

Abstract
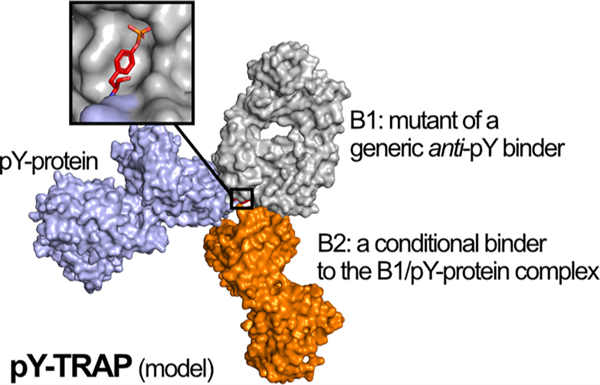
Engineering sequence-specific antibodies (Abs) against phosphotyrosine (pY) motifs embedded in folded polypeptides remains highly challenging because of the stringent requirement for simultaneous recognition of the pY motif and the surrounding folded protein epitope. Here, we present a method named phosphotyrosine Targeting by Recombinant Ab Pair, or pY-TRAP, for in vitro engineering of binders for native pY proteins. Specifically, we create the pY protein by unnatural amino acid misincorporation, mutagenize a universal pY-binding Ab to create a first binder B1 for the pY motif on the pY protein, and then select against the B1−pY protein complex for a second binder B2 that recognizes the composite epitope of B1 and the pY-containing protein complex. We applied pY-TRAP to create highly specific binders to folded Ub-pY59, a rarely studied Ub phosphoform exclusively observed in cancerous tissues, and ZAP70-pY248, a kinase phosphoform regulated in feedback signaling pathways in T cells. The pY-TRAPs do not have detectable binding to wild-type proteins or to other pY peptides or proteins tested. This pY-TRAP approach serves as a generalizable method for engineering sequence-specific Ab binders to native pY proteins.
GRAPHICAL ABSTRACT:
INTRODUCTION
Protein tyrosine phosphorylation (pY) regulates numerous cellular functions in eukaryotes.1,2 Recent advances in pY-peptide proteomics have allowed the identification of thousands of pY modifications especially enabled by broadly specific pY binding antibodies (Abs) that do not depend on flanking peptide sequence.3,4 Crystal structures show that these Abs exquisitely bind the pY residue without contacting neighboring side chains.5 In addition to generic pY binders, there is a need for specific pY binding probes to follow these events in complex cellular settings and tissues.6,7 Most commercial phospho-specific Abs were generated using animal immunization with the disordered phosphopeptides.8 However, this method is low-throughput and expensive, often generates low-affinity, low-specificity, and nonrenewable reagents, and does not apply to nonimmunogenic antigens.7,8
In vitro display methods based on phage or yeast display have provided powerful renewable and nonanimal derived alternatives for next-generation pY binders with higher specificity and affinity.6,9,10 For example, several laboratories have reported the engineering of Src Homology 2 (SH2) domains as anti-pY peptide binders to linear peptides with KD values in the mid- to high-nM range.11,12 We have previously generated phage libraries derived from a peptide binding Ab that permitted selection of antiphosphoserine and antiphosphothreonine Abs that bind linear peptides with similar affinities and specificities.7 Higher-affinity pY binders with KD values in the low-nM range have been identified using an approach called pY-clamps.13 This utilized a circularly permuted Grb2 SH2 domain linked to an evolvable fibronectin type III (FN3) domain. This biparatopic approach utilized a class of linear pY antigens having a specific peptide binding motif governed by the natural Grb2 SH2 domain.
While linear pY signaling motifs are common in nature, pY sites are also commonly embedded in three-dimensional structural domains.14 A systematic analysis of phosphorylation sites banked in the mtcPTM database found that pY modifications do not occur more frequently in loops than α- helices or β-strands.15 Bioinformatics studies presented here further confirm the high frequency of pY modifications in folded epitopes. However, generating highly specific and high-affinity binders to pY sites in tertiary folded protein domains presents significant challenges. While producing a pY peptide immunogen is readily achieved by peptide synthesis, until recently it has been challenging to generate site-specific pY proteins for selections. Furthermore, the recombinant selection approaches using natural motif-based pY binders11–13 are well-suited for linear pY epitopes, but these are not easily re-engineered for tertiary folds due to steric hindrance.
Here, we address the challenge of making pY binders in two folded protein domains, ubiquitin (Ub) and Zeta-chain-associated protein kinase 70 (ZAP70), using a protein engineering platform we call pY Targeting by Recombinant Ab Pairs, or pY-TRAP. In this method, we first generate the site-specifically modified pY protein using a recently described unnatural amino acid misincorporation method.16 We then identify a first modest affinity binder, B1, that covers the pY modification and the nearby amino acids by selection from a generic 4G10 anti-pY antibody library. We next identify a second, conditional binder, B2, that only binds to the folded pY antigen/B1 complex at a composite site using a Fab-phage library geared toward protein antigens. The identified pY-TRAP binders have high affinity and specificity to the pY proteins.
RESULTS
A Large Proportion of pY Modifications Occur within a Rigid Secondary Structure.
pY-containing sites are often in disordered regions or loops creating linear epitopes for binding partners such as SH2, which are widespread mediators of cell signaling. However, pY modifications can also be embedded in nonlinear, three-dimensional protein structures for which the functions are far less understood (Figure 1A). We sought to determine the prevalence of pY modifications that occurred within different secondary structures. The pY-containing sites were retrieved from PhosphoSitePlus (phosphosite.org),4 a broadly used database that provides comprehensive information on protein PTMs (Figure S1A). In total, 6063 pY sites in 2076 human proteins were identified, along with full-length sequence information from the UniProt database.17 527 of the proteins (covering 1501 pY sequences) have available three-dimensional structures in the PDB database for the parental, nonphosphorylated form. We used a standard Dictionary of Secondary Structure of Proteins (DSSP) algorithm18 to extract secondary structure information from the 1501 sequences from the PDB structures (Figure 1B). We found that pY modifications occur in the helix and sheet more than 50% of the time, and this increased to 70% when centered on the exact tyrosine residue. We broadened the analysis to all 2076 pY-containing proteins to predict the secondary structure from the sequence only using the Garnier−Osguthorpe−Robson (GOR) algorithm (Figure 1C)19 and obtained a similar result. As controls, the same algorithms were applied to randomly selected sequences in the studied proteins to determine the average secondary structure, which showed that roughly 55% are helix and sheet (Figure S1B,C). These results highlighted that a large proportion of tyrosine phosphorylation modifications occur in folded epitopes. Developing Abs targeting these three-dimensional pY sites is critical for studying their biological functions.
Figure 1.
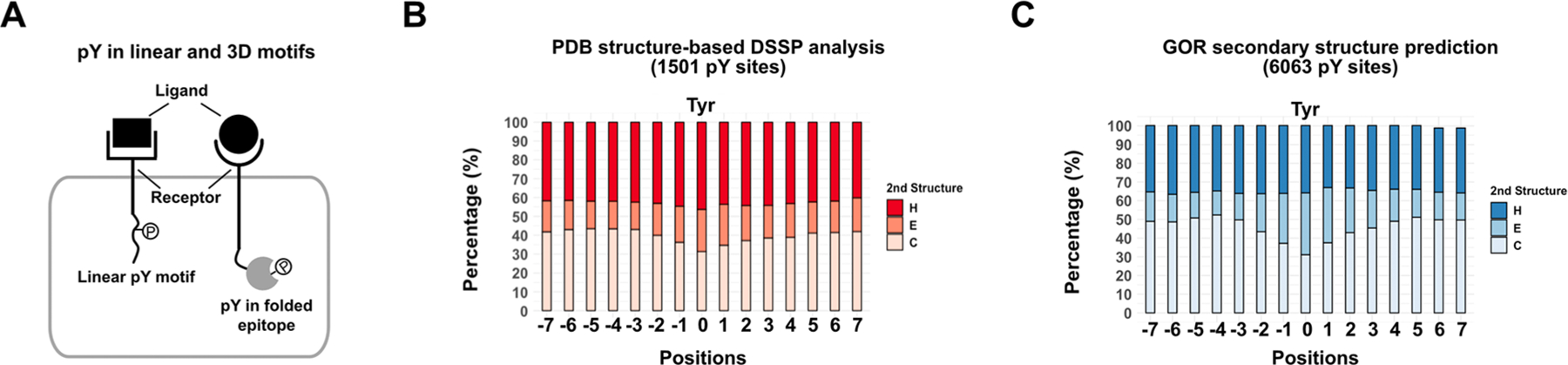
Secondary structure analyses of pY sequences suggest that pY epitopes are commonly embedded in 3D epitopes containing a helix or sheet structure. (A) Schematic illustration of linear and three-dimensional (3D) pY motifs in the receptor proteins in a cell. (B) Secondary structure analysis of 1501 pY sites using the DSSP algorithm based on available PDB structures. (C) Secondary structure prediction of 6063 pY sequences using the GOR algorithm. Position 0 is the pY residue; positions −7 to −1 or 1 to 7 are the seven residues upstream or downstream of the pY motif. H, helices (3,4,5- turn helix); E, strand (ß-sheet, ß-bridge); C, loop (bend, turn, coil).
Generation of Recombinant Biotinylated/Phosphorylated Ubiquitin.
Our first native pY protein targeted a largely uncharacterized pY site at position 59 in human Ub. While serine/threonine phosphorylation of Ub is well-known to modulate protein ubiquitination,20,21 little is known about how phosphorylation of Y59 impacts Ub function. Ub-pY59 is observed almost exclusively in cancerous tissues by proteomics studies, but its biological relevance remains unclear.16,22
Methods to prepare proteins with native pY introduced site-specifically have not been available until recently.16,23 We generated natively folded Ub-pY59 using an unnatural amino acid (Uaa) misincorporation strategy with a biotinylated Avi-tag for immobilization during phage selection (Figure S2A,B).16,24 Protein biotinylation and pY generation via Uaa deprotection were confirmed by SDS-PAGE gel and liquid chromatography− mass spectrometry (LC−MS) (Figure S2C,D).
Engineering a Generic Anti-pY Ab for Binding to the pY Moiety in Ub-pY59.
The overall strategy for pY-TRAPs is shown in Figure 2A. To engineer the first pY binder (B1), we started with the commercially available pan-specific recombinant anti-pY Ab, 4G10.25 We previously grafted the CDRs onto the highly stable and high expressing trastuzumab scaffold known as 4D5 (4G104D5) and determined the structure (Figure 2B).5 This antibody fully covers the pY residue, and we found that it bound weakly to Ub-pY59 (KD of 1.8 μM) as determined by biolayer interferometry (BLI) experiments (Tables S1 and S2). As a specificity control, we tested a generic pY peptide GGG-pY-GGG, with a highly accessible pY and without side chains that may interfere with binding. Indeed, the generic peptide binds to the parental 4G104D5 (KD = 297 nM) about 5- fold tighter than does Ub-pY59 to the parental (KD of 1.8 μM). We believe that this reflects the focused contacts with the pY peptide and steric issues for Ub-pY59.
Figure 2.
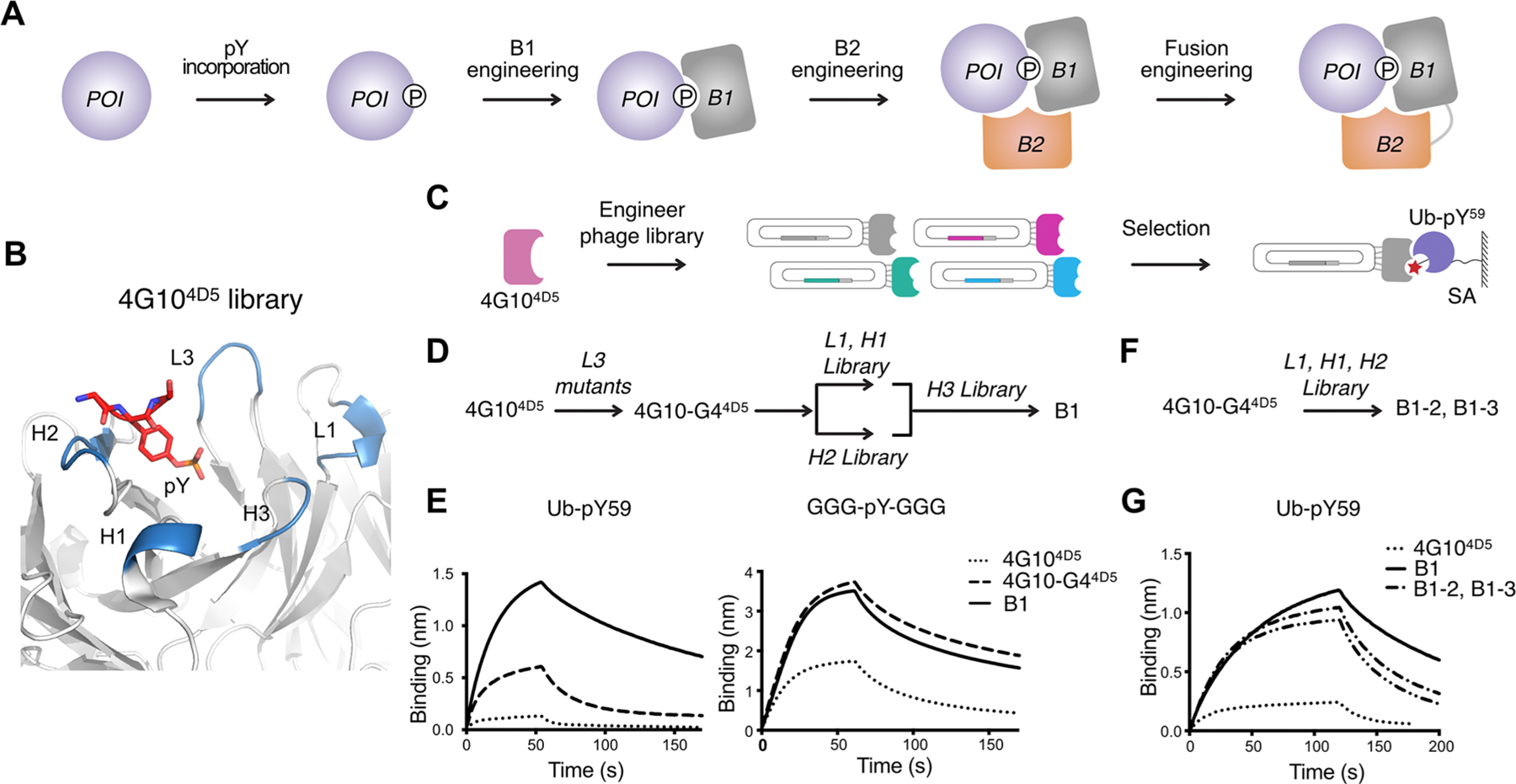
Anti-Ub-pY59 B1 engineering. (A) Four-step process to create pY-TRAPs to pY-native proteins: first, generate a protein of interest (POI) with the single pY modification; second, identify B1 that interacts with the pY motif and the surrounding sequence; third, identify B2 that conditionally binds to the complex of B1 and the POI at a composite site which includes both the POI and B1, and not either alone; and finally, engineer B1−B2 fusions to further improve binding affinity. (B) Structure of the 4G104D5 scaffold (PDB: 6DF1) showing the CDR loops (blue) and the pY residue (red). (C) Phage display workflow for anti-Ub-pY59 B1 selection. (D) CDR-walking approach to engineer anti-Ub-pY59 B1. (E) BLI characterization of 4G104D5, 4G10-G44D5, and B1 to Ub-pY59 and GGG-pY-GGG. (F) Single library approach where CDRs L1, H1, and H2 are simultaneously mutated to engineer anti-Ub-pY59 B1 binders. (G) BLI characterization of 4G104D5, B1, B1−2, and B1−3. The KDs of the interactions are summarized in Table S1.
We embarked on further optimization of the 4G104D5 and displayed it on phage. We first tested phage selection mutants of CDR L3 which is in closest contact with the pY to optimize pY recognition and specificity of Ub-pY59.5 We identified one mutant L3, 4G10-G44D5, that was about 5-fold improved in affinity (KD of 357 nM) relative to the parental 4G104D5 (Figure S3A,B and Table S1). However, the 4G10-G44D5 Fab also bound 2-fold tighter (KD = 160 nM) to the generic peptide, GGG-pY-GGG, suggesting that the enhanced affinity was partially due to strengthened binding specifically to the pY group.5 Next, we used a sequential CDR-walking approach26 to further improve the binding affinity to Ub-pY59 (Figure 2B,D, Figure S3C). We first created a phage library with mutations in L1/H1 and another library with mutations in H2 (Lib-L1-H1, Lib-H2, Table S3). We immobilized the biotinylated Ub-pY59 antigen on streptavidin (SA) magnetic beads and used it for phage selections using a catch and release selection strategy (Figure 2C).5 Interestingly, the sequences of the selected phage clones showed that the last three residues in H2 are highly conserved, suggesting that these amino acids may play a critical role for binding to the pY group (Figure S3D,E). Next, we combined the mutations from the Lib-L1-H1 and Lib-H2 selections and generated an H3 library based on this new mutant. In addition to varying the amino acid compositions, the length of H3 was also engineered (Table S3). Usually CDR-H3 plays the dominant role among the six CDRs.27,28 However, in this case, none of the H3 mutants we generated showed strengthened binding to Ub-pY59, suggesting that H3 does not play a dominant role in the interaction of 4G104D5 with Ub-pY59 (Figure S3C). The tightest-binding anti-Ub-pY59 B1 has a KD of 42 nM to Ub-pY59 and 127 nM to the generic control peptide (Figure 2E, Table S1). The 8.5-fold increase in binding affinity from 4G10- G44D5 to B1 is specific to Ub-pY59 but not to the GGG-pY-GGG peptide, indicating that the gained affinity toward Ub-pY59 resulted from interactions with tertiary sequences surrounding pY and not the pY residue itself.
To increase the efficiency for B1 engineering, based on the knowledge learned from these CDR-walking experiments, we generated a phage library by randomizing sequences in CDR L1, H1, and H2 using 4G10-G44D5 as the parental sequence (Lib- L1-H1-H2, Figure 2F, Figure S3A and Table S3). Using this single-step library, we identified two Abs, B1−2 and B1−3, that interacted with Ub-pY59 with KD values of 63 or 79 nM, respectively, which is ∼5-fold tighter than the parental clone and only slightly weaker than the variant identified using the more laborious sequential CDR-walking approach (KD = 42 nM) (Figure 2G, Table S1). Although these B1 selectants bind significantly tighter to Ub-pY59 than the parental 4G104D5 Fab, they also bind only 2-fold weaker to the GGG-pY-GGG control peptide (KD of 127 nM), indicating that B1 would likely recognize other pY modifications.
Engineering B2 for Binding to the Ub-pY59/B1 Complex.
