

Abstract
Amajor challenge in the development of a gene therapy for hemophilia A is the selection of cell type- or tissue-specific promoters to ensure factor VIII (FVIII) expression without eliciting an immune response. As liver sinusoidal endothelial cells are the major FVIII source, understanding the transcriptional F8 regulation in these cells would help to optimize the minimal F8 promoter (pF8) to efficiently drive FVIII expression. In silico analyses predicted several binding sites (BS) for the E26 transformation-specific (Ets) transcription factors Ets-1 and Ets-2 in the pF8. Reporter assays demonstrated a significant up-regulation of pF8 activity by Ets-1 or Ets- 1/Est-2 combination, while Ets-2 alone was ineffective. Moreover, Ets-1/Ets- 2-DNA binding domain mutants (DBD) abolished promoter activation only when the Ets-1 DBD was removed, suggesting that pF8 up-regulation may occur through Ets-1/Ets-2 interaction with Ets-1 bound to DNA. pF8 carrying Ets-BS deletions unveiled two Ets-BS essential for pF8 activity and response to Ets overexpression. Lentivirus-mediated delivery of green fluorescent protein (GFP) or FVIII cassettes driven by the shortened promoters, led to GFP expression mainly in endothelial cells in the liver and to longterm FVIII activity without inhibitor formation in HA mice. These data strongly support the potential application of these promoters in hemophilia A gene therapy.
Introduction
Hemophilia A (HA) is a recessive X-linked bleeding disorder caused by a number of F8 gene mutations which associate with deficiencies of the coagulation factor VIII (FVIII).1,2 As HA is a monogenic disease with a lifelong elevated bleeding risk with no conclusive therapeutic options, it is an ideal candidate for gene therapy.3 In the last years many forward steps in the development of new therapeutic strategies have been made, thus rapidly changing the therapeutic landscape of HA. The introduction of the extended half-life FVIII concentrates, of the bi-specific monoclonal antibody Emicizumab and the start of gene therapy clinical trials have remarkably contributed to the improvement of patient care.4
The development of effective gene therapeutic approaches for HA, has seen little progression when compared to hemophilia B5. This is due to the complexity of FVIII synthesis, the size of the FVIII protein (2,351 amino acids) and its high immunogenic properties, with the development of neutralizing antibodies in 25% of patients when FVIII is exogenously administered.6,7 As it stands, the goal is to provide a single dose therapy that is effective and offers a lasting cure with sustained FVIII activity to HA patients.
When considering gene therapy approaches aimed at restoring and sustaining FVIII activity, the liver is considered the primary target organ, as it is the principal site of FVIII synthesis and possesses the necessary tolerogenic properties.8 The identity of liver cells capable of synthesizing and releasing FVIII has generated an extensive debate over the years.9 This has significantly influenced the understanding of the regulatory elements involved in promoting the preferential expression of FVIII in liver cells. The F8 promoter (pF8), reported to be a 1.2 kb region upstream of the F8 translation start site, was first described in 1984.10 With hepatocytes originally considered the major source of FVIII,11,12 the first in vitro studies aimed at elucidating the transcription factors (TF) responsible for pF8 activation, were performed using hepatocyte- derived cell lines. In hepatocytes, Figueiredo and McGlynn described the region from -279 and -64 to be responsible for maximal promoter activity.13,14 They identified and confirmed the binding of several hepatic TF, such as CCAAT/enhancer-binding proteins (C/EBPα and C/EBPb), and hepatocyte nuclear factor 1 (HNF-1) and 4 (HNF-4). Other TF binding sites (TFBS) on pF8 were also identified in this study, however, to date their involvement has never been thoroughly investigated.
While heavily debated, it has recently been demonstrated that liver FVIII production predominantly occurs in the liver sinusoidal endothelial cells (LSEC),15–18 which represent a principal but not exclusive source.19–22 In fact, detection of FVIII mRNA in many tissues, suggests that a highly complex and likely tissue-specific transcriptional regulation exists. Recently, our group described pF8’s ability to direct a specific and long-term FVIII expression in LSEC after a lentiviral vector (LV)-delivery in HA mice.23 Importantly, this targeted restoration of FVIII did not trigger an immune response, one of the major obstacles for the successful treatment of HA patients. In the present study, we used data from an in silico analysis of the pF8 region,23 to extrapolate and assess the role of the most represented endothelial-specific TFBS on F8 transcriptional regulation. Understanding the stimuli and the TF required for maximal promoter activity in endothelial cells (EC), offers an inportant forward step in the development of gene therapeutic approaches for HA. To date, several clinical trials using the adeno-associated viral vectors (AAV) to delivery FVIII in HA patients have started. Despite the promising results, some concerns have been raised, like the vector dose, the variability of FVIII activity among the different subjects and the decline of FVIII expression overtime. 24 Our optimization of the minimal pF8 size opens up the possibility to explore new perspectives in the field the HA gene therapy by introducing for the first time a pF8 suitable for vectors with a limited expression cassette, like the AAV which, to date, are the only ones successfully used in clinical trials for HA.
Methods
Animal studies
Experiments, described in the Online Supplementary Appendix, were performed according to an approved protocol by the Animal Care and Use Committee of the University of Eastern Piedmont and the Italian Health Ministry, Italy (Project n. DB064.5, date of approval n°492/2016-PR 17/05/2016).
Identification of putative endothelial transcription factors binding sites on the F8 promoter
In silico prediction of TFBS distribution on pF8 was retrieved from a previous analysis performed by Merlin et al.23 using PROMO 3.0.25 In order to identify potential endothelial TFBS, two parameters were considered: the number of consensus sites identified on pF8 using a stringent dissimilarity rate (<3) and the expression and functional role of TF in EC.
