

Abstract
Decreases in energy metabolism following traumatic brain injury (TBI) are attributed to impairment of glycolytic flux and oxidative phosphorylation. Glucose utilization post-TBI is decreased while administration of alternative substrates has been shown to be neuroprotective. Changes in energy metabolism following TBI happens in two phases; a period of hyper-metabolism followed by prolonged hypo-metabolism. It is not understood how different cerebral metabolic states may impact substrate metabolism and ultimately mitochondrial function. Adult male or female Sprague Dawley rats were given sham surgery or controlled cortical impact (CCI) and were assigned one of two administration schemes. Glucose, lactate or beta-hydroxybutyrate (BHB) were infused i.v. either starting immediately after injury or beginning 6 h post-injury for 3 h to reflect the hyper- and hypometabolic stages. Animals were euthanized 24 h post-injury. The peri-contusional cortex was collected and assayed for mitochondrial respiration peroxide production, and citrate synthase activity. Tissue acetyl-CoA, ATP, glycogen and HMGB1 were also quantified. Sex differences were observed in injury pattern. Administration based on cerebral metabolic state identified that only early lactate and late BHB improved mitochondrial function and peroxide production and TCA cycle intermediates in males. In contrast, both early and late BHB had deleterious effects on all aspects of metabolic measurements in females. These data stress there is no one optimal alternative substrate, but rather the fuel type used should be guided by both cerebral metabolic state and sex.
Keywords: Traumatic brain injury, Alternative substrate, Metabolism, Mitochondria, Sex
1. Introduction
Traumatic brain injury (TBI) is now recognized as a serious health issue for which there are no treatments available. Impacts to the head initiate multiple cascades that contribute to ongoing secondary injuries and dysfunction. One of the well described cascades was first described by Yoshino et al., who was the first to define the different stages and recovery trajectory of cerebral glucose metabolism following TBI in rodents (Yoshino et al., 1991). It was shown that cerebral metabolism undergoes two specific metabolic stages after TBI that occur in both experimental models of TBI and humans. The first is an acute and transient hypermetabolic stage that is associated with an increase in extracellular potassium, glutamate and accumulation of calcium resulting in increased glucose utilization (Bergsneider et al., 1997; Hutchinson et al., 2009; Prins et al., 2013a). This is followed by a prolonged hypometabolic stage starting at 6 h post-injury with the duration being injury severity dependent (Gardner et al., 2014; Maudsley et al., 2017; Yeo et al., 2011). This phase has been associated with both inhibition of glycolysis and mitochondrial dysfunction (Hutchinson et al., 2009; Li et al., 2012; Verweij et al., 2000; Vink et al., 1994).
The role of glucose in metabolism cannot be addressed without consideration of the mitochondria, which have a prominent role in both phases of cerebral metabolic crisis and dysfunction has been observed from 30 mins to 7 days post-injury (Hill et al., 2017; Signoretti et al., 2001; Singh et al., 2006). The increases in cellular calcium and oxidative stress during the hypermetabolic period result in mitochondrial dysfunction that include inhibition of electron transport chain complexes and permeability transition pore opening (Buki et al., 1999; Greco et al., 2016; Kilbaugh et al., 2011; Kilbaugh et al., 2015; Mbye et al., 2008; Sullivan et al., 1999). In addition to damaged mitochondria, inhibition of glycolysis during the hypometabolic period can lead to insufficient entry of acetyl CoA into the TCA cycle resulting in decreased oxidative phosphorylation (Casey et al., 2008; Vagnozzi et al., 2007). Studies have shown that administration of exogenous substrates including ketones (BHB), lactate and pyruvate and additional administration of glucose to be neuroprotective following TBI (Carteron et al., 2018; Davis et al., 2008; Glenn et al., 2015; Greco et al., 2016; Maalouf et al., 2007; Prins et al., 2005; Prins and Hovda, 2009; Shi et al., 2015; Shijo et al., 2017). Exogenous substrates are typically thought to bypass glycolysis and enter directly into the TCA cycle though the conversion to acetyl CoA and also reduce oxidative stress through direct scavenging, while additional glucose may support local tissue glucose insufficiency. Despite two very different patterns of cerebral metabolism, it has yet to be determined whether each of these substrates have preferential actions during different cerebral metabolic states.
In addition to supporting neuronal metabolism and viability, alternative substrates can promote glial health. In the adult brain, glycogen is stored in astrocytes, which can serve as a supplemental fuel for neurons in crisis (Cali et al., 2019). Increases in metabolic demand are typically met with increased blood flow to provide glucose and O2 (Yellen, 2018), but intense neural activity can quickly deplete local glucose and as glucose concentrations decrease towards the Km of hexokinase, phosphorylation becomes rate limiting (Brown and Ransom, 2007). However, glial glycogen enzymes can catabolize glycogen into lactate and pyruvate rapidly and without ATP to provide an alternative substrate to neurons (Carpenter et al., 2015). Glycogen catabolism is biochemically favored under high metabolic conditions and low glucose and without neighboring glia, there will be increased neuronal death and decreased repair and plasticity. This supported by work from Shijo et al. that showed exogenous sodium pyruvate administered within 24 h of injury significantly improved astrocytic metabolism (Shijo et al., 2017). Astrocytes contain receptors for advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4) receptors and are vulnerable to increased high mobility group box 1 (HMGB1). HMGB1 is a DNA binding protein that facilitates binding of other proteins to DNA. Binding of HMGB1 to either receptor promotes neuroinflammation and binding to astrocytic RAGE receptors results in a proinflammatory phenotype (Okuma et al., 2014; Pedrazzi et al., 2007). Following TBI, HMGB1 translocates from the nucleus to the cytoplasm and is released from dead and/or dying neurons (Gao et al., 2012; Okuma et al., 2014). Further from the injury core, HMGB1 intracellular expression is increased with concurrent increases in plasma HMGB1 (Okuma et al., 2012). Recently, plasma HMGB1 has also been investigated as a marker of injury severity (Au et al., 2012). The role of alternative substrates improving glial health is absolutely vital and has not been examined during the different metabolic stages after TBI.
While alternative substrates have been shown to be neuroprotective in both our lab and others, a criterion for administration has not yet been established. It also remains unclear which of these alternative substrates is optimal, how they impact neuronal and glial metabolism and how they are metabolized during the different metabolic stages of TBI. It is hypothesized that alternative substrate metabolism will be dependent on the cerebral metabolic state at the time of administration.
2. Methods
2.1. Subjects
Young adult male (n = 84) or female (n = 96) Sprague-Dawley rats were received from Charles River Breeding Labs (Hollister, CA) and maintained in standard temperature and lighting conditions (23 ± 2 °C, 12 h/12 h lighting cycle (06:00–18:00 h) with food and water available ad libitum. Rats were acclimated to their environment for 1wk prior to experimentation. Rats were randomized to drug and experimental groups listed in Table 1. Sample size was estimated using G*Power 3.1 (Universität Düsseldorf). Experimenters were blinded to conditions. All procedures were approved by the UCLA Chancellor’s Committee for Animal Research and the manuscript prepared according to ARRIVE guidelines for reporting animal research.
Table 1.
Experimental groups.
| MITO (E/L) | Irradiated (E/L) | |
|---|---|---|
| Veh-SHAM | (3/3) | (3/3) |
| Veh-CCI | (6/6) | (6/6) |
| GLC | (6/6) | (6/6) |
| BHB | (6/6) | (6/6) |
| LAC | (6/6) | (6/6) |
| COMBO** | (6) | (6) |
Experimental groups and n for either male or females. Veh, vehicle; GLC, glucose; BHB, beta-hydroxybutyrate; LAC, lactate; COMBO, early LAC + late BHB; E, early administration; L, late administration
in males only.
2.2. Cannulations
Anesthesia was induced with 3% isoflurane vaporized in 100% O2 and then maintained with 2% isoflurane during surgery. The femoral artery and vein were cannulated with a polyethylene tube (PE-50) on the day of study and animals were restrained to a piece of cardboard during the length of infusion. After completion of infusion, the cannula was tied off and the area sutured closed and animals were returned to their home cage.
2.3. CCI injury
Anesthesia was induced with 3% isoflurane vaporized in 100% O2 and then maintained with 2% isoflurane during surgery. The head was positioned in a stereotaxic frame, a midline incision was made, and a 6-mm craniotomy was drilled centered at −4 mm anterior-posterior (AP), 5 mm midlateral (ML) relative to bregma. A CCI injury was produced on the exposed left cortex using an electronically controlled pneumatic piston cylinder (Hydraulics Control, Inc., Emeryville, CA) as previously described (Prins et al., 2004b). In the present study, the 5 mm diameter flat rod tip was angled at 22.5° away from vertical and compressed the cortex at 1.9 m/s to a depth of 2 mm. After injury, a small piece of gel foam was placed over the craniotomy site to reduce bleeding and the wound sutured closed. Sham injured rats received only a craniotomy but no cortical injury.
2.4. Substrate administration and infusion
Groups received a 3 h i.v. infusion of either saline (0.9%), lactate (100 mM), beta-hydroxybutyrate (BHB) (2 M) or glucose (30%) at a flow rate of 10.8 μl/s based on previous publications (Prins et al., 2004a, 2004b; Rice et al., 2002; Shijo et al., 2015). To best target treatment during hyper- or hypo-metabolic periods there were two distinct infusion start times. To target the hyper-metabolic state, early infusion was started immediately following injury, while late infusion began 6 h post-injury during the hypo-metabolic phase. Immediately prior to the start of infusion and immediately after, venous blood was collected in EDTA collection tubes and spun at 3000 g for 10 min and plasma was collected. Glucose and lactate were measured using Stat Profile Prime (Nova Biomedical) while BHB while measured using BHB Stat Site BHB test strips and meter (Stanbio).
2.5. Tissue fixation
Tissue was fixed using microwave irradiation in order to effectively reduce postmortem changes of energy-related metabolites/enzymes in the nervous tissues (Sharpless and Brown, 1978). All animals were anesthetized with isoflurane before a beam of microwave irradiation at 2450 MHz and nominal output of 3.5 kW was focused for 2.45–2.90 s directly on the head (Thermex-Thermatron Model 4104 Microwave Fixation System, Louisville, KY). Individual irradiation time was adjusted based on the minimal time necessary for the temperature of the core to reach 80 °C, which is sufficient to denature enzymes (Guidotti et al., 1974). Rats were quickly decapitated and the brains were dissected hemispherically, placed into 1.5 ml Eppendorf tubes, and ipsilateral cortex was submerged into methylbutane on dry ice before storing at −70 °C prior to metabolite quantification.
2.6. Mitochondrial isolation
Rats were briefly restrained within a decapicone (Braintree Scientific, Braintree, MA) and euthanized by decapitation and the ipsilateral peri-contusional cortex was rapidly dissected and homogenized in ice-cold isolation buffer (225 mM mannitol, 25 mM sucrose, 10 mM Hepes, 1 mM EGTA, pH 7.4 at 4 °C). The homogenate was centrifuged at 4000 rpm for 3 min. The pellet was discarded and the supernatant centrifuged at 14000 rpm for 8 min. The pellets were resuspended in 1.5 ml isolation media and 4 ul of 10% digitonin was added. The tubes were gently inverted 6 times and left to incubate on ice for 4 min and then spun at 14000 rpm for 8 min. The pellets were resuspended in 1 ml of isolation buffer and 10 mg/ml de-fatted bovine serum albumin was added. After a final centrifugation at 14000 rpm for 8 min, the mitochondria were resuspended in 30 ul of EGTA free isolation media.
2.7. Protein determination
Protein concentrations were measured using a Lowry DC kit (BioRad, Hercules, CA) with bovine serum albumin used as concentration standards.
2.8. Mitochondrial respiration
The respiratory activities of isolated brain mitochondria were measured polarimetrically with a Clark-type oxygen electrode apparatus (Hansatech Instruments, Norfolk, England). Mitochondria were suspended at a protein concentration of 0.5 mg/ml in buffer containing 125 mM KCl, 20 mM HEPES, 2 mM K2HPO4, 0.01 mM EGTA, 1 mM MgCl2 (pH 7.0) at 37 °C, plus glutamate (5 mM) and malate (0.1 mM). Addition of ADP (0.5 mM) was used to initiate State 3 (phosphorylating) respiration. Oligomycin (2.5 μg/ml), an inhibitor of the mitochondrial ATP synthase, was used to induce State IV (resting) respiration. Maximal respiration was initiated with the addition of the protonophore uncoupler, FCCP (54 nM). Rates of oxygen consumption are expressed as nmol O2/mg mitochondrial protein/min). The respiratory control ratio (RCR) is defined as the rate of ADP-stimulated oxygen consumption (State III) divided by the rate of respiration determined in the presence of oligomycin (State IV).
2.9. Mitochondrial H2O2 production
Adapted from Starkov (Starkov, 2010), deenergized mitochondria (0.5 mg/ml) were suspended in 200ul of respiration buffer containing 4/U of horseradish peroxidase and 10 μM Amplex red. The background fluorescent signal was read for 120 s (555ex/581em). NAD+ linked respiratory substrates were added and baseline H2O2 production was recorded for 120 s. 10 μM rotenone was added to stimulate H2O2 production and production was again recorded for 120 s.
2.10. Western blots
Tissue homogenates were lysed in RIPA buffer (25 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, pH 7.6 at 4 °C) containing a cocktail of protease inhibitors (Calbiochem). Equal amounts of protein were separated by SDS-PAGE (4–12% Bis-Tris gels, Invitrogen) and transferred to PVDF membranes (Invitrogen), blocked in TBST plus 5% non-fat milk and then incubated with the following primary antibody: HMGB-1 (Abcam, ab18256) overnight at 4 °C. The membranes were then washed and TBST and incubated for 1 h at room temperature. The washed membranes were then treated with enhanced chemiluminescence detection reagent (Thermo Scientific). All Blots were developed using ChemiDoc XRS+ Molecular Imager (BioRad) and analyzed using Quantity One software (BioRad). Band densities were normalized to the total amount of protein loaded per lane using Sypro Ruby (BioRad).
2.11. Citrate synthase activity
Citrate synthase activity was quantified in isolated mitochondria according to manufacturer’s instructions (Sigma-Aldrich).
2.12. Metabolic quantification
Microwave irradiated tissue was utilized to prevent endogenous breakdown of glycogen (Sigma-Aldrich), ATP (Abcam) and acetyl CoA (Sigma-Aldrich), while mitochondria were deproteinated with perchloric acid. Metabolites were quantified according to manufacturer’s instructions.
2.13. Statistical analysis
Data are expressed as means ± SEM of n different experiments or a five-number summary of the data set. Differences between independent groups were assessed using either Student’s t-test or one-way ANOVA, with Tukey’s test for comparison between groups using SPSS Software 25. (SPSS, Inc., Chicago, IL). P < .05 was considered to be statistically significant.
3. Results
3.1. Timing of metabolic intervention matters
The cerebral metabolic crisis observed following TBI involves both the disruption of cellular respiration (glycolysis) and bioenergetics (oxidative phosphorylation). Exogenous delivery of alternative substrates including ketones (BHB), lactate and pyruvate have been shown to improve different outcome measures after TBI. While each of these compounds have shown differential abilities to act as free radical scavengers and improve mitochondrial function, it is yet unknown if perceived benefits are dependent on the cerebral metabolic state at the time of delivery and if this is further influenced by sex.
3.2. Plasma levels of alternative substrates
Plasma levels of BHB, glucose and lactate were measured immediately prior to infusion and after as demonstrated by (Fig. 1A/F). There were no significant effects of early or late glucose administration of plasma glucose in injured female animals. In glucose treated males, plasma glucose was significantly reduced by 28% at 9 h post-injury (F(2, 11), f = 4.612, p = .035) compared to pre-infusion levels (Fig. 1B). This decrease may represent either changes in plasma glucose in relation to food restriction during infusion (Wang et al., 2010) or increased cerebral uptake. Activity of the pentose phosphate pathway has been shown to increase by 6 h post-injury and increased glucose shunting to this pathway may be responsible for the decrease. This supports the decreased ROS production observed in males 24 h post-injury that received late glucose administration (Fig. 4A). Plasma levels of lactate remained unchanged in males (Fig. 1C) while improving mitochondrial function and could represent immediate utilization as a substrate or conversion to pyruvate. Interestingly despite having no effect on mitochondrial function, plasma lactate was significantly decreased by early administration ((F(2, 29), f = 4.316, p = .023) (Fig. 1H). Differences in uptake of lactate may be sex-specific as actions of estrogen receptor-α on mitochondria can regulate lactate dehydrogenase activity (Nagai et al., 1988). Fasting has also been shown to increase monocarboxylic transporter expression in female mice (Schutkowski et al., 2014). Significant changes were found in plasma BHB in both male and female animals. In BHB treated males, both early and late administration of BHB resulted in significant plasma increases above 100% (F(2,24), f = 5.076, p = .015) (Fig. 1D). While baseline plasma glucose and lactate did not differ between males and females, baseline female plasma BHB was significantly lower compared to baseline male BHB. 0.28 ± 0.04 vs. 0.53 ± 0.12 mmol/L, respectively, p = .008 (data not shown). Further BHB was significantly increased by over 500% in females (F(2, 33), f = 163.8, p = .000) (Fig. 1I), potentially suggesting different mechanisms of metabolism that can result in supraphysiological levels that may have deleterious effects on the brain and organ function. Indeed, it is not yet understood how the brain is able to differentiate between what is a beneficial vs. detrimental level of circulating ketones (Fedorovich et al., 2018).
Fig. 1.
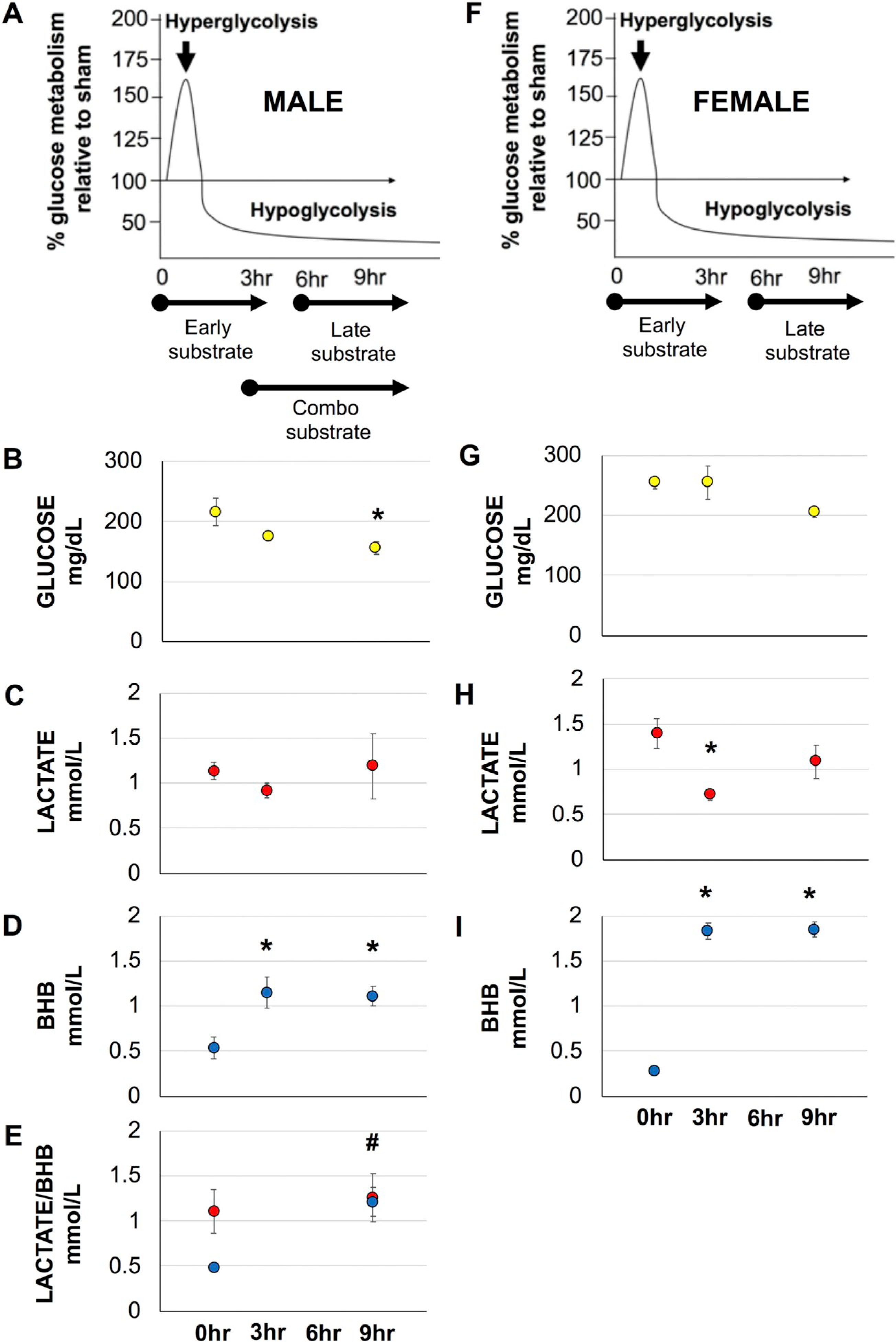
Blood plasma concentrations of Glucose, Lactate and BHB in injured animals prior to and 3 and 9 h post-injury. A-E, male animals. F–I, female animals. Average (±SEM) for blood concentrations. # for BHB, compared to pre-BHB only. *p < .05.
Fig. 4.
