR, Becher B, Antel JP, Prat A, Quintana FJ, 2020. MAFG-driven astrocytes promote CNS inflammation. Nature 578 (7796), 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, Colton CA, 2009. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 159 (3), 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M, 2004. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci 24 (21), 5016–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV, 2011. Central nervous system pericytes in health and disease. Nat. Neurosci 14 (11), 1398–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher MR, Kirov SA, Harris KM, 2007. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 55 (1), 13–23. [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H, 2004. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J. Biol. Chem 279 (6), 4929–4940. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT, 2010. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci 30 (7), 2636–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Bie B, Foss JF, Naguib M, 2020. Amyloid fibril-induced astrocytic glutamate transporter disruption contributes to complement C1q-Mediated microglial pruning of glutamatergic synapses. Mol. Neurobiol 57 (5), 2290–2300. [DOI] [PubMed] [Google Scholar]
- Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B, 2011. In vivo evidence for lactate as a neuronal energy source. J. Neurosci 31 (20), 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M, 2013. Sleep drives metabolite clearance from the adult brain. Science 342 (6156), 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgrafe K, Mandelkow EM, Holtzman DM, 2014. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med 211 (3), 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA, 2007. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab 27 (4), 697–709. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhang R, Shi C, Mao C, Yang Z, Suo Z, Torp R, Xu Y, 2017. AQP4 association with amyloid deposition and astrocyte pathology in the Tg-ArcSwe mouse model of alzheimer’s disease. J. Alzheimers Dis 57 (1), 157–169. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ji WG, Zhang YJ, Zhou LP, Chen H, Yang N, Zhu ZR, 2020. Riluzole ameliorates soluble Abeta1–42-induced impairments in spatial memory by modulating the glutamatergic/GABAergic balance in the dentate gyrus. Prog. Neuropsychopharmacol. Biol. Psychiatry, 110077. [DOI] [PubMed] [Google Scholar]
- Yao J, Hamilton RT, Cadenas E, Brinton RD, 2010. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim. Biophys. Acta 1800 (10), 1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Wu Y, Gao Y, Li Z, Li W, Zhang C, 2016. The’ selfish brain’ is regulated by aquaporins and autophagy under nutrient deprivation. Mol. Med. Rep 13 (5), 3842–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Mei X, Ezan P, Mato S, Matias I, Giaume C, Koulakoff A, 2016. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 23 (10), 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, Xu J, Hsu CY, Mills JC, Holtzman DM, Lee JM, 2006. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci 26 (43), 10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Yao J, Sancheti H, Feng T, Melcangi RC, Morgan TE, Finch CE, Pike CJ, Mack WJ, Cadenas E, Brinton RD, 2015. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging 36 (7), 2282–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M, 2018. Two classes of secreted synaptic organizers in the central nervous system. Annu. Rev. Physiol 80, 243–262. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA, 2012. Genomic analysis of reactive astrogliosis. J. Neurosci 32 (18), 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nani S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G, 2015. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med 21 (8), 880–886. [DOI] [PubMed] [Google Scholar]
- Zeppenfeld DM, Simon M, Haswell JD, D’Abreo D, Murchison C, Quinn JF, Grafe MR, Woltjer RL, Kaye J, Iliff JJ, 2017. Association of perivascular localization of Aquaporin-4 with cognition and alzheimer disease in aging brains. JAMA Neurol. 74 (1), 91–99. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Zhou Y, Qiu LB, Ding GR, Pang XF, 2012. Altered expression of matrix metalloproteinases and tight junction proteins in rats following PEMF-induced BBB permeability change. Biomed. Environ. Sci 25 (2), 197–202. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV, 2015. Establishment and dysfunction of the blood-brain barrier. Cell 163 (5), 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Chen Z, Yun W, Ren J, Li C, Wang H, 2015. Extrasynaptic NMDA receptor in excitotoxicity: function revisited. Neuroscientist 21 (4), 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, 2011. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci 12 (12), 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Pucci S, Vilella A, Gotti C, 2018. Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol 6 (4), 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G, 2003. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci 6 (1), 43–50. [DOI] [PubMed] [Google Scholar]
- Zumkehr J, Rodriguez-Ortiz CJ, Cheng D, Kieu Z, Wai T, Hawkins C, Kilian J, Lim SL, Medeiros R, Kitazawa M, 2015. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 36 (7), 2260–2271. [DOI] [PubMed] [Google Scholar]