

Abstract
The complex [FeIII2(μ-O2)(L3)4(S)2]4+ (L3 = 2-(4-thiazolyl)benzimidazole, S = solvent) forms upon reaction of [FeII(L3)2] with H2O2 and is a functional model of peroxo-diiron intermediates invoked during the catalytic cycle of oxidoreductases. The spectroscopic properties of the complex are in line with those of complexes formed with N-donor ligands. [FeIII2(μ-O2)(L3)4(S)2]4+ shows both nucleophilic (aldehydes) and electrophilic (phenol, N,N-dimethylanilines) oxidative reactivity and unusually also electron transfer oxidation.
A bidentate ligand based iron complex shows nucleophillic and electrophillice reactivity in the oxidation of organic substrates.
Dinuclear nonheme iron enzymes, including soluble methane monooxygenase (sMMO),1 toluene monooxygenases (TMO),2 steraoyl-ACP Δ9-desaturase (Δ9D),3 ribonucleotide reductases (RNR-R2),4 cyanobacterial aldehyde deformylase oxygenase (cADO),5 arylamine N-oxygenase (CmII),6p-aminobenzoate oxygenase (AurF),7 and deoxyhypusine hydroxylase (hDOHH)8 are responsible for a broad range of oxidative reactions such as hydrogen atom transfer (HAT), oxygen atom transfer (OAT) and C–C bond cleavage. For such enzymes, catalytic (μ-1,2-peroxo)diiron(iii) (P) intermediates have been postulated as key intermediates during the formation of mixed valent iron(iii)iron(iv) (X) or diiron(iv) (Q) intermediates to react via electrophilic HAT and OAT reactions. P intermediates show nucleophilic reactivity in deformylation of aldehydes.9
Several (μ-oxo)(μ-1,2-peroxo)diferric,10 (μ-OH or μ-OR)(μ-1,2-peroxo)diferric,11 and (μ-1,2-peroxo)diferric species have been characterized12 spectroscopically (resonance Raman, UV-vis absorption, Mössbauer, etc.) as structural models, however only a few functional models have been reported where the peroxodiiron(iii) species is capable of direct electrophilic and/or nucleophilic reactions.13 We reported the first examples for the reactivity of peroxodiiron(iii) species in H2O2 disproportionation,13a RNR-R2 type phenol oxidation and as mimics for ADO reactivity towards aldehydes.13a,c Ambiphilic behaviour in these complexes was rationalised by the involvement of two distinct oxidants, namely functional models of the nucleophilic P, and the electrophilic Q species.
The spectroscopic characterization of complex with [FeIII2(μ-O2)(L1,2)4(S)2]4+ cores (S = solvent), as synthetic models of non-heme iron oxygenases, generated from the trishomoleptic complexes [FeII(L1)3](CF3SO3)2 (1) (L1 = 2-(2′-pyridyl)benzimidazole) and [FeII(L2)3](CF3SO3)2 (2) (L2 = 2-(2′-pyridyl)-N-methylbenzimidazole) was reported earlier (Scheme 1).12c
Scheme 1. Structures of ligands and complexes discussed in the text.

Here we report the formation of the peroxo-diiron(iii) species [FeIII2(μ-O2)(L3)4(S)2]4+ (5) from the N-heterocyclic ligand, 2-(4-thiazolyl)benzimidazole ligand (L3) (Scheme 1). This complex shows electrophilic and nucleophilic reactivity in the oxidation of O–H bonds (H2O2, phenols), aldehyde deformylation, and oxidative N-demethylation of DMA via electrophilic C–H activation. The broad range of reactivity makes the complex a good functional model for diiron oxidoreductase enzymes.
The hetero-bidentate 2-(4-thiazolyl)benzimidazole ligand (L3) reacts spontaneously with Fe(ii) salts to form thermodynamically stable yellow [FeII(L3)3]2+ (5) complex (Scheme 1), which was characterized by UV-visible spectroscopy, electrospray mass spectroscopy (ESI-MS) (Fig. S1 and S2†), 1H NMR spectroscopy Fig. S3†), and X-ray single crystal analysis of [FeII(L3)3](ClO4)2 (5) (Fig. 1, Fig. S4, Tables S1 and S2†)). The Fe–N bond distances of 5 are longer than 2.1 Å and are typical of high-spin Fe(ii) complexes in contrast to the analogous low-spin Fe(ii) complex 2 in which the Fe–N bond distances are 2.0 Å (Fig. S4†).