To further improve binding specificity and affinity, we sought to identify a second binder (B2) that conditionally binds the complex of B1/Ub-pY59, but not to B1 nor Ub-pY59 alone, nor to the B1/GGG-pY-GGG complex (Figures 2A and 3A). To select for such a conditional B2 Fab, we utilized a synthetic Fab-phage library extensively used for selection of globular protein antigens.29 We first removed Fabs that bind to Ub-pY59 using a negative selection by incubating with immobilized Ub-pY59 on SA beads (Figure 3A). The B1/Ub-pY59 complex positive selection bait was generated by mixing SA-immobilized Ub-pY59 with 5 μM of B1-Fab and incubating for 30 min at room temperature (RT). This B1/Ub-pY59 complex was then incubated with the Ub-pY59-cleared phage library to pull down complex-specific binders. The B1-Fab (5 μM) was added both to the phage library/beads mixture as well as to the wash buffers to maintain the formation of the B1/Ub-pY59 complex. Having excess amounts of B1-Fab in solution also prevented enrichment of binders that interact with B1 alone on the SA beads. Phage titer experiments showed that binders were specific to the B1/Ub-pY59 complex, as they enriched as a function of round of selection (Figure 3B).
Figure 3.
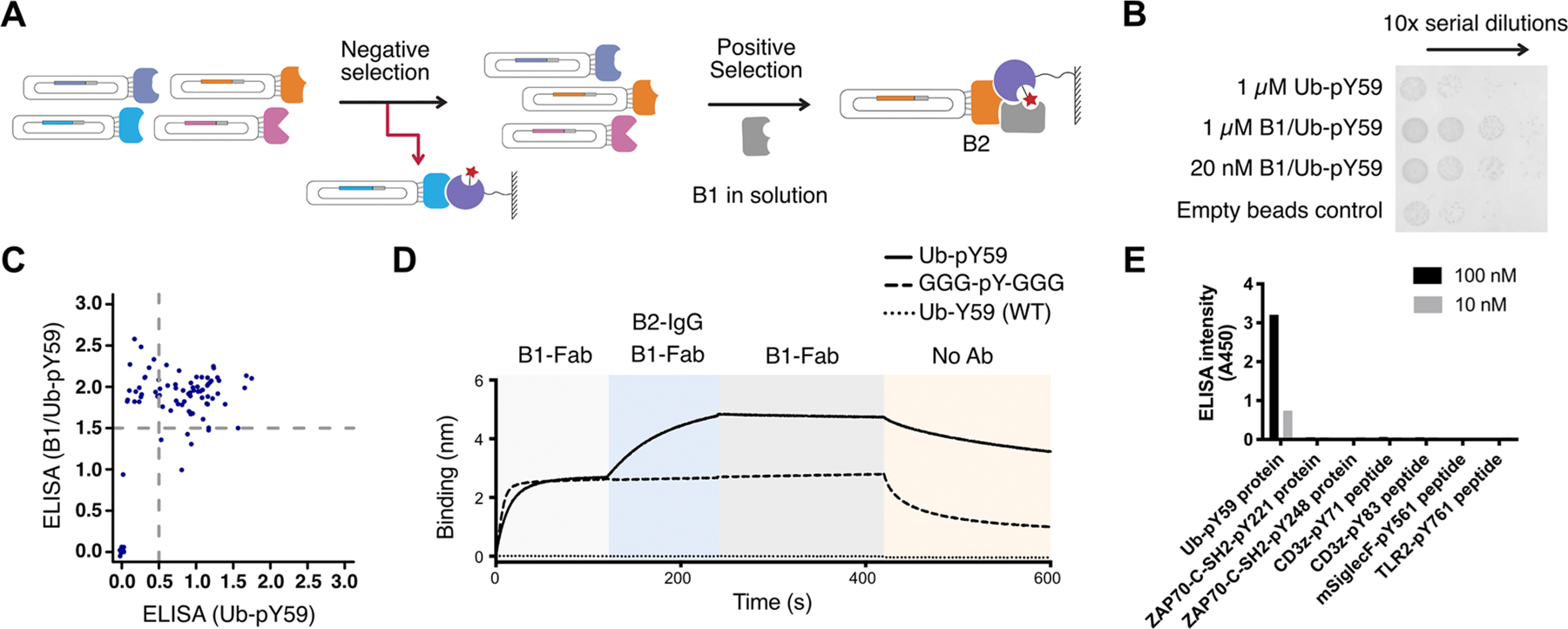
Anti-Ub-pY59 B2 engineering dramatically enhances affinity and specificity. (A) Phage display workflow for negative and positive selection for anti-Ub-pY59 B2. (B) Enrichment phage binders as a function of round by phage titer experiments. Binders that strongly interact with the B1/Ub-pY59 complex were more enriched than binders against Ub-pY59 as seen by higher numbers of phagemid colonies. (C) Characterization of binding of Fab-phage to Ub-pY59 or B1/Ub-pY59 in ELISA revealed clones that interact with both the Ub-pY59 and the B1/Ub-pY59 complex (upper right quadrant), and clones that selectively bind to the B1/Ub-pY59 complex (upper left quadrant). (D) Sequential BLI experiments show that B1 binds both Ub-pY59 (solid curve) and the GGG-pY-GGG control (dashed curve), while B2 added subsequently only recognizes the B1/Ub-pY59 complex (solid curve) but not the B1/GGG-pY-GGG complex (dashed curve); neither B1 nor B2 binds to the WT Ub-Y59 protein (dotted curve). (E) ELISA experiment showing that the anti-Ub-pY59 TRAP binders are highly selective toward Ub-pY59 and not other pY proteins or peptides such as the human ZAP70 SH-2 domain modified at two sites, CD3ζ modified at two sites, and TLR2 and murine SiglecF (mSiglecF) modified at one site.
Next, we characterized the binding of 96 phage clones to the B1/Ub-pY59 complex or Ub-pY59 alone using a phage enzyme-linked immunosorbent assay (ELISA). A group of Fab phage clones were identified that selectively interact with the complex but not Ub-pY59 alone (Figure 3C). These Fab-phage clones were recombinantly expressed as Fabs or IgGs for further characterization. A sequential BLI experiment was performed to identify the tightest binding B2 against the B1/Ub-pY59 complex (Table S2). The B1-Fab bound to both Ub-pY59 and GGG-pY-GGG, but remarkably the B2 only interacted with the B1/Ub-pY59 complex (KD = 5 nM for Fab and 0.6 nM for IgG) and not detectably to the B1/GGG-pY-GGG complex (Figure 3D). In addition, neither B1-Fab nor B2-Fab recognized the nonphosphorylated Ub-Y59 (WT) (Figure 3D). These results demonstrate that the B2 TRAP binder is highly selective to the B1/Ub-pY59 complex. In effect, the pY-TRAP is a conditional biparatopic binder that depends on B1 binding the pY protein before B2 can engage it and the native protein.
In the phage ELISA experiment, we also observed a number of nonconditional clones that interacted with both the Ub-pY59 and the B1/Ub-pY59 complex (Figure 3C). The enrichment of these binders was likely due to the incomplete negative clearance of the library with Ub-pY59. One of these clones, designated as BWT, was expressed and characterized in BLI experiments. Unlike B2, BWT not only bound to the B1/Ub-pY59 complex but also bound to unmodified Ub-Y59 (WT) (Figure S4).
We further interrogated the specificity of the anti-Ub-pY59 TRAP binders by testing the binders against a panel of pY protein or peptide antigens in an ELISA assay (Figure 3E). The pY proteins or peptides (100 or 10 nM) were immobilized on an ELISA plate, followed by incubation with a mixture of anti-Ub-pY59 B1-Fab and B2-IgG. An anti-IgG-HRP secondary Ab that binds to B2-IgG was used to detect binding of B2-IgG to the pY-antigen in complex with B1-Fab. We observed the ELISA signal only with the Ub-pY59 antigen, which further highlighted the specificity and utility of the identified TRAP binders.
The engineered anti-Ub-pY59 B1 binder has a binding affinity of 42 nM to the antigen. Of note, when we tried to select for a B2 binder against the parental 4G10-G44D5/Ub-pY59 complex that interacts with almost 10-fold weaker affinity (KD = 357 nM), no enrichment in the phage titer measurement was observed. This suggests that a moderate binding affinity between B1 and pY antigen is important for the successful enrichment of B2 binders by phage display.
Engineering and Characterization of B1−B2 Fusions.
We next explored how a B1 and B2 fusion performs as an anti-Ub-pY59 binder. B1 was converted to a single-chain variable fragment (B1-scFv) and fused to the C terminus of B2-Fab heavy chain (HC) with a 26-aa linker (Methods section, Figure 4A). The scFv-HC plasmid was coexpressed with the B2-Fab light chain (LC) plasmid to generate the B1−B2 fusion. We found that the B1−B2 fusion bound Ub-pY59 with a very high affinity (KD = 0.5 nM), while it interacted with GGG-pY-GGG with a KD of 133 nM. We further tuned the linker length and found that a 14-residue linker resulted in the greatest difference in KDs for Ub-pY59 and GGG-pY-GGG (0.5 vs 147 nM) (Figure 4A,B, Table S1). This fusion protein does not interact with Ub-Y59(WT), Y59A, or Y59E variants showing strong dependence of pY59 (Figure S5A).
Figure 4.