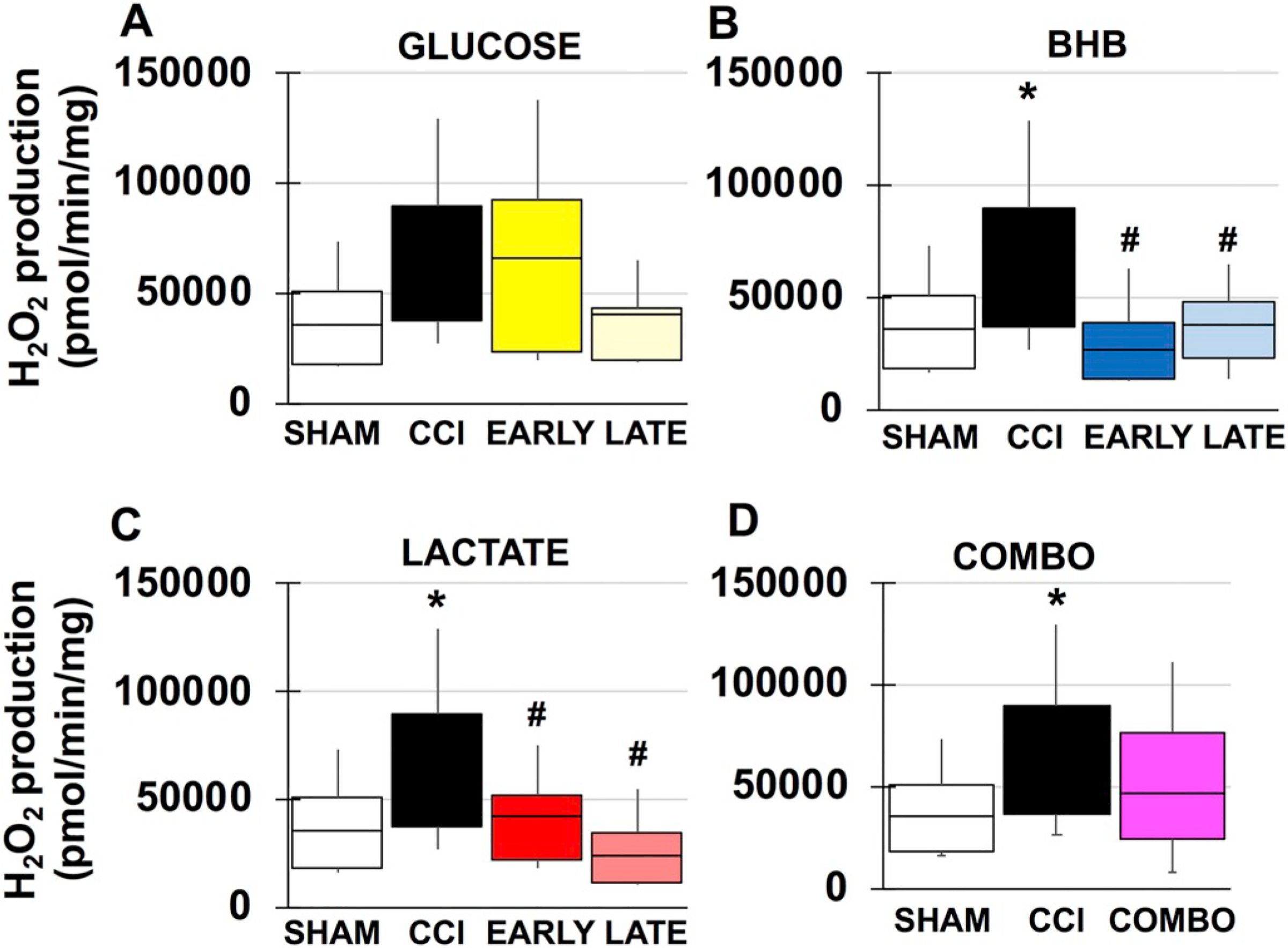
Mitochondrial peroxide production in males. A. Sham and vehicle CCI compared to glucose. B. Sham and vehicle CCI compared to BHB. C. Sham and vehicle CCI compared to lactate. D. Sham and vehicle CCI compared to combination. *compared to sham, #compared to CCI. p < .05.
3.3. Mitochondrial respiration
Results for the changes in mitochondrial respiration 24 h after TBI in males and females are shown in Figs. 2 and 3, respectively, while statistical information is located in Supplemental Tables 1–7. The results will be presented by each alternative substrate. Glucose: While glucose has been shown to improve learning, potential benefits have not been explored acutely and exogenous glucose may prove beneficial during hyperglycolysis. In sham males, as previously demonstrated, there is a significant decrease in state 3 dependent respiration and respiratory control ratio (RCR) after CCI (Fig. 2). In contrast, while there was a significant effect on state 3 respiration in females there were no overall changes in RCR (Fig. 3). Although glycolysis has been shown to be inhibited at later time points after TBI, acute glucose administration may support higher metabolic needs and thereby delay and/or lessen the magnitude of the hypo-metabolic period. In males, both early and late glucose administrated significantly increased state 3 compared to vehicle treated injured animal and respectively (Fig. 2A). Further, late glucose doubled state 3 respiration compared to sham and early glucose treated animals (Fig. 2A). Glucose also increased mitochondrial uncoupling (state 4). Early glucose increased rates compared to sham and vehicle CCI animals (Fig. 2E). Late glucose had similar effects compared to sham and vehicle CCI (Fig. 2A/E). Early and late glucose in males also significantly increased maximal respiratory capacity (Fig. 2L) compared to vehicle CCI and respectively. Early and late glucose had the opposite effect in females, resulting in reduced maximal rate compared to shams (Fig. 3G). While glucose treated mitochondria were more uncoupled, there were no differences in RCR compared to females where late glucose caused a significant decrease in RCR compared to vehicle CCI (Fig. 3J). BHB: One of the benefits afforded by BHB is in its ability to bypass the glycolytic blockade via conversion to acetyl CoA and direct entry into the TCA cycle. In males, early and late BHB significantly increased state 3 respiration compared to vehicle CCI) and (Fig. 2B). Early and late BHB had opposite effects in females. Early BHB significantly decreased state 3 respiration while late BHB increased it compared to vehicle CCI (Fig. 3B). In females, early BHB decreased uncoupling while late BHB increased uncoupling compared to shams and vehicle CCI and early BHB in females and respectively (Fig. 3E). In females, early BHB reduced maximal respiration compared to shams while late BHB significantly decreased maximal respiration compared to sham it was improved compared to vehicle CCI and early BHB (Fig. 3H). While no significant changes in RCR were observed in males from BHB treatment (Fig. 2M), in females, decreases in state 3 from early BHB and increases in state 4 respiration from late BHB significantly worsened RCR compared to vehicle CCI (Fig. 3K). Lactate: Similar to BHB, lactate’s main advantage is its role as a fuel source independent of glycolysis. Exogenous lactate is thought to be converted to pyruvate, further supplementing the acetyl CoA pool. In males, early lactate prevented injury induced changes in state 3 respiration while maintaining RCR (Fig. 2C). Conversely, late lactate did not improve state 3 respiration compared to vehicle CCI and reduced maximal respiratory capacity although RCR was not changed compared to shams. RCR was unchanged in female lactate treated animals despite significant improvement in state 3 respiration from late lactate and changes in maximal respiration from both early and late lactate treatment (Fig. 3C/I). Combination: The intention of this study was to identify which alternative fuel type may best suit different metabolic needs and to additionally address whether a combination of treatment may have an additive effect on improving outcome. Candidate substrates were chosen based on mitochondrial respiration and were only identified in males. Subsequent experiments to determine mechanism of action were only done in candidate substrates in males. Despite no effect of injury at 24 h in females, the surprising finding that substrate addition following injury impaired normal mitochondrial function and given the importance nature of understanding the basis of sex differences, necessitated the need to identify mechanism of action. In males, both early lactate and late BHB were chosen as candidates for combination treatment. Combination treatment significantly increased state 3 respiration while also increasing uncoupling (fig. D/H). A separate analysis was done to determine if combination treatment improved outcome compared to early lactate or late BHB. State 4 uncoupled respiration was significantly increased compared to both early lactate (F(4, 29), f = 4.818, p = .043) and late BHB (F(4, 29), f = 4.818, p = .007).
Fig. 2.

Mitochondrial respiration from male peri-contusional ipsilateral cortex 24 h post-injury in sham or treated animals. *compared to sham, #compared to CCI, & compared to early vs. late administration, p < .05. Statistical values available in Supplemental Tables 1–4.
Fig. 3.
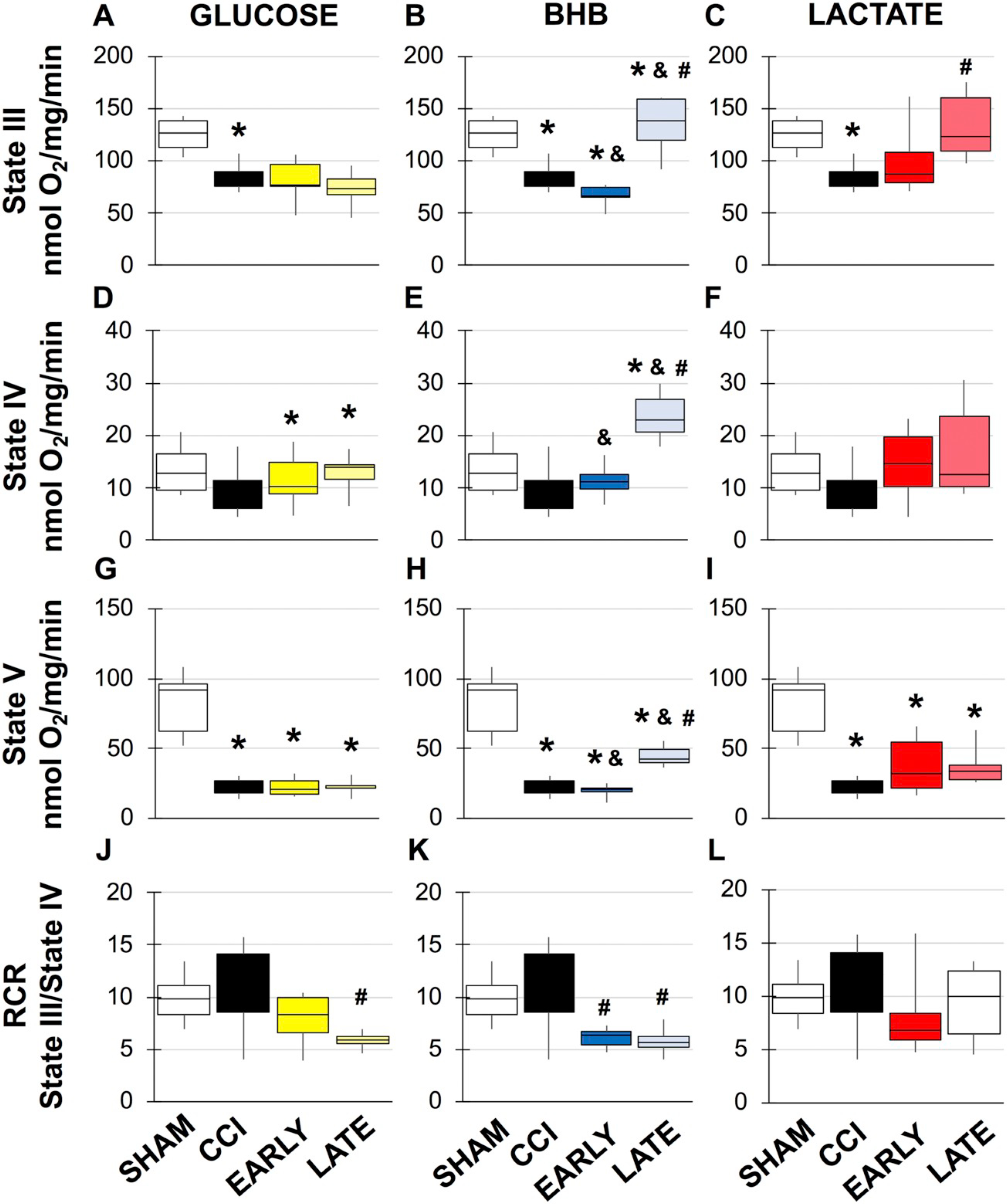
Mitochondrial respiration from female peri-contusional ipsilateral cortex 24 h post-injury in sham or treated animals. *compared to sham, #compared to CCI, &compared to early vs. late administration, p < .05. Statistical values available in Supplemental Tables 5–7.
