

Abstract
Neutrophil extracellular traps (NET) induce a procoagulant response linking inflammation and thrombosis. Low levels of miR-146a, a brake of inflammatory response, are involved in higher risk of cardiovascular events, but the mechanisms explaining how miR-146a exerts its function remain largely undefined. The aim of this study was to explore the impact of miR-146a deficiency in NETosis both in sterile and non-sterile models in vivo, and to investigate the underlying mechanism. Two models of inflammation were used: (i) Ldlr-/- mice transplanted with bone marrow from miR-146a-/- or wild-type mice were fed a high-fat diet, generating an atherosclerosis model; and (ii) an acute inflammation model was generated by injecting lipopolysaccharide (1 mg/kg) into miR-146a-/- and wildtype mice. miR-146a deficiency increased NETosis in both models. Accordingly, miR-146a-/- mice showed significantly reduced carotid occlusion time and elevated levels of NET in thrombi following FeCl3-induced thrombosis. Infusion of DNAse I abolished arterial thrombosis in both WT and miR-146a-/- mice. Interestingly, miR-146a-deficient mice have aged, hyperreactive and pro-inflammatory neutrophils in their circulation which are more prone to form NET independently of the stimulus. Furthermore, we demonstrated that patients with community-acquired pneumonia with reduced miR-146a levels associated with the T variant of the functional rs2431697 had an increased risk of cardiovascular events due, in part, to an increased generation of NET.
Introduction
Neutrophils play a crucial role in immunity and injury repair but also contribute to the development of several thrombo-inflammatory diseases.1 The pathophysiological role of neutrophils has become more evident following the discovery of neutrophil extracellular traps (NET).2 NET are large web-like structures released upon neutrophil activation, comprising a matrix of DNA and histones which are decorated with antimicrobial proteins, such as myeloperoxidase or neutrophil elastase (NE).3 These structures were first identified as a novel innate defense mechanism;3 however, NET also promote thrombosis through activation of platelets and the coagulation cascade.4 Therefore, NET interconnect immunity, inflammation, and thrombosis within a physiological process known as immunothrombosis.5 Thus, uncontrolled or aberrant activation of immunothrombosis promotes thrombotic pathologies such as stroke, myocardial infarction, deep vein thrombosis, or disseminated intravascular coagulation during sepsis.5,6
A large variety of infectious and sterile inflammatory stimuli have been described as triggers of NETosis. Interestingly, their consequences on neutrophil activation vary depending on the nature and duration of the stimulus. 7 Two mechanisms resulting in NET formation have been described: lytic and vital NETosis.8 Specific microorganisms or lipopolysaccharides (LPS) induce vital NETosis directly or indirectly through toll-like receptors (TLR), via a TLR4-activated platelet interaction, by an oxidant- independent mechanism.9,10 This strategy aims to limit the spread of bacteria and keeps neutrophils alive for further functions.8 Alternatively, lytic NETosis can be induced by diverse sterile agonists such as phorbol 12- myristate 13-acetate,3,11 cholesterol crystals11 or cytokines (interleukin-6,12 interleukin-8,3,13 interferon-14). This drives NADPH oxidase activation via protein kinase C and Raf- MEK-ERK, which generates reactive oxygen species (ROS) inducing neutrophil death.15
MicroRNA (miRNA or miR) are a family of small noncoding RNA that regulate gene expression.16 Since their discovery, miRNA have been implicated as key modulators of numerous physiological and pathological processes. 17-20 In particular, miR-146a directly mediates thromboinflammatory processes as: (i) it inhibits several proinflammatory elements of the TLR-NF (nuclear factor)-B pathway21,22 and (ii) it is predominantly expressed in cells that promote thrombogenesis (monocytes/macrophages, platelets, neutrophils, and endothelial cells).23,24 Our group demonstrated that reduced levels of miR-146a, guided by the T variant of the functional miR-SNP rs2431697, are predictors of adverse cardiovascular events in patients with atrial fibrillation.24 Interestingly, in vitro activation of rs2431697 TT neutrophils exacerbated NET release in accordance with elevated plasma NE levels found in T carriers, who also had a higher risk of adverse cardiovascular events.25 Overall, our data suggest that miR-146a is crucially involved in linking inflammation, thrombosis, and NETosis. However, the role of miR-146a in immunothrombosis and NETosis is unknown. Here, we investigated the involvement of miR-146a in NET formation leading to arterial thrombosis in both sterile and nonsterile inflammatory murine models. Thus, we characterized the neutrophil phenotype to explain the exacerbated NETosis. Finally, we evaluated the association between low miR-146a levels, NETosis markers and the occurrence of thrombosis in septic patients.
Methods
Mice models
Atherosclerosis model
Irradiation and bone marrow (BM) transplantation were performed as previously described.26 Briefly, Ldlr-/-mice were irradiated and transplanted with BM cells obtained from wild-type (WT) or miR-146a-/- mice. After 4 weeks on a standard diet, transplanted mice were fed an atherogenic diet for 8 weeks. Blood was collected from the facial vein into 3.2% citrate and plasma samples were stored at -80ºC until analysis. Heart and aorta were extracted from euthanized mice after in situ perfusion with phosphatebuffered saline and fixed with 4% paraformaldehyde overnight at 4ºC. Tissue was embedded in paraffin and cross-sections from the aortic root were made.
Endotoxemia model
WT and miR-146a-/- mice were injected intraperitoneally with a sublethal dose of LPS (1 mg/kg) (E. coli 0111/B4, Sigma-Aldrich, Madrid, Spain). For morphological analysis, lungs were fixed with 4% paraformaldehyde overnight at 4ºC, embedded in paraffin and sectioned. Lung damage was evaluated in one section/ mouse using a semi-quantitative score via a blind method following the pathologist’s criteria.