Fig. 1. X-ray structure of 5. Thermal ellipsoids are plotted at 50% probability level.
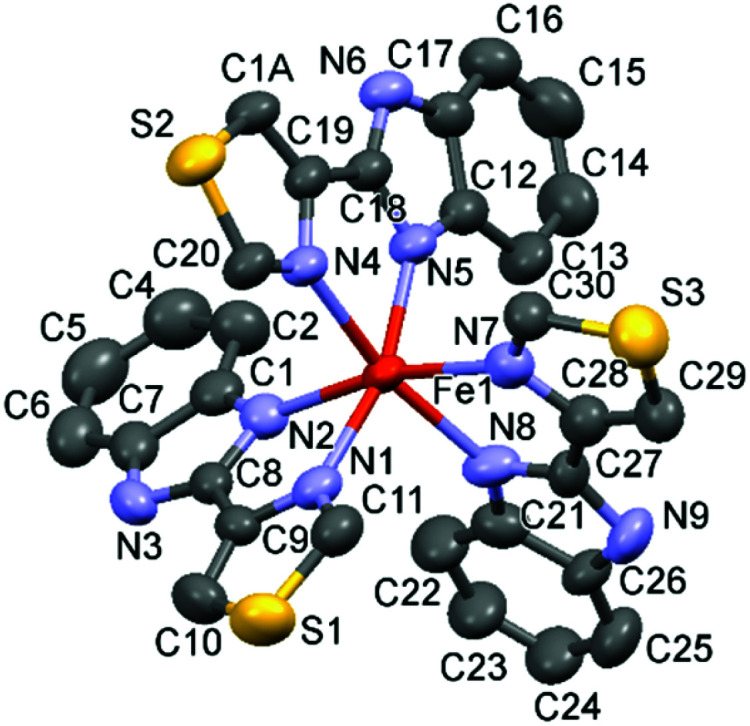
Raman spectroscopy in the solid state and solution show that the structure is retained in solution, i.e. that unbound ligand is not present (Fig. S5†). However, 1H NMR spectroscopy (Fig. S3†) indicates that more than one complex is present in solution, with distinct sets of signals corresponding to several coordination isomers, e.g., mer and fac. In contrast to the low spin complexes 1 and 2,12c in solution, 5 does not show absorption in the visible region, which is consistent with a high-spin electronic configuration of the latter (Fig. 2). These data highlight the sensitivity of the spin-state to the heteroaromatic unit in the ligand.
Fig. 2. (Left) UV-vis absorption spectra of FeII(L1,2,3)3]2+ (1, 2, 5) in MeCN and their corresponding complexes [FeIII2(μ-O2)(L1,2,3)2(S)2]4+ (3, 4 and 6) generated by addition of 4 equiv. H2O2 at 25 °C (inset). (Right) Raman spectrum of 6 at 785 nm generated from 5 (10 mM) by addition of 2 equiv. H2O2, with solvent signals subtracted.

In acetonitrile, a green (μ-1,2-peroxo)diiron(iii) intermediate (λmax = 705 nm, ε = 1200 M−1 cm−1) forms upon addition of H2O2 to 5 at room temperature, that is similar to those formed from 1, 2 (λmax = 720 nm (ε = 1360 M−1 cm−1) and λmax = 685 nm (ε = 1400 M−1 cm−1), respectively12c), Fig. 2. These characteristic absorption bands can be ascribed to the charge transfer between Fe(iii) and the O22− ligand (Fig. 2, inset). Similarly, the resonance Raman spectrum of 6 obtained with λexc = 785 nm shows features at 877 and 463 cm−1, which can be assigned to ν(O–O) and ν(Fe–O) stretching modes, respectively (Fig. 2). The formation of a Fe–O–Fe bridge in the peroxide complex is possible and cannot be excluded; however, in the absence of observable bands for such an Fe–O–Fe motif in the resonance Raman spectra at 785 nm (ref. 14) and the correlations drawn earlier by Que and co-workers between O–O stretching frequency and Fe–Fe separation, the detailed structure of the complex cannot be assigned and is therefore indicated as [FeIII2(μ-O2)(L1,2,3)2(S)2]4+.
