

SUMMARY
A significant increase in dietary fructose consumption has been implicated as a potential driver of cancer. Metabolic adaptation of cancer cells to utilize fructose confers advantages for their malignant growth, but compelling therapeutic targets have not been identified. Here, we show that fructose metabolism of leukemic cells can be inhibited by targeting the de novo serine synthesis pathway (SSP). Leukemic cells, unlike their normal counterparts, become significantly dependent on the SSP in fructose-rich conditions as compared to glucose-rich conditions. This metabolic program is mediated by the ratio of redox cofactors, NAD+/NADH, and the increased SSP flux is beneficial for generating alpha-ketoglutarate from glutamine, which allows leukemic cells to proliferate even in the absence of glucose. Inhibition of PHGDH, a rate-limiting enzyme in the SSP, dramatically reduces leukemia engraftment in mice in the presence of high fructose, confirming the essential role of the SSP in the metabolic plasticity of leukemic cells.
eTOC
Excessive fructose intake has been implicated in cancer progression, and Jeong et al. show an unexpected metabolism of fructose in cancer cells. Acute myeloid leukemic cells upregulate the serine synthesis pathway to metabolize fructose-derived carbons, and targeting PHGDH, a rate limiting enzyme in the serine synthesis pathway, significantly reduces the tumor burden in the presence of high fructose.
Graphical Abstract
INTRODUCTION
Dietary fructose consumption, which has increased by >100-fold over the past two centuries, now accounts for xs~10% of total caloric intake in the United States (Marriott et al., 2009). As fructose is more palatable but less able to promote satiety than glucose, its increased consumption often leads to the development of obesity and metabolic syndrome (Dhingra et al., 2007; Johnson et al., 2007). Recently, multiple studies have demonstrated that cancer cells are able to utilize fructose as an additional fuel for their proliferation and metastasis. For example, pancreatic cancer cells increased flux through the non-oxidative pentose phosphate pathway in fructose-rich conditions, which led to the preferential utilization of fructose for nucleotide synthesis as compared to glucose (Liu et al., 2010). An elevated expression of aldolase-B, a key enzyme in fructose metabolism, potentially facilitates liver metastasis of breast cancer and colon cancer cells under a high-fructose diet (Bu et al., 2018). GLUT5, a potent transporter of fructose, has been shown to be significantly upregulated in non-small-cell lung cancer samples compared to normal lung tissues, which aids cancer cells to metabolize fructose and proliferate in glucose-limited conditions (Weng et al., 2018). Also, its elevated expression has been shown to accelerate glycolysis and fatty acid synthesis in murine colorectal cancer cells under treatment with high-fructose corn syrup (Goncalves et al., 2019). Considering that the metabolic plasticity of cancer cells enables them to exploit any available carbon sources to adapt and proliferate in diverse environments, high fructose intake might lead to accelerated tumor growth (Aune et al., 2012; Carreño et al., 2019; Strober and Brady, 2019). While much is known regarding fructose metabolism in the liver and small intestine, a deeper understanding of fructose metabolism in cancer is necessary to shed light on effective diagnostic and therapeutic strategies.
Risk of hematologic malignancy, including acute myeloid leukemia (AML), has been shown to increase in obese populations (Mazzarella et al., 2020; Poynter et al., 2016), and systematic metabolic alterations, which favor disease progression, have been observed in AML mouse models and patients with AML (Ye et al., 2018). In particular, an abnormal level of fructose in the bone marrow of individuals with AML has been reported, suggesting that fructose metabolism could be dysregulated (Chen et al., 2016). Based on these previous studies, we therefore sought to uncover mechanistic evidence that would link a therapeutic target to the altered fructose level in the bone marrow. More broadly, we aimed to fundamentally understand to what degree AML cells could utilize fructose for their malignant growth.
Here, we provide an underlying mechanism of how AML cells metabolize fructose in glucose-limited conditions. We have identified that AML cells exhibit a higher metabolic flux through the de novo serine synthesis pathway (SSP) in fructose-rich conditions compared to glucose-rich conditions, and their utilization of fructose can be inhibited by targeting phosphoglycerate dehydrogenase (PHGDH), a rate-limiting enzyme in the SSP. The SSP diverts the glycolytic intermediate, 3-phosphoglycerate (3-PG), to make serine and provide a precursor for 1-carbon metabolism (Ducker and Rabinowitz, 2017). Multiple studies have shown that the SSP plays a role in generating an anaplerotic substrate, alpha-ketoglutarate (aKG), for tricarboxylic acid (TCA) cycle, providing precursors for nucleotide synthesis and maintaining redox balance in multiple cancer cells, and therefore targeting the SSP has emerged as a promising therapeutic approach (Ducker and Rabinowitz, 2017; Pacold et al., 2016; Possemato et al., 2011; Sullivan et al., 2019a; Wei et al., 2019). We have found that fructose metabolism through the SSP in AML cells is mediated by redox cofactors, NAD+ and NADH, whose ratio is significantly higher in fructose-rich conditions than glucose-rich conditions. Importantly, this dependency on SSP was not observed in normal hematopoietic CD34+ cells, indicating a therapeutic index for targeting this pathway in AML. Leveraging this metabolic mechanism, we demonstrate that not only do leukemic cells increase the SSP flux in vivo but inhibition of PHGDH in the presence of high fructose dramatically reduces leukemia engraftment. Taken together, this work demonstrates that the SSP plays a critical role in the metabolic adapation of leukemic cells in the setting of elevated fructose and the SSP inhibition could be a promising therapeutic approach for leukemia.
RESULTS
Leukemic cells even with different characteristics of fructose metabolism proliferate well in fructose-rich conditions.
To investigate the fructose metabolism of leukemic cells, we cultured four leukemic cell lines, MOLM13 (FLT3-ITD AML), K562 (BCR-ABL CML), THP1 (MLL-AF9 AML) and KASUMI1 (AML1-ETO AML), in media with 10 mM glucose or fructose. Surprisingly, their growth rates in the fructose-rich condition were similar or higher than in the glucose-rich condition (Figures 1A and 1B). Further studies would be necessary to explain the outgrowth of THP1 cells in the fructose-rich condition, but we identified that the metabolic features of the four cell lines could be divided into two groups: MOLM13 and K562 cells consumed fructose much slower than glucose, whereas THP1 and KASUMI1 cells consumed those sugars with a similar rate (Figure 1C; Figure S1A). As expected, the former two cell lines generated less lactate in the fructose-rich condition than in the glucose-rich condition, and the latter two cell lines generated a similar amount of lactate in both conditions. To identify the metabolic fate of fructose in those cell lines, we conducted isotope tracing experiments with [2-13C] glucose or fructose, analyzing the metabolites in media using 13C-nuclear magnetic resonance (NMR) spectroscopy. Intriