

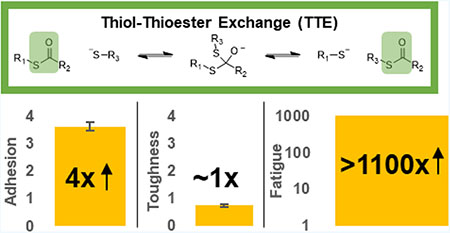
Abstract
Of importance for adhesive materials, particularly pressure-sensitive adhesive (PSA) systems, is the ability to increase bulk toughness without reduction of adhesion. Previous approaches for increasing PSA durability sacrifice permanent cross-linking or adhesive potential, limiting performance. In this work, covalent adaptable networks (CANs) derived from thiol-thioester exchange (TTE) are utilized as a basis for adhesive films. Tensile and single-lap shear tests were conducted for adhesive materials containing no filler, 15 wt % nanoparticles functionalized with thioester-containing acrylate, or 15 wt % nanoparticles functionalized with nonthioester-containing acrylate. Additionally, fatigue experiments were conducted on unfilled adhesives. Results indicate that TTE improves toughness, adhesion, and fatigue in unfilled materials. Filled adhesives with activated TTE showed a nearly fourfold increase in adhesion with slightly reduced toughness compared to uncatalyzed filled specimens. This work has implications in many industries, from biomedical to automotive, as toughness and fatigue resistance are important considerations for adhesive applications.
Keywords: adhesive, covalent adaptable network, dynamic covalent chemistry, fatigue, pressure-sensitive adhesive, toughness
Graphical Abstract
INTRODUCTION
Adhesives are an attractive approach both in combination with and in place of other methods of material bonding (such as mechanical fastening, welding, or soldering) due to their low cost, ease of use, low specific weight, ability to bond a variety of materials, and limited geometrical or thermal restrictions1,2. One important class of these versatile materials is pressure-sensitive adhesives (PSAs), which are ubiquitous for applications that require rapid and/or temporary binding, including adhesive tapes, permanent and removeable labels, medical bandages, and sticky notes. Moreover, PSAs require little-to-no substrate surface preparation, easily bind to high-energy surfaces, and are readily tuned for adhesion to a variety of low-energy surfaces. Key properties including low modulus and viscoelasticity provide good surface contact between a PSA and the desired substrate after the application of light pressure, enabling the physical interactions that are responsible for adhesion—such as hydrogen bonding, van der Waals forces, and chain diffusion—to occur without additional heat or curing.1–3 However, the same properties that make PSAs attractive materials (most notably low modulus) also lead to poor cohesive strength and toughness, limiting their use to low stress applications. Two important strategies for improving toughness in PSAs that have been previously implemented are (i) inclusion of sacrificial or labile bonds and (ii) reinforcement with inorganic fillers.
Sacrificial or labile bonds are weak or temporary moieties that are deliberately designed to break or dissociate in response to sufficiently large stresses, alleviating stress buildup to prevent or delay failure. Approaches for incorporating sacrificial or labile bonds include designing systems with hydrogen bonding3,6, coordination bonds,7,8 double or triple networks9,12, and dynamic covalent chemistries13,15. In many of these systems, the physical and/or supramolecular interactions that dissipate energy via bond dissociation do so at the cost of ultimate mechanical strength, due to the often-weaker physical bonds that make up the material. Double or triple networks incorporate weak chemical bonds in secondary and/or tertiary networks to dissipate energy and improve toughness without reducing properties like modulus and strength in the virgin material. However, destruction of the secondary and/or tertiary network(s) is irreversible, accumulating both molecular and material damage that ultimately leads to permanent loss of mechanical properties and eventual failure of the adhesive bond12,16–19.
In conventional polymer networks, permanent covalent cross-linking precludes many of the aforementioned toughening mechanisms. However, a great deal of effort has gone into the development of covalent adaptable networks (CANs) as a class of cross-linked materials capable of actively reshuffling covalent bonds. In these cross-linked networks, covalent bond reorganization occurs by incorporation of one of several dynamic covalent chemistries (DCCs), which are activated by light, elevated temperature, or the presence of a catalyst.20 When DCCs are leveraged, CANs promote advantageous behaviors otherwise not associated with cross-linked polymer networks, such as intrinsic self-healing, stress relaxation, fatigue resistance, and toughening. This rich field of inquiry consists of a variety of dynamic moieties, including Diels–Alder linkages,21 transesterification,22,23 disulfide exchange,24 addition–fragmentation,25–29 and thiol-thioester exchange (TTE),30–33 among others, which make up an ever-broadening toolbox for CANs design.
While DCCs generally are of interest for materials science applications, the TTE is particularly appealing for PSAs because the resulting network may display dynamic behavior for the duration of the material service life without significant changes in cross-linking density and without the need for an external user to activate a healing mechanism. TTE requires a basic or nucleophilic catalyst and an excess of thiols in the network to promote exchange, increasing the rate of exchange as the basicity or nucleophilicity of the catalyst increases.30 While the exchange mechanism differs for basic and nucleophilic catalysts, excess thiols are desirable for any dynamic behavior to occur.30 In either case, exchange occurs in the network as long as catalyst is present, and the rate of bond exchange is highly tunable by the choice and concentration of the base or nucleophile catalyst.
Inorganic fillers are high modulus additives that toughen composite materials. They are employed in a vast range of materials from structural materials to dental restoratives to car tires, enabling energy dissipation at the filler–matrix interface by mechanisms such as crazing,34 cavitation,35 and desorption of polymer chains36 from the filler surface. Even low filler loadings have been found to significantly improve stiffness and ultimate strength with minimal adverse effects on elongation, resulting in a substantial increase in toughness.37–39 For adhesives, an increase in modulus is often accompanied by a decrease in adhesive properties. This trade-off is particularly relevant in PSAs because tack (immediate “stickiness” of the adhesive) is directly tied to rapid surface contact and surface interactions, which are adversely affected by an increased modulus.
In this work, thiol-thioester exchange and functionalized inorganic fillers, specifically silica nanoparticles (SNPs), are implemented in tandem to improve the performance of pressure sensitive adhesives with the goal of using the strengths of one approach to complement the weaknesses of the other. Silica nanoparticles were chosen due to their prevalence of use, ease of functionalization, and potential for dynamic bond exchange. Although on much longer time scales than the DCCs proposed here with thiol-thioester exchange, it has been shown that siloxane bonds are also dynamic and may contribute to dynamic viscoelastic behavior in networks.40 In prior work, the incorporation of DCCs in bulk resin, SNP filler surface, or both in glassy nanocomposites has been shown to enhance the mechanical properties of the bulk composite through the reduction of stress both in the resin and at the particle interface41,42 as well as introduce reprocessability in these materials.43,44 While a key driving force for toughening in such glassy materials arises from reducing stress due to volumetric shrinkage (which is far less prominent in low modulus networks), a significant additional improvement in toughness arises when the dynamic chemistry is active during mechanical testing. One of the factors driving this increase in toughness is relaxing stresses and even healing of damage sustained at the filler–resin interface, which is a site of stress concentration45 in these materials.