The partial order in H2O2 (1st) and complex 5 (1st) was determined with a second order rate constant k = 6.54 ± 0.28 M−1 s−1 at 293 K (Table S3†) that is close to that for complexes 1 and 2 (5.38 M−1 s−1 and 6.6 M−1 s−1 at 293 K, respectively).12c The half-lives (t1/2) for complexes 3, 4 and 6 are 1200 s, 4740 s, and 400 s at 288 K, respectively, demonstrating that complex 6 is more reactive (kdecay = 0.95 × 10−3 s−1) compared to complex 3 and 4 (kdecay = 1.066 × 10−4 s−1) (Fig. S6 and Table S5†), and the large ΔH‡ of 84(6) kJ mol−1 with a small ΔS‡ of −2(21) J mol−1 K−1 (Fig. S7†) suggests a unimolecular decay process. Overall, the change from a pyridyl to a thiazolyl moiety in the ligand is sufficient to switch spin state in the iron(ii) state and while it does not affect rate of formation of the biomimetically relevant diiron(iii) complexes, it does affect the stability of the species. The presence of excess ligand does not affect the rate of formation of the peroxide bridged species and has only a modest effect on its rate of self-decay (Fig. S6†) Hence, it is of interest whether the reduced stability is translated into increased reactivity in the oxidation of organic substrates. The series of complexes [FeIII2(μ-O2)(L2)2(CH3CN)2]4+,12c [FeIII2(μ-O2)(L3)2(CH3CN)2]4+ and [FeIII2(μ-O)(μ-O2)(Py)2-indH)2(CH3CN)2]2+ ‡ 10c allows their reactivity towards benzaldehyde to be compared.13b,c
The rate of the decomposition of 6 (loss of absorbance at 705 nm) in CH3CN in the presence of substrates was compared with that for 3 and 4 (Scheme 2). The second-order rate constants in the oxidation of benzaldehyde (BA) with 6 is 2.86 M−1 s−1 at 288 K (Fig. 3, Table S6†), which is three times that for 4 (0.934 M−1 s−1) but identical to 3 (2.39 M−1 s−1), Fig. S8†).
Scheme 2. Reactions discussed in text.

Fig. 3. Reaction of [FeIII2(μ-O2)(L3)2(S)2] (6) (S = solvent) with benzaldehyde. UV-vis absorption spectrum over time (inset absorbance at 705 nm over time) following addition of (Upper) benzaldehyde and (Lower) phenylacetaldehyde to in situ generated 6 and corresponding plots of k′obs (= kobs − kselfdecay) versus [aldehyde] at 278 K.

The activity of 6 in nucleophilic and electrophilic oxidation reactions at 278 K was explored with benzaldehyde (BA), phenylacetaldehyde (PAA), and with 4-Me-DMA, 2,6-DTBP, respectively. The nucleophilic Baeyer–Villiger reactions with BA and PAA resulted in the formation of benzoic acid and benzaldehyde (∼85% and ∼65%, respectively based on 6), and the second order rate constants (k2) were 0.61 M−1 s−1 for BA and 0.022 M−1 s−1 for PAA at 288 K (Fig. 3, Table S7†). H-atom abstraction from the 2,6-DTBP, a model electrophilic substrate, resulted in the formation of 3,3′,5,5′-tetra-tert-butyl-4,4′-diphenoquinone (∼80%), with a k2 = 0.008 M−1 s−1 (Fig. 4, Table S8†). Notably, 6 reacted with 2,6-DTBP an order of magnitude more slowly than does [FeIII2(μ-O)(μ-O2)(Py)2-indH)2(CH3CN)2]2+ ‡ with a k2 = 0.028 M−1 s−1 at 278 K.13c
Fig. 4. Reactions of [FeIII2(μ-O2)(L3)2(S)2]4+ (6) (S = solvent) with 4-methyl-N,N-dimethylaniline (DMA). A) Time course of the decay of 6 monitored at 705 nm with DMA. B) Plot of k′obs (= kobs − kselfdecay) versus [substrate] for reactions of 6 with DMA at 278 K. C) Time course of the decay of 6 monitored at 705 nm with 2,6-di-tert-butylphenol (2,6-DTBP). D) Plot of k′obs (= kobs − kselfdecay) versus [substrate] for reactions of 6 with 2,6-DTBP at 278 K.