Ferric chloride-induced arterial thrombosis
Animals were anesthetized by intraperitoneal injection (xylazine hydrochloride 10 mg/kg + ketamine hydrochloride 100 mg/kg). The carotid artery was isolated and the injury was generated by applying a piece of filter paper (5x1 mm) (GE Healthcare Whatman 1003917, Fisher, Madrid, Spain) soaked in a 7.5% ferric chloride (FeCl3) solution for 2 min (Sigma-Aldrich, Madrid Spain). After washing the arterial surface with saline solution and removing residual FeCl3, a Doppler ultrasound flow probe (Model 0.5 PSB, Transonic Systems, Ithaca, NY, USA) was applied. The probe was connected to a flow meter (Model TS420, Transonic Systems, Ithaca, NY, USA) that continuously registered the blood flow. Occlusion time was defined as the time elapsed from the withdrawal of FeCl3 to the lack of blood flow (≤0.01 mL/min) for at least 3 consecutive minutes. Experiments were stopped after 30 min if no occlusion occurred. For the experiments with DNase I, mice were injected intravenously (tail vein) with 10 g of Pulmozyme® (Roche Farma, Madrid, Spain) diluted in saline, 15 min before FeCl3 treatment.
Patients and controls
The study (NEUMONAC study #2011/0219 [05/07/2011]) was approved by the Ethical Committee (ceic@iislafe.es) of our institution and performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its subsequent amendments. Consecutive patients admitted with communityacquired pneumonia (CAP) were included from October 2015 to June 2018 at Hospital Universitario y Politécnico La Fe (Valencia, Spain). The patients’ details are provided in the Online Supplementary Methods.
Healthy subjects were randomly selected among blood donors from our Transfusion Center and gave their informed consent to enrollment into the study.
Statistical analysis
All statistical analyses were conducted using GraphPad Prism 3.8 (GraphPad Software, Inc., La Jolla, CA, USA) and SPSS 22.0 for Windows (SPSS, Inc., Chicago, IL, USA). Differences between two groups were assessed by an unpaired Student t-test or Mann- Whitney U test, where appropriate. For multiple comparisons, one-way analysis of variance on ranks with the Bonferroni posthoc test was used. Data are shown as mean ± standard error of mean or as median (95% confidence interval), as appropriate. A P-value <0.05 was considered statistically significant.
Further details on the materials and methods are available in the Online Supplementary Methods.
Results
miR-146a deficiency promotes NET formation in an atherosclerosis model
Previous data from our laboratory demonstrated that miR-146a deficiency upregulates NETosis in both murine and human in vitro models.25 Thus, we aimed to extrapolate this finding to different in vivo models. We first utilized an atherosclerosis mouse model previously generated by our group.26 Briefly, Ldlr-/- mice were transplanted with BM from miR-146a-/- or WT animals and fed a highfat diet for 8 weeks (Figure 1A). As reported, transplant efficiency, body weight, circulating blood cell counts, plasma lipid profile, and atherosclerotic burden (atheroma, lesion, and necrotic core areas) were similar between the experimental groups.26 No differences were found in cell-free (cf)DNA and NE plasma levels between the two groups of mice before they were fed the high-fat diet (data not shown). As expected, after 8 weeks of the atherogenic diet, notable differences in NET components between the two groups were observed. As shown in Figure 1B, plasma cfDNA levels were significantly higher in Ldlr-/- BM miR-146a-/- mice than in Ldlr-/- BM WT littermates (348.1 ± 73.0 vs. 177.3 ± 39.4 ng/mL, respectively; P<0.05). Similarly, plasma NE was almost 2-fold higher in BM miR-146a-/- mice than in BM WT mice (228.6 ± 32.6 vs. 113.0± 14.2 ng/mL, respectively; P<0.05) (Figure 1C). In addition, immunofluorescence analysis of aortic valves revealed that, although intact neutrophils were found in both cases (Online Supplementary Figure S1), there were substantial differences in the size and the amount of NET. DNA, and NE, and H2B staining revealed an increase of large NET in the atherosclerotic lesions and adhered to the vascular wall of BM miR-146a-/- mice compared to BM WT mice, in which we only identified scattered NET throughout all the sections (Figure 1D). Quantification of NET in whole sections within aortic roots confirmed that miR-146a-/- mice had more NET than had WT mice (ICorr values 0.78 ± 0.09 vs. 0.53 ± 0.07, respectively; P<0.01) (Figure 1E). In addition, zooming within NETotic areas demonstrated that H2B and NE co-localization was near to 100% (R=0.97; Costes P-value=1), in contrast to the low co-localization found in intact neutrophils (R=0.13; Costes P-value=1) (Figure 1F, Online Supplementary Movies S1 and S2). Collectively, these results demonstrated a role for miR-146a in NET formation during the process of atherosclerosis.
miR-146a mediates NETosis and lung damage in an lipopolysaccharide-induced, sublethal model of inflammation
The molecular mechanisms leading to NETosis differ depending on the triggering stimulus.7 In order to investigate whether miR-146a may link different pathways provoking NETosis, we next examined the implication of miR-146a on NET formation using a non-sterile inflammatory mouse model generated by sublethal injection of LPS (1 mg/kg) for 4 h and 24 h. As expected, there was a significant progressive reduction in circulating leukocyte and platelet counts following induction of endotoxemia, although no relevant differences between miR-146a-/- and WT animals (Online Supplementary Figure S2) were observed. As shown in Figure 2, basal plasma NET markers were similar between miR-146a-/- and WT mice. However, following LPS administration at 4 h and 24 h, plasma cfDNA levels were higher in miR-146a-/- mice than in WT mice, with the difference being statistically significant at 4 h (1653.0 ± 216.5 vs. 845.6 ± 294.4 ng/mL, respectively; P<0.01) (Figure 2A). Similarly, LPS injection resulted in significantly higher levels of plasma NE in miR- 146a-/- mice when compared to WT mice, after 4 h (1671.3 ± 95.6 vs. 1206.1 ± 99.2 ng/mL, respectively; P<0.01) and 24 h (2458.0 ± 57.1 vs. 1524.6 ± 61.2 ng/mL, respectively; P<0.001) (Figure 2B). Higher NETosis in miR- 146a-/- mice following LPS challenge was confirmed by western blotting. Plasma citrullinated histone 3 (citH3) levels were higher in miR-146a-/- mice than in WT mice at both 4 h and 24 h (Figure 2C). Finally, additional inflammatory and coagulation markers were analyzed. LPSdependent ROS production at 24 h was significantly higher compared with basal levels only in miR-146a-/- mice (178%; P<0.05) (Figure 2D). Additionally, the amounts of thrombin-antithrombin (TAT) complexes were significantly greater 4 h after LPS injection than at baseline only in miR-146a-/- mice (43%; P<0.05) (Figure 2E).
Although all mice indistinctly survived sublethal LPS injection, we explored whether LPS challenge could differentially damage mice lungs. Staining of lung sections showed a notable increase of reticulin in samples from miR-146a-/- mice compared with their WT littermates after 4 h of LPS (Figure 2F). According to the pathologist’s criteria, the analysis showed that both reticulin score and global lung injury were increased in miR-146a-/- mice compared to WT mice (Figure 2G). Therefore, miR-146a-deficient mice had higher NETosis, oxidative stress and activation of coagulation and consequently, greater lung damage after LPS injection.
miR-146a deficiency confers an aged, overactive, and pro-inflammatory phenotype to neutrophils
Although neutrophils have been considered to be a relatively homogeneous population, evidence demonstrating their heterogeneity is emerging.1 To investigate whether the increased ability of miR-146a-/- neutrophils to form NET in the above-mentioned models was related to a particular phenotype determined by miR-146a deficiency, we explored the aged/activated state of neutrophils by flow cytometry. miR-146a deficiency did not alter the total percentage of neutrophils in the circulation (Online Supplementary Figures S2 and S3). Analysis of neutrophil surface markers revealed that miR-146a-/- neutrophils exhibited significantly higher levels of Cxcr4 and CD11b, and lower levels of CD62L than WT neutrophils (Figure 3A). Our results also showed that neutrophils from miR-146a-/- mice had significantly lower Cxcr1 levels than neutrophils from their WT littermates (Figure 3B), suggesting an additional pro-inflammatory ability for the miR-146a-/- neutrophil phenotype.27-29 Tlr4 has been implicated in the process of aging30 and is a target of miR-146a.31 Thus, we measured and compared its levels in miR-146a-/- and WT neutrophils from whole blood and found that, as described before, aged neutrophils (defined by Cxcr4high and CD62Llow surface expression) expressed higher levels of Tlr4 than the rest. Interestingly, aged miR-146-/- neutrophils expressed significantly more Tlr4 than aged WT neutrophils (Figure 3C). Moreover, miR-146a deficiency in isolated neutrophils produced significantly higher ROS levels (Figure 3D) and elevated oxygen consumption rate (Figure 3E) at basal state compared with those in neutrophils from WT mice. Thus, miR-146a deficiency seems to promote aging and hyperreactivity to neutrophils, a pro-inflammatory phenotype that could contribute to the exacerbated NETosis of these cells.
Figure 1.

miR-146a deficiency enhances neutrophil extracellular trap formation in atherosclerosis. (A) Ldlr-/- mice were transplanted with bone marrow (BM) from miR-146a-/- or wild-type (WT) mice. After 4 weeks of recovery, mice were fed a high-fat diet for 8 weeks. Blood and aortic tissues were extracted for quantification of the formation of neutrophil extracellular traps (NET) (n=10-12/group). (B) Plasma cell-free (cf)DNA was measured using Sytox Green. (C) Plasma neutrophil elastase (NE) activity was quantified by enzyme-linked immunosorbent assay. (D) Representative confocal microscopy images of an aortic valve leaflet, an atherosclerotic area, and a NET detail from Ldlr-/- BM WT, and Ldlr-/- BM miR-146a-/ mice immunostained for DNA (DAPI, blue), NE (green), and H2B (red). The dashed yellow line denotes an atherosclerotic lesion (L) boundary; the lumen (Lu), and valves (V) are also marked. White arrowheads point to NET. (E) NET quantification in aortic valve sections from Ldlr-/- BM WT, and Ldlr-/- BM miR-146a-/- mice performed with the Colocalization Colormap Fiji plugin. The correlation index (ICorr) represents the fraction of positively correlated (co-localized) H2B and NE pixels in one representative section per mouse in six WT and six miR-146a-/- BM-transplanted Ldlr-/- mice. (F) Fluorescence intensity plots of H2B and NE in a region of interest (yellow line) of a NET and a neutrophil found in the aortic valve from a Ldlr-/- BM miR-146a-/- and a Ldlr-/- BM WT mouse, respectively. The correlation of H2B and NE fluorescence intensity was determined using the Pearson correlation coefficient and Costes method. In the Pearson correlation test, the R value ranges between -1 and 1, with 1 being a perfect correlation, 0 no linear correlation, and −1 a perfect negative linear correlation. Costes analysis compares pixel correlation for no-randomized with randomized images and calculates significance. The Costes P-value was 1 in both cases, indicating that the probability of random images correlating to real images is 0. P-value calculations were performed using the Mann-Whitney U test. Data represent mean ± standard error of mean, *P<0.05, **P<0.01.
Figure 2.

miR-146a deficiency mediates NETosis in endotoxemia. miR- 146a-/- and wild-type (WT) mice were injected intraperitoneally with a sublethal dose of lipopolysaccharide (LPS) (1 mg/kg). Plasma markers of neutrophil extracellular traps (NET) were measured 4 h and 24 h after LPS treatment (n=9/group). (A) Cell-free (cf)DNA levels were quantified by Sytox Green. (B) Neutrophil elastase (NE) levels were measured by enzymelinked immunosorbent assay (ELISA). (C) Citrilluniated histone 3 (CitH3) plasma levels were analyzed by western blot. (D) Reactive oxygen species (ROS) were quantified by fluorometry 24 h after treatment with LPS. (E) Thrombin-antithrombin (TAT) complex levels were detected by ELISA 4 h after LPS treatment. (F) Morphology of lungs from WT and miR-146a-/- mice after LPS stimulation. Lung sections from WT and miR-146a-/- mice treated 4 h with LPS (1 mg/kg) were stained for reticulin (n=9/group) to observe tissue structure and global injury. Representative images of reticulin staining from WT and miR-146a-/- lungs after LPS at 20x and 40x magnification. (G) Semi-quantitative analysis for reticulin and global lung damage according to the pathologist’s criteria. One section/mouse was scored in a blinded fashion into four grades from 0 to 3 (0=normal, 1=mild, 2=moderate, 3=severe). P-value calculations were performed using one-way analysis of variance on ranks with the Bonferroni post-hoc test or the Mann-Whitney U test, where appropriate. Data represent mean ± standard error of mean, *P<0.05, **P<0.01, ***P<0.001.
Figure 3.

miR-146a determines neutrophil phenotype. (A) Flow cytometry analysis of CD11b, CD62L, and Cxcr4 (P<0.05 for all markers) on neutrophils (Ly6G-positive population) from blood of wild-type (WT) and miR-146a-/- mice (n=6 mice in both groups). Contour plots from flow cytometry analysis of CD62L and CD11b on neutrophils from a representative WT mouse and an miR-146a-/- mouse (bottom left panel). Comparison of Cxcr4 expression between WT and miR-146a-/- representative mice (bottom right panel). (B) Flow cytometry analysis of Cxcr1 on neutrophils from blood of WT and miR-146a-/- mice (left panel; n=5 mice in both groups; P<0.01). Comparison of Cxcr1 expression on a representative mouse with each genotype (right panel). (C) Tlr4 levels were measured in Cxcr4high and CD62Llow (aged) neutrophil subpopulation versus the rest (n=6 mice in both groups; P<0.05). Contour plots from flow cytometry analysis of CD62L and Cxcr4 on peripheral blood pool neutrophils from six WT and six miR-146a-/- mice (right panel). (D) Bone marrow (BM) isolated neutrophils were incubated with 10 M H2DCFDA for 30 min at 37°C and analyzed by flow cytometry (n=3 mice in both groups). (E) BM neutrophils were seeded on the plate and the oxygen consumption rate (OCR) was measured by a Seahorse Analyzer. (mean ± standard deviation, n=3 mice for each group, samples in quadruplicate). Statistical analyses between groups of mice were performed using the Mann- Whitney test (A-C) or t-test (D, E); *P<0.05, **P<0.01.
Figure 4.

miR-146a deficiency accelerates carotid thrombotic occlusion. (A) Mouse model of FeCl3-induced carotid arterial thrombosis. The carotid artery from wildtype (WT) and miR-146a-/- mice was isolated and exposed to 7.5% FeCl3 for 2 min (WT n=24, miR-146a-/- n=17). A Doppler ultrasound flow probe registered the blood flow continuously. (B) Basal blood flow. (C) Time to carotid thrombotic occlusion. (D) Alternatively, WT (n=7) and miR-146a-/- (n=7) mice were treated with DNase I (10 g, i.v.) 15 min before vessel injury and the carotid occlusion time was measured for a maximum of 30 min. (E) Representative images of neutrophil extracellular traps (citrullinated histone 3 [citH3]-positive nuclei) in the carotid thrombi from WT and miR-146a-/- mice analyzed by immunofluorescence. For each genotype the upper row shows the complete cross-section of the thrombosed carotid artery (bar corresponds to 100 m) and the lower row a higher magnification of the artery wall (bar corresponds to 20 m) and the detail of a neutrophil (bar corresponds to 5 m) (F-H) Quantification of total infiltrated nucleated cells, citH3-positive cells and the ratio of total nucleated to citH3-positive cells in the thrombi and the adventitial layer of carotid sections from WT and miR-146a-/- mice (n=10/group). P-value calculations were performed using the unpaired Student t-test and Mann-Whitney U-test. Data represent mean ± standard error of mean, *P<0.05.
Loss of miR-146a accelerates time to carotid thrombotic occlusion by increasing NET release
Thus, we hypothesized that under a thrombotic stimulus, these neutrophils may favor the production of NET, thereby strongly contributing to thrombosis. To test this hypothesis, we induced carotid endothelial injury with FeCl3 in miR-146a-/- and WT mice (Figure 4A) and determined time to arterial occlusion. Although basal blood flow was similar between the two groups of animals (Figure 4B), miR-146a-/- mice showed a significant reduction in time to occlusion compared to the time in WT littermates (374.5 ± 21.15 vs. 452.5 ± 26.14 s, respectively; P<0.05) (Figure 4C). The occlusion times of WT and miR- 146a-/- mice were similar upon treatment with DNAse I (Figure 4D). Thus, DNAse I treatment abolished arterial occlusion, supporting the concept that NET are essential for thrombus formation and stability upon FeCl3 damage. To further analyze the association between thrombosis and NETosis, thrombi within carotids were isolated and immunofluorescence for citH3 was performed. FeCl3- induced injury promoted similar infiltration of nucleated cells in both groups (Figure 4E-F); however, the number of DNA-citH3-positive cells within the carotid thrombi was significantly higher in miR-146a-/- mice than in WT ones (19.3 ± 3.3 vs. 10.0 ± 3.6 cells, respectively; P<0.05) (Figure 4G). The ratio of citH3-positive to total nucleated cells found in the thrombi of carotid sections was significantly higher in miR-146a-/- mice than in WT mice (Figure 4H, Online Supplementary Figure S4).
Of note, no relevant differences in F12 coagulation factor, recently implicated in NETosis,32 was observed between the two groups (data not shown). Thus, miR-146a deficiency may accelerate vessel occlusion due, in part, to increased NETosis associated with the presence of hyperreactive neutrophils.