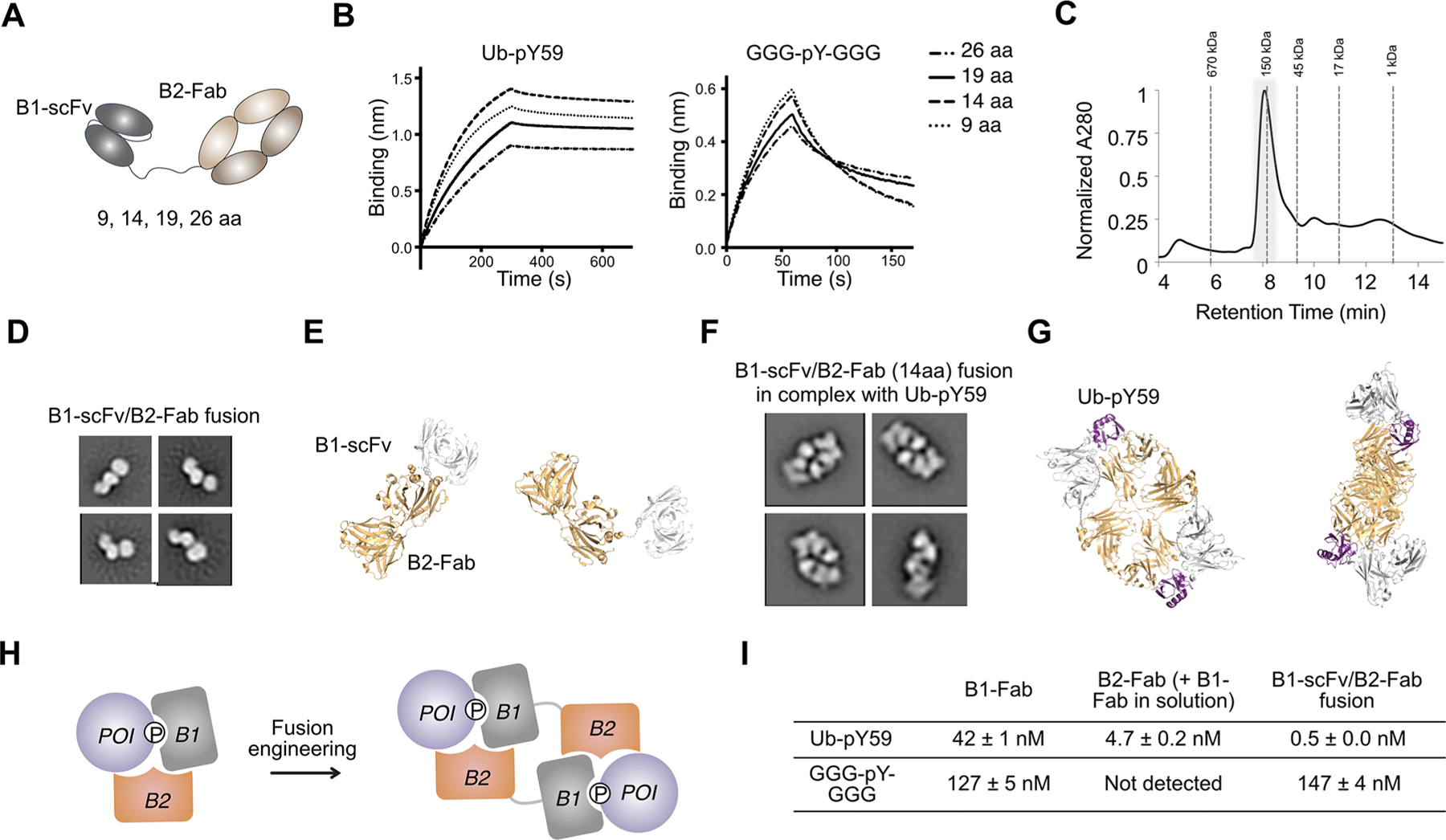
B1-scFv/B2-Fab fusion engineering. (A) Cartoon illustration of the B1-scFv/B2-Fab fusion. Various linker lengths (9, 14, 19, and 26 residues) were tested in the fusion protein between B1 and B2. (B) BLI experiments show that B1-scFv/B2-Fab with a 14-aa linker has the biggest difference in binding affinity for Ub-pY59 and GGG-pY-GGG. (C) The SEC analysis of the complex of B1-scFv/B2-Fab and Ub-pY59 shows that the complex has a molecular weight ∼150 kDa. The peak (highlighted in gray background) is collected for SDS-PAGE and NS-EM analysis. (D) Representative 2D class averages of NS-EM data for the B1-scFv/B2-Fab fusion alone. (E) Structural models of the B1-scFv/B2-Fab fusion. Light orange, Fab, PDB 1N8Z; gray, scFv, PDB 6DF1. (F) Representative 2D class averages of NS-EM data for B1-scFv/B2-Fab in complex with Ub-pY59. (G) Structure models of a 2:2 dimer of B1-scFv/B2-Fab and Ub-pY59. B1-scFv and B2-Fab in one polypeptide chain interact with two different Ub-pY59 molecules. Light orange, Fab, PDB 1N8Z; gray, scFv, PDB 6DF1; purple, Ub, PDB 5XK5. (H) Cartoon illustration of the formation of a 2:2 dimer of B1-scFv/B2-Fab and Ub-pY59. (I) Summary of KD values for B1-Fab, B2-Fab, and B1-scFv/B2-Fab to Ub-pY59 and the GGG-pY-GGG peptide.
To explore the molecular mechanism driving the enhanced affinity of the B1−B2 fusion to Ub-pY59, we used negative stain electron-microscopy (NS-EM) to study the structure of the protein complex. The complex of the 14-residue B1-scFv/B2- Fab fusion and Ub-pY59 was prepared by mixing equal molar amounts of the two proteins, which were then purified by size exclusion high-performance liquid chromatography (HPLC) (Figure 4C). The peak of the protein complex was collected, and a fraction was run on SDS-PAGE gel to validate the formation of the equal stoichiometric complex (Figure S5B). However, the retention time of the complex on HPLC indicated that the size of the B1−B2/Ub-pY59 complex was ∼150 kDa, which is twice the size of a 1:1 complex (∼88 kDa) (Figure 4C). Furthermore, while NS-EM imaging showed that the B1-scFv/B2-Fab fusion has an expected scFv-Fab monomeric structure (Figure 4D,E), NS-EM 2D averages of the complex showed that it is much bigger than B1-scFv alone. The structure clearly contains two “donut-shape” Fab structures in each molecule (Figure 4F). Together, these results indicated that B1−B2 and Ub-pY59 formed a 2:2 complex.
To further study the 2:2 complex, we modeled the structures in PyMOL and found that the B1 and B2 could easily accommodate domain swapping within a single B1−B2 fusion polypeptide and two different Ub-pY59 molecules to produce a 2:2 complex (Figure 4G,H). This 2 on 2 cooperative binding model results in intermolecular avidity contributing to higher affinity (Figure 4B, Table S1). Furthermore, based on this structural model, we estimated that a long linker (>100 Å, approximately 50 residues30) would be necessary for B1 and B2 intramolecular association to simultaneously bind a single Ub-pY59 molecule and form a 1:1 complex (Figure S5C). We tried to express a B1-scFv/B2-Fab fusion with longer linkers but obtained very little protein with a linker as long as 40 aa. This result suggested that a long linker fusion is not an ideal molecular format (Figure S5D). Further studies will explore if a differing fusion design, such as reversing the order of B1/B2 fusion, using two scFvs instead of a scFv and a Fab, making a bispecific Fc linked parallel construct, or fusing each of B1 and B2 to an interaction domain, would lead to a binder that can form a 1:1 complex with Ub-pY59.
The B1−B2 fusion to Ub-pY59 represents one of the tightest synthetic binders (KD = 0.5 nM) to PTMs to our knowledge (Figure 4I). It has great selectivity against the GGG-pY-GGG peptide which is a very stringent off-target control for binding generic pY because of its high accessibility (KD = 147 nM). As an additional control that binding depends on surrounding protein structure, we incorporated pY using the described Uaa approach into E64 which is highly exposed in Ub, to produce Ub-pY64. The anti-Ub-pY59 B1−B2 fusion did not show any detectable binding to Ub-pY64 further confirming its high specificity for Ub-pY59 (Figure S5E).
Phosphoproteomics with 4G104D5 Variants Reveal a Large pY-Target Landscape for pY-TRAPs.
To explore the scope of targetable pY antigens using the 4G104D5 phage library, we performed phosphoproteomics on lysates of Jurkat cells using WT 4G10, 4G10-G64D5, and an SH2 variant, called Src- Superbinder, another broadly used pan-specific anti-pY binding domain.11 Briefly, pY peptides were first immunoprecipitated from trypsin-treated cell lysates (Figure 5A) with Ni-NTA resin-immobilized binders, followed by TiO2 enrichment. Using a standard pY peptide enrichment workflow, we identified 4824 pY-sequences with 4G10, 4775 with 4G10-G64D5, and 4446 with Src-Superbinder (Figure 5A,B, Appendix Table S5).
Figure 5.