Generation of the constructs
The full-length human pF8 (1,175 basepairs [bp]) was excised from a plasmid already available in our laboratory and cloned with XhoI-HindIII into a promoterless pNL1.1 vector at the 5’ of NanoLuc® luciferase reporter gene. Serial deletions of pF8 were generated via polymerase chain reaction using diverse primer sets carrying restriction sites, while E26 transformation-specific (Ets)-core sequence (GGAA) deletions were performed using site-directed mutagenesis, according to the manufacturer’s instructions (Stratagene, San Diego, CA, USA).
Human Ets-1 cDNA was obtained from Origene (#RC227466) while Ets-2 cDNA expressing vector (CMV-Ets-2 in pCDNA 3.1) was available in our laboratory. Ets-1 and Ets-2 DNA binding domains were removed by mutagenesis according to the protocol described by Follo et al.26 All mutagenesis primers are listed in the Online Supplementary Tables S2 to S4.
The single guide RNA (sgRNAF8.1, sgRNAF8.2 and the control sgRNAF7.5) were designed using ZiFit web tool, as described by Pignani et al.27, scanning for the S. Pyogenes PAM sequence (NGG) both in the sense and antisense strands. dCas9- VPR was a gift from Dr George Church (Addgene plasmid # 63798).
Transient transfection and luciferase assays
For the luciferase reporter assay, ECV-304 and HEK293T cells were seeded in a 24-well plate at a density of 5x104 cells/well 24 hours (h) prior to transfection. Luciferase pF8 reporter plasmids along with Ets-1 and/or Ets-2 expressing constructs, or dCas9- VPR and gRNA, were transfected (1 mg) into cells using the Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific), according to the manufacturer’s instructions. Firefly luciferase vector pGL4.54 (TK-Firefly; #E5061, Promega) was used as an internal control. In order to maintain the amount of total DNA, the pUC19 vector was used to transfect cells. After 24 h, the cells were lysed, and luciferase activity assayed performed using the Dual-Luciferase Reporter Assay System (Promega). Both firefly and NanoLuc signals were measured at 562 nm using a Victor X microplate reader (PerkinElmer). Results are expressed as mean ± standard deviation (SD) of the fold change, calculated as the average of the ratio of stimulated to non-stimulated promoter.
Statistical analysis
All data were continuous and expressed as an average ± SD. Parametric analysis was used because the groups were balanced with the same number of observations. One-way analysis of variance (ANOVA) was performed to compare changes in promoter activity among promoter variants and to separately evaluate the difference in Ets-response of each promoter tested. Two-way ANOVA was carried out to compare changing in FVIII activity over time and among the mice groups. P-values less than 0.05 were considered statistically significant for the overall test while Bonferroni’s adjustment was used for multiple comparisons. The statistical analyses were performed with GraphPad Prism 5.0 (GraphPad Software).
Results
In silico prediction of endothelial transcription factors involved in F8 promotor regulation
By taking advantage of the in silico analysis on pF8 previously performed in our laboratory,23 we mapped all the TFBS on pF8 recognized by TF expressed by endothelial cells (Figure 1A). In total, we identified 16 TFBS which interestingly all belonged to the Ets family. Further, each was distributed in the first -600 bp of pF8 (Figure 1A). In order to improve the accuracy of the analysis, we reduced the dissimilarity rate parameter from 5 to 3, opting to assess the role of these TF in pF8 regulation (Figure 1A, black asterisk). Using this approach, we identified seven endothelial BS, four recognized by Ets-1, two by Ets-2 and one by Elk-1, with two of the Ets-1 sites overlapping those of Ets-2 and Elk-1 (Figure 1B).
Both Ets-1 and Ets-2 are well-known TF, being downstream effectors of the RAS/ERK signaling pathways and regulating several genes involved in endothelial functions including angiogenesis.28–31 Based on their redundant contribution in crucial endothelial cell processes,28 we focused our studies on the potential co-operative role of Ets-1 and Ets-2 in regulating FVIII expression.
Figure 1.

F8 promoter showing several putative E26 transformation-specific (Ets) binding motifs for Ets-1 and Ets-2 transcription factors. (A) Table showing the endothelial binding sites (BS) on F8 promoter (pF8) identified by in silico analysis. Black asterisks indicate BS with a dissimilarity score <3 selected to evaluate their role in promoter regulation. (B) Schematic representation of the distribution of the selected Ets-BS on the proximal pF8 (-1,175 basepairs). (C) Reverse transcriptase polymerase chain reaction (RT-PCR) analysis of factor VIII (FVIII), Ets-1 and Ets-2 expression in ECV-304 and HEK293T cell lines. Blood outgrowth endothelial cells (BOEC), were used as positive control. Base pairs on the left indicate the expected PCR products size. (D) Western immunoblot analysis on ECV-304 and HEK293T whole cell lysates using the anti-Ets-1 and anti-Ets-2 antibodies. Black arrows indicate the major Ets-1 isoforms p51 (51 kDa) and p42 (42 kDa). Black lines indicated the expected weight of protein detected (for antibodies and reagents see the Online Supplentary Appendix).