Low miR-146a levels in patients with community-acquired pneumonia are associated with an increased risk of cardiovascular events
In order to test the clinical impact of our in vivo results, we recruited 259 patients with CAP with a 30-day follow- up period. Online Supplementary Table S1 shows the demographic and clinical characteristics of all 259 patients, as well as the frequencies and distribution of the rs2431697 genotype. We found no significant differences between the main cardiovascular risk factors and genotype (Online Supplementary Table S1). In addition, both Sepsis-related Organ Failure Assessment (SOFA) score and neutrophil count at entry were similar in all patients independently of their genotype (Online Supplementary Table S1). One hundred and twelve plasma samples were available for DNA-citH3 measurement. From these, 81 (72.3%) were considered as positive (optical density [OD] values >0.200, the maximum OD value yielded for 30 healthy controls) (Online Supplementary Figure S5, Online Supplementary Table S2). As described for septic patients, plasma levels of DNA-citH3 correlated with clinical outcomes. 33,34 Thus, plasma DNA-citH3 levels were significantly higher in septic patients (n=58) than in non-septic patients (n=23) (P<0.001). Furthermore, DNA-citH3 levels were significantly higher in patients in whom treatment failed or cardiovascular events occurred (n=18) than in the rest of the patients (n=63) (P=0.028) (Online Supplementary Table S2).
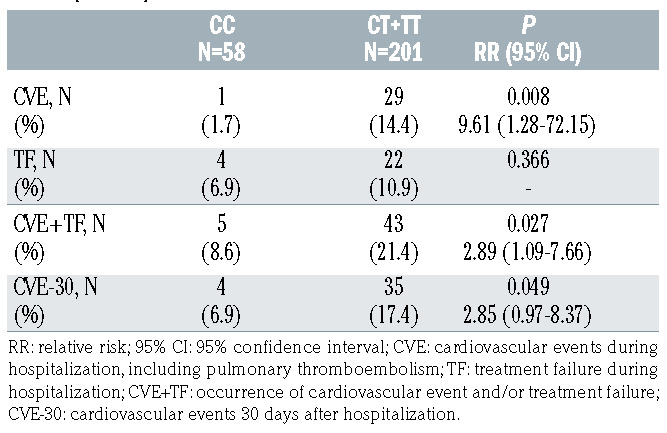
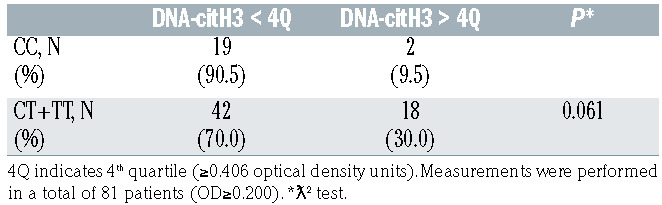
Interestingly, we observed significant differences between the occurrence of cardiovascular complications and rs2431697 genotype. As shown in Table 1, 30 of the 259 patients suffered from cardiovascular events when hospitalized and 29 of them were carriers of the T allele (relative risk [RR]=9.61, [95% CI]: 95% confidence interval: 1.28-72.15, P=0.008). The risk of cardiovascular events remained significantly higher for CT+TT patients 30 days after hospitalization (RR=2.85, 95% CI: 0.97-8.37, P=0.049) (Table 1). In addition, 21.4% of the carriers of the T allele developed a cardiovascular event and failed to respond to therapy (RR=2.89, 95% CI: 1.09-7.66, P=0.027) (Table 1). Finally, among patients with the highest DNA-citH3 plasma levels (≥0.406 OD units) we found a 3-fold higher frequency of T carriers individuals (30%) compared to CC homozygotes (9.5%; P=0.061) (Table 2).
Table 1.
Relationship between hospital cardiovascular complications and rs2431697 genotype in patients with community-acquired pneumonia (n=259).
Discussion
In the last years, the functional versatility of neutrophils has moved towards a fascinating area of research in which the inflammatory capacity of these cells is tightly linked to the development of cardiovascular diseases.35 On the other hand, the pathophysiological role of NET in thrombogenesis is now firmly established.36 In this process, known as immunothrombosis, the mechanisms leading to NETosis differ depending on the triggering stimulus.7 Identifying key regulators of NETosis37 is a challenge to further contribute to the development of anti-thrombotic therapeutic tools that would not affect the essential role of NET fighting germs. In this work, we show that miR-146a, a well-known molecular brake to the NF-B pathway, underlies NETosis-mediated thrombosis after both sterile and non-sterile stimulation.
First, we demonstrated that sterile inflammation in a model of miR-146a deficiency in the hematopoietic lineage caused greater NET formation. However, we previously showed that miR-146a deficiency in the hematopoietic lineage had no effect on atherogenesis after 8 weeks of an atherogenic diet.26 Here, our results suggest that miR-146a deficiency may participate in thrombosis rather than in atherogenesis, through NETosis. This hypothesis is in accord with results published by Franck et al. demonstrating that NETosis does not alter atherosclerotic plaque formation but rather increases thrombosis in a model of plaque erosion.38 Supporting this hypothesis, previous reports have shown that thrombosis induction increases NET deposition.39,40 Consistently, when arterial thrombosis was induced by FeCl3 injury, we observed an increased generation of NET in the thrombi of miR-146a-/- mice compared to WT mice, with a shorter time to vessel occlusion. The abolition of thrombosis by removal of NET with DNAse I in miR- 146a-/-and WT mice, further supports the essential role of miR-146a in NET production. In this sense, Massberg et al. also found in a murine model of FeCl3 injury that the inhibition of NET components, by infusion of anti-H2AH2B- DNA antibody, prolonged the occlusion time and generated low stability thrombi in the carotid.39
Table 2.
rs2431697 genotype frequencies according to DNA-citrullinated H3 plasma levels in patients with community-acquired pneumonia.