3.4. ROS production
TBI has previous been shown to increase mitochondrial ROS production in male injury models and the current results are consistent with these studies (Kumar Sahel et al., 2019) (F(3, 19), f = 8.363, p = .014) (Fig. 4). Both BHB and lactate have varying degrees of free radical scavenging properties that reduce oxidative stress, assist mitochondrial function and potentially prevent energy collapse. Both timepoints of administration of BHB (F(3, 22), f = 8.363, p = .001), (F(3, 22), f = 8.363, p = .004) and lactate (F(3, 22), f = 9.237, p = .010), (F(3, 22), f = 9.237, p = .000), significantly reduced mitochondrial peroxide production, respectively (Fig. 4B/C). Although late glucose had a 102% decrease in peroxide production and may be due to pentose phosphate pathway shunting, neither treatment timepoint nor combination had a significant effect (Fig. 4A). In females, there was no observed injury effect. Although both glucose and BHB treatment impaired mitochondrial function, only late BHB increased peroxide production compared to sham (F(3, 21), f = 4.614, p = .044) and early BHB (F(3, 21), f = 4.614, p = .014) (Fig. 5B) and is likely due to increased mitochondrial uncoupling.
Fig. 5.
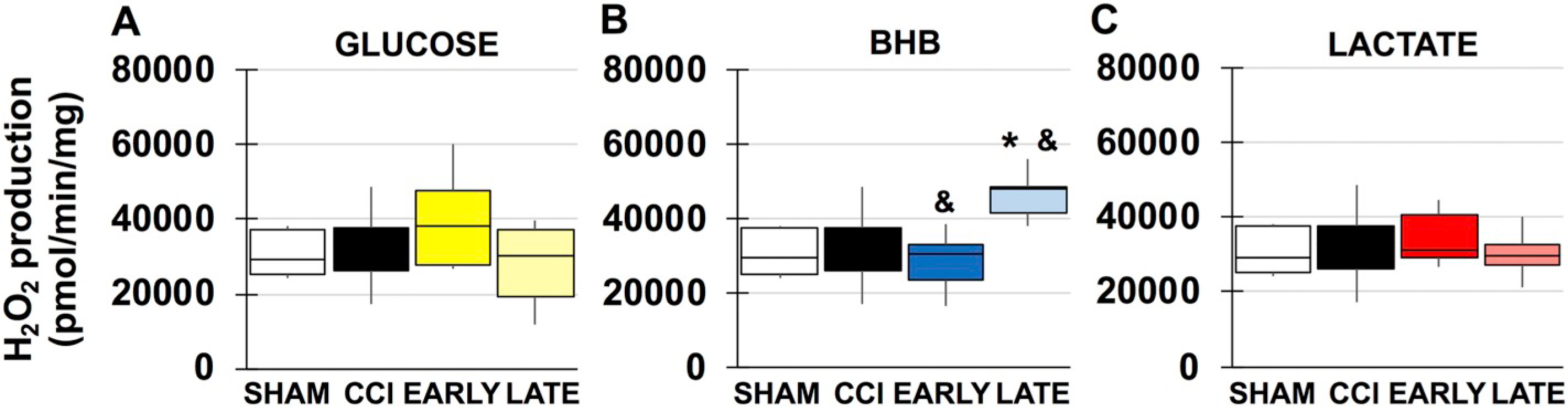
Mitochondrial peroxide production in females. A. Sham and vehicle CCI compared to glucose. B. Sham and vehicle CCI compared to BHB. C. Sham and vehicle CCI compared to lactate. *compared to sham, #compared to CCI. &compared to early vs. late administration p < .05.
3.5. Metabolic intermediates/TCA cycle
Changes in cerebral metabolism following TBI have overwhelmingly been attributed to either disruptions of glycolysis or end point mitochondrial function. Less is known of how alterations in the initiation and progression of the TCA cycle may contribute to mitochondrial function and ultimately cerebral metabolism. Acetyl CoA: Under normal glucose conditions, acetyl CoA is produced by the oxidative decarboxylation of pyruvate or through ß-oxidation of fatty acids within the mitochondrial matrix. Acetyl CoA can also be produced extramitochondrially through the breakdown of citrate. In males, acetyl CoA was reduced by half though not significant. Both BHB (F(4, 28), f = 4.064, p = .013) and lactate (F(4, 28), f = 4.064, p = .030) significantly increased tissue acetyl CoA, likely due to increased conversion of BHB to acetyl CoA and lactate to pyruvate (Fig. 6). There was no injury effect in females although early glucose increased acetyl CoA (F(3, 20), f = 3.36, p = .023) and could represent increased glycolysis (Fig. 7A). Similar to males, early BHB significantly increased acetyl CoA (F(3, 22), f = 7.765, p = .040) while late BHB actually reduced tissue content (F(3, 22), f = 7.765, p = .05) (Fig. 6B). Lactate did not result in any significant changes. Citrate Synthase: Citrate synthase is the first step of the TCA cycle and limits its rate. It is dependent on levels of oxaloacetate and acetyl CoA as well as ratios of ATP:ADP and NADH:NAD+. Citrate Synthase activity was significantly reduced in male injured animals (F(4, 29), f = 3.587, p = .036) (Fig. 6B). There was a trend towards improvement with lactate and combination treatments, but they were not significant. There was also an injury affect within the female group (F(3, 16), f = 8.031, p = .008) (Fig. 7D–F). Activity continued to be decreased with either late glucose (F(3, 14), f = 4.411, p = .034) (Fig. 4D) and early (F(3, 16), f = 8.031, p = .003) and late (F(3, 16), f = 8.031, p = .005) BHB treatment (Fig. 7E).
Fig. 6.
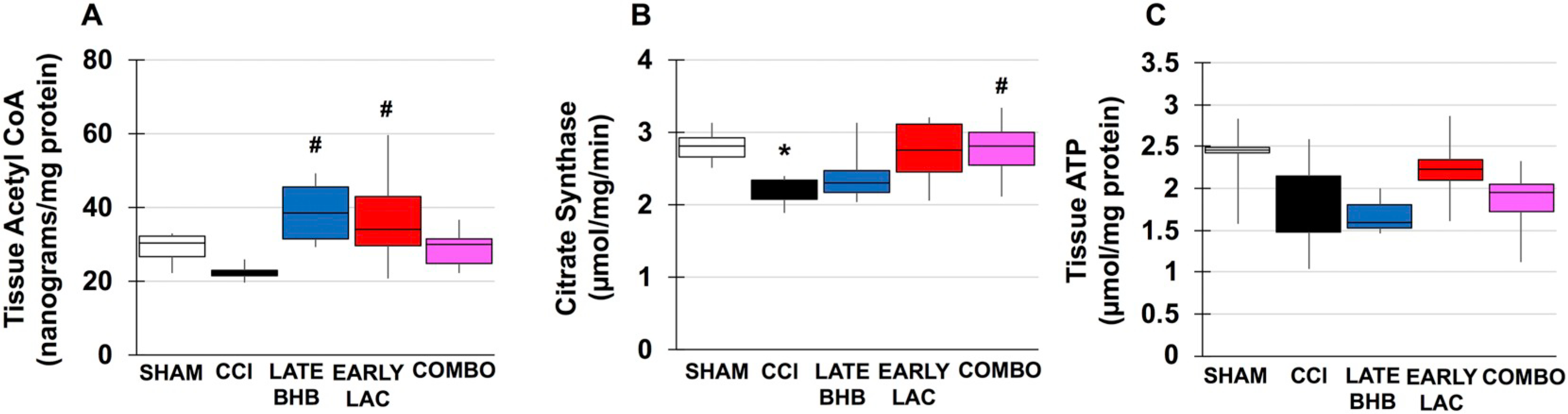
TCA Cycle intermediates and ATP content in males. A. Acetyl CoA content in injured vs. optimal treatment groups. B. Citrate synthase activity in injured vs. optimal treatment groups. C. ATP content in injured vs. optimal treatment groups *compared to sham, #compared to CCI. p < .05.
Fig. 7.
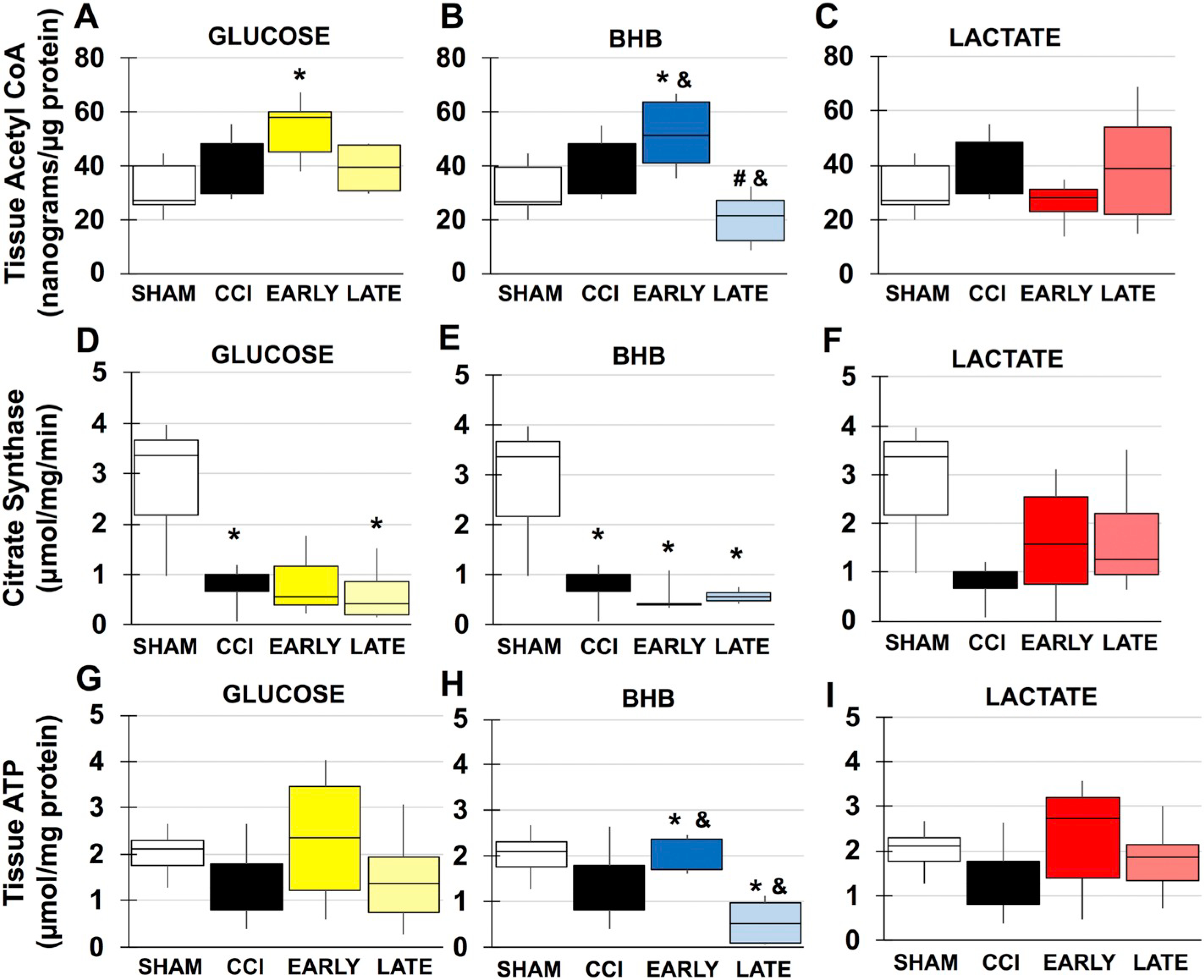
TCA Cycle intermediates and ATP content in females. A-C. Acetyl CoA content in A. Glucose B. BHB and C. lactate. D-F. Citrate synthase activity in D. glucose B. BHB C. lactate. Groups. G-I. ATP content in A. glucose B. BHB C. lactate *compared to sham, #compared to CCI &compared to time. p < .05.
3.6. Stress signals
During periods of metabolic demand, local blood glucose utilization increases as well as breakdown of glycogen to provide lactate as fuel via the lactate shuttle. TBI places further increased demands on brain metabolism and may lead to dissociation between glycolysis and oxidative phosphorylation, resulting in a type of “stress-signal” for energy need and activation of damage-associated molecular patterns (DAMPs). ATP: Despite evidence of mitochondrial dysfunction and disruption of the TCA cycle, no significant differences were seen between sham, vehicle CCI or drug treated male animals (Fig. 6C). In female animals, there was no significant impact of injury or glucose or lactate treatment (Fig. 7G/I). In late BHB animals however, ATP was significantly reduced by 273% compared to sham (F(3, 20), f = 5.982, p = .007) or 263% in early BHB (F(3, 20), = 5.982, p = .013) treated animals (Fig. 7H). Glycogen: In the adult brain, glycogen is predominantly found in astrocytes. Its main proposed function is to act as an energy substrate when transient local tissue energy demands exceed available blood glucose. In male animals, there was no injury effect of significantly decreased glycogen. BHB (F(4, 29), f = 4.992, p = .023), lactate (F(4, 29), f = 4.992, p = .010 and combination (F(4, 29), f = 4.992, p = .006) however, significantly increased tissue glycogen content (Fig. 8A). In females, similar results were found. Early glucose was found to increase tissue content compared to both sham (F(3, 22), f = 4.595, p = .038) and late treatment (F(3, 22), f = 4.595, p = .015) (Fig. 9A). Both early (F(3, 21), f = 2.480, p = .035) and late lactate (F(3, 21), f = 2.480, p = .035) had significant increases as well (Fig. 9C). HMGB1: Release of HMGB1 from injured and necrotic cells can serve as a “danger-signal” to neighboring microglia and contributes to overactivation of the immune system, mediated through the RAGE receptor. Although there were no significant injury differences in males or females, HMBG1 was reduced by 77% (Fig. 8B) and 76% (Fig. 9D–F), respectively, within the ipsilateral cortex, consistent with studies observing immediate decreases in HMGB1 within the contusion area.
Fig. 8.
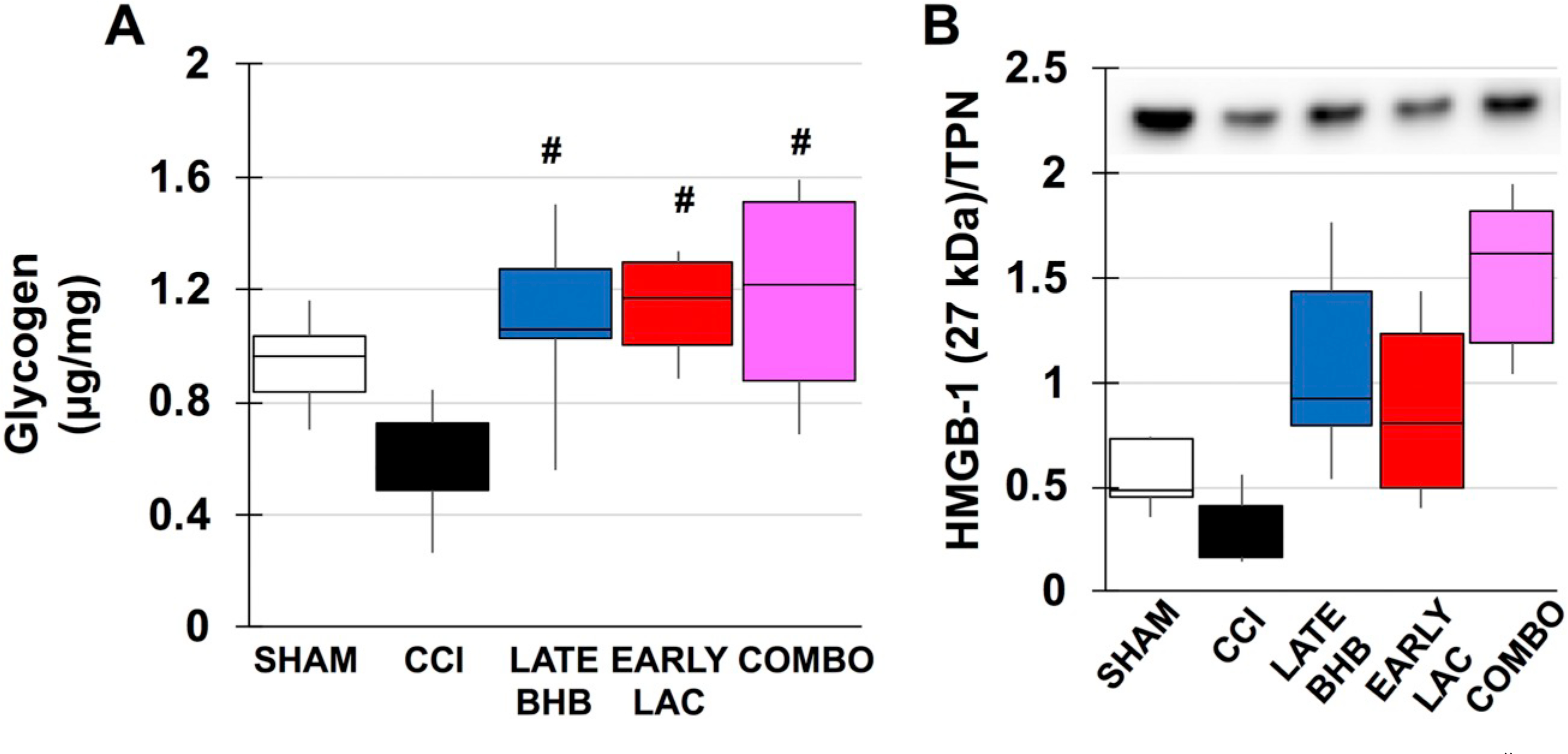
Markers of astrocytic health in males. A. Glycogen content B. Densitometric ratios for HMGB1/total protein. #compared to CCI. p < .05.
Fig. 9.
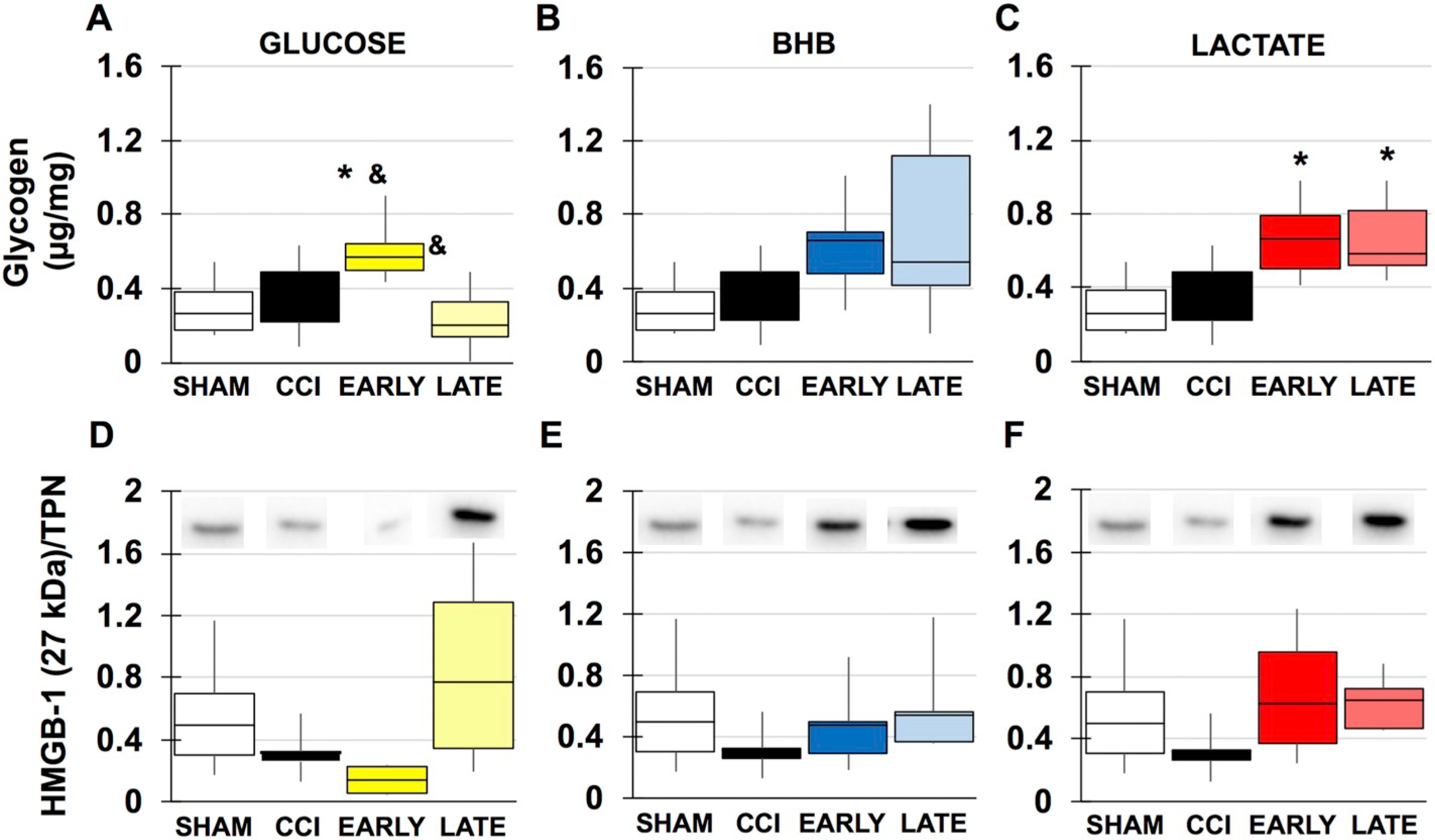
Markers of astrocytic health in females. A-C Glycogen content. A. glucose B. BHB C. lactate. D-F Densitometric ratios for HMGB1/total protein D. glucose E. BHB F. lactate. ⁎compared to sham. &compared to time. p < .05.
4. Discussion
The current study shows that neuroprotective properties of alternative substrates are time and sex dependent. Several studies have identified alternative substrates that confer neuroprotection, but have not established a rationale for timing of administration. We have established two distinct criteria of administration, either during the hyper- or hypo-metabolic stage following TBI. We have shown that alternative substrates deemed to be neuroprotective are metabolized differently at different post-injury metabolic time points and that combination of treatments within a short period may not be advantageous. More so, nuances in substrate behavior are also outcome measure specific. Further, the vast majority of alternative substrate work has been completed in male animals. We show that in our female animals, candidate substrates identified as neuroprotective in males actually worsen outcomes in females and switching substrate types can act as a stressor following injury. This establishes that alternative substrate administration following TBI should be matched by both sex and metabolic stage.
4.1. Metabolic state and sex determine alternative substrate metabolism males
Both early and late treatment of each substrate stimulated ADP-driven O2 consumption (state 3 respiration), implying increased glycolytic flux and mitochondrial demand, however only early administration of lactate prevented mitochondrial uncoupling (proton leak, state 4) (Fig. 2). Both late lactate and early BHB attenuated mitochondrial uncoupling as reflected in improved mitochondrial function/coupling (RCR). Lack of improvement of early glucose on mitochondrial function and sustained ROS production suggests just the addition of substrates is inefficient in preventing mitochondrial collapse and suggest other means of neuroprotection are necessary.
In addition to acting as a fuel source, lactate and BHB have both been shown to protect against glutamate excitotoxicity (Jourdain et al., 2016; Llorente-Folch et al., 2016; Maalouf et al., 2007; Youssef, 2015). Despite both early BHB and lactate reducing ROS (Fig. 4B/C), mitochondria from early BHB remained uncoupled (Fig. 4B) and may be due to differences in neuroprotective strategies against excitotoxicity. BHB’s actions are likely mediated through its scavenging abilities, while lactate has direct action on the mitochondria via the aspartate-glutamate carrier and could potentially prevent changes in mitochondrial membrane potential that would prevent uncoupling (Llorente-Folch et al., 2016). Pyruvate is a known glutamate scavenger and increased conversion of lactate to pyruvate could enhance this capability (Li et al., 2014; Zlotnik et al., 2012).
A controversial finding is the significant stimulation of state 3 respiration by late glucose (Fig. 2). This suggests that despite known decreases in NAD+ (Clark et al., 2007; Deng-Bryant et al., 2011; Tavazzi et al., 2005) and GAPDH activity (Izumi and Zorumski, 2010), glycolytic flux can be stimulated and decreases in glycolytic flux during hypometabolism may instead represent a coping strategy to reduce secondary damage and promote repair, but retains the capacity to deal with a second insult. Late glucose also significantly reduced ROS production (Fig. 4A) that can likely be attributed to increased shunting of glucose to pentose phosphate pathway, production of NADPH and thus increased antioxidant capacity. Despite the capacity to reduce ROS production early impairment of mitochondrial function by initial calcium rise and ROS was not reversible. Increased blood glucose can also initiate the polyol pathway, resulting in increased inflammation that can affect mitochondrial function (Chung et al., 2003; Xu et al., 2016). We were also interested in effects of combining substrates that appeared to best increase mitochondrial function. Based on this, we combined early lactate with late BHB. Animals were infused with lactate 0–3 h, saline 3–6 h and finally BHB 6–9 h post-injury. Similarly, combination treatment stimulated state 3 respiration, but surprisingly mitochondrial were uncoupled and ROS production was not reduced compared to using BHB and lactate separately (Fig. 4). We think this could imply that while the brain is prepared to alternate to one other fuel source and adapt, adding a second within a short period of time may act a metabolic stressor. Increased cerebral ketone concentrations may displace lactate oxidation, resulting in lactate accumulation and further placing demand on already impaired glucose utilization (Pan et al., 2000; Rolleston and Newsholme, 1967).
4.2. Females
Unlike males, despite an injury-induced significant decrease in state 3 respiration, overall RCR was not significantly impacted by injury (Fig. 3) and changes were not observed in ROS production (Fig. 5A–C). This signifies a different injury timeline compared to males that may impact that has been previously observed. Neither glucose nor lactate had significant impact on RCR, but both early and late BHB significantly reduced RCR (Fig. 3). Studies on exogenous BHB supplementation overall are lacking, but especially in women. Exogenous BHB is known to affect blood glucose and the hormones ghrelin and insulin (Stubbs et al., 2018; Sumithran et al., 2013). In women it lowers estrogen and can increase cortisol (Bergendahl et al., 1999; Estacio et al., 1996; Rosen and Petty, 1974; Ryan et al., 2018). Differential effects on mitochondrial function could be due to interference in signaling to estrogen receptors located on the mitochondria. In the single study observing exogenous ketones on female rats and TBI, BHB did not improve tissue extravasation in injured animals and increased basal tissue extravasation in healthy naïve shams (Orhan et al., 2016). As the females do not appear to be as metabolically impacted as males, additional glucose and lactate may not be perceived as stressors as they are both routinely metabolized by the brain.
4.3. Somewhere in the middle (of the TCA cycle)
Despite numerous studies on glycolytic flux and mitochondrial function following TBI, the pathway that links the two, the TCA cycle, remains understudied. The TCA cycle is regulated by its first enzyme, citrate synthase. Citrate synthase is homeostatically regulated by the energy conditions of the cell, specifically the ratios of ATP:ADP and NADH:NAD+ that signal the cell’s energy needs are being met (Pietrocola et al., 2015). Other conditions that increase amounts of acetyl CoA and succinyl CoA can inhibit enzyme activity as well leading to decreased electron flux into the mitochondria.
4.4. Males
We compared injury against our most effective treatment groups. While injury alone reduced acetyl CoA by 108% (Fig. 6A), it was not significant, though it does suggest glycolytic flux has decreased and is consistent with current literature. This may also suggest inhibition of the pyruvate dehydrogenase complex and/or shunting of pyruvate to other pathways. No significant differences in mitochondrial acetyl CoA were found in males or females. This may be due to limitations of the assay. Acetyl CoA is quickly converted in highly metabolic tissues and requires the use of irradiation fixation to measure. Although mitochondrial samples were immediately deproteinated, this may not have been able prevent acetyl CoA degradation. We found that both lactate and BHB increased acetyl CoA content (Fig. 6A). Given the stimulation of state 3 respiration this was expected as both BHB and lactate should be converted into acetyl CoA. Injury significantly reduced citrate synthase activity and was only improved by combination treatment. Lack of acetyl CoA in injured animals likely contributes to the decline in enzymatic activity; however, acetyl CoA was significantly increased in both lactate and BHB indicating while enzymatic activity was not rescued indicating another mechanism of inhibition. As shown in Fig. 6C, ATP was not significantly decreased and is not a candidate. Continued inhibition may instead be driven to changes in the NADH:NAD+ ratio. NAD+ is consumed through many mechanisms following TBI, including degradation by the DNA repair enzyme, PARP that can lead to redox imbalance (Clark et al., 2007; Won et al., 2012).
4.5. Females
In contrast to males no injury effect was seen on tissue acetyl CoA content (Fig. 7A–C). Although there was no injury effect, early glucose increased acetyl CoA (Fig. 7A) and is likely due to stimulation of glycolysis from additional glucose, implying though there is no significant change in RCR, there is still a high metabolic need immediately following injury in females. Demand appears to be further increased in early BHB animals as acetyl CoA is increased (Fig. 7B). In contrast, late BHB significantly decreased compared to injured animals and raises the possibly that demand is so high, glycolysis cannot keep up with acetyl CoA production such that it is measurable. Alternatively, it could be due to mitochondrial inhibition and thus negative feedback on citrate synthase and glycolysis. Unsurprisingly, as lactate had no significant effects on RCR, acetyl CoA was not affected. Despite adequate acetyl CoA, citrate synthase activity was significantly reduced in injured animals (Fig. 7D–F). Similarly, this could be due changes in NADH:NAD+ ratio. In addition to degradation of NAD+ by PARP, mitochondrial inhibition at complex I will prevent conversion of NADH to NAD+ (Blacker and Duchen, 2016; Pryde and Hirst, 2011), thus maintaining a high NADH:NAD+ ratio as may be evidenced by decreased RCR in late glucose and continued citrate synthase inhibition. Though activity is decreased in injury and glucose groups (Fig. 7D), it does not appear a limiting step as no significant changes in ATP present (Fig. 7G–I). Both BHB groups also had significantly diminished activity (Fig. 7E) that could be further compounded by BHB metabolism. Conversion of BHB to acetyl CoA requires NAD+ as a cofactor. In addition, elevated levels of acetyl CoA production can directly inhibit citrate synthase. BHB metabolism also results in the production of succinate that interfere with enzyme activity as well (Newman and Verdin, 2014). This may be more applicable to early BHB as late BHB ATP content was significantly reduced compared to shams. The combination of low acetyl CoA, decreased citrate synthase activity and uncoupled mitochondria likely all contribute to this decrease. Acetyl CoA was not affected by lactate treatment (Fig. 7B) and appears to attenuate decreases in enzyme activity. In addition to its role as a fuel source, lactate may contribute to repletion of NAD+. Following TBI, nicotinamine phosphoribosyltransferase (NAMPT) is upregulated and increases production of nicatinamine mononucleotide (NMN) (Chen et al., 2012). In the presence of abundant oxaloacetate (OAA), lactate and OAA are converted into malate and pyruvate via the lactate-malate transhydrogenase and requires NMN as a cofactor (Kane, 2014). Excessive pyruvate under uncoupled glycolysis and oxidative phosphorylation, which mimics anaerobic conditions, favors the conversion of pyruvate to lactate, creating NAD+ in the process (Burns and Manda, 2017). Inhibition of citrate synthase (Fig. 7D–F) could lead to build up of OAA and in combination with excess lactate lead to conversion of both to pyruvate and malate.
4.6. Alternative substrates prevent cellular stress and promote glial health
The timeline of glycogen breakdown and replenishment has not been established in the brain following TBI. Glycogen was decreased by 62% in injured animals, but was not significant (Fig. 8A). This may reflect ongoing replenishment processes and require looking at immediate time points to capture adequate glycogen breakdown. All treatment groups had significantly higher glycogen content compared to injured animals (Fig. 8A), although the mechanisms are likely different. Early excess lactate is proposed to prevent glycogen breakdown by decreasing energetic “stress-signals” from neurons to astrocytes. In addition to glucose, lactate can also use glycogenesis to replace glycogen stores (Stevenson et al., 1987). In the case of BHB, by providing an alternative fuel, ingested glucose could be shunted to glycogenesis, while combination treatment may be driven by both or either treatments. HMGB1 has been shown to decrease in contusional tissue (Gao et al., 2012), while increasing amounts are show in peri-contusional and non-injured tissues (Gao et al., 2012). While our data show a pattern that in is alignment with published studies, it was not significant (Fig. 8B). This may be due to limitations in technique. Homogenates were obtained from the entire ipsilateral irradiated cortex and thus, based on regional differences in expression, our effect may be washed out.
5. Conclusions
While many studies have shown alternative substrates can have neuroprotective properties, their effects on the entire metabolic pathway remain unclear. This is the first study to examine the contribution of cerebral metabolic state on substrate metabolism. We have shown that neuroprotective qualities of alternative substrates are dependent on cerebral metabolic state (Fig. 10). Despite being assumed to be neuroprotective, substrates administered at a suboptimal time can have deleterious effects on metabolic pathways. We also addressed how alternative substrates primarily explored in male rodents are metabolized in females. Our data show that at 24 h post-injury males and females have different injury patterns. In addition, exogenous BHB significantly worsened metabolic outcomes in female rats compared to males and implores the need to study both sexes and study them independently of each other. Our also open the possibility of revisiting “failed” alternative substrates as they may have just not been administered at the correct time. Exogenous glucose has been shown to improve performance in the spontaneous alternation task in naïve animals and could have benefits later during recovery. Our study is currently limited by only analyzing a single acute timepoint. Other groups have shown different aspects of injury that peak at different times in males vs. females that includes cytoskeletal damage (Kupina et al., 2003) and inflammatory patterns (Villapol et al., 2017). To date, no one has yet characterized female cerebral metabolism or mitochondrial function following as has been completed in male animals (Hall et al., 2004; Prins et al., 2013b; Singh et al., 2006; Yoshino et al., 1991). Future studies are needed such that we understand what the injury profile means in regard to outcome and allows the ability to design interventions as appropriately needed. Further work is needed to determine the long-term effectiveness of a single acute infusion on metabolism. While in the rat changes in cerebral metabolism are chronologically similar between animals, this is not observed clinically. Hyper- and hypo-metabolism happen in differing ranges between patients and currently imaging techniques such as fludeoxyglucose-positron emission tomography (FDG-PET) is the only way to confirm cerebral metabolic state. While informative, these techniques are not feasible for every patient and often are not able to be done repeatedly to track recovery. Finding a minimally invasive correlate to cerebral metabolism is necessary to determine what fuel type a patient should receive and provides a method to track recovery. In addition, this study shows that substrates may act partially imperfect depending on the outcome variable. This provides justification to begin making decisions of what substrate types may be the most beneficial, while also exposing sex-differences in metabolism that could potentially worsen outcomes and emphasizes the need for clarity in understanding of injury mechanism and metabolism between male and female TBI patients.