Ets-1 and Ets-2 in vitro co-operation in F8 promotor regulation
In order to explore the role of Ets-1 and Ets-2 in pF8 regulation, the -1,175 bp region of pF8 was cloned into the Nanoluc vector, and pF8 activity assessed in the presence of Ets-1, Ets-2 or in combination. Two human cell lines were transfected: ECV-304 and HEK293T. The ECV-304, previously considered as spontaneously transformed human umbilical endothelial cells,32 were selected as a model expressing FVIII, Ets-1 and Ets-2 at the mRNA and protein level, while HEK293T as cells expressing low levels of Ets-1 and Ets-2 (Figure 1C and D). The luciferase assays showed different but consistent results between the two cell types tested. A 3- and 5-fold upregulation was observed in ECV-304 upon co-transfection of pF8 with Ets-1 and Ets-1/Ets-2, respectively (Figure 2A). No significant effect was observed with Ets-2 alone. This data highlights that Ets-1 appears to play a major role on pF8 transactivation. In HEK293T cells where Ets-1 is expressed at negligible levels, we observed a 7-fold up-regulation of pF8 only in response to Ets-1 overexpression, with no further increase observed in presence of Ets-2 (Figure 2B). These results reinforce the central role of Ets-1 in regulating pF8 and highlight the presence of cell-specific regulatory mechanisms.
In order to elucidate whether the pF8 up-regulation observed in ECV-304 in response to Ets-1 and Ets-2 is mediated by a direct DNA binding of Ets, we generated Ets mutants carrying an in-frame deletion of their DNA binding domain (DBD) (Figure 2C). We evaluated their cooperation with the wild-type (WT) counterparts in the regulation of pF8 activity. Ets-1 without DBD failed to upregulate the pF8, while Ets-1 co-expressed with the Ets2-DBD mutant, preserved the 5-fold increase in pF8 activity, as observed with the WT proteins (Figure 2D). These results suggest that in the regulation of pF8, Ets-1 may directly bind the DNA, while Ets-2 modulates pF8 through its interaction with Ets-1.33
Figure 2.

In vitro Ets-1 and Ets-2 co-operation in F8 promotor transactivation. (A and B) Histograms report the fold increase of luciferase activity after transfection of F8 promotor (pF8) alone (set as 1) or in combination with Ets-1, Ets-2 or both in (A) ECV-304 or (B) HEK293T cell lines. (C) Schematic representation of Ets-1 and Ets-2 protein structure, with highlighted amino acid position of the DNA binding domain (DBD). For both proteins, TAD indicates the transactivation domain while ID indicates the inhibitory domain. (D) Histograms report the fold increase of luciferase activity after transfection of pF8-1175 alone (set as 1) or with the combination of Ets-DBD with the non-mutated counterparts. Results are expressed as mean ± standard deviation from three independent experiments performed in triplicate. *P<0.05.
Identification of key Ets-responsive elements in the F8 proximal promoter region
In order to decipher the importance of each Ets BS in pF8 activity, we generated several pF8 mutants and tested their impact. We adopted two different strategies. The first aimed at eliminating progressively the Ets BS by performing deletions of pF8 (Figure 3A and B). Using this approach in ECV-304 cells, we observed that the basal activity of the full-length promoter (pF8-1,175) was maintained in pF8-600 and pF8-446, where one Ets-2 BS was eliminated. Further shortening of the promoter (pF8-342) caused a minimal reduction of its activity with respect to pF8-1175, while a significant reduction (<20-40% pF8-1,175) was observed with the pF8-246 (Figure 3C). Analysis of the shortened pF8 promoters in response to Ets overexpression, confirmed that pF8-600 and pF8-446 maintain the same response as pF8-1175, demonstrating a 3- and 5-fold upregulation with Ets-1 and Ets-1/Ets-2 combined (Figure 3D). Despite a clear reduction in its promoter activity, pF8-342 interestingly maintained a ~3- to 5- fold activation with Ets-1 or Ets-1/Ets-2 combined, indicating that a specific response is maintained despite the elimination of one Ets-1 and one Ets-2 BS. When the pF8 was shortened further to 246 bp, a dramatic reduction in promoter activity was observed and was sustained despite Ets overexpression (Figure 3D). This was also validated in HEK293T cells (Online Supplementary Figure S1A and B). Overall, these results suggest that pF8 regulation may depend on the number of available Ets BS.
Figure 3.

Crucial E26 transformation-specific-responsive elements are localized in the first 342 bases of F8 promoter region. (A) Table summarizing the Ets-1 and Ets-2 binding sites distributed across the F8 promotor (pF8) and scheme of mutagenesis strategies used to investigate the role of each E26 transformation-specific binding site (Ets-BS). Ets-BS destroyed by mutagenesis are highlighted in black and are indicated with E followed by a number. Only Ets-BS with a dissimilarity score <3 were disrupted by mutagenesis. (B) Schematic representation of mutagenesis strategies used to perform single or multiple deletions of Ets-BS on pF8. Colored boxes highlight the Ets-BS which underwent to a single deletion. Colored scissors indicate the points on pF8 where it was cut to generate the shortened forms with multiple ETS-BS deletions. (C and D) Histograms representing fold change in luciferase activity of shortened promoters at (C) basal level or after overexpression of (D) Ets-1, Ets-2 or both. (E and F) Histograms showing changing in luciferase activity of Ets-BS single deleted promoters at (E) basal level or after overexpression of (F) Ets-1, Ets-2 or both. All experiments were performed in ECV-304 cells. Results are expressed as mean ± standard deviation from three independent experiments performed in triplicate. *P<0.05.