Next, we created a non-sterile model of inflammation by LPS injection into miR-146a-/- mice. Indeed, miR-146a-/- mice exhibit hyperreactivity to LPS, with an exacerbated inflammatory response, demonstrating miR-146a as a regulator of autoimmunity, myeloproliferation, and cancer. 23 Our results showed that miR-146a deficiency provokes higher rates of NETosis in LPS-challenged mice. Of note, no differences were found in the capacity of phagocytosis between miR-146a-/- and WT neutrophils in vitro after LPS challenge (data not shown). In addition, miR-146a deficiency promotes a pro-coagulant phenotype with increased levels of thrombin-antithrombin complexes and ROS production and regulates the extent of organ damage following infection, as miR-146a-/- mice developed greater lung damage than did WT mice. Although the use of murine models is considered useful for understanding the pathophysiology of sepsis, there are notable differences between these models and sepsis in humans.41 We therefore examined NETosis markers and their relationship with thrombosis in CAP patients. Plasma NETosis markers correlated with the incidence of sepsis in accordance with previously reported data,42 further supporting the concept that sepsis is a relevant model for investigating the role of NET in humans. Interestingly, we found a correlation between NETosis and thrombotic complications in CAP patients. Our group has previously described that the miR-SNP rs2431697 of MIR146A is associated with an increased risk of cardiovascular events in patients with atrial fibrillation.24 Here, we have verified the role of rs2431697 genotype in cardiovascular events in patients with CAP. Supporting our findings, Xie et al. reported that activation of miR-146a expression decreased markers of myocardial injury in rats treated with LPS.43 In addition, the injection of miR-146a has been described to play a protective role in the cardiac dysfunction induced by sepsis in a murine model, with longer survival of the mice.44 Results from our cohort indicate that NETosis could be a functional path by which miR-146a levels lead to thrombosis in septic patients. Of note, we found that among patients with higher levels of NETosis markers, carriers of the T allele were 3-fold more frequent than CC patients. The concept of sepsis and cardiovascular diseases sharing a common pathophysiology has been explored recently.45 Common gene signaling pathways for both sepsis and cardiovascular diseases were investigated and genetic variations in miR-146a targets such as IL6 or IRAK1 were found to be shared in patients with both diseases.45 Although the etiology of sepsis and cardiovascular disease is not identical, the two conditions share the same end-points of inflammation, coagulation, and endothelial activation.46,47 Overall our results demonstrate that miR-146a is involved in thrombogenesis of two diseases with cardiovascular events, atrial fibrillation and sepsis, a priori in two different models. Accordingly, miR-146a may be yet another regulator of immunothrombosis.
The mechanisms leading to the increased NET formation by miR-146a are still elusive. Interestingly, proinflammatory activity of neutrophils has been shown to correlate positively with aging.30 Aged neutrophils are the first line of defense in the context of acute inflammation since they do not return to the bone marrow, arrive first at the inflammatory tissues and infiltrate them in a TLR4/CD11b-dependent manner.48 Thus, we investigated neutrophil phenotype in miR-146a-/- mice in order to explain the greater NET formation observed in our different models. We found that miR-146a-/- resting neutrophils have an aging phenotype CD62Llow CD11bhigh Cxcr4high, and a lower expression of Cxcr1.1
Figure 5.
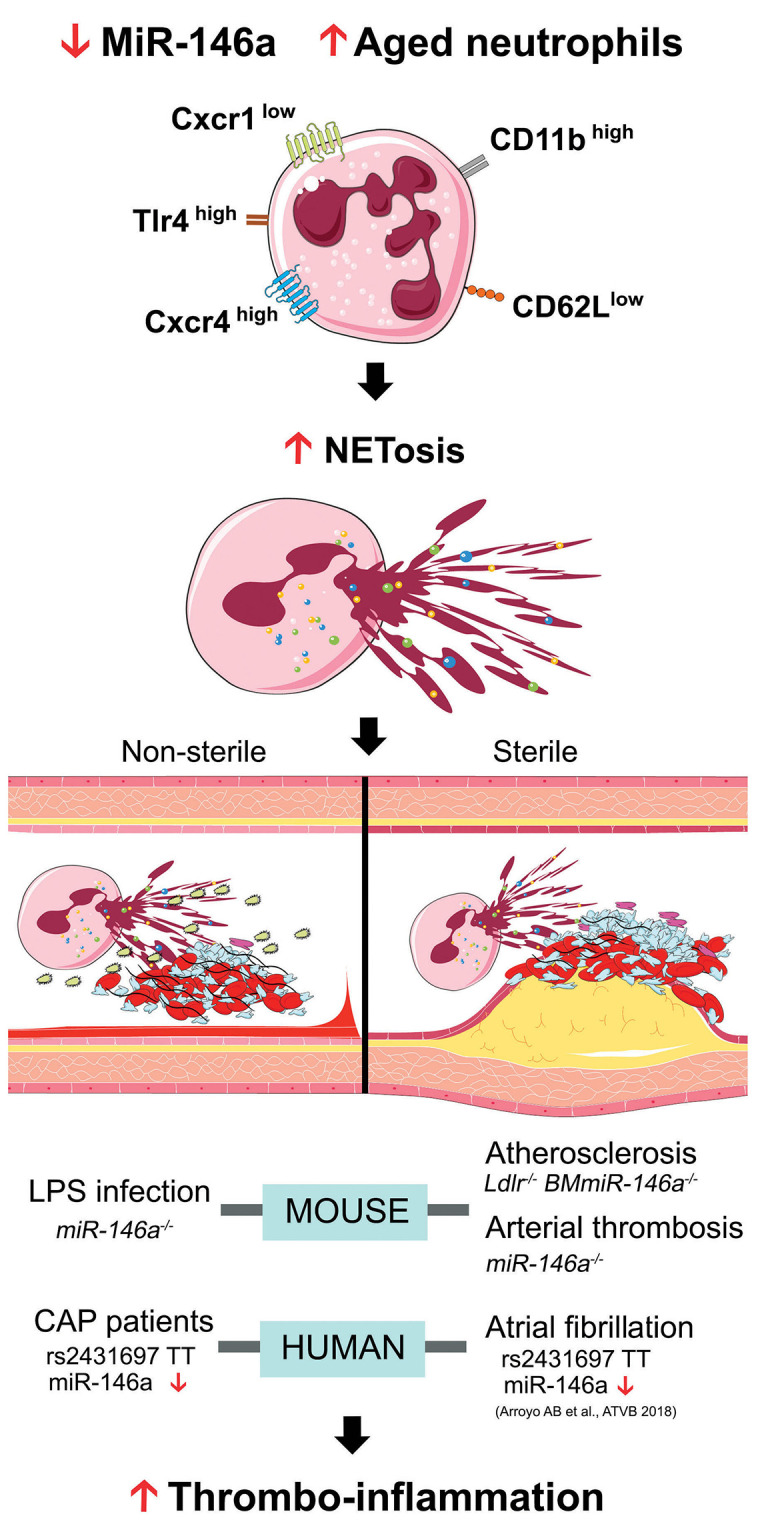
miR-146a plays a relevant role in thrombo-inflammation after sterile and non-sterile stresses. Low miR-146a levels promote an aging phenotype in neutrophils (CD62low CD11bhigh Cxcr4high Tlr4high) which primes these cells. Upon sterile or non-sterile stimuli, primed neutrophils would be more prone to NETosis leading to a thrombo-inflammatory process. MiR-146a: microRNA- 146a; LPS: lipopolysaccharide; CAP: community-acquired pneumonia.