Additionally, H-atom abstraction from 4-Me-DMA proceeded at a similar rate with a second-order rate constant k2 = 0.008 M−1 s−1 at 278 K is noticeably faster than with [FeIV(N4Py*)(O)]2+ ‡/4Me-DMA system (9.8 ± 0.4) × 10−2 M−1 s−1 at 298 K) reported recently (Table S9†).15 The relative reactivity of 6 toward the electrophilic and nucleophilic model compounds shows the order BA > 4Me-DMA > PAA > 2,6-DTBP (Fig. 5).
Fig. 5. Decay of absorbance of [FeIII2(μ-O2)(L3)2(S)2] (6) (S = solvent) at 705 nm in CH3CN upon and in the presence of 4Me-DMA, 2,6-DTBP or PAA at 278 K with 2nd order rate constants for comparison.

Complex 6 is able to oxidize the N,N-dimethylanilines under mild anaerobic conditions with formation of MA with CH2O (80% yield w.r.t. 6) as the main product (see ESI†). Hammett plot analysis shows that the rate constant for the N-demethylation of DMA by 6 is sensitive to changes in the electronic properties of the DMA (Fig. 6A), with a ρ value that is identical to that for the [FeIV(N4Py)(O)]2+/DMA system (ρ = −2.1), where electron transfer (ET) and concomitant proton transfer (PT) was proposed (Fig. 6B, Tables S10 and S11†).16 The correlation of log k2 values with the one-electron oxidation potentials of DMAs (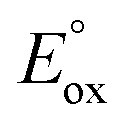 ) is good, and the slope (−3.13) is comparable to those obtained for the [FeIV(N4Py)(O)]2+/DMA (−3.3), [FeIV(TMC)(O)]2+/DMA (−4.0) and [FeIV(TPFPP)(O)]2+/DMA (−5.0) systems (Fig. 6C). ‡ 16 The magnitude of these values is also in a good agreement with the ET-PT mechanism proposed elsewhere and together these data provide strong evidence for the electrophilic feature of the active oxidant.
) is good, and the slope (−3.13) is comparable to those obtained for the [FeIV(N4Py)(O)]2+/DMA (−3.3), [FeIV(TMC)(O)]2+/DMA (−4.0) and [FeIV(TPFPP)(O)]2+/DMA (−5.0) systems (Fig. 6C). ‡ 16 The magnitude of these values is also in a good agreement with the ET-PT mechanism proposed elsewhere and together these data provide strong evidence for the electrophilic feature of the active oxidant.
Fig. 6. Hammett plot of log krel against the σp of para-substituted DMAs (A and C) at 278 K and 4R-BAs (D) at 288 K. (B) Plot of log k2 against the 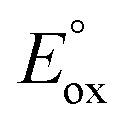 of para-substituted DMAs.
of para-substituted DMAs.

An opposite trend can be observed for the oxidation of BA derivatives with a large positive Hammett ρ value of +2.34, suggesting a nucleophilic attack of the peroxide on the aldehyde C-atom in the rate-determining step (Fig. 6D). Similar values were obtained for mononuclear peroxo complexes,16 but they are slightly lower than those with [FeIII2(μ-O2)(L2)2(CH3CN)2]4+ (+0.67)13b and [FeIII2(μ-O)(μ-O2)(Py)2-indH)2(CH3CN)2]2+ (+0.48)13c complexes. However, the strength of the O–O bond in 6 (νO–O tretch 877 cm−1), which is close to that of H2O2, indicates that formation of Fe(iv) O species except transiently possible is unlikely and instead electron transfer oxidation of DMA precedes proton transfer.