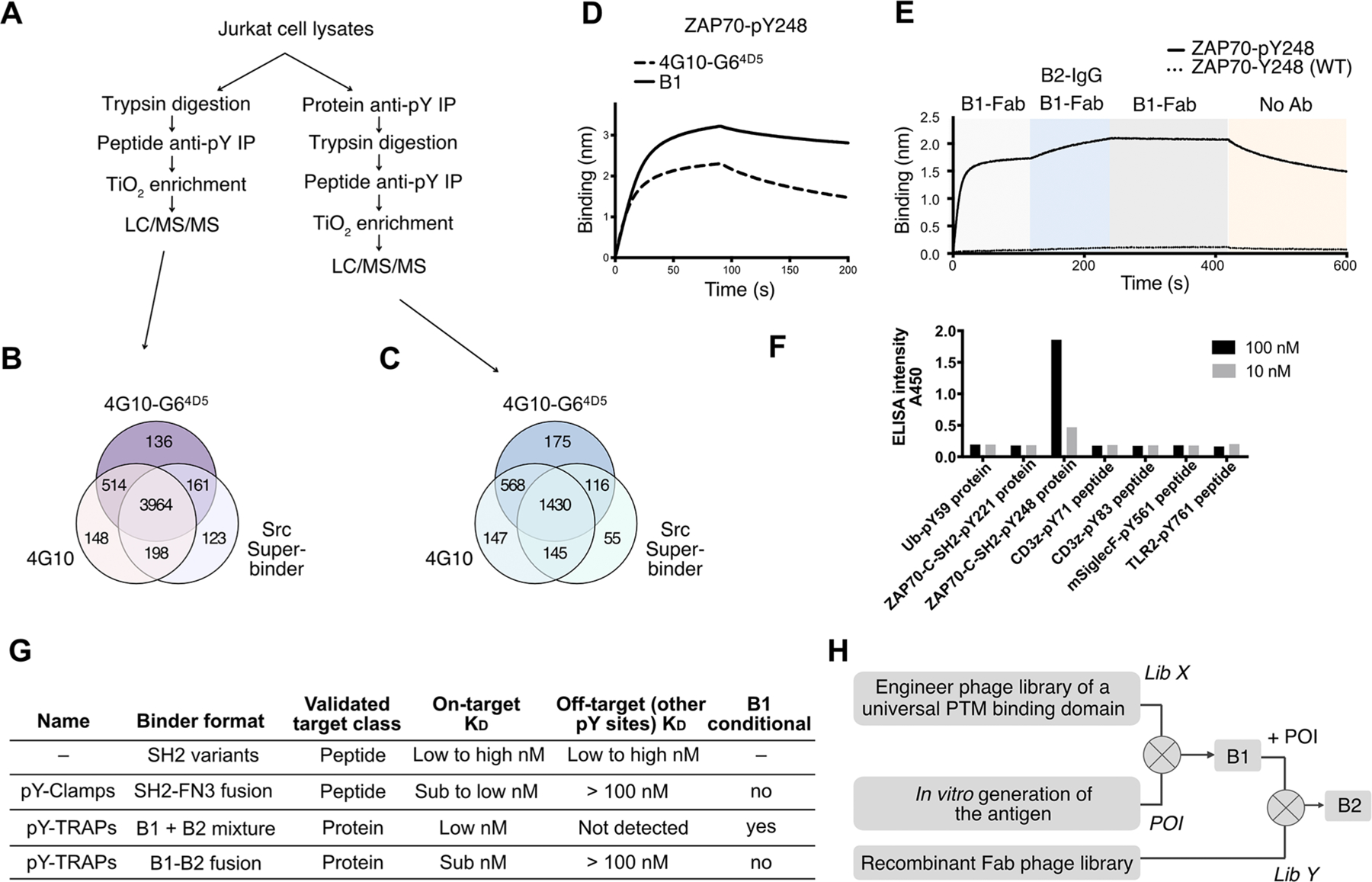
The TRAP platform has the potential to generate binders to thousands of other pY sequences and different protein PTMs. (A) Phosphoproteomics workflow to globally identify pY sequences from Jurkat cell lysates by pY peptide immunoprecipitation (IP) or pY protein +peptide IP. (B) Number of pY sequences identified in LC/MS/MS following peptide IP and TiO2 enrichment by 4G10, 4G10-G64D5, or Src- Superbinder SH2 domain. (C) Number of pY sequences identified in LC/MS/MS following protein IP, peptide IP, and TiO2 enrichment by 4G10, 4G10-G64D5, or Src-Superbinder SH2 domain. (D) The BLI characterization shows that ZAP70-pY248 binds tighter with the identified B1 than with parental 4G10-G64D5. (E) Sequential BLI experiments show that B1 binds ZAP70-pY248 (solid curve) but not ZAP70-Y248 (WT) (dotted curve), and B2-IgG added subsequently recognizes the B1/ZAP70-pY248 complex (solid curve) but not the ZAP70-Y248 (WT) (dotted curve). (F) The ELISA experiment showed that the anti-ZAP70-pY248 TRAP binders are highly selective against ZAP70-pY248 and not other pY proteins/peptides. (G) Comparison of methods for engineering sequence-specific pY binders. pY-TRAPs can be used in two formats. The B1 plus B2 format provided the highest selectivity while the B1−B2 fusion format resulted in a tighter binder. Except for pY-TRAPs, the other methods generated binders against the linear form of a pY antigen. (H) Proposed workflow to engineer TRAP binders against other protein PTMs.
To further explore the accessibility of pY on folded proteins, we repeated this experiment with an additional step of immunoprecipitation from Jurkat whole cell lysates (Figure 5A) prior to trypsinization to capture native pY proteins. Using the pY protein+peptide enrichment workflow, we identified 2290 pY-sequences with 4G10, 2289 with 4G10-G64D5, and 1746 with Src-Superbinder (Figure 5C, Appendix Table S4). The ∼50% fewer pY sequences identified using the protein +peptide enrichment workflow confirmed that a significant proportion of native pY motifs are not readily accessible to generic pY binders due to steric hindrance of the folded protein epitope. However, the thousands of native pY sequences identified from this proteomics experiment indicated that 4G10 has substantial initial binding affinities to a large number of candidate proteins suited for pY-TRAP engineering. The Src- Superbinder enriched a fewer set of pY sequences. There was not complete overlap suggesting that both types of generic pY binders could allow a better pY sequence coverage.31 These data also suggested candidates for pY-TRAPs using SH2 domain libraries to generate potential B1 binders.
Generalizing the pY-TRAP Method to ZAP70-pY248.
It is not uncommon for proteins to have multiple phosphotyrosine sites, and we wished to test a protein in this class both for robustness of pY misincorporation and pY-TRAP generation. Within our Jurkat cell proteomics data set, we identified 11 phosphorylated sites in the ZAP70 kinase known to play a role in T cell activation and feedback signaling mechanisms (Appendix Tables S4 and S5).32,33 Three of these sites (Y178, Y221, Y248) are within the second SH2 domain (residues 167−259). Most of these sites do not have commercially available pY-Abs, preventing us from gaining a more comprehensive understanding of their functions. Using the described Uaa approach16 we achieved robust expression of pY221 and pY248 within the SH2 domain but not for pY178 (Figure S6). This is not surprising as it is known that expression of proteins by unnatural amino acid misincorporation can vary depending on target sequence possibly due to ribosome stalling and other effects.
We subjected the pY221 and pY248 antigens to phage selection using a 4G10-G64D5 Lib-L1-H1-H2 phage library to identify B1 binders (Table S3). An anti-ZAP70-pY248 B1 binder was identified that was ∼3-fold tighter (KD = 21 nM) to ZAP70-pY248 than the parental 4G10-G64D5 (KD = 56 nM) (Figure 5D, Table S1). Separately, we selected for B1s to ZAP70-pY221, but none of the isolated B1s bound significantly tighter. The affinities of B1-Fabs to Ub-pY59 and ZAP70-pY248 were higher (KD = 42 nM and 21 nM, respectively) than the binding of the B1 for ZAP70-pY221 (KD = 75 nM).
We further optimized the B2 engineering workflow (Figure S7). Instead of providing a high concentration of B1-Fab in the previous process (Figure 2A), we coimmobilized both pY protein and B1 on SA beads to create a high local concentration of the two interacting partners. We hypothesized that this would stabilize the pY protein/B1 complex avoiding the need for high concentrations of B1 in solution. To test this hypothesis, we performed a B2-IgG pulldown experiment with biotinylated Ub-pY59 and biotinylated B1 immobilized on SA beads. Supplementing additional B1-Fab in solution did not result in an increased level of B2 pulldown, suggesting that immobilizing both Ub-pY59 and B1 on the beads had created a stable complex (Figure S7A).
Based on this result, we performed B2 selections for the pY ZAP70 antigen-B1 complexes by immobilizing both biotinylated ZAP70-pY antigens and biotinylated B1 on SA beads (Figure S7B). Briefly, a Fab-phage library was negatively selected against the immobilized ZAP70 pY antigens, followed by another negative selection against immobilized B1 binders. Next, a positive selection was performed with preincubated, SA-immobilized pY protein/B1 complexes. After four rounds of selection, we identified a B2 binder highly specific to B1/ ZAP70-pY248 (Figure 5E, KD = 4 nM). Of note, no complex specific binders were identified for the weaker B1/ZAP70- pY221 complex. It is possible that there is a threshold affinity to select B2s to this pY protein-B1 complex. BLI and ELISA experiments demonstrated that the B2 binder is highly specific to the B1/ZAP70-pY248 complex (Figure 5E,F, Figure S8).
DISCUSSION
Engineering high-affinity recombinant binders using multiple binding arms has been used extensively to increase affinity and specificity: for example, the generation of bispecific Abs where two independently selected Abs bind two separate-linked or membrane bound proteins, or biparatopic Abs where two independent binders with nonoverlapping but neighboring epitopes are linked together on the same protein.34 The pY-TRAP represents a novel class in that B2 binding was conditional to the presence of the complex of the pY protein and B1. During our B2 selections, we did find independent binders (we called BWT), some of which we could have used as biparatopic constructs but did not as BWT could bind independent of phosphorylation. Phosphotyrosine forms can have a range of stoichiometry, and we deliberately did not want B2 to bind independently to the pY protein or B1 as this would reduce specificity for the complex.
We specifically chose the 4G104D5 as a broadly specific B1 scaffold because of its exquisite dependence on pY, ability to bind pY in tertiary epitopes, high level expression in Escherichia coli, and single selection library to further enhance potency surrounding the pY residue. Although this starting scaffold will certainly not bind all pY proteins, the pY proteomics here provides a list of about 2300 pY sites (from Jurkat extracts alone) that were pulled down with the 4G10 Ab which could be possible candidate pY targets for pY-TRAP selections. Another limitation is that not all pY protein domains will be amenable to the unnatural pY incorporation due to large size, acid sensitivity, or other expression issues. Fortunately, both pY sites in Ub, and two of three sites tested in ZAP70, did express. An alternative method to generating pY proteins was recently published using a different Uaa and synthetase which provides another option for generating the pY antigens.23 Also, not all B2 selections worked, and this may be because the B1−pY protein affinity was not sufficient to present the complex. It is possible that coimmobilizing higher concentrations of B1 and pY protein will mitigate this by enforcing high complex formation. In addition, the current B1 library may not be the most optimal for all pY proteins. Further exploration on how each of the CDRs contribute to binding to a broader panel of pY antigens will promise a further improved, second-generation library for engineering tighter B1 binders.
The pY-TRAP approach enabled us to generate high-affinity and specific binders against native Ub-pY59 and ZAP70-pY248, for which there are no commercially available Ab reagents. We believe that building pY protein binders to tertiary pY epitopes nicely complements and expands approaches for linear pY epitopes using diversified SH2 or PTB domains and pY-Clamps (Figure 5G).11–13 Although protein kinase substrate specificity is typically defined by linear consensus motifs,35,36 recent evidence suggests that protein-folding creates structure-based motifs for specific kinase recognition.14,15,37,38 Consistent with these findings, our bioinformatics analysis revealed that tyrosine occurs very frequently in rigid tertiary structures. pY-TRAPs serve as vital reagents for assigning the biological functions, determining the upstream kinases, and studying signaling regulations of this class of pY modifications.
We envision that this conditional two-binder TRAP method may be generalizable to other PTMs if generic B1 binding scaffolds are developed to PTMs (Figure 5H). In this regard, recombinant anti-methylated (Me)-lysine Abs that independently bound two neighboring Me-lysine sites on histone H3 were recently reported.39 Interestingly when the Abs were mixed, they discovered that they bound cooperatively with higher affinity. Additionally, natural domains that recognize a different protein PTM could be used as B1, for example, SH2 variants for sulfotyrosine,40,41 and lectins for glycosylation,42,43 while B2 domains bind the complex of B1 and PTM-containing antigen. Overall, there is a great potential to generalize this TRAP platform to additional protein modifications for making a wide variety of PTM-TRAPs to serve as research agents, biomarkers, or therapeutics.
METHODS
Secondary Structure Analysis.
Secondary structures for pY sequences were performed using an in-house informatics pipeline written in R. Scripts are available for download from https://github.com/crystaljie/pY_2rd_structure_solvent_accessibility_analysis.git. The protein phosphorylation site information used for this analysis was downloaded from PhosphoSitePlus (Appendix Table S1). Only the pY sequences in human proteins were used for analysis. Based on the full-length protein sequences from Uniprot,17 the GOR algorithm19 was applied to predict the secondary structure of the pY sites (6063 pY sites in 2076 proteins) or randomly picked sequences from these proteins (Appendix Table S2). For the sequences that have PDB structures available (1501 pY sites in 527 proteins), DSSP algorithm18 was used to obtain the exact secondary structure information for the pY sites or randomly picked sequences from these proteins (Appendix Table S3).
Plasmid Construction.
Plasmids were constructed by standard molecular biology methods, and complete plasmid sequences are available upon request. The DNA fragment of human Ub or ZAP70 was synthesized by integrated DNA technologies (IDT). The TAG mutation for pY incorporation was introduced by overlap-extension PCR. The antigens were subcloned into the inducible bacterial expression pTak16,44 vector with a Tobacco Etch Virus (TEV) protease cleavage site, an AviTag, and a six-consecutive-histidines tag (His-tag) at the C-terminus. The plasmid pBK-MmNpYRS encoding the Uaa-specific synthetase was provided by the Lei Wang lab (University of California, San Francisco16). All the Fabs were constructed in a dual-expression vector that expresses the light chain and the heavy chain with the pelB and the stII signal peptides, respectively, for the periplasm expression.5 A C-terminal 6× His tag was put in the heavy chain for purification. All the IgGs were constructed in the pFUSE-hIgG1 vector (InvivoGen), with the heavy chain genetically fused to the hIgG1-Fc on one vector, and the light chain on a separate copy of the vector. The B1-scFv/B2-Fab fusion was also constructed in the pFUSE-hIgG1 vector (InvivoGen), with B2-Fab light chain on one vector, and B2 Fab heavy chain fused to the N-terminus of B1-scFv on another copy of the vector. The sequences of the linker between the heavy chain and scFv are as follows: 26 aa, GGSGSAGGLNDIFEAQKIEWHESSGS; 19 aa, AGGLNDIFEAQKIEWHEGS; 14 aa, AGGLNDIFEAQKGS; 9 aa, AGGLNDIGS.
Fab Expression.
C43 (DE3) Pro+ E. coli containing Fab expression vectors were grown in 2xYT at 37 °C to an OD-600 of 0.4−0.8, and then, Fab expression was induced by the addition of 0.5 mM IPTG. Incubation temperature was subsequently reduced to 30 °C, and the cultures were allowed to shake for 16−20 h. Cells were harvested by centrifugation and lysed using B-PER lysis buffer or sonication. The lysate was incubated at 60 °C for 20 min and centrifuged to remove the inclusion body. The Fabs were purified by Ni2+-NTA resin and buffer exchanged in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.5) for further characterization.
IgG and B1-scFv/B2-Fab Fusion Expression.
The IgGs were expressed by cotransfection of the pFuse-light chain and the pFuse-heavy chain-Fc vectors. The B1-scFv/B2-Fab fusion proteins were expressed by cotransfection of the pFUSE vector encoding B2 Fab light chain and another pFUSE vector encoding B2 Fab heavy chain fused to the N-terminus of B1-scFv. Expi293 (Life Technologies) cells were transiently cotransfected with two vectors at a mass ratio of 1:1. The ExpiFectamine 293 transfection kit (Life Technologies) was used for transfections as per manufacturer’s instructions. Cells were incubated for 5 days at 37 °C in a 5% CO2 environment before the supernatants were harvested by centrifugation. The IgGs and the B1-scFv/B2-Fab fusion were purified by Protein A and Protein L affinity chromatography, respectively.
pY Misincorporation.
pY was incorporated into Ub and ZAP70 according to a previous protocol.16 Briefly, BL21 cells were cotransformed with the pTak-antigen plasmids (chloramphenicol resistant) and pBK-MmNpYRS (kanamycin resistant). 35 μg/mL chloramphenicol and 50 μg/mL kanamycin were added to the bacterial culture to select the double transformants. The cells were grown in 2xYT at 37 °C to an OD-600 of 0.4−0.8, and then, 1 mM Uaa 1 (Figure S1) was added. Protein expression was then induced by the addition of 0.5 mM IPTG. The incubation temperature was subsequently reduced to 18 °C, and the cultures were allowed to shake for 16−20 h. Cells were harvested by centrifugation and lysed by sonication. The lysate was centrifuged to remove cell debris and the inclusion body. The proteins were purified by Ni2+-NTA resin and exchanged into TBS buffer.
To cleave the protection group on the Uaa, to a diluted protein solution (0.1−1.0 mg mL−1) in TBS buffer, HCl (4 M) was added to reach a final concentration of 0.4 M. The reactions were incubated at 4 °C for 24−72 h, and the cleavage efficiency was monitored by mass spectrometry on a Xevo G2-XS mass spectrometer (Waters).
In Vitro Biotinylation.
The purified proteins were biotinylated on their AviTags using the standard protocol provided by Avidity. Biotinylation was monitored by intact protein mass spectrometry on a Xevo G2-XS mass spectrometer (Waters). The biotinylated proteins were then separated into aliquots, analyzed by SDS-PAGE, snap-frozen, and stored at −80 °C for later use.
Phage Library Construction.
The phagemids that encode the 4G104D5 gene with the stop codon within the CDRs were used as templates for Kunkel mutagenesis with oligonucleotides designed to correct the stop codons and introduce the designed mutations at each site.7,45 The library designs are listed in Table S3. The resulting mutagenesis reactions were electroporated into SS320 electro-competent cells (Lucigen). After 1 h of recovery at 37 °C, the cells were expanded into 500 mL of 2xYT cultures with 50 μg/mL carbenicillin and 1010/mL M13KO7 helper phage. 50 μg/mL kanamycin was added to the culture after 1 h of shaking at 37 °C. The culture was grown for approximately 20 h with 250 rpm shaking at 37 °C. The next day, the cells were pelleted by centrifugation. The phage was precipitated from the supernatant by adding 1/5 volume of 20% PEG8000 and 2.5 M NaCl. The phage library was resuspended in TBS buffer with 50% glycerol and 2 mM EDTA and stored at −20 °C. Template phagemid plasmid, mutagenesis oligos, and a small quantity of library stocks can be shared with the academic community upon request.
Phage Display.
All phage selections were done according to previous protocols.5,46 For B1 selection, the 4G104D5 libraries were incubated with biotinylated pY protein immobilized on SA-coated magnetic beads (Promega). Empty beads were used for library clearance and enrichment tests. In total, four rounds of selections were performed with a decreasing concentration of pY-antigen (300, 100, 30, 10 nM). Starting from round 2, the phage library was first enriched by protein L magnetic beads to deplete nondisplayed or truncated Fab phage before each round of the selection.