The second mutagenesis approach focused on the importance of each Ets site identified using a dissimilarity score <3. This approach selectively deleted the GGAA core from E1, E2, E3, E4, E5 Ets-1 and Ets-2 BSs (Figure 3B). As the Ets BSs are in the first 600 bp of pF8, we used pF8-600 to normalize the results as it preserves all the identified endothelial TFBS. Using this approach, we identified two important BS: -189 to -198 (E1) and -217 to -223 (E2). The disruption of E1 drastically reduced pF8 basal activity by approximately 60%, while the others failed to alter its activation (Figure 3E; Online Supplementary Figures S1C and S2A). Of note, the disruption of E1 site even preserving a response to Ets-1 overexpression abrogated the co-operative up-regulation mediated by Ets-1/2, thus agreeing with the elimination of the only Ets-2 site overlapping that of Ets-1. On the other hand, disruption of the E2 site decreased exclusively the pF8 up-regulation in response to Ets overexpression (Figure 3F; Online Supplementary Figures S1D and S2B). Overall, the E1 and E2 sites appear to be central to pF8 regulation with E2 maintaining the basal pF8 activity in the absence of an Etsinduced upregulation.
Enhancement of F8 promoter activity by delivery of CRISPR/VPR activation system to E2 and E3 E26 transformation-specific binding sites
CRISPR activation system (CRISPRa) is an emerging tool that exploits deactivated Cas9, single guide RNA (sgRNA) and transcription activators for gene activation.34 We have recently described the potential of the CRISPR activation system in transactivating and upregulating pF8 activity by using two specific sgRNA targeting the first 300 bp of the promoter region.27 Based on our in silico sanalysis, these sgRNA guides, F8.1 and F8.2, mediate the recruitment of deactivated Cas9 fused to a tripartite transcriptional activator (VPR)34 to the two promoter regions encompassing E3 (-313 to -319) and E2 (-219 to -223) Ets- 1 BS, respectively (Figure 4A). In order to further demonstrate the central role of E2 in the modulation of pF8 activity, we transfected both ECV-304 and HEK293T cells with CRISPRa system using sgRNA F8.1 and F8.2 as reference guides covering the essential E2 and the E3 Ets-1 BS. As a negative control guide we performed the same experiment using a sgRNA targeting coagulation Factor VII promoter (pF7) (Online Supplementary Figure S3). By applying this approach to pF8-1175, pF8-600 and pF8-446, we confirmed the ability of CRISPRa to upregulate the promoter activity. Importantly, the efficiency of the system was dependent on the cell type, the sgRNA used and the promoter length. Specifically, in ECV-304, where Ets-1 is expressed, we observed a 3- to 6-fold increase in activity of each pF8 using the sgRNA F8.2, while no significant changes were observed with the sgRNA F8.1 (Figure 4B). In HEK293T cells, however, where Ets-1 is insignificant, we observed an upregulation in pF8 activity, ranging from a 20- to 40-fold, likely explained by the lack of competition for the BS involved (Figure 4C). Taken together, our results, highlight a central role of the sgRNA F8.2 in pF8 transactivation confirming the powerful role of the E2 site in promoter regulation.
Figure 4.

Delivery of CRISPRa to E2 and E3 E26 transformation-specific binding sites induces a significant increase in F8 promoter activity. (A) Schematic representation of the two single guide RNA (sgRNA) guides used to drive the recruitment of the CRISPR activation system (CRISPRa) system to pF8. (B and C) Graphs showing the transactivation effects of the sgRNA F8.1 and F8.2 on pF8-1175, pF8-600 and pF8-342 in (B) ECV-304 and (C) HEK293T cells. Results are expressed as mean ± standard deviation from two independent experiments performed in triplicate. *P<0.05
Figure 5.

F8 shortened promoters drive green fluorescent protein expression in hepatic endothelial cells. Immunofluorescence analyses of C57BL/6 livers 2 weeks after lentiviral vector (LV) LV.pF8-600.GFP, LV.pF8-446.GFP or LV.pF8.342.GFP delivery, compared to the full length LV.pF8-1175.GFP. (A) Representative pictures of liver sections stained for the endothelial marker (Lyve-1, in red) and the green fluorescent protein (GFP) using 400x magnification. (B) Liver of LV-injected mice decorated with the macrophage marker F4/80 (red) in combination with GFP (green). Scale bars, 50 mM. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). n=3-5 (mice); sections=3-6 per mouse. (for antibodies specifications refers to the Online Supplementary Appendix).
In vivo maintenance of high endothelial specificity by shortened promoters
In order to assess the activity and specificity of the newly identified shortened pF8 sequences in vivo, we generated LV expressing GFP under the control of pF8-600, pF8-446 or pF8-342, and we administrated 5x108 TU to C57BL/6 mice. pF8-1175 was used as a control. Two weeks after injection, hepatic GFP expression was detected primarily in endothelial Lyve-1+ cells in all mice (Figure 5A), with rare F4/80+ macrophages GFP positive (GFP+) seen with the pF8-446 promoter (Figure 5B). Co-staining with Lyve-1 was still visible 1 month post-injection, with a few F4/80+GFP+ cells appearing in the pF8-1175 and pF8-600 injected mice (Online Supplementary Figure S4A and B).
Figure 6.