In contrast, the approach used here is to implement surface-functionalized SNPs in combination with TTE to improve toughness and adhesion in low modulus, rubbery films to produce functional PSAs derived from thiol–ene monomers. It is hypothesized that incorporation of DCCs into rubbery networks will enhance adhesion, with greater and more rapid adhesion predicted to scale with more rapid TTE kinetics, and that this bond rearrangement will facilitate not only enhanced adhesion but also improved fatigue behavior as a result of the effective self-healing ability in CANs. Further, given the importance of the interface between the particles and resin, the effects of having or not having the TTE reaction at the interface are evaluated.
EXPERIMENTAL METHODS
Materials.
Pentaerytritol tetrakis(3-mercaptopropionate) (PETMP), 1,4-diazabicyclo[2.2.2]octane (DABCO), and Irgacure 819 (I819) were obtained from Sigma-Aldrich and used without additional purification. The thioester-diene (TE-diene) was synthesized using methods reported elsewhere.31 OX-50 silica nanoparticles (average diameter of the agglomerates was approximately 40 μm) were generously donated by Evonik Silicas and used for the inorganic fillers. These fillers were functionalized using methods reported elsewhere.41
General Procedure for Sample Preparation.
One equivalent of TE-diene was mixed with 0–15 mol % DABCO relative to TE-diene depending on the sample, 0.3 wt % MEHQ as a radical inhibitor during mixing, followed by 1 wt % I819 as a visible light photoinitiator and 0 or 15 wt % SNPs in filled samples. One equivalent PETMP (2:1 ratio of thiol to alkene) was added, mixed well, then the mixture was placed under vacuum for several minutes to remove air bubbles due to mixing. For SNP-filled samples, SNPs were dispersed in the TE-diene mixture via sonication, vortex mixing, and manual stirring to ensure homogeneous mixing of the SNPs. PETMP was then added, and the mixture was fully homogenized. Samples were cured with 400–500 nm visible light at 70 mW/cm2 for 5 min on each side and allowed to sit at room temperature for 24 h. For tensile testing, resins were cured directly in silicon rubber molds between glass slides. For all other testing, laminated films were initially created using silicon rubber spacers placed between two glass plates.
Stress Relaxation.
Stress relaxation was measured by a TA Q800 dynamic mechanical analyzer. Samples were held at a constant 5% strain and allowed to relax for 60 min at 25 °C. The relaxation time was determined from a plot of the normalized stress vs time. The characteristic relaxation time was taken to be τ* and found by fitting these curves to the stretched exponential function e−(t/τ*)β (τ* and β values are reported in Table S1).
Tensile Testing.
Tensile testing was performed on an Exceed Model E42 MTS with a 500 N load cell. Specimens were cast directly into dog bone-shaped molds with testing dimensions (L × W × Th) of 15 × 3 × 0.5 mm after the preparation described above. Tensile tests were performed at 0.067 min−1 until failure, in triplicate at minimum. Raw data was smoothed using Lowess smoothing, and the toughness was taken as the total area under the stress–strain curve until failure for each sample. The results stated in the following sections were determined to be statistically significant by unpaired Student’s t tests with an alpha of 0.05 and a minimum of three replicates.
Lap Shear Adhesion.
Adhesive strength was determined using a tensile single-lap shear experiment. Adhesive samples with dimensions (L × W × Th) of 20 × 2.5 × 0.5 mm were prepared and supported by a thin, strong elastic backing. Substrate samples consisting of filler-free and catalyst-free versions of the adhesive samples were prepared in an identical manner. Adhesive and substrate samples were adhered via light pressure with an overlap of 2.5 × 2.5 mm. Staples brand invisible tape was added to both sides of the bonded specimens, spaced 2.5 mm away from the overlap region, for additional grip support. A schematic of the prepared specimens can be found in Figure S4. Prepared specimens were evaluated at different dwell times and tested in tension until failure at a constant crosshead speed of 0.1 mm/s with a load gap of 15 mm in a TA RSA-G2 dynamic mechanical analyzer. Failure mode (adhesive, cohesive, or mixed) was noted for each specimen. The results stated in the following sections were determined to be statistically significant by unpaired Student’s t tests with an alpha of 0.05 and a minimum of three replicates.
Fatigue.
Specimens for fatigue testing were prepared in the same manner as for lap shear adhesion but with larger dimensions (20 × 5 × 0.5mm, (L × W × Th), with a 5 × 5mm overlap and Staples brand invisible tape grip supports 5 mm away from the overlap region). Samples were loaded into one grip of a TA RSA-G2 analyzer with a load gap of 20 mm before zeroing the instrument force measurement. The other end of the specimen was secured to the second grip, and the load gap was adjusted manually to return to a reading of zero axial force on the instrument. Specimens were cyclically loaded under uniaxial tension with 6% strain at 10 Hz or 2% strain at 5 Hz for a maximum of 100 000 cycles or until failure.
Characterization.
Dynamic Mechanical Analysis.
The glass transition temperature was measured by a TA RSA-G2 dynamic mechanical analyzer. Measurements were collected at a frequency of 1 Hz and a temperature ramp rate of 3 °C/min. Specimens were cut into a rectangular shape (L × W × Th, 10 mm × 4 mm × 0.5 mm).
RESULTS AND DISCUSSION
The monomers used to produce adhesive films are depicted in Figure 1a. In all cases, a CAN was produced with TE-diene and PETMP via the thiol–ene reaction initiated by Irgacure 819 with 400–500 nm light. To accommodate the need for excess thiols to promote TTE (Figure 1b), one equivalent of PETMP with one equivalent TE-diene was used, resulting in a 2:1 ratio of thiol-to-ene functional groups. The nucleophilic catalyst DABCO was selected because: (i) it shows excellent activity in the TTE reaction and (ii) nucleophilic catalysts have been shown to have a minimal effect on the final conversion in the radical-mediated thiol–ene reaction used to polymerize these materials.30 Although a small decrease in rubbery modulus was observed with the addition of catalyst due to the ability of the nucleophilic catalyst to add across a thioester and temporarily lower the local cross-linking density, the glass transition temperature was not affected by catalyst loading over the range of DABCO concentration used in this study (0–15 mol %) (Figure S1).
Figure 1.
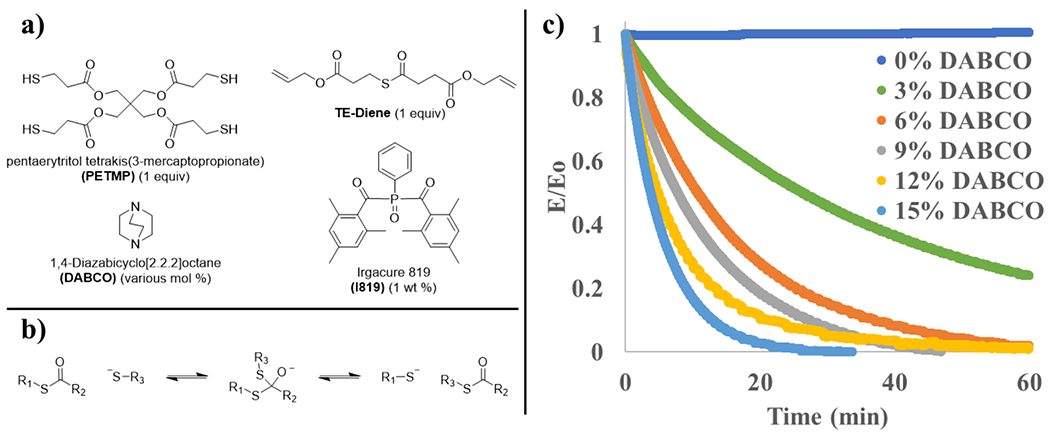
(a) Structures of the thiol, thioester containing alkene, and photoinitiator used to synthesize adhesive films as well as the nucleophilic thiol-thioester exchange catalyst. All samples contain 1 equiv of PETMP, 1 equiv of TE-diene, 1 wt % I819 relative to the monomers, and various mol % DABCO relative to TE-diene. (b) General scheme of thiol-thioester exchange. (c) Stress relaxation curves for thiol–ene adhesives with various mol % DABCO as the nucleophilic catalyst at an applied strain of 5%.
Unfilled Adhesives.
Stress relaxation was performed to characterize the effect of the TTE rate as controlled by the catalyst concentration on the time scale of macroscopic material behavior for films containing 0, 3, 6, 9, 12, and 15 mol % DABCO (Figure 1c). Herein, the use of a 0% catalyst loading sample serves as an effective negative molecular control on the impact of the TTE reaction on the material behavior. In the absence of the catalyst, despite the remainder of the network structure being identical to the other samples, only a minimal amount of TTE will occur. As expected, no relaxation is observed in specimens that do not contain catalyst. As the catalyst concentration increases, a nonlinear decrease in relaxation time occurs from 40.6 min at 3 mol % catalyst to 4.5 min at 15 mol % catalyst (Figure S2 and Table S1). This decreasing relaxation time indicates that the more rapid rearrangement of covalent bonds with increasing catalyst concentration is manifest in more efficient stress dissipation. This observation is consistent with previously reported relaxation times for increasingly basic and/or nucleophilic catalysts, and it serves as a useful baseline to compare adhesive performance and mechanical properties at varying catalyst concentrations.
Toughness was calculated from tensile stress–strain curves (Figure S3), tested at 0.067 min−1, for each of the above catalyst concentrations (Figure 2a). Even a small amount of DABCO results in a decrease in modulus and ultimate tensile stress while simultaneously increasing the elongation at break. This behavior is attributed to energy dissipation from covalent bond rearrangement, which reduces the maximum stress buildup while allowing greater displacement and rearrangement of the polymer chains, even in the otherwise cross-linked network. For lower (3 and 6 mol %) catalyst loadings, the decrease in modulus and ultimate tensile stress compared to the uncatalyzed system is offset by the increase in elongation, resulting in nearly identical toughness to the catalyst-free control. The 9 mol % DABCO samples exhibit some recovery of the tensile strength in combination with greater elongation, resulting in a peak toughness value over the range of concentrations measured. There is a subsequent decrease in the ultimate tensile strength at the highest catalyst loading (15 mol %) that decreases the toughness below the peak value at this strain rate, although the toughness remains greater than the uncatalyzed control. There is some indication as to why this occurs when toughness is shown as a function of the characteristic relaxation time in Figure 2b. Here, it is clear that there is not an appreciable net effect on toughness for samples with relaxation times longer than approximately 15 min. However, materials with relaxation times under 15 min (9, 12, and 15 mol % DABCO) display toughness values greater than that of the catalyst-free formulation. This behavior suggests a relationship between the relaxation time, which is intrinsic to the material, and productive bond rearrangement, that improves the material performance. In the context of these tensile experiments, although 9 mol % represents an optimal loading for achieving toughness in these unfilled films, a sufficiently high rate of TTE is required to increase the toughness in these soft materials.
Figure 2.
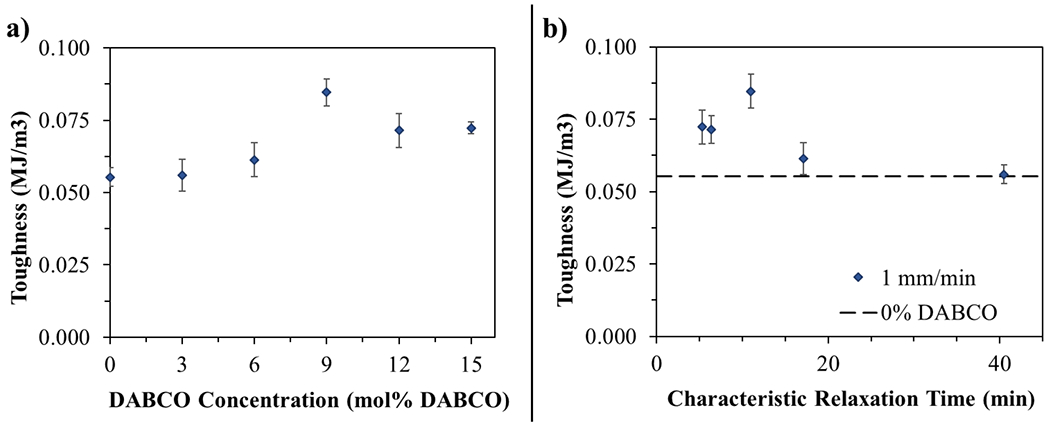
(a) Toughness measured by tensile test as a function of mol % DABCO tested at a strain rate of 1 mm/min. (b) Toughness as a function of the characteristic relaxation time as measured by stress relaxation for each catalyst concentration. Toughness values were calculated by taking the area under the stress–strain curve. Error bars are the standard error at each catalyst concentration with a minimum of five replicates.
Adhesive strength measured by lap shear for this range of catalyst concentrations is shown in Figure 3a. The substrate chosen for these tests is the same thiol–ene material as the adhesive but without the presence of the catalyst. Lap-shear strength was measured at two dwell times, or durations that the substrate and adhesive were in contact: 120 min and less than 5 min (as short of time as practical to prepare the sample and run the test). At all catalyst concentrations, the longer dwell time improves lap shear strength by a factor of 2 or greater. For the short dwell time, the strength reaches a peak around 9 mol % DABCO, although this value is not statistically different at the 95% confidence level from the lap shear strength values of the other catalyst-containing samples. When allowed to dwell for 120 min, the adhesive strength increases with catalyst concentration, reaches a maximum at 9 mol %, and then decreases as catalyst loading is increased further. In materials where no catalyst is present, it is expected that longer dwell times provide more time for physical interactions and better surface contact between the adhesive and the substrate to develop. When TTE is also active, even at low catalyst concentrations, bond exchange at or near the interface likely has two notable effects at short dwell times. First, any covalent bonds that form in that time are inherently stronger than physical entanglements and van der Waals forces. At the same time, those same covalent bonds promote additional surface contact and physical interactions. Both effects strengthen adhesion at long times.
Figure 3.
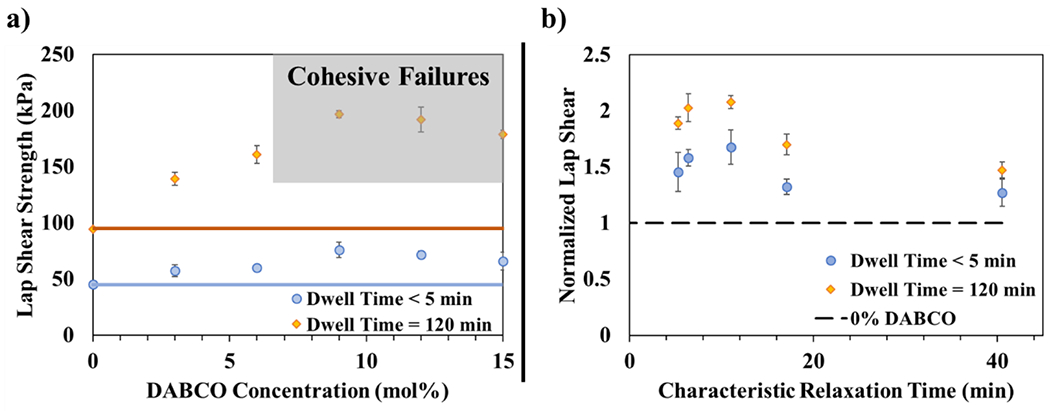
(a) Lap shear strength as a function of catalyst concentration for short dwell times (<5 min) and after 120 min dwell time. The solid lines indicate the lap shear strength with no catalyst. (b) Normalized lap shear strength (normalized to the strength measured for the 0% catalyst sample to indicate the increase over TTE-inactive control) as a function of characteristic relaxation time. The lap shear values are normalized to the samples that do not contain catalyst, which is indicated by the dashed line. All error bars represent the standard error of the data.
The peak in lap shear strength after 120 min can be attributed to a shift in failure mechanism as catalyst concentration increases (shown in Figure 3a). Specimens containing 0, 3, and 6 mol % DABCO fail adhesively, indicating that the interfacial adhesive strength is lower than the cohesive strength of the material itself. However, at and above 9 mol % catalyst, the failure mechanism switches from adhesive failure solely at the interface to cohesive failure of the adhesive material on either side of the lap joint, suggesting that performance is limited by bulk material properties rather than interfacial bonding. This conclusion is strengthened by the fact that toughness and lap shear strength exhibit similar behavior with respect to characteristic relaxation time (Figure 2b and Figure 3b, respectively), reaching a maximum within the same range of relaxation times. When taken together, these observations indicate that although shorter relaxation times may well promote better adhesion through covalent bond exchange, care must be taken to avoid deterioration in cohesive strength at relaxation times lower than the optimal range as cohesive strength is the limit of strength in this regime. Despite this issue, the decrease in lap shear strength associated with increased catalyst loading (and decreased relaxation time) is sufficiently small that it is unlikely to be realized in practice by the user. Efforts could be taken to increase the cohesive strength of the materials in this short relaxation time and cohesive failure regime as a method to increase PSA performance.
Fatigue Testing of Unfilled Adhesives.
Although simple mechanical toughness and adhesive strength are crucial to the performance of PSAs and have been shown to improve with addition of optimal TTE-based formulations, the ability to maintain performance after repeated use is often even more important to consider. To evaluate the long-term durability of adhesive films with DCCs under cyclic loading, fatigue testing was performed on unfilled specimens with 0, 3, 6, and 9 mol % DABCO loading. Conditions for testing were derived from lap shear tests, with a 0.5 mm thick test sample adhered with a 5 × 5 mm overlap onto a 0.5 mm thick catalyst-free TE-diene and PETMP substrate, both supported by a strong elastic backing. The single-lap joint was fatigued under uniaxial tension and strain-control at 6% strain and a frequency of 10 Hz until failure or 100 000 cycles. A strain of 6% was chosen because it corresponds to 65–75% of the failure strain for unfilled lap shear samples with higher DABCO loading (6 and 9%), as reported above for similar lap shear specimens with an overlap region smaller than these fatigue samples. The cycles-to-failure for adhesive films containing at these catalyst concentrations are shown in Figure 4a.
Figure 4.