Fig. 10.
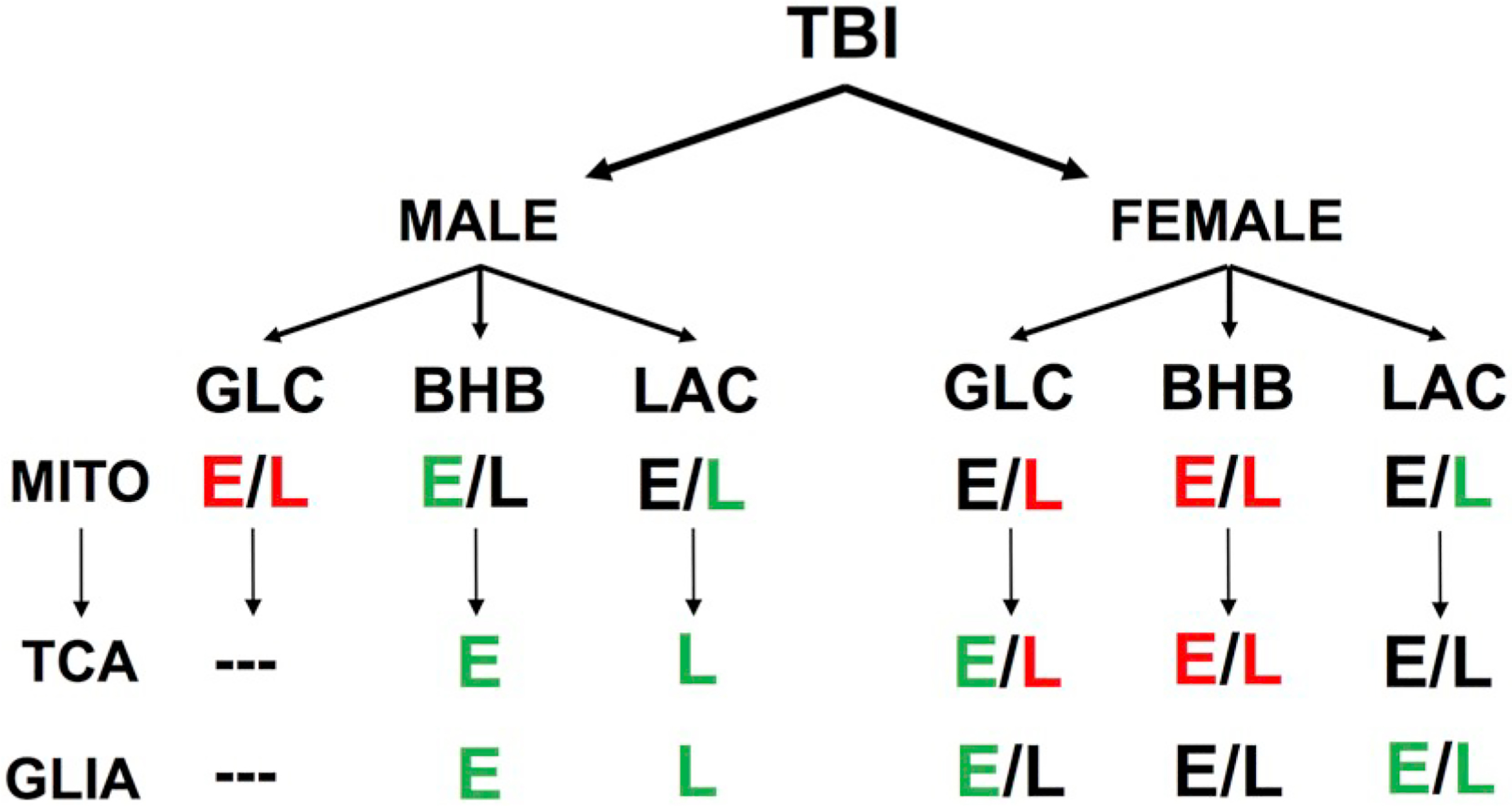
Differential effects of substrate administration based on early (E) or late (L) administration and sex. Positive effects are represented in green, negative effects in red and neutral in black.
Supplementary Material
Funding
This work was supported by the National Institutes of Health: NS058489, NS104311; UCLA Brain Injury Research Center, Marilyn and Austin Anderson Fellowship, Easton Labs for Brain Health and Steve Tisch BrainSPORT Program. Funding sources had no involvement in study design; or in the collection, analysis or interpretation of data.
Abbreviations:
- BHB
Beta-hydroxybutyrate
- CCI
Controlled cortical impact
- FDG-PET
fludeoxyglucose-positron emission tomography
- Glc
Glucose
- HMGB1
High Mobility Group Box 1
- NAMPT
Nicotinamide phosphoribosyltransferase
- NMN
Nicotinamide mononucleotide
- RCR
Respiratory control ratio
- TLR4
Toll-like receptor 4
- TBI
Traumatic brain injury
Footnotes
Declaration of Competing Interest
The authors report no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.expneurol.2020.113289.
References
- Au AK, Aneja RK, Bell MJ, Bayir H, Feldman K, Adelson PD, Fink EL, Kochanek PM, Clark RS, 2012. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J. Neurotrauma 29, 2013–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergendahl M, Evans WS, Pastor C, Patel A, Iranmanesh A, Veldhuis JD, 1999. Short-term fasting suppresses leptin and (conversely) activates disorderly growth hormone secretion in midluteal phase women–a clinical research center study. J. Clin. Endocrinol. Metab 84, 883–894. [DOI] [PubMed] [Google Scholar]
- Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, Phelps ME, McArthur DL, Caron MJ, Kraus JF, Becker DP, 1997. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J. Neurosurg 86, 241–251. [DOI] [PubMed] [Google Scholar]
- Blacker TS, Duchen MR, 2016. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med 100, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Ransom BR, 2007. Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Povlishock JT, 1999. Postinjury cyclosporin a administration limits axonal damage and disconnection in traumatic brain injury. J. Neurotrauma 16, 511–521. [DOI] [PubMed] [Google Scholar]
- Burns JS, Manda G, 2017. Metabolic pathways of the Warburg effect in health and disease: perspectives of choice, chain or chance. Int. J. Mol. Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali C, Tauffenberger A, Magistretti P, 2019. The strategic location of glycogen and lactate: from body energy reserve to brain plasticity. Front. Cell. Neurosci 13, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter KL, Jalloh I, Hutchinson PJ, 2015. Glycolysis and the significance of lactate in traumatic brain injury. Front. Neurosci 9, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carteron L, Solari D, Patet C, Quintard H, Miroz JP, Bloch J, Daniel RT, Hirt L, Eckert P, Magistretti PJ, Oddo M, 2018. Hypertonic lactate to improve cerebral perfusion and glucose availability after acute brain injury. Crit. Care Med 46, 1649–1655. [DOI] [PubMed] [Google Scholar]
- Casey PA, McKenna MC, Fiskum G, Saraswati M, Robertson CL, 2008. Early and sustained alterations in cerebral metabolism after traumatic brain injury in immature rats. J. Neurotrauma 25, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Weng JF, Hong WC, Luo LF, Yu W, Luo SD, 2012. Change in plasma visfatin level after severe traumatic brain injury. Peptides 38, 8–12. [DOI] [PubMed] [Google Scholar]
- Chung SS, Ho EC, Lam KS, Chung SK, 2003. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol 14, S233–S236. [DOI] [PubMed] [Google Scholar]
- Clark RS, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabo C, 2007. Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD + depletion and improves water maze performance after traumatic brain injury in mice. J. Neurotrauma 24, 1399–1405. [DOI] [PubMed] [Google Scholar]
- Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG, 2008. Fasting is neuroprotective following traumatic brain injury. J. Neurosci. Res 86, 1812–1822. [DOI] [PubMed] [Google Scholar]
- Deng-Bryant Y, Prins ML, Hovda DA, Harris NG, 2011. Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J. Neurotrauma 28, 1813–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacio MA, Yamada S, Tsukamura H, Hirunagi K, Maeda K, 1996. Effect of fasting and immobilization stress on estrogen receptor immunoreactivity in the brain in ovariectomized female rats. Brain Res. 717, 55–61. [DOI] [PubMed] [Google Scholar]
- Fedorovich SV, Voronina PP, Waseem TV, 2018. Ketogenic diet versus ketoacidosis: what determines the influence of ketone bodies on neurons? Neural Regen. Res 13, 2060–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao TL, Yuan XT, Yang D, Dai HL, Wang WJ, Peng X, Shao HJ, Jin ZF, Fu ZJ, 2012. Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J. Trauma Acute Care Surg 72, 643–649. [DOI] [PubMed] [Google Scholar]
- Gardner A, Iverson GL, Stanwell P, 2014. A systematic review of proton magnetic resonance spectroscopy findings in sport-related concussion. J. Neurotrauma 31, 1–18. [DOI] [PubMed] [Google Scholar]
- Glenn TC, Martin NA, Horning MA, McArthur DL, Hovda DA, Vespa P, Brooks GA, 2015. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J. Neurotrauma 32, 820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco T, Glenn TC, Hovda DA, Prins ML, 2016. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cerebr. Blood Flow Metab 36, 1603–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Cheney DL, Trabucchi M, Doteuchi M, Wang C, 1974. Focussed microwave radiation: a technique to minimize post mortem changes of cyclic nucleotides, dopa and choline and to preserve brain morphology. Neuropharmacology 13, 1115–1122. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC, 2004. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J. Neurotrauma 21, 9–20. [DOI] [PubMed] [Google Scholar]
- Hill RL, Singh IN, Wang JA, Hall ED, 2017. Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem. Int 111, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson PJ, O’Connell MT, Seal A, Nortje J, Timofeev I, Al-Rawi PG, Coles JP, Fryer TD, Menon DK, Pickard JD, Carpenter KL, 2009. A combined microdialysis and FDG-PET study of glucose metabolism in head injury. Acta Neurochir. 151, 51–61 (discussion 61). [DOI] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF, 2010. Neuroprotective effects of pyruvate following NMDA-mediated excitotoxic insults in hippocampal slices. Neurosci. Lett 478, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Allaman I, Rothenfusser K, Fiumelli H, Marquet P, Magistretti PJ, 2016. L-lactate protects neurons against excitotoxicity: implication of an ATP-mediated signaling cascade. Sci. Rep 6, 21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane DA, 2014. Lactate oxidation at the mitochondria: a lactate-malate-aspartate shuttle at work. Front. Neurosci 8, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS, 2011. Cyclosporin a preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J. Neurotrauma 28, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbaugh TJ, Karlsson M, Byro M, Bebee A, Ralston J, Sullivan S, Duhaime AC, Hansson MJ, Elmer E, Margulies SS, 2015. Mitochondrial bioenergetic alterations after focal traumatic brain injury in the immature brain. Exp. Neurol 271, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Sahel D, Kaira M, Raj K, Sharma S, Singh S, 2019. Mitochondrial dysfunctioning and neuroinflammation: recent highlights on the possible mechanisms involved in traumatic brain injury. Neurosci. Lett 710, 134347. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Detloff MR, Bobrowski WF, Snyder BJ, Hall ED, 2003. Cytoskeletal protein degradation and neurodegeneration evolves differently in males and females following experimental head injury. Exp. Neurol 180, 55–73. [DOI] [PubMed] [Google Scholar]
- Li J, Gu L, Feng DF, Ding F, Zhu G, Rong J, 2012. Exploring temporospatial changes in glucose metabolic disorder, learning, and memory dysfunction in a rat model of diffuse axonal injury. J. Neurotrauma 29, 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hou X, Qi Q, Wang L, Luo L, Yang S, Zhang Y, Miao Z, Zhang Y, Wang F, Wang H, Huang W, Wang Z, Shen Y, Wang Y, 2014. Scavenging of blood glutamate for enhancing brain-to-blood glutamate efflux. Mol. Med. Rep 9, 305–310. [DOI] [PubMed] [Google Scholar]
- Llorente-Folch I, Rueda CB, Perez-Liebana I, Satrustegui J, Pardo B, 2016. L-lactate-mediated neuroprotection against glutamate-induced excitotoxicity requires ARALAR/AGC1. J. Neurosci 36, 4443–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM, 2007. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264 (Epub 2007 Jan 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley AA, Govind V, Saigal G, Gold SG, Harris L, Sheriff S, 2017. Longitudinal MR spectroscopy shows altered metabolism in traumatic brain injury. J. Neuroimaging 27, 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED, 2008. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp. Neurol 209, 243–253. [DOI] [PubMed] [Google Scholar]
- Nagai MA, Sonohara S, Brentani MM, 1988. Estrogen control of lactate dehydrogenase isoenzyme-5 in human breast cancer. Int. J. Cancer 41, 10–16. [DOI] [PubMed] [Google Scholar]
- Newman JC, Verdin E, 2014. Beta-hydroxybutyrate: much more than a metabolite. Diabetes Res. Clin. Pract 106, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuma Y, Liu K, Wake H, Zhang J, Maruo T, Date I, Yoshino T, Ohtsuka A, Otani N, Tomura S, Shima K, Yamamoto Y, Yamamoto H, Takahashi HK, Mori S, Nishibori M, 2012. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol 72, 373–384. [DOI] [PubMed] [Google Scholar]
- Okuma Y, Liu K, Wake H, Liu R, Nishimura Y, Hui Z, Teshigawara K, Haruma J, Yamamoto Y, Yamamoto H, Date I, Takahashi HK, Mori S, Nishibori M, 2014. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 85, 18–26. [DOI] [PubMed] [Google Scholar]
- Orhan N, Ugur Yilmaz C, Ekizoglu O, Ahishali B, Kucuk M, Arican N, Elmas I, Gurses C, Kaya M, 2016. Effects of beta-hydroxybutyrate on brain vascular permeability in rats with traumatic brain injury. Brain Res. 1631, 113–126. [DOI] [PubMed] [Google Scholar]
- Pan JW, Rothman TL, Behar KL, Stein DT, Hetherington HP, 2000. Human brain beta-hydroxybutyrate and lactate increase in fasting-induced ketosis. J. Cerebr. Blood Flow Metab 20, 1502–1507. [DOI] [PubMed] [Google Scholar]
- Pedrazzi M, Patrone M, Passalacqua M, Ranzato E, Colamassaro D, Sparatore B, Pontremoli S, Melloni E, 2007. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J. Immunol 179, 8525–8532. [DOI] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G, 2015. Acetyl coenzyme a: a central metabolite and second messenger. Cell Metab. 21, 805–821. [DOI] [PubMed] [Google Scholar]
- Prins ML, Hovda DA, 2009. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J. Neurotrauma 26, 1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Lee SM, Fujima LS, Hovda DA, 2004a. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J. Neurochem 90, 666–672. [DOI] [PubMed] [Google Scholar]
- Prins ML, Lee SM, Fujima LS, Hovda DA, 2004b. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J. Neurochem 90, 666–672. [DOI] [PubMed] [Google Scholar]
- Prins ML, Fujima LS, Hovda DA, 2005. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res 82, 413–420. [DOI] [PubMed] [Google Scholar]
- Prins M, Greco T, Alexander D, Giza CC, 2013a. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech 6, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ML, Alexander D, Giza CC, Hovda DA, 2013b. Repeated mild traumatic brain injury: mechanisms of cerebral vulnerability. J. Neurotrauma 30, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde KR, Hirst J, 2011. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem 286, 18056–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AC, Zsoldos R, Chen T, Wilson MS, Alessandri B, Hamm RJ, Bullock MR, 2002. Lactate administration attenuates cognitive deficits following traumatic brain injury. Brain Res. 928, 156–159. [DOI] [PubMed] [Google Scholar]
- Rolleston FS, Newsholme EA, 1967. Effects of fatty acids, ketone bodies, lactate and pyruvate on glucose utilization by guinea-pig cerebral cortex slices. Biochem. J 104, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen EF, Petty LC, 1974. Food deprivation effects on some estrogen-sensitive responses in female rats. Physiol. Behav. 12, 767–770. [DOI] [PubMed] [Google Scholar]
- Ryan KK, Packard AEB, Larson KR, Stout J, Fourman SM, Thompson AMK, Ludwick K, Habegger KM, Stemmer K, Itoh N, Perez-Tilve D, Tschop MH, Seeley RJ, Ulrich-Lai YM, 2018. Dietary manipulations that induce ketosis activate the HPA Axis in male rats and mice: a potential role for fibroblast growth factor21. Endocrinology 159, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutkowski A, Wege N, Stangl GI, Konig B, 2014. Tissue-specific expression of monocarboxylate transporters during fasting in mice. PLoS One 9, e112118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NS, Brown LL, 1978. Use of microwave irradiation to prevent postmortem catecholamine metabolism: evidence for tissue disruption artifact in a discrete region of rat brain. Brain Res. 140, 171–176. [DOI] [PubMed] [Google Scholar]
- Shi H, Wang HL, Pu HJ, Shi YJ, Zhang J, Zhang WT, Wang GH, Hu XM, Leak RK, Chen J, Gao YQ, 2015. Ethyl pyruvate protects against blood-brain barrier damage and improves long-term neurological outcomes in a rat model of traumatic brain injury. CNS Neurosci. Ther 21, 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shijo K, Ghavim S, Harris NG, Hovda DA, Sutton RL, 2015. Glucose administration after traumatic brain injury exerts some benefits and no adverse effects on behavioral and histological outcomes. Brain Res. 1614, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shijo K, Sutton RL, Ghavim SS, Harris NG, Bartnik-Olson BL, 2017. Metabolic fate of glucose in rats with traumatic brain injury and pyruvate or glucose treatments: a NMR spectroscopy study. Neurochem. Int. 102, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Lazzarino G, Beaumont A, Vagnozzi R, 2001. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J. Neurotrauma 18, 977–991. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED, 2006. Time course of posttraumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J. Cereb. Blood Flow Metab 26, 1407–1418 (Epub 2006 Mar 1415). [DOI] [PubMed] [Google Scholar]
- Starkov AA, 2010. Measurement of mitochondrial ROS production. Methods Mol. Biol 648, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson RW, Mitchell DR, Hendrick GK, Rainey R, Cherrington AD, Frizzell RT, 1987. Lactate as substrate for glycogen resynthesis after exercise. J. Appl. Physiol 62, 2237–2240. [DOI] [PubMed] [Google Scholar]
- Stubbs BJ, Cox PJ, Evans RD, Cyranka M, Clarke K, de Wet H, 2018. A ketone Ester drink lowers human ghrelin and appetite. Obesity (Silver Spring) 26, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW, 1999. Cyclosporin a attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol 160, 226–234. [DOI] [PubMed] [Google Scholar]
- Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, Proietto J, 2013. Ketosis and appetite-mediating nutrients and hormones after weight loss. Eur. J. Clin. Nutr 67, 759–764. [DOI] [PubMed] [Google Scholar]
- Tavazzi B, Signoretti S, Lazzarino G, Amorini AM, Delfini R, Cimatti M, Marmarou A, Vagnozzi R, 2005. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 56, 582–589 (discussion 582–589). [DOI] [PubMed] [Google Scholar]
- Vagnozzi R, Tavazzi B, Signoretti S, Amorini AM, Belli A, Cimatti M, Delfini R, Di Pietro V, Finocchiaro A, Lazzarino G, 2007. Temporal window of metabolic brain vulnerability to concussions: mitochondrial-related impairment–part I. Neurosurgery 61, 379–388 (discussion 388–379). [DOI] [PubMed] [Google Scholar]
- Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP, 2000. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J. Neurosurg 93, 815–820. [DOI] [PubMed] [Google Scholar]
- Villapol S, Loane DJ, Burns MP, 2017. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 65, 1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, Golding EM, Headrick JP, 1994. Bioenergetic analysis of oxidative metabolism following traumatic brain injury in rats. J. Neurotrauma 11, 265–274. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang Y, Xiang X, Zhu Y, Men J, He M, 2010. Estimation of the normal range of blood glucose in rats. Wei Sheng Yan Jiu 39 (133–137, 142). [PubMed] [Google Scholar]
- Won SJ, Choi BY, Yoo BH, Sohn M, Ying W, Swanson RA, Suh SW, 2012. Prevention of traumatic brain injury-induced neuron death by intranasal delivery of nicotinamide adenine dinucleotide. J. Neurotrauma 29, 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Begley P, Church SJ, Patassini S, McHarg S, Kureishy N, Hollywood KA, Waldvogel HJ, Liu H, Zhang S, Lin W, Herholz K, Turner C, Synek BJ, Curtis MA, Rivers-Auty J, Lawrence CB, Kellett KA, Hooper NM, Vardy ER, Wu D, Unwin RD, Faull RL, Dowsey AW, Cooper GJ, 2016. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: metabolic basis for dementia. Sci. Rep 6, 27524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G, 2018. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol 217, 2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo RA, Gasparovic C, Merideth F, Ruhl D, Doezema D, Mayer AR, 2011. A longitudinal proton magnetic resonance spectroscopy study of mild traumatic brain injury. J. Neurotrauma 28, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP, 1991. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 561, 106–119. [DOI] [PubMed] [Google Scholar]
- Youssef FF, 2015. Ketone bodies attenuate excitotoxic cell injury in the rat hippocampal slice under conditions of reduced glucose availability. Neurol. Res 37, 211–216. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Sinelnikov I, Gruenbaum BF, Gruenbaum SE, Dubilet M, Dubilet E, Leibowitz A, Ohayon S, Regev A, Boyko M, Shapira Y, Teichberg VI, 2012. Effect of glutamate and blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome and pathohistology of the hippocampus after traumatic brain injury in rats. Anesthesiology 116, 73–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.