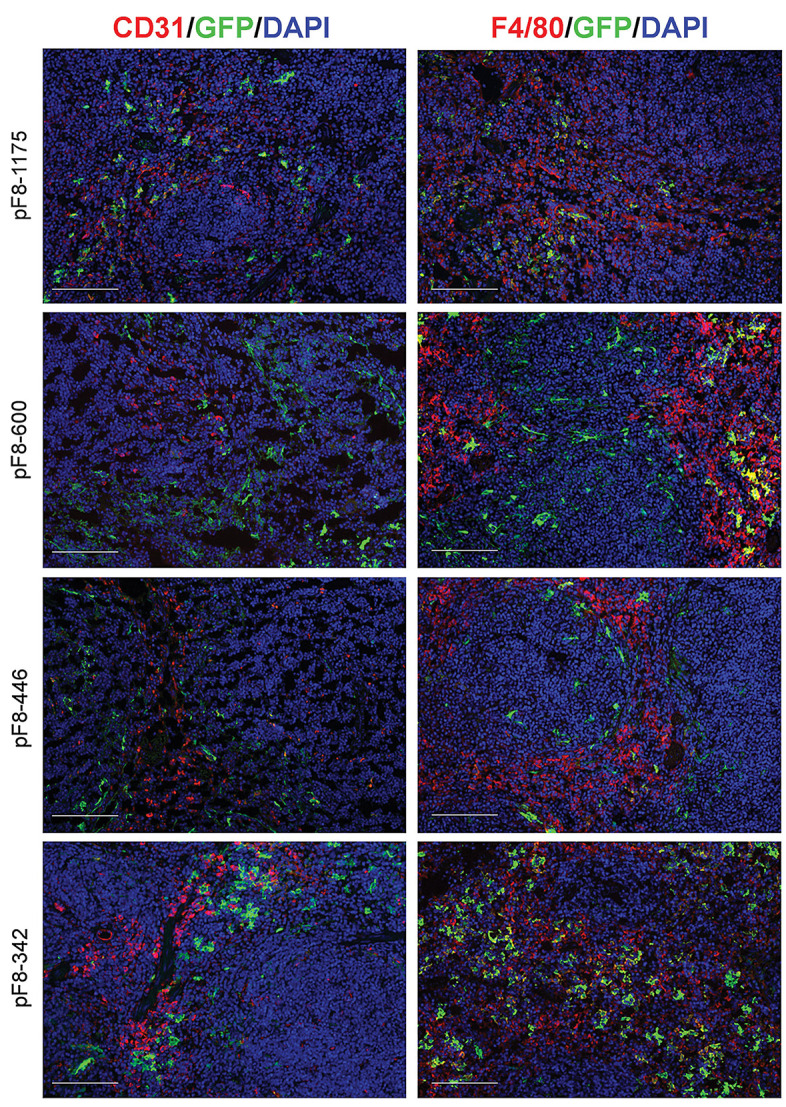
Green fluorescent protein expression in spleens under the control of pF8 shortened promoter sequences. Representative immunofluorescence images from mice spleens, 2 weeks following the delivery of lentiviral vector (LV) carrying different promoters. In red the endothelial marker CD31 (left side) or the macrophage marker F4/80 (right side) with green fluorescent protein (GFP) (green). Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bars, 100 mM. n=3-5 (mice); sections=5-7 per mouse.
As previously observed for pF8-117523 in the spleen at 2 weeks, all pF8 short forms resulted in very low co-staining with the putative endothelial marker CD31 (Figure 6, left panel), while pF8-1,175, pF8-600 and pF8-446 showed a partial GFP expression in F4/80+ cells (Figure 6, right panel). Interestingly, in these mice several GFP+ cells were evident around the germinal centers (GC), resembling the EC lining in the marginal sinus of the spleen GC.35 Unlike other promoters, the spleens of mice receiving the LV.pF8-342, showed a higher percentage of F4/80+/GFP+ cells (Figure 6, right panel) 2 weeks after the LV injection. The same GFP distribution pattern, but with a reduced expression, was confirmed in all treated mice at 1 and 2 months post LVdelivery. In particular, the LV.pF8-342.GFP-injected mice showed the most relevant reduction of GFP expression in F4/80+ cells over time (Online Supplementary Figures S5 and S6). Taken together, these results demonstrate that a significant reduction in pF8 size does not compromise its activity or specificity, suggesting that pF8-342 is enough for maintaining an endothelial-specific expression.
In vivo long-term recovery of factor VIII activity in a hemophilia A mouse model using the shortened F8 promotor
One of the major obstacles in HA gene therapy is represented by the large size of the FVIII expression cassette. As such, the choice of a suitable promoter to drive its expression is fundamental for success. In order to explore the ability of our new shortened promoters in rescuing FVIII expression in vivo, we generated LV carrying FVIII under their control and injected 1x109 TU in B6/129 HA mice (n=4-6). pF8-1175 was used as a control. The FVIII activity promoted by pF8-600, pF8-446 resulted in a long-term (28 weeks) therapeutic correction (~10%), with no differences seen among them, and with respect to pF8-1175 (Figure 7A). Unexpectedly, despite the reduced response to Ets showed by luciferase assays, also the pF8-342 resulted in long-term therapeutic correction comparable to the other promoters tested. Importantly, independent of the size of the promoter used in the transfer construct, no anti-FVIII antibodies were detected in all treated mice, indicating the maintenance of specificity and, then, of an immunological profile similar to those observed for pF8-117523 (Figure 7B). High levels of anti-FVIII antibodies were instead detected in HA mice receiving FVIII under the control of the ubiquitous 3-phosphoglycerate kinase promoter, reinforcing the importance of a targeted expression of FVIII to avoid a specific immune response.
Figure 7.

In vivo long-term correction of factor VIII activity with no immune response by F8 shortened promoters. (A) Graphic representation of factor VIII (FVIII) activity of B6/129 HA mice injected with lentiviral vecor (LV) LV.pF8-600.FVIII (grey square, n=6), LV.pF8-446.FVIII (light blue rhombus, n=4) or LV.pF8-342.FVIII (green triangle, n=4). pF8-1175.FVIII (black circle, n=4) were used as a control, while untreated hemophilia A (HA) mice were used as the negative control (violet X, n=3). No statistical differences in FVIII activity levels were observed among the four LV-injected experimental groups. (B) Enzyme-linked immunosorbent assay showing the absence of anti-FVIII antibodies in plasma of HA mice after LV delivery. Anti-FVIII positive HA mice injected with LV.PGK-FVIII served as positive control, while untreated HA mice were used as the negative control. Plasma dilution 1:2,000. Color bars were maintained according to the color scale used for FVIII activity graph.