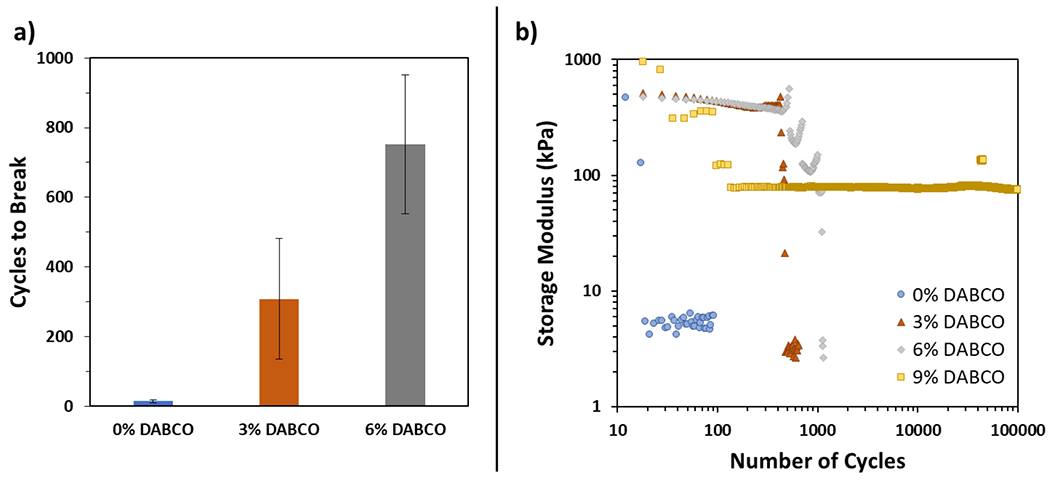
(a) Cycles to break during fatigue testing at 6% strain and 10 Hz for 0, 3, and 6 mol % DABCO. The 9 mol % DABCO specimens are not depicted here because they did not fail after the full duration of 100 000 cycles. Error bars represent the standard error of the data. (b) Representative log–log plot of the storage modulus as a function of number of cycles during fatigue testing for 0, 3, 6, and 9 mol % DABCO samples.
Despite large variability in total number of cycles until failure, activation of TTE in these networks demonstrates fatigue resistance, as evidenced by an increase in the number of cycles-to-break and a shift in failure mechanism (Figure 4b). For samples without TTE (0 mol % catalyst), failure is rapid, smooth, and interfacial after an average of 15 cycles. When thioester exchange is active, the lap joint demonstrates dramatically enhanced resistance to fatigue, breaking after an average of 300 and 750 cycles with 3 and 6 mol % catalyst, respectively. At these concentrations, a stick–slip failure mechanism is observed. As the samples fatigue, repeated “slipping” and “sticking” occurs, during which the adhesive film first slides across then catches the substrate. This mechanism, which is consistent with the DCC-based self-healing in these materials, allows the material to delay catastrophic failure, increasing the total number of cycles sustained before complete interfacial separation. This stick–slip phenomenon was observed in some but not all 3 and 6 mol % samples, contributing to the large variability in cycles-to-break for these groups. For the highest catalyst loading tested (9 mol % DABCO), the sample survived the duration of the 100 000 cycle tests with an initial drop in storage modulus but no obvious change beyond the first 200 cycles, suggesting a lap joint whose strength is maintained for the majority of the fatiguing process evaluated here.
To quantify better the fatigue behavior of the unfilled material without catalyst, milder fatigue conditions were also applied. Fatigue at a strain of 2% with a frequency of 5 Hz was conducted on 0 and 3 mol % DABCO specimens. Under these conditions, the control failed after approximately 20 000 cycles, while the 3 mol % DABCO sample survived without change in storage modulus after 100 000 cycles. This demonstrates that with fatigue conditions that are more favorable to the control group, even relatively low concentrations of TTE-enabling catalyst significantly enhance material fatigue resistance.
Filled Adhesives.
OX-50 SNPs were surface functionalized with acrylate moieties that either contain thioester (TE-SNP) or do not contain thioester (C-SNP) (Figure 5a). All filled adhesive samples contained 15 wt % particles. A catalyst loading of 9 mol % was chosen as the optimal catalyst concentration based on the unfilled PSAs as described above. The addition of either the control or thioester SNPs did not change the glass transition temperature as compared to the unfilled samples, as would be expected (Figure S6), although a small decrease in cross-linking density with catalyst presence was observed here as it was for the unfilled films. Additionally, there were no differences observed in characteristic relaxation time between films made with either SNP type (Figure S6), indicating that the primary source of stress relaxation in this test occurs within the matrix.
Figure 5.
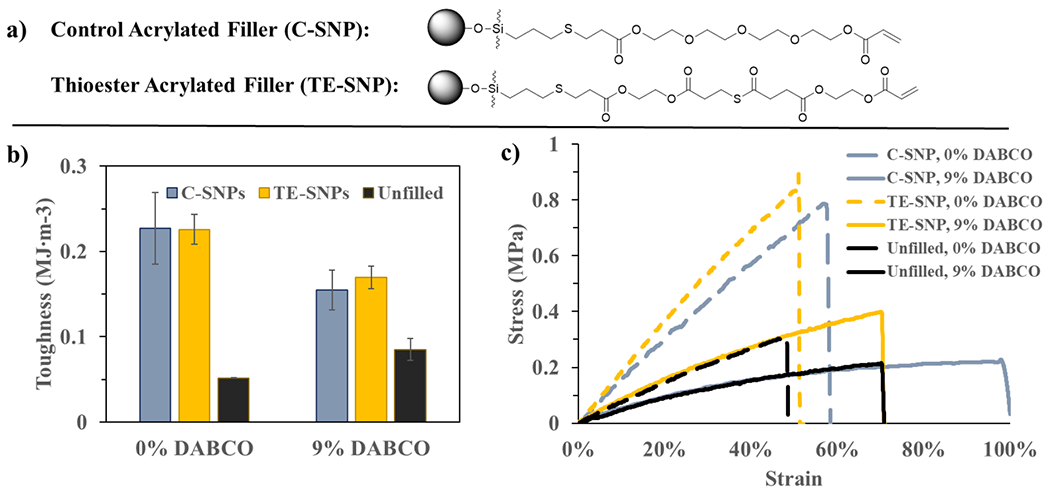
(a) General structures for SNPs without thioester-containing acrylate (C-SNP, control) or with thioester-containing acrylate (TE-SNP). (b) Toughness measured in tensile testing with 15 wt % control SNPs, 15 wt % TE-SNPs, or without filler when TTE is inactive (0 mol % DABCO) or active (9 mol % DABCO). (c) representative stress–strain curves for tensile testing films with C-SNPs, with TE-SNPs, or without filler. Variation in mechanical properties is clearly seen with the addition of catalyst (solid lines) compared to catalyst-free formulations (dashed lines). All error bars represent the standard error.
Toughness was evaluated for each nanocomposite adhesive film (Figure 5b) based on the tensile stress strain curves (Figure 5c) under the same conditions as their unfilled counterparts at the same 9 mol % catalyst loading. Addition of filler into the adhesive system increased toughness by nearly 340% in catalyst-free formulations, largely because of increases in material stiffness caused by the inorganic filler. This effect is less pronounced for catalyst-containing films, where C-SNP and TE-SNP adhesives displayed an 82 and 99% increase, respectively, in toughness relative to unfilled material at the same catalyst loading, noting that the difference in toughness between samples filled with C-SNPs and TE-SNPs is not statistically significant. It is important to stipulate that the unfilled system with catalyst is already toughened significantly by TTE. The increase in toughness for filled-TE adhesives is 200–230% compared to the unfilled, non-TE control. Irrespective of the point of reference, TTE does not further increase toughness in filled samples at this filler loading, in contrast to what is observed for unfilled samples. This suggests that, given 15 wt % filler and 9 mol % DABCO, the increase in toughness is driven by filler reinforcement.