Conclusions
We have shown the complex [FeIII2(μ-O2)(L3)4(CH3CN)2]4+6, where L3 is not pyridine based, forms rapidly with H2O2 as terminal oxidant. The peroxy species serves as a functional model of the peroxo-diiron intermediates considered central to the catalytic cycle of oxidoreductases, in particular in showing both nucleophilic and electrophilic oxidation of substrates. The data show that a move towards ligand sets that are closer in electronic character to those available in metalloenzymes is key to emulating their reactivity.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
Financial support from The Netherlands Ministry of Education, Culture and Science (Gravity Program 024.001.035 to W.R.B.), the Hungarian National Research Fund (OTKA K108489 to JK), GINOP-2.3.2-15-2016-00049 and the European Research Council (279549 to WRB) are gratefully acknowledged.
Electronic supplementary information (ESI) available: Additional spectroscopic and kinetic data, synthetic procedures and characterisation. CCDC 2053805. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt01502h
Footnotes
((Py)2-indH = 1,3-bis(2′-pyridylimino)-isoindoline, N4Py = 1,1-di(pyridin-2-yl)-N,N-bis(pyridin-2-ylmethyl)methanamine, TPFPP = 5,10,15,20-tetrakis-pentafluorophenylporphyrin, TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane.
Notes and references
- (a) Wallar B. J. Lipscomb J. D. Chem. Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]; (b) Tinberg C. E. Lippard S. J. Acc. Chem. Res. 2011;44:280. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Bochevarov A. D. Li J. Song W. J. Friesner R. A. Lippard S. J. J. Am. Chem. Soc. 2011;133:7384. doi: 10.1021/ja110287y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Newman L. M. Wackett L. P. Biochemistry. 1995;34:14066. doi: 10.1021/bi00043a012. [DOI] [PubMed] [Google Scholar]; (c) Studts J. M. Mitchell K. H. Pikus J. D. McClay K. Steffan R. J. Fox B. G. Protein Expression Purif. 2000;20:58. doi: 10.1006/prep.2000.1281. [DOI] [PubMed] [Google Scholar]; (d) Song W. J. Behan R. K. Naik S. G. Huynh B. H. Lippard S. J. J. Am. Chem. Soc. 2009;131:6074. doi: 10.1021/ja9011782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox B. G. Lyle K. S. Rogge C. E. Acc. Chem. Res. 2004;37:421. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- Nordlund P. Reichard P. Annu. Rev. Biochem. 2006;75:681. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- (a) Schirmer A. Rude M. A. Li X. Popova E. del Cardayre S. B. Science. 2010;329:559. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]; (b) Li N. Chang W.-C. Warui D. M. Booker S. J. Krebs C. Bollinger J. M. Biochemistry. 2012;51:7908. doi: 10.1021/bi300912n. [DOI] [PubMed] [Google Scholar]; (c) Krebs C. Bollinger Jr. J. M. Booker S. J. Curr. Opin. Chem. Biol. 2011;15:291. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jia C. Li M. Li J. Zhang J. Zhang H. Cao P. Pan X. Lu X. Chang W. Protein Cell. 2015;6:55. doi: 10.1007/s13238-014-0108-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris T. M. Vu V. V. Meier K. K. Komor A. J. Rivard B. S. Münck E. Que Jr. L. Lipscomb J. D. J. Am. Chem. Soc. 2015;137:1608. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y. S. Zhang H. Brunzelle J. S. Nair S. K. Zhao H. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6858. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Han Z. Sakai N. Boettger L. H. Klinke S. Hauber J. Trautwein A. X. Hilgenfeld R. Structure. 2015;23:882. doi: 10.1016/j.str.2015.03.002. [DOI] [PubMed] [Google Scholar]; (b) Vu V. V. Emerson J. P. Martinho M. Kim Y. S. Münck E. Park M. H. Que Jr. L. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14814. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park M. H. J. Biochem. 2006;139:161. doi: 10.1093/jb/mvj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Trehoux A. Mahy J.-P. Avenier F. Coord. Chem. Rev. 2016;322:142. [Google Scholar]; (b) Jasniewski A. J. Que Jr. L. Chem. Rev. 2018;118:2554. doi: 10.1021/acs.chemrev.7b00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kryatov S. V. Taktak S. Korendovych I. V. Rybak-Akimova E. V. Kaizer J. Torelli S. Shan X. Mandal S. MacMurdo V. L. Mairata i Payeras A. Que Jr. L. Inorg. Chem. 2005;44:85. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]; (b) Cranswick M. A. Meier K. K. Shan X. Stubna A. Kaizer J. Mehn M. P. Münck E. Que Jr. L. Inorg. Chem. 2012;51:10417. doi: 10.1021/ic301642w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pap J. S. Cranswick M. A. Balogh-Hergovich E. Barath G. Giorgi M. Rohde G. T. Kaizer J. Speier G. Que Jr. L. Eur. J. Inorg. Chem. 2013:3858. doi: 10.1002/ejic.201300162. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fiedler A. T. Shan X. Mehn M. P. Kaizer J. Torelli S. Frisch J. R. Kodera M. Que Jr. L. J. Phys. Chem. A. 2008;112:13037. doi: 10.1021/jp8038225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ookubo T. Sugimoto H. Nagayama T. Masuda H. Sato T. Tanaka K. Maeda Y. Okawa H. Hayashi Y. Uehara A. Suzuki M. J. Am. Chem. Soc. 1996;118:701. [Google Scholar]; (b) Dong Y. Menage S. Brennan B. A. Elgren T. E. Jang H. G. Pearce L. L. Que Jr. L. J. Am. Chem. Soc. 1993;115:1851. [Google Scholar]; (c) Hayashi Y. Kayatani T. Sugimoto H. Suzuki M. Inomata K. Uehara A. Mizutani Y. Kitagawa T. Maeda Y. J. Am. Chem. Soc. 1995;117:11220. [Google Scholar]; (d1) Dong Y. Yan S. Young V. G. Que Jr. L. Angew. Chem., Int. Ed. 1996;35:618. [Google Scholar]; (d2) Frisch J. R. Vu V. V. Martinho M. Münck E. Que Jr. L. Inorg. Chem. 2009;48:8325. doi: 10.1021/ic900961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Brunold T. C. Tamura N. Kitajima N. Moro-Oka Y. Solomon E. I. J. Am. Chem. Soc. 1998;120:5674. [Google Scholar]; (b) Kim K. Lippard S. J. J. Am. Chem. Soc. 1996;118:4914. [Google Scholar]; (c) Pap J. S. Draksharapu A. Giorgi M. Browne W. R. Kaizer J. Speier G. Chem. Commun. 2014;50:1326. doi: 10.1039/c3cc48196d. [DOI] [PubMed] [Google Scholar]; (d) Kitajima N. Tamura N. Amagai H. Fukui H. Moro-oka Y. Mizutani Y. Kitagawa T. Mathur R. Heerwegh K. J. Am. Chem. Soc. 1994;116:9071. [Google Scholar]; (e) Duboc-Toia C. Ménage S. Ho R. Y. N. Que Jr. L. Lambeaux C. Fontecave M. Inorg. Chem. 1999;38:1261–1268. doi: 10.1021/ic980958j. [DOI] [PubMed] [Google Scholar]
- (a) Szávuly M. I. Surducan M. Nagy E. Surányi M. Speier G. Silaghi-Dumitrescu R. Kaizer J. Dalton Trans. 2016;45:14709. doi: 10.1039/c6dt01598k. [DOI] [PubMed] [Google Scholar]; (b) Kripli B. Csendes F. V. Török P. Speier G. Kaizer J. Chem. – Eur. J. 2019;25:14290. doi: 10.1002/chem.201903727. [DOI] [PubMed] [Google Scholar]; (c) Kripli B. Szávuly M. Csendes F. V. Kaizer J. Dalton Trans. 2020;49:1742. doi: 10.1039/c9dt04551a. [DOI] [PubMed] [Google Scholar]
- Fiedler A T. Shan X. Mehn M. P. Kaizer J. Torelli S. Frisch J. R. Kodera M. Que Jr. L. J. Phys. Chem. A. 2008;112:13037. doi: 10.1021/jp8038225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakk-Bogáth D. Kripli B. Meena B. I. Speier G. Kaizer J. Polyhedron. 2019;169:169. [Google Scholar]
- Nehru K. Seo M. S. Kim J. Nam W. Inorg. Chem. 2007;46:293. doi: 10.1021/ic0614014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.