Discussion
One of the major challenges in the design of gene transfer vectors over the years, has involved the selection of cell type- or tissue-specific promoters suitable for restoring transgene expression in a defined disease setting. In gene therapy, transcriptional targeting represents a way to restrict mRNA and protein expression to a specific cell type, even if transduction of viral vectors is likely to occur in multiple cell types.36 In the context of F8 gene transfer, several attempts have been made to achieve a selective and stable gene expression in various cell types. This has been challenging due to the complexity of FVIII synthesis, its size and immunogenic behavior, therefore highlighting the need for novel approaches. Using previous in silico analyses performed on the native pF8,23 we investigated in the present study the profile of TF potentially involved in promoting the expression of FVIII in EC. We identified and demonstrated in vitro that the Ets family of TF plays a fundamental role in the modulation of pF8 activity, offering regulatory elements that can be explored to promote FVIII expression in a targeted manner. Further, we defined a minimal pF8 required to efficiently drive FVIII expression in vivo, potentially overcoming a number of the current obstacles associated with F8 gene delivery.37
With LSEC now accepted as the main FVIII producing cells,15–18 understanding the key EC FVIII-specific regulatory elements is essential in promoting FVIII expression. The Ets-family of TF met our restricted criteria, with Ets-1 and -2 having the greatest number of BS across the pF8. More importantly, they are known to have essential roles in embryonic endothelial cell survival and in adult angiogenesis. 28 In adults the expression of Ets-1 is maintained at very low levels in the resting endothelium and is induced after specific stimuli. In particular, Ets-1 is reported to be up-regulated in response to pro-angiogenic and proinflammatory stimuli such as TNF-α, hepatocyte growth factor and platelet-derived growth factor.38 In addition, hypoxia has been demonstrated to regulate Ets-1 expression through the hypoxia inducible factor-1.29 Further, LSEC are known to be physiologically exposed to decreasing oxygen pressure along the liver lobule.39
In our case, by overexpressing Ets-1 and Ets-2, both independently and collectively, we clearly demonstrate the involvement of Ets-1 in the regulation of pF8 activity. Interestingly, the transactivation efficiency of Ets-1 appeared to be potentiated by the presence of Ets-2, likely via protein-protein interactions. Co-operation between Ets-1 and -2 has been shown to be involved in the reactivation of the TERT gene in cancer.40 In this context, the cooperative interaction of Ets-1 and Ets-2 promotes the recruitment of p52 to a mutant form of the TERT promoter enhancing TERT expression when the non-canonical NF-κB signaling is activated. In our case, we observed this phenomenon in ECV-304 but not in HEK293T cells, suggesting the existence of cell-specific regulation with respect to F8 expression. Both Ets-1 and Ets-2 are tightly regulated transcription factors,41 and it might be that in specific cells, like HEK293T, the forced expression of one is able to bypass the establishment of a co-operative interaction. A similar regulation is not surprising for F8 due to the necessity of a tight control, since it has been demonstrated that high expression of FVIII can cause cellular stress and increased immunogenicity.42
In order to clarify the relevance of the Ets-BS for the regulation of pF8, we opted to generate several reduced sized pF8 progressively removing single or multiple Ets sites. This approach demonstrated that the -342 bp region, in which three Ets-1 and one Ets-2 sites were maintained, represented a minimal functional promoter in vitro. Further reduction of the pF8 to 246 bp, which excluded an additional Ets-BS, caused a strong suppression of basal pF8 activity and preserved a minimal response to Ets-1 and Ets-2 overexpression. Of note, we identified that the -223 to -217 Ets-1 (E2) site is the most relevant regulatory element needed to achieve the maximal up-regulation of pF8 activity. In recent years, several mutations targeting the nucleotide positions ranging from -218 to -221, encompassing the E2 site, have been described to be responsible for different degrees of HA.43 It has been clearly shown that these nucleotides are part of a highly conserved region where any change compromises the functionality of the pF8. Our in vitro studies highlight the importance of the -218 to -221 sequence, identifying it as the most essential Ets-1 responsive element on pF8. The use of a CRISPRa system to transactivate pF8, reinforces this finding, highlighting the E2 site as being essential for pF8 regulation. While only a modest up-regulation of pF8 was detectable in ECV-304, it is likely that the physiological expression of Ets-1 hampers the efficient binding of the sgRNA to the E2 site. The use of CRISPRa also supports the concept of the binding of Ets to the E2 site.
The in vivo delivery of LV expressing the GFP or FVIII transgene under the control of each described shortened promoter, reinforces the results obtained in vitro. All LVtreated mice displayed GFP expression in liver Lyve-1+ cells, most likely LSEC,44 resembling the pattern detectable using pF8-1175. Nevertheless, a few F4/80+ macrophages expressed GFP, especially with respects to the two longer pF8. This is not surprising because we previously described that the complete absence of macrophage targeting using an endothelial promoter, was only obtained by inserting a multiple miRNA target (mirT) sequence recognized by the hematopoietic-specific miRNA 142-3p (mirT-142-3p).45
F8 gene transfer in HA mice confirmed the power of each tested promoter to drive long-term and stable FVIII expression without the appearance of inhibitors. Importantly, therapeutic levels of FVIII activity were detectable in all treated mice at similar levels to those observed in mice delivered with the control pF8-1,175 promoter. Such results are encouraging with respect to the current obstacles associated with the achievement of an efficient F8 gene delivery.