This apparent decrease in effectiveness of DCCs and filler incorporation to increase toughness likely is due to differences inherent to filled versus unfilled networks. For soft unfilled materials, the bulk of the network is allowed to deform under tensile forces. At appropriate catalyst concentrations, covalent bond rearrangement throughout the network reduces stress buildup and promotes further elongation before failure, offsetting the associated decrease in modulus and tensile strength. In soft filled materials, most of the deformation takes place in the low modulus organic matrix, which no longer occupies the entire material volume and is constrained by the filler. Consequently, when DCCs are activated, stresses are still continuously relaxed by covalent bond rearrangement, but the increase in elongation no longer offsets the decrease in modulus and ultimate tensile strength. This observation may serve to explain why activation of TTE with the addition of catalyst displays a decrease in toughness for filled materials, which is unexpected given the substantial increase TTE activation supplies for the unfilled materials. The effect of DCCs lowering the modulus may limit the performance in the filled systems while the effect of DCCs greatly increasing elongation enhances the performance for unfilled systems.
With respect to tensile lap shear strength, filled PSAs exhibit similar behavior to their unfilled counterparts (Figure 6b). In all cases the adhesive strength of the unfilled materials is stronger than that of the filled materials (shown in Figure 6a), but to different degrees. Without catalyst, a longer dwell time (120 min) increases the adhesive strength by roughly 50% for both filler types. This similar increase for both systems is expected, given that without catalyst there should be no difference between the C-SNP and TE-SNP filled networks. When DABCO is present to activate TTE, both filled materials experience an increase in adhesive strength, but with two differing outcomes, depending on dwell time: (1) At short dwell times (<5 min), there is still no discernible difference in adhesive strength between the two SNP types, while (2) at the longer dwell time, the increase in adhesive strength for TE-SNP-containing films is statistically higher (p-value <0.001) than the increase in strength associated with C-SNP-containing films, which do not contain a TTE-capable filler–resin interface. The addition of a dynamic filler–resin interface leads to roughly twice the increase in adhesive strength than the same material without the dynamic interface.
Figure 6.
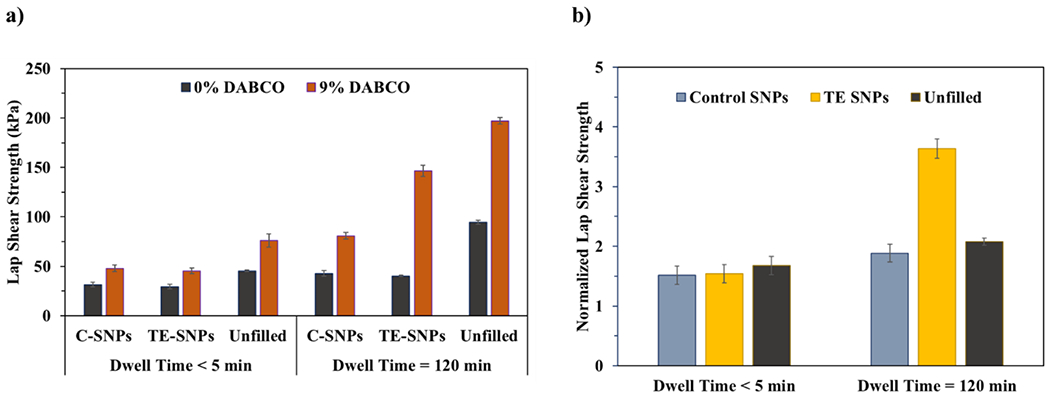
(a) Tensile lap shear strength as a function of dwell time (<5 or 120 min), filler type (C-SNPs, TE-SNPs, or Unfilled), and catalyst loading (0 and 9 mol % DABCO). (b) Tensile lap shear strengths of DABCO-containing films normalized to their corresponding DABCO-free formulations for samples containing C-SNPs, TE-SNPs, or no filler. Dashed line indicates baseline value given no change from uncatalyzed controls. All error bars represent the standard error.
As stated above, unfilled materials reach a higher tensile lap shear strength in each case. However, the normalized lap shear strength values depicted in Figure 6b shows that the relative amount by which TTE improves adhesion differs both by material and dwell time. For short dwell times (small time scales), physical and chemical interactions between the substrate and adhesive have little opportunity to fully develop. As a result, the fractional increase in adhesive strength due to TTE is similar for the unfilled (67%), C-SNP (51%), and TE-SNP (55%) materials. At longer dwell times, physical and chemical association between substrate and adhesive have sufficient time to develop, enhancing tensile lap shear strength significantly for all three materials (107, 88, and 260% for unfilled, C-SNP, and TE-SNP films, respectively) compared to the same material without catalyst.
There are likely two reasons for this dramatic increase in strength after long dwell times when TTE is active. First, TTE increases creep in cross-linked networks. As discussed previously, increased TTE exchange rates that lead to material flow facilitate surface contact at the macroscopic and microscopic scales to improve adhesive strength over time. Second, because both substrate and adhesive contain thioester and free thiols, TTE can occur across the interface. Covalent bonding at the interface, in addition to physical bonding mechanisms, greatly increases binding strength over time, effectively blending adhesive and substrate where they meet. These effects are enhanced by TE surface-functionalized fillers, likely because (i) the dynamic interface allows for more facile motion of the organic matrix around the SNPs, increasing adhesive flow to the substrate surface, and (ii) the surface functionalization of the TE-SNPs enable the filler particles themselves to engage in dynamic exchange with the substrate, possibly reinforcing the network not only in the bulk of the material but also directly at the substrate–adhesive interface.
CONCLUSIONS
An investigation of how TTE and functionalized silica nanoparticles affect the performance of pressure-sensitive adhesives composed of thiol–ene films was carried out. Analysis demonstrated that for unfilled films, an optimal dynamic exchange catalyst loading existed that significantly increased film toughness and lap shear strength compared to a control without DCCs. It is hypothesized that there is a relationship between the DCC reaction rate, the characteristic relaxation time of the material, and the maximum properties. Inclusion of functionalized nanofiller into these films further increased material toughness and modulus but with a slight sacrifice of tensile lap shear strength. This sacrifice in lap shear strength could be largely mitigated via activation of thiol-thioester exchange. This effect was especially pronounced for specimens containing thioester-functionalized filler, where tensile lap shear strength more than tripled upon activation of the DCC at the interface.