Thus, we speculate that miR-146a deficiency could act as a priming effector on resting neutrophils so that, under any inflammatory stimulus, (sterile or non-sterile) these cells would reach higher activation rates than WT neutrophils. Our results show that Tlr4, which is a validated target of miR-146a, is overexpressed in aged miR-146a-/- neutrophils in comparison with WT ones. One possible explanation for this finding may be that the deficiency of miR-146a favors a higher deregulation of the Tlr4/Nf-B axis in aged neutrophils than in non-aged ones. Thus, this hypothesis would also explain in part why circulating aged neutrophils are more prone to make NET, a previous indolent signal being necessary to prime these cells.49,50 Indeed, it has been shown that Tlr4-/- mice or germ-free mice have less aged neutrophils than WT littermates.30
In accord with our hypothesis, we observed higher ROS levels and oxygen consumption rate in neutrophils from miR146-/- mice than WT mice, which may further suggest a primed status in miR-146a-/- neutrophils. Importantly, the aging process is promoted in large part by ROS that produce cellular damage.51 ROS (mainly produced by NADPH oxidase) are also crucial to initiate the process of NET formation.52
There are a few limitations in this study. First, for the in vivo inflammation models, we did not use a complex marker such as a granulocytic enzyme bound to DNA to measure NET. NET were quantified using cfDNA, NE, and citH3 for the LPS model and tissue immunofluorescence in the atherosclerotic model. Thus, we cannot rule out that part of the cfDNA and NE measured in plasma was due to neutrophil secretion or to apoptotic processes. Another point is that our study was performed in full miR-146a-/- mice or BM transplanted mice. Given the interplay of other cells with neutrophils to promote NETosis, in particular platelets,9,10 it would be of interest in future studies to dissect the mechanisms implicating miR-146a in the formation of NET using conditional models of miR-146a deficiency.
In summary, our results demonstrate that miR-146a deficiency promotes an aged neutrophil phenotype priming these cells for activation. Upon both sterile and nonsterile stimuli, such sensitized neutrophils are more prone to form NET, leading to thrombosis when physiological control is overwhelmed (Figure 5). Indeed, we demonstrate this hypothesis in a human disease, sepsis, in which patients with low miR-146a levels due to rs2431697 have increased plasma NET markers and a higher risk of cardiovascular events. These data not only strengthen the proposed interconnection between inflammation and thrombosis but also open new perspectives for the development of therapeutic tools against NETosis in different thrombo-inflammatory contexts.
Supplementary Material
Acknowledgments
The authors thank Dr Juan José Cerón, Alberto Martínez, and Dr. Antonio Moscardó for technical assistance and Clive Drakeford and Javier Corral for reviewing the manuscript.
Funding Statement
Funding This work was supported by research grants from Instituto de Salud Carlos III (ISCIII), Fondo Europeo de Desarrollo Regional “Investing in your future” (PI17/00051 y PI17/01421) (PFIS18/0045 to AMdlR-G) (CD18/00044: to SA), and Fundación Séneca (19873/GERM/15). The CNIC is supported by the ISCIII, the Ministerio de Ciencia, Innovación y Universidades (MCIU), and the Fundación Pro CNIC, and is a Severo Ochoa Center of Excellence (SEV-2015-0505). ABA has a research fellowship from Sociedad Española de Trombosis y Hemostasia (SETH). The MCIU supported AdM. (predoctoral contract BES-2014-067791).