For B2 selection for Ub-pY59, the synthetic phage library was first incubated with pY protein immobilized on SA beads to deplete any binders to the pY protein alone. Subsequently, the cleared library was incubated with immobilized pY-antigen with 5 μM B1-Fab in solution to enrich binders against the pY protein/B1 complex. In total, four rounds of selections were performed with 500, 500, 500, and 100 nM of pY antigen, respectively. B1-Fab (5 μM) was supplemented to both the selection reaction and the wash buffers.
For B2 selection for ZAP70-pY221 and pY248, the synthetic phage library was first incubated with immobilized pY protein and B1, respectively, to deplete any binders to pY protein or B1 alone. Subsequently, the cleared library was incubated with the B1/pY-antigen complex (with both B1 and pY-antigen immobilized on the SA beads) to enrich binders against the pY protein/B1 complex. In total, four rounds of selections were performed with 500, 500, 500, and 100 nM of pY antigen, respectively.
ELISA.
ELISAs were performed according to standard protocols. Briefly, 384-well Maxisorp plates were coated with NeutrAvidin (10 μg/ mL) overnight at 4 °C and subsequently blocked with BSA (2% w/v) for 1 h at 20 °C. 10−100 nM pY antigens were captured on the NeutrAvidin-coated wells for 30 min followed by the addition of various concentrations of phage or recombinant antibodies for 30 min. The secondary Abs were either a horseradish peroxidase (HRP)-conjugated anti-M13 phage antibody (Sino Biological) for phage ELISA or an antihuman-IgG antibody (Sigma-Aldrich) for recombinant protein ELISA. The ELISA plates were washed three times after each incubation, and Ab binding was detected by TMB substrate (VWR) and read at 450 nm.
Binding Kinetics Analysis.
BLI experiments were performed at room temperature using an Octet RED384 instrument (ForteBio). Biotinylated pY antigens were immobilized to an optically transparent SA biosensor (ForteBio). Different concentrations of antibodies in kinetics buffer (TBS, pH 7.5, 0.05% Tween-20, 0.2% BSA) were used as the analyte in a 384-well microplate (Greiner Bio-One). Antibody CDR sequences are listed in Table S2. Affinities (KDs) were calculated from a global fit (1:1) of the data using the Octet RED384 Data Analysis HT software. Measurements at the single antibody concentration were first used to estimate binding strengths of different selectants, and measurements at a series of antibody concentrations (covering concentrations below and above the estimated KD) were used to determine KD values (Table S1). The measurements were performed at least twice (single- or multiconcentration), and representative Octet traces are shown in Figure S9.
Negative Stain Electromicroscopy.
The samples B1-scFv/B2- Fab fusion and its complex with Ub-pY59 were negatively stained and observed on a Tecnai T12 microscope and a Tecnai T20 microscope (FEI Company) using the discharged continuous carbon grids as previously described,47,48 respectively. Images were acquired at room temperature with a pixel size of 2.21 Å/pixel (T12, operated at 120 kV) or 3.319 Å/pixel (T20, operated at 200 kV) on the level of specimen using a 4K × 4K CCD camera (UltraScan 4000, Gatan Inc.). After all micrographs were visually screened, the contrast transfer function (CTF) was estimated for each micrograph by Gctf.49 The particle was selected using the Gautomatch (https://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch) without template. Individual particles were extracted from the raw images with the 100 × 100 pixel window for T12 images or 80 × 80 pixel window for T20 images and were subjected to 25 cycles of 2D classification with a mask diameter of 200 Å for B1-scFv/B2-Fab or 220 Å for its complex with Ub-pY59 in Relion 3.0.50 The output 2D averages after several rounds were analyzed by comparing with the available protein structures utilized in this design.
pY Phosphoproteomics.
The phosphoproteomics experiment was performed according to the standard pY phosphoproteomics protocol31 and manufacture protocols. Briefly, Jurkat cells were pretreated with 0.1 mM freshly activated pervanadate for 15 min at 37 °C. The cells were spun down and washed with prechilled (4 °C) TBS (20 mM Tris, 150 mM NaCl), and lysed in prechilled (4 °C) lysis buffer (0.5% Triton-100, 50 mM Tris, 150 mM NaCl) with phosphatase inhibitor cocktail (Sigma-Aldrich, 1:100) and protease inhibitor cocktail (Sigma-Aldrich, 1:100). After rotating at 4 °C for 15 min, cell lysates were spun at 16000g to remove cell debris. 10 nmol his-tagged 4G10, 4G10-G64D5, or Src-Superbinder was loaded to 100 μL of prewashed Ni2+-NTA resin (Thermo Fisher Scientific) and washed twice with TBS buffer. For phosphoprotein IP, the cell lysates were added to the Ni2+-NTA slurry and rotated for >2 h at 4 °C. For phosphopeptide IP, proteins were first denatured, reduced, alkylated, digested to peptides by MS-compatible, sequencing grade trypsin (Promega, 1:50 w/w), and then added to the Ni2+-NTA resin for pulling down pY peptides. The Ni2+-NTA resin was washed 6 times with TBS buffer and eluted with 100 mM phenyl phosphate in 500 mM ammonium bicarbonate buffer. The elution was desalted using a SOLA column, and phosphopeptides were further enriched by the TiO2 phosphopeptide enrichment kit (Thermo Fisher Scientific). The elution from TiO2 enrichment was resuspended in 2% ACN + 0.1% FA and analyzed by a Q Exactive Plus (Thermo Fisher) mass spectrometer.
Immunoprecipitation with Anti-Ub-pY59-TRAP Binders.
2.5 μM biotinylated B1-Fab and biotinylated Ub-pY59 were incubated for 30 min at RT. This B1/Ub-pY59 complex or 2.5 μM biotinylated Ub-pY59 only was loaded onto the SA beads. 0 or 5 μM of B1-Fab was supplied in solution. Subsequently, 5 μM B2-IgG was added to the reaction mixtures and incubated for 30 min at RT. The beads were washed with TBS buffer or TBS+5 μM B1-Fab. Proteins were eluted with 0.1 M acidic acid.
Supplementary Material
Appendix Table S5 (XLSX)
ACKNOWLEDGMENTS
We gratefully acknowledge members of the Wells lab for helpful discussions and support. We gratefully thank Nicholas Young for help with ZAP70 phosphoprotein generation and characterization. We also valued helpful discussions with Dr. Sachdev Sidhu.
Funding
J.A.W. is grateful for funding from the Harry and Dianna Hind Endowed Professorship in Pharmaceutical Sciences, NCI P41CA196276, the UH3 CA246635–01 from the HubMAP consortium, and Celgene Corporation that helped support this work. Postdoctoral Fellowship support included a Merck Postdoctoral Fellowship from the Damon Runyon Research Foundation (DRG-2297–17 to X.X.Z.), a Postdoctoral Fellowship from Human Frontier Science Program (to K.Z.), and a Postdoctoral Fellowship from NIH (F32CA236151–02 to J.Z.). L.W. acknowledges the support of the NIH (R01GM118384). Y.C. acknowledges the support of the NIH (P50AI150476).
ABBREVIATIONS
- Ab
antibodies
- pY-TRAP
pY-Targeting by Recombinant Antibody Pairs
- scFv
single-chain variable fragment
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c08458.
Additional data and figures including bioinformatics analysis, Ub-pY59 generation, engineering of anti-Ub-pY59 B1, sequential BLI experiments, characterization, generation of ZAP70-C-SH2 pY antigens, optimization of the B2 engineering workflow, and BLI measurements (PDF)
Appendix Table S1 (XLS)
Appendix Table S2 (XLS)
Appendix Table S3 (XLS)
Appendix Table S4 (XLSX)
Appendix Table S5 (XLSX)
The authors declare the following competing financial interest(s): X.X.Z. and J.A.W. declare that a provisional patent application related to this work is under consideration.
Contributor Information
Xin X. Zhou, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Colton J. Bracken, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States
Kaihua Zhang, Department of Biochemistry and Biophysics, University of California, San Francisco, California 94158, United States.
Jie Zhou, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Yun Mou, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Lei Wang, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Yifan Cheng, Department of Biochemistry and Biophysics and Howard Hughes Medical Institute, University of California, San Francisco, California 94158, United States.
Kevin K. Leung, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
James A. Wells, Department of Pharmaceutical Chemistry and Department of Cellular and Molecular Pharmacology, University of California, San Francisco, California 94158, United States; Chan Zuckerberg Biohub, San Francisco, California 94158, United States.