Furthermore, the fatigue properties of unfilled pressure-sensitive adhesives were evaluated, showing that specimens with active thiol-thioester exchange could withstand cycles-to-break several orders of magnitude higher than specimens without dynamic bonding. Together, these results demonstrate that both the toughness and the adhesive performance of pressure-sensitive adhesives are improved and tailored based on inclusion of dynamic chemistries in the bulk phase and/or at the filler interface. These conclusions have implications in design strategies for adhesive systems for use in a variety of fields, including aerospace, automotive, and dental industries, among others.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health (1 F31 DE027861-01A1) and by the National Science Foundation (NSF CHE 1808484). Additional thanks to Su Sie Park for assistance in performing lap shear experiments, Nancy Sowan for helpful discussion, and to Dr. Benjamin Fairbanks for his input with respect to editing of the manuscript.
ABBREVIATIONS
- CANs
covalent adaptable networks
- DCCs
dynamic covalent chemistries
- DMA
dynamic mechanical analysis
- SNPs
silica nanoparticles
- C-SNPs
control silica nanoparticles
- TE-diene
thioester diene
- TE-SNPs
thioester silica nanoparticles
- TTE
thiol-thioester exchange
- PSAs
pressure-sensitive adhesives
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsapm.9b00992.
Synthetic procedures, DMA characterization, example stress–strain curves, IR spectra of functionalized SNPs, and NMR spectra of monomers (PDF)
The authors declare the following competing financial interest(s): C.N.B. is an inventor on patents that address the formation of composites with dynamic covalent interfaces and has the potential to receive royalties from these patents.
REFERENCES
- (1).Figueiredo JCP; Campilho R; Marques EAS; Machado JJM; da Silva LFM Adhesive thickness influence on the shear fracture toughness measurements of adhesive joints. Int. J. Adhes. Adhes 2018, 83, 15–23. [Google Scholar]
- (2).Jojibabu P; Ram GDJ; Deshpande AP; Bakshi SR Effect of carbon nano-filler addition on the degradation of epoxy adhesive joints subjected to hygrothermal aging. Polym. Degrad. Stab 2017, 140, 84–94. [Google Scholar]
- (3).Tiu BDB; Delparastan P; Ney MR; Gerst M; Messersmith PB Enhanced Adhesion and Cohesion of Bioinspired Dry/Wet Pressure-Sensitive Adhesives. ACS Appl. Mater. Interfaces 2019, 11 (31), 28296–28306. [DOI] [PubMed] [Google Scholar]
- (4).Cheng SJ; Zhang MQ; Dixit N; Moore RB; Long TE Nucleobase Self-Assembly in Supramolecular Adhesives. Macromolecules 2012, 45 (2), 805–812. [Google Scholar]
- (5).Fan HL; Wang JH; Jin ZX Tough, Swelling-Resistant, Self-Healing, and Adhesive Dual-Cross-Linked Hydrogels Based on Polymer-Tannic Acid Multiple Hydrogen Bonds. Macromolecules 2018, 51 (5), 1696–1705. [Google Scholar]
- (6).Han L; Lu X; Wang MH; Gan DL; Deng WL; Wang KF; Fang LM; Liu KZ; Chan CW; Tang YH; Weng LT; Yuan HP A Mussel-Inspired Conductive, Self-Adhesive, and Self-Healable Tough Hydrogel as Cell Stimulators and Implantable Bioelectronics. Small 2017, 13 (2), 1601916. [DOI] [PubMed] [Google Scholar]
- (7).Lin P; Ma SH; Wang XL; Zhou F Molecularly Engineered Dual-Crosslinked Hydrogel with Ultrahigh Mechanical Strength, Toughness, and Good Self-Recovery. Adv. Mater 2015, 27 (12), 2054–2059. [DOI] [PubMed] [Google Scholar]
- (8).Heinzmann C; Coulibaly S; Roulin A; Fiore GL; Weder C Light-Induced Bonding and Debonding with Supramolecular Adhesives. ACS Appl. Mater. Interfaces 2014, 6 (7), 4713–4719. [DOI] [PubMed] [Google Scholar]
- (9).Ducrot E; Chen YL; Bulters M; Sijbesma RP; Creton C Toughening Elastomers with Sacrificial Bonds and Watching Them Break. Science 2014, 344 (6180), 186–189. [DOI] [PubMed] [Google Scholar]
- (10).Tanaka Y; Kuwabara R; Na YH; Kurokawa T; Gong JP; Osada Y Determination of fracture energy of high strength double network hydrogels. J. Phys. Chem. B 2005, 109 (23), 11559–11562. [DOI] [PubMed] [Google Scholar]
- (11).Gong JP Why are double network hydrogels so tough? Soft Matter 2010, 6 (12), 2583–2590. [Google Scholar]
- (12).Haque MA; Kurokawa T; Gong JP Super tough double network hydrogels and their application as biomaterials. Polymer 2012, 53 (9), 1805–1822. [Google Scholar]
- (13).Lossada F; Guo JQ; Jiao DJ; Groeer S; Bourgeat-Lami E; Montarnal D; Walther A Vitrimer Chemistry Meets Cellulose Nanofibrils: Bioinspired Nanopapers with High Water Resistance and Strong Adhesion. Biomacromolecules 2019, 20 (2), 1045–1055. [DOI] [PubMed] [Google Scholar]
- (14).Tang J; Wan L; Zhou Y; Pan H; Huang F Strong and efficient self-healing adhesives based on dynamic quaternization crosslinks. J. Mater. Chem. A 2017, 5 (40), 21169–21177. [Google Scholar]
- (15).Michal BT; Spencer EJ; Rowan SJ Stimuli-Responsive Reversible Two-Level Adhesion from a Structurally Dynamic Shape-Memory Polymer. ACS Appl. Mater. Interfaces 2016, 8 (17), 11041–11049. [DOI] [PubMed] [Google Scholar]
- (16).Yu QM; Tanaka Y; Furukawa H; Kurokawa T; Gong JP Direct Observation of Damage Zone around Crack Tips in Double-Network Gels. Macromolecules 2009, 42 (12), 3852–3855. [Google Scholar]
- (17).Tanaka Y; Kawauchi Y; Kurokawa T; Furukawa H; Okajima T; Gong JP Localized yielding around crack tips of double-network gels(a). Macromol. Rapid Commun 2008, 29 (18), 1514–1520. [Google Scholar]
- (18).Tanaka Y A local damage model for anomalous high toughness of double-network gels. Epl 2007, 78 (5), 56005. [Google Scholar]