References
- 1.Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. 2016;127(18):2173-2181. [DOI] [PubMed] [Google Scholar]
- 2.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2017;18(2):134-147. [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532-1535. [DOI] [PubMed] [Google Scholar]
- 4.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34-45. [DOI] [PubMed] [Google Scholar]
- 6.Laridan E, Martinod K, De Meyer S. Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost. 2019;45(01):86-93. [DOI] [PubMed] [Google Scholar]
- 7.Kenny EF, Herzig A, Krüger R, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6:e24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23(3):279-287. [DOI] [PubMed] [Google Scholar]
- 9.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463-469. [DOI] [PubMed] [Google Scholar]
- 10.Maugeri N, Campana L, Gavina M, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074-2088. [DOI] [PubMed] [Google Scholar]
- 11.Desai J, Kumar S V, Mulay SR, et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol. 2016;46(1):223-229. [DOI] [PubMed] [Google Scholar]
- 12.Joshi MB, Lad A, Bharath Prasad AS, Balakrishnan A, Ramachandra L, Satyamoorthy K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013;587(14):2241-2246. [DOI] [PubMed] [Google Scholar]
- 13.Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190(3):1217-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinelli S, Urosevic M, Daryadel A, et al. Induction of genes mediating interferondependent extracellular trap formation during neutrophil differentiation. J Biol Chem. 2004;279(42):44123-44132. [DOI] [PubMed] [Google Scholar]
- 15.Ley K, Hoffman HM, Kubes P, et al. Neutrophils: new insights and open questions. Sci Immunol. 2018;3(30):eaat4579. [DOI] [PubMed] [Google Scholar]
- 16.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7):421-433. [DOI] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2): 215-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barwari T, Joshi A, Mayr M. MicroRNAs in cardiovascular disease. J Am Coll Cardiol. 2016;68(23):2577-2584. [DOI] [PubMed] [Google Scholar]
- 19.Essandoh K, Fan G-C. Role of extracellular and intracellular microRNAs in sepsis. Biochim Biophys Acta. 2014;1842(11):2155-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J-Q, Papp G, Szodoray P, Zeher M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev. 2016;15(12):1171-1180. [DOI] [PubMed] [Google Scholar]
- 21.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481-12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saba R, Sorensen DL, Booth SA. MicroRNA- 146a: a dominant, negative regulator of the innate immune response. Front Immunol. 2014;5:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boldin MP, Taganov KD, Rao DS, et al. miR- 146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208(6):1189-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roldan V, Arroyo AB, Salloum-Asfar S, et al. Prognostic role of MIR146A polymorphisms for cardiovascular events in atrial fibrillation. Thromb Haemost. 2014;112(4):781-788. [DOI] [PubMed] [Google Scholar]
- 25.Arroyo AB, de los Reyes-García AM, Rivera-Caravaca JM, et al. MiR-146a regulates neutrophil extracellular trap formation that predicts adverse cardiovascular events in patients with atrial fibrillation. Arterioscler Thromb Vasc Biol. 2018;38(4):892-902. [DOI] [PubMed] [Google Scholar]
- 26.del Monte A, Arroyo AB, Andrés-Manzano MJ, et al. miR-146a deficiency in hematopoietic cells is not involved in the development of atherosclerosis. PLoS One. 2018;13(6): e0198932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckley CD, Ross EA, McGettrick HM, et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol. 2006;79(2):303-311. [DOI] [PubMed] [Google Scholar]
- 28.Hu N, Westra J, Rutgers A, et al. Decreased CXCR1 and CXCR2 expression on neutrophils in anti-neutrophil cytoplasmic autoantibody-associated vasculitides potentially increases neutrophil adhesion and impairs migration. Arthritis Res Ther. 2011;13(6):R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu R, Bao C, Huang H, et al. Low expression of CXCR1/2 on neutrophils predicts poor survival in patients with hepatitis B virusrelated acute-on-chronic liver failure. Sci Rep. 2016;6:38714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang D, Chen G, Manwani D, et al. Neutrophil ageing is regulated by the microbiome. Nature. 2015;525(7570):528-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Cui Z, Liu L, et al. MiR-146a mimic attenuates murine allergic rhinitis by downregulating TLR4/TRAF6/NF-κB pathway. Immunotherapy. 2019;11(13):1095-1105. [DOI] [PubMed] [Google Scholar]
- 32.Stavrou EX, Fang C, Bane KL, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. 2018;128(3):944-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashiba M, Huq A, Tomino A, et al. Neutrophil extracellular traps in patients with sepsis. J Surg Res. 2015;194(1):248-254. [DOI] [PubMed] [Google Scholar]
- 34.Hirose T, Hamaguchi S, Matsumoto N, et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS One. 2014;9(11):e111755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonaventura A, Montecucco F, Dallegri F, et al. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc Res. 2019;115(8):1266-1285. [DOI] [PubMed] [Google Scholar]
- 36.Jiménez-Alcázar M, Kim N, Fuchs T. Circulating extracellular DNA: cause or consequence of thrombosis? Semin Thromb Hemost. 2017;43(06):553-561. [DOI] [PubMed] [Google Scholar]
- 37.Khan MA, Palaniyar N. Transcriptional firing helps to drive NETosis. Sci Rep. 2017;7:41749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franck G, Mawson TL, Folco EJ, et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury. Circ Res. 2018;123(1):33-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Massberg S, Grahl L, von Bruehl M-L, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16(8):887-896. [DOI] [PubMed] [Google Scholar]
- 40.Knight JS, Luo W, O’Dell AA, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. 2014;114(6): 947-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liew PX, Kubes P. The neutrophil’s role during health and disease. Physiol Rev. 2019;99(2):1223-1248. [DOI] [PubMed] [Google Scholar]
- 42.Ebrahimi F, Giaglis S, Hahn S, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur Respir J. 2018;51(4):1701389. [DOI] [PubMed] [Google Scholar]
- 43.Xie J, Zhang L, Fan X, Dong X, Zhang Z, Fan W. MicroRNA-146a improves sepsisinduced cardiomyopathy by regulating the TLR-4/NF-κB signaling pathway. Exp Ther Med. 2019;18(1):779-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao M, Wang X, Zhang X, et al. Attenuation of cardiac dysfunction in polymicrobial sepsis by microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J Immunol. 2015;195(2):672-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakada T, Takahashi W, Nakada E, Shimada T, Russell JA, Walley KR. Genetic polymorphisms in sepsis and cardiovascular disease. Chest. 2019;155(6):1260-1271. [DOI] [PubMed] [Google Scholar]
- 46.Montecucco F, Liberale L, Bonaventura A, Vecchiè A, Dallegri F, Carbone F. The role of inflammation in cardiovascular outcome. Curr Atheroscler Rep. 2017;19(3):11. [DOI] [PubMed] [Google Scholar]
- 47.Ayoub KF, Pothineni NVK, Rutland J, Ding Z, Mehta JL. Immunity, inflammation, and oxidative stress in heart failure: emerging molecular targets. Cardiovasc Drugs Ther. 2017;31(5-6):593-608. [DOI] [PubMed] [Google Scholar]
- 48.Uhl B, Vadlau Y, Zuchtriegel G, et al. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood. 2016;128(19):2327-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. 2018;9:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ortmann W, Kolaczkowska E. Age is the work of art? Impact of neutrophil and organism age on neutrophil extracellular trap formation. Cell Tissue Res. 2018;371(3):473-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, cell senescence, and novel molecular mechanisms in aging and agerelated diseases. Oxid Med Cell Longev. 2016; 2016:3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2): 231-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.