There are some limitations in our study. While Ets TF seem to be involved in pF8 regulation, the direct binding of Ets-1 and Ets-2 to pF8, and their specific role in LSEC remains to be confirmed. Further, the difficulty to isolate and maintain primary LSEC for extended periods in culture, 46 limits the use of luciferase assays.
Taken together, our in vitro and in vivo results have identified the region from 0 to -342 as a minimal pF8 which preserves its activity and Ets-response and is comparable to the full-length pF8. Evidence for a role of Ets-1 and the Ets-2 in pF8 regulation offers a new insight into the molecular mechanisms promoting FVIII expression in specific cell types. To date, F8 has not been reported to be amongst the large number of genes shown to be direct targets of Ets-1 and Ets-2 regulation, such as KDR, FLT1, ANGPT2, TEK, VWF47 and CDH5.48 Interestingly, all these genes are normally expressed at different stages in EC, suggesting a common regulation with FVIII.
Establishing the minimal promoter sequence required for the maintenance of a regulated FVIII expression restricted to LSEC, offers new perspectives for developing novel approaches to cure HA. For example, it provides the possibility of inserting a pF8 into an AAV-FVIII construct. Currently, these classes of vectors are the safest and most extensively used for HA gene therapy,49 however, they are limited by the size of the expression cassette, making it impossible to introduce both F8 and its full-length promoter. The efficient pF8 size reduction (~70%) described herein, opens up the possibility of engineering shortened chimeric pF8 by including sequences enriched in TFBS (like Ets-1) which can enhance the pF8 activity, as has been described for the transthyretin promoter in hepatocytes. 50
Supplementary Material
Aknowledgments
The authors thank Dr. Chiara Borsotti for help in revising the manuscript. Dr. Diego Zanolini for the contribution in planning some initial cloning experiments. Dr. Diego Cotella for providing the promoterless pNL1.1 plasmid and suggestions for luciferase experiments setting. Dr. Daniela Capello and Prof. Fabrizio Faggiano for contribution in luciferase assays data analysis. Dr. Silvia Buzzi for performing mice maintenance and coagulation assays.
Funding Statement
Funding This work was supported partly by European Research Council (ERC) #261178 and Horizon 2020 (HemAcure project #66742) to AF.
References
- 1.Hoyer LW. Hemophilia A. N Engl J Med. 1994;330(1):38-47. [DOI] [PubMed] [Google Scholar]
- 2.Bolton-Maggs PHB, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801-1809. [DOI] [PubMed] [Google Scholar]
- 3.Mannucci PM, Tuddenham EG. The hemophilias-- from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773-1779. [DOI] [PubMed] [Google Scholar]
- 4.Weyand AC, Pipe SW. New therapies for hemophilia. Blood. 2019;133(5):389-398. [DOI] [PubMed] [Google Scholar]
- 5.Pipe SW. Gene therapy for hemophilia. Pediatr Blood Cancer. 2018;65(2). [DOI] [PubMed] [Google Scholar]
- 6.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A - safety, efficacy, and development of inhibitors. N Engl J Med. 2002;328(7):453-459. [DOI] [PubMed] [Google Scholar]
- 7.Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983-3996. [PubMed] [Google Scholar]
- 8.Aravalli RN, Belcher JD, Steer CJ. Liver-targeted gene therapy: approaches and challenges. Liver Transplant. 2015;2(6):718-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arruda VR. The search for the origin of factor viii synthesis and its impact on therapeutic strategies for hemophilia A. Haematologica. 2015;100(7):849-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature. 1984;312(5992):326-330. [DOI] [PubMed] [Google Scholar]
- 11.Ingerslev J, Christiansen BS, Heickendorff L, Petersen CM. Synthesis of factor VIII in human hepatocytes in culture. Thromb Haemost. 1988;60(3):387-391. [PubMed] [Google Scholar]
- 12.Wion KL, Kelly D, Summerfield JA, Tuddenham EGD, Lawn RM. Distribution of factor VIII mRNA and antigen in human liver and other tissues. Nature. 1985;317(6039):726-729. [DOI] [PubMed] [Google Scholar]
- 13.Figueiredo MS, Brownlee GG. cis-acting elements and transcription factors involved in the promoter activity of the human factor VIII gene. J Biol Chem. 1995;270(20):11828-11838. [DOI] [PubMed] [Google Scholar]
- 14.McGlynn LK, Mueller CR, Begbie M, Notley CR, Lillicrap D. Role of the liver-enriched transcription factor hepatocyte nuclear factor 1 in transcriptional regulation of the factor V111 gene. Mol Cell Biol. 1996;16(5): 1936-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118(3):935-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahani T, Covens K, Lavend’homme R, et al. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost. 2014;12(1):36-42. [DOI] [PubMed] [Google Scholar]
- 17.Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123(24): 3706-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everett LA, Cleuren ACA, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123(24):3697-3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zanolini D, Merlin S, Feola M, et al. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica. 2015;100(7): 881-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madeira CL, Layman RE, De Vera ME, Fontes PA, Ragni M V. Extrahepatic factor VIII production in transplant recipient of hemophilia donor liver. Blood. 2009;113(21): 5364-5365. [DOI] [PubMed] [Google Scholar]