- (19).Webber RE; Creton C; Brown HR; Gong JP Large strain hysteresis and mullins effect of tough double-network hydrogels. Macromolecules 2007, 40 (8), 2919–2927. [Google Scholar]
- (20).Scheutz GM; Lessard JJ; Sims MB; Sumerlin BS Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets. J. Am. Chem. Soc 2019, 141 (41), 16181–16196. [DOI] [PubMed] [Google Scholar]
- (21).Chen XX; Dam MA; Ono K; Mal A; Shen HB; Nutt SR; Sheran K; Wudl F A thermally re-mendable cross-linked polymeric material. Science 2002, 295 (5560), 1698–1702. [DOI] [PubMed] [Google Scholar]
- (22).Montarnal D; Capelot M; Tournilhac F; Leibler L Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334 (6058), 965–968. [DOI] [PubMed] [Google Scholar]
- (23).Capelot M; Montarnal D; Tournilhac F; Leibler L Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets. J. Am. Chem. Soc 2012, 134 (18), 7664–7667. [DOI] [PubMed] [Google Scholar]
- (24).Green MS; Tobolsky AV A NEW APPROACH TO THE THEORY OF RELAXING POLYMERIC MEDIA. J. Chem. Phys 1946, 14 (2), 80–92. [Google Scholar]
- (25).Fairbanks BD; Singh SP; Bowman CN; Anseth KS Photodegradable, Photoadaptable Hydrogels via Radical-Mediated Disulfide Fragmentation Reaction. Macromolecules 2011, 44 (8), 2444–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kloxin CJ; Scott TF; Bowman CN Stress Relaxation via Addition-Fragmentation Chain Transfer in a Thiol-ene Photo-polymerization. Macromolecules 2009, 42 (7), 2551–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Park HY; Kloxin CJ; Abuelyaman AS; Oxman JD; Bowman CN Stress Relaxation via Addition-Fragmentation Chain Transfer in High T-g, High Conversion Methacrylate-Based Systems. Macromolecules 2012, 45 (14), 5640–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Scott TF; Schneider AD; Cook WD; Bowman CN Photoinduced plasticity in cross-linked polymers. Science 2005, 308 (5728), 1615–1617. [DOI] [PubMed] [Google Scholar]
- (29).Hall DN; Oswald AA; Griesbaum K Allene Chemistry. 4. Unsymmetrical terminal thiol-allene diadducts. Effect of allylic reversal. J. Org. Chem 1965, 30 (11), 3829. [Google Scholar]
- (30).Worrell BT; Mavila S; Wang C; Kontour TM; Lim CH; McBride MK; Musgrave CB; Shoemaker R; Bowman CN A user’s guide to the thiol-thioester exchange in organic media: scope, limitations, and applications in material science. Polym. Chem 2018, 9 (36), 4523–4534. [Google Scholar]
- (31).Worrell BT; McBride MK; Lyon GB; Cox LM; Wang C; Mavila S; Lim CH; Coley HM; Musgrave CB; Ding YF; Bowman CN, Bistable and photoswitchable states of matter. Nat. Commun 2018, 9. DOI: 10.1038/s41467-018-05789-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Brown TE; Carberry BJ; Worrell BT; Dudaryeva OY; McBride MK; Bowman CN; Anseth KS Photopolymerized dynamic hydrogels with tunable viscoelastic properties through thioester exchange. Biomaterials 2018, 178, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bracher PJ; Snyder PW; Bohall BR; Whitesides GM The Relative Rates of Thiol-Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water. Origins Life Evol Biospheres 2011, 41 (5), 399–412. [DOI] [PubMed] [Google Scholar]
- (34).Sultan JN; Laible RC; McGarry FJ Microstructure of two-phase polymers. Appl. Polym. Symp 1971, 16, 127–136. [Google Scholar]
- (35).Fu JF; Shi LY; Yuan S; Zhong QD; Zhang DS; Chen Y; Wu J Morphology, toughness mechanism, and thermal properties of hyperbranched epoxy modified diglycidyl ether of bisphenol A (DGEBA) interpenetrating polymer networks. Polym. Adv. Technol 2008, 19 (11), 1597–1607. [Google Scholar]
- (36).Bhowmick AK Effect of Model Fillers on the Adhesive Strength of Pressure-Sensitive Tapes. J. Adhes. Sci. Technol 1989, 3 (5), 371–381. [Google Scholar]
- (37).Lei F; Zhang CT; Cai ZP; Yang JL; Sun HY; Sun DZ Epoxy toughening with graphite fluoride: Toward high toughness and strength. Polymer 2018, 150, 44–51. [Google Scholar]
- (38).Xia T; Ye Y; Qin WL Acrylonitrile-butadiene-styrene colored with a nanoclay-based filler: mechanical, thermal and colorimetric properties. Polym. Bull 2019, 76 (7), 3769–3784. [Google Scholar]
- (39).Park YT; Qian YQ; Chan C; Suh T; Nejhad MG; Macosko CW; Stein A Epoxy Toughening with Low Graphene Loading. Adv. Funct. Mater 2015, 25 (4), 575–585. [Google Scholar]
- (40).Zheng PW; McCarthy TJ A Surprise from 1954: Siloxane Equilibration Is a Simple, Robust, and Obvious Polymer Self-Healing Mechanism. J. Am. Chem. Soc 2012, 134 (4), 2024–2027. [DOI] [PubMed] [Google Scholar]
- (41).Sowan N; Cox LM; Shah PK; Song HB; Stansbury JW; Bowman CN Dynamic Covalent Chemistry at Interfaces: Development of Tougher, Healable Composites through Stress Relaxation at the Resin-Silica Nanoparticles Interface. Adv. Mater. Interfaces 2018, 5 (18), 1800511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Legrand A; Soulie-Ziakovic C Silica-Epoxy Vitrimer Nano-composites. Macromolecules 2016, 49 (16), 5893–5902. [Google Scholar]
- (43).Chen X; Li LQ; Wei T; Venerus DC; Torkelson JM Reprocessable Polyhydroxyurethane Network Composites: Effect of Filler Surface Functionality on Cross-link Density Recovery and Stress Relaxation. ACS Appl. Mater. Interfaces 2019, 11 (2), 2398–2407. [DOI] [PubMed] [Google Scholar]
- (44).Spiesschaert Y; Guerre M; Imbernon L; Winne JM; Du Prez F Filler reinforced polydimethylsiloxane-based vitrimers. Polymer 2019, 172, 239–246. [Google Scholar]
- (45).Fu SY; Feng XQ; Lauke B; Mai YW Effects of particle size, particle/matrix interface adhesion and particle loading on mechanical properties of particulate-polymer composites. Composites, Part B 2008, 39 (6), 933–961. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.