REFERENCES
- (1).Hunter T Tyrosine phosphorylation: thirty years and counting. Curr. Opin. Cell Biol 2009, 21, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lim WA; Pawson T Phosphotyrosine signaling: evolving a new cellular communication system. Cell 2010, 142, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hornbeck PV; Kornhauser JM; Tkachev S; Zhang B; Skrzypek E; Murray B; Latham V; Sullivan M PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 2012, 40, D261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hornbeck PV; Zhang B; Murray B; Kornhauser JM; Latham V; Skrzypek E PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 2015, 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mou Y; Zhou XX; Leung K; Martinko AJ; Yu J-Y; Chen W; Wells JA Engineering Improved Antiphosphotyrosine Antibodies Based on an Immunoconvergent Binding Motif. J. Am. Chem. Soc 2018, 140, 16615–16624. [DOI] [PubMed] [Google Scholar]
- (6).Hattori T; Koide S Next-generation antibodies for post-translational modifications. Curr. Opin. Struct. Biol 2018, 51, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Koerber JT; Thomsen ND; Hannigan BT; Degrado WF; Wells JA Nature-inspired design of motif-specific antibody scaffolds. Nat. Biotechnol 2013, 31, 916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brumbaugh K; Johnson W; Liao WC; Lin MS; Houchins JP; Cooper J; Stoesz S; Campos-Gonzalez R Overview of the generation, validation, and application of phosphosite-specific antibodies. Methods Mol. Biol 2011, 717, 3–43. [DOI] [PubMed] [Google Scholar]
- (9).Kehoe JW; Velappan N; Walbolt M; Rasmussen J; King D; Lou J; Knopp K; Pavlik P; Marks JD; Bertozzi CR; Bradbury AR Using phage display to select antibodies recognizing post-translational modifications independently of sequence context. Mol. Cell. Proteomics 2006, 5, 2350–2363. [DOI] [PubMed] [Google Scholar]
- (10).Mann JK; Wood JF; Stephan AF; Tzanakakis ES; Ferkey DM; Park S Epitope-guided engineering of monobody binders for in vivo inhibition of Erk-2 signaling. ACS Chem. Biol 2013, 8, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kaneko T; Huang H; Cao X; Li X; Li C; Voss C; Sidhu SS; Li SS Superbinder SH2 domains act as antagonists of cell signaling. Sci. Signaling 2012, 5, No. ra68. [DOI] [PubMed] [Google Scholar]
- (12).Malabarba MG; Milia E; Faretta M; Zamponi R; Pelicci PG; Di Fiore PP A repertoire library that allows the selection of synthetic SH2s with altered binding specificities. Oncogene 2001, 20, 5186–5194. [DOI] [PubMed] [Google Scholar]
- (13).Yasui N; Findlay GM; Gish GD; Hsiung MS; Huang J; Tucholska M; Taylor L; Smith L; Boldridge WC; Koide A; Pawson T; Koide S Directed network wiring identifies a key protein interaction in embryonic stem cell differentiation. Mol. Cell 2014, 54, 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Durek P; Schudoma C; Weckwerth W; Selbig J; Walther D Detection and characterization of 3D-signature phosphorylation site motifs and their contribution towards improved phosphorylation site prediction in proteins. BMC Bioinf 2009, 10, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jimenez JL; Hegemann B; Hutchins JR; Peters JM; Durbin R A systematic comparative and structural analysis of protein phosphorylation sites based on the mtcPTM database. Genome Biol 2007, 8, R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hoppmann C; Wong A; Yang B; Li S; Hunter T; Shokat KM; Wang L Site-specific incorporation of phosphotyrosine using an expanded genetic code. Nat. Chem. Biol 2017, 13, 842–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Consortium, U. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kabsch W; Sander C Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (19).Garnier J; Gibrat J-F; Robson B GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol 1996, 266, 540–553. [DOI] [PubMed] [Google Scholar]
- (20).Swatek KN; Komander D Ubiquitin modifications. Cell Res 2016, 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yau R; Rape M The increasing complexity of the ubiquitin code. Nat. Cell Biol 2016, 18, 579–586. [DOI] [PubMed] [Google Scholar]
- (22).Chong RA; Wu K; Spratt DE; Yang Y; Lee C; Nayak J; Xu M; Elkholi R; Tappin I; Li J; Hurwitz J; Brown BD; Chipuk JE; Chen ZJ; Sanchez R; Shaw GS; Huang L; Pan ZQ Pivotal role for the ubiquitin Y59-E51 loop in lysine 48 polyubiquitination. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 8434–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Luo X; Fu G; Wang RE; Zhu X; Zambaldo C; Liu R; Liu T; Lyu X; Du J; Xuan W; Yao A; Reed SA; Kang M; Zhang Y; Guo H; Huang C; Yang PY; Wilson IA; Schultz PG; Wang F Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat. Chem. Biol 2017, 13, 845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fairhead M; Howarth M Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol 2015, 1266, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Eisinger D; Stiles L; LaMarche A; Jelinek T Recombinant monoclonal antibody to phosphotyrosine-containing proteins. US patent US 6824989 B1, September 1, 2000. [Google Scholar]
- (26).Yang W-P; Green K; Pinz-Sweeney S; Briones AT; Burton DR; Barbas CF III CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into the picomolar range. J. Mol. Biol 1995, 254, 392–403. [DOI] [PubMed] [Google Scholar]
- (27).Almagro JC Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: implications for the rational design of antibody repertoires. J. Mol. Recognit 2004, 17, 132–143. [DOI] [PubMed] [Google Scholar]
- (28).Xu JL; Davis MM Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity 2000, 13, 37–45. [DOI] [PubMed] [Google Scholar]
- (29).Persson H; Ye W; Wernimont A; Adams JJ; Koide A; Koide S; Lam R; Sidhu SS CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J. Mol. Biol 2013, 425, 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).George RA; Heringa J An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng., Des. Sel 2002, 15, 871–879. [DOI] [PubMed] [Google Scholar]
- (31).Bian Y; Li L; Dong M; Liu X; Kaneko T; Cheng K; Liu H; Voss C; Cao X; Wang Y Ultra-deep tyrosine phosphoproteomics enabled by a phosphotyrosine superbinder. Nat. Chem. Biol 2016, 12, 959–966. [DOI] [PubMed] [Google Scholar]
- (32).Szabo M; Czompoly T; Kvell K; Talaber G; Bartis D; Nemeth P; Berki T; Boldizsar F Fine-tuning of proximal TCR signaling by ZAP-70 tyrosine residues in Jurkat cells. Int. Immunol 2012, 24, 79–87. [DOI] [PubMed] [Google Scholar]
- (33).Helou YA; Nguyen V; Beik SP; Salomon AR ERK positive feedback regulates a widespread network of tyrosine phosphorylation sites across canonical T cell signaling and actin cytoskeletal proteins in Jurkat T cells. PLoS One 2013, 8, No. e69641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Spiess C; Zhai Q; Carter PJ Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol 2015, 67, 95–106. [DOI] [PubMed] [Google Scholar]
- (35).de Oliveira PS; Ferraz FA; Pena DA; Pramio DT; Morais FA; Schechtman D Revisiting protein kinase-substrate interactions: Toward therapeutic development. Revisiting protein kinase-substrate interactions: Toward therapeutic development. Sci. Signaling 2016, 9, No. re3. [DOI] [PubMed] [Google Scholar]
- (36).Kaneko T; Joshi R; Feller SM; Li SSC Phosphotyrosine recognition domains: the typical, the atypical and the versatile. Cell Commun. Signaling 2012, 10, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Duarte ML; Pena DA; Nunes Ferraz FA; Berti DA; Paschoal Sobreira TJ; Costa-Junior HM; Abdel Baqui MM; Disatnik MH; Xavier-Neto J; Lopes de Oliveira PS; Schechtman D Protein folding creates structure-based, noncontiguous consensus phosphorylation motifs recognized by kinases. Sci. Signaling 2014, 7, No. ra105. [DOI] [PubMed] [Google Scholar]
- (38).Trost B; Kusalik A Computational prediction of eukaryotic phosphorylation sites. Bioinformatics 2011, 27, 2927–2935. [DOI] [PubMed] [Google Scholar]
- (39).Hattori T; Lai D; Dementieva IS; Montano SP; Kurosawa K; Zheng Y; Akin LR; Swist-Rosowska KM; Grzybowski AT; Koide A; Krajewski K; Strahl BD; Kelleher NL; Ruthenburg AJ; Koide S Antigen clasping by two antigen-binding sites of an exceptionally specific antibody for histone methylation. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 2092–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ju T; Niu W; Guo J Evolution of Src Homology 2 (SH2) Domain to Recognize Sulfotyrosine. ACS Chem. Biol 2016, 11, 2551–2557. [DOI] [PubMed] [Google Scholar]
- (41).Lawrie J; Niu W; Guo J Engineering of a sulfotyrosine-recognizing small protein scaffold for the study of protein tyrosine O-sulfation. Methods Enzymol 2019, 622, 67–89. [DOI] [PubMed] [Google Scholar]
- (42).Cummings RD; Etzler ME Antibodies and Lectins in Glycan Analysis. In Essentials of Glycobiology; Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler, Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2009; Chapter 45. [PubMed] [Google Scholar]
- (43).Zhang L; Luo S; Zhang B The use of lectin microarray for assessing glycosylation of therapeutic proteins. MAbs 2016, 8, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Takimoto JK; Dellas N; Noel JP; Wang L Stereochemical basis for engineered pyrrolysyl-tRNA synthetase and the efficient in vivo incorporation of structurally divergent non-native amino acids. ACS Chem. Biol 2011, 6, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kunkel TA; Bebenek K; McClary J Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol 1991, 204, 125–139. [DOI] [PubMed] [Google Scholar]
- (46).Hornsby M; Paduch M; Miersch S; Saaf A; Matsuguchi T; Lee B; Wypisniak K; Doak A; King D; Usatyuk S; Perry K; Lu V; Thomas W; Luke J; Goodman J; Hoey RJ; Lai D; Griffin C; Li Z; Vizeacoumar FJ; Dong D; Campbell E; Anderson S; Zhong N; Graslund S; Koide S; Moffat J; Sidhu S; Kossiakoff A; Wells J A High Through-put Platform for Recombinant Antibodies to Folded Proteins. Mol. Cell. Proteomics 2015, 14, 2833–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ohi M.; Li Y.; Cheng Y.; Walz T. Negative staining and image classification powerful tools in modern electron microscopy. Biol. Proced. Online 2004, 6, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Booth DS; Avila-Sakar A; Cheng Y Visualizing proteins and macromolecular complexes by negative stain EM: from grid preparation to image acquisition. J. Visualized Exp 2011, No. e3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhang K Gctf: Real-time CTF determination and correction. J. Struct. Biol 2016, 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Scheres SHW RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 2012, 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix Table S5 (XLSX)