- 21.Follenzi A, Raut S, Merlin S, Sarkar R, Gupta S. Role of bone marrow transplantation for correcting hemophiliaA in mice (supplemental methods). Blood. 2012;4(23):1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollestelle MJ, Thinnes T, Crain K, et al. Tissue distribution of factor VIII gene expression in vivo--a closer look. Thromb Haemost. 2001;86(3):855-861. [PubMed] [Google Scholar]
- 23.Merlin S, Famà R, Borroni E, et al. FVIII expression by its native promoter sustains long-term correction avoiding immune response in hemophilic mice. Blood Adv. 2019;3(5):825-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peyvandi F, Garagiola I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia. 2019;25(5):738-746. [DOI] [PubMed] [Google Scholar]
- 25.Farré D, Roset R, Huerta M, et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003; 31(13):3651-3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Follo C, Isidoro C. A fast and simple method for simultaneous mixed site-specific mutagenesis of a wide coding sequence. Biotechnol Appl Biochem. 2008;49(Pt 2):175-183. [DOI] [PubMed] [Google Scholar]
- 27.Pignani S, Zappaterra F, Barbon E, et al. Tailoring the CRISPR system to transactivate coagulation gene promoters in normal and mutated contexts. Biochim Biophys Acta Gene Regul Mech. 2019;1862(6):619-624. [DOI] [PubMed] [Google Scholar]
- 28.Wei G, Srinivasan R, Cantemir-Stone CZ, et al. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood. 2009;114(5):1123-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dittmer J. The biology of the Ets1 protooncogene. Mol Cancer. 2003;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oettgen P. Functional redundancy of Ets1 and Ets2. Blood. 2009;114(5):934-935. [DOI] [PubMed] [Google Scholar]
- 31.Sato Y, Teruyama K, Nakano T, et al. Role of transcription factors in angiogenesis: Ets-1 promotes angiogenesis as well as endothelial apoptosis. Ann N Y Acad Sci. 2001;947:117-123. [PubMed] [Google Scholar]
- 32.Takahashi K, Sawasaki Y, Hata J-I, Mukai K, Goto T. Spontaneous transformation and immortalization of human endothelial cells. Vitr Cell Dev Biol. 1990;26(3):265-274. [DOI] [PubMed] [Google Scholar]
- 33.Basuyaux JP, Ferreira E, Stéhelin D, Butticè G. The Ets transcription factors interact with each other and with the c-Fos/c-Jun complex via distinct protein domains in a DNAdependent and -independent manner. J Biol Chem. 1997;272(42):26188-26195. [DOI] [PubMed] [Google Scholar]
- 34.Chavez A, Scheiman J, Vora S, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015; 12(4):326-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Pikkarainen T, Elomaa O, et al. Defective microarchitecture of the spleen marginal zone and impaired response to a thymus-independent type 2 antigen in mice lacking scavenger receptors MARCO and SR-A. J Immunol. 2005;175(12):8173-8180. [DOI] [PubMed] [Google Scholar]
- 36.Nicklin SA, Reynolds PN, Brosnan MJ, et al. Analysis of cell-specific promoters for viral gene therapy targeted at the vascular endothelium. Hypertension. 2001;38(1):65-70. [DOI] [PubMed] [Google Scholar]
- 37.Pierce GF, Iorio A. Past, present and future of haemophilia gene therapy: from vectors and transgenes to known and unknown outcomes. Haemophilia. 2018;24 Suppl 6:60-67. [DOI] [PubMed] [Google Scholar]
- 38.Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta. 2007;1775(2):298-312. [DOI] [PubMed] [Google Scholar]
- 39.Poisson J, Lemoinne S, Boulanger C, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. 2017;66(1):212-227. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Zhou QL, Sun W, et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol. 2015;17(10):1327-1338.0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poon GMK, Kim HM. Signatures of DNA target selectivity by ETS transcription factors. Transcription. 2017;8(3):193-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lange AM, Altynova ES, Nguyen GN, Sabatino DE. Overexpression of factor VIII after AAV delivery is transiently associated with cellular stress in hemophilia A mice. Mol Ther Methods Clin Dev. 2016;3:16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nougier C, Roualdes O, Fretigny M, et al. Characterization of four novel molecular changes in the promoter region of the factor VIII gene. Haemophilia. 2014;20(2):149-156. [DOI] [PubMed] [Google Scholar]
- 44.Arimoto J, Ikura Y, Suekane T, et al. Expression of LYVE-1 in sinusoidal endothelium is reduced in chronically inflamed human livers. J Gastroenterol. 2010;45(3): 317-325. [DOI] [PubMed] [Google Scholar]
- 45.Merlin S, Cannizzo ES, Borroni E, et al. A novel platform for immune tolerance induction in hemophilia A mice. Mol Ther. 2017;25(8):1815-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeLeve L, Maretti-Mira A. Liver sinusoidal endothelial cell: an update. Semin Liver Dis. 2017;37(04):377-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwachtgen JL, Janel N, Barek L, et al. Ets transcription factors bind and transactivate the core promoter of the von Willebrand factor gene. Oncogene. 1997;15(25):3091-3102. [DOI] [PubMed] [Google Scholar]
- 48.Lelièvre E, Mattot V, Huber P, Vandenbunder B, Soncin F. ETS1 lowers capillary endothelial cell density at confluence and induces the expression of VE-cadherin. Oncogene. 2000;19(20):2438-2446. [DOI] [PubMed] [Google Scholar]
- 49.George LA, Ragni MV, Samelson-Jones BJ, et al. Spk-8011: preliminary results from a phase 1/2 dose escalation trial of an investigational AAV-mediated gene therapy for hemophilia a. Blood. 2017;130(Suppl 1):S604. [Google Scholar]
- 50.Chuah MK, Petrus I, De Bleser P, et al. Liverspecific transcriptional modules identified by genome-wide in silico analysis enable efficient gene therapy in mice and nonhuman primates. Mol Ther. 2014;22(9): 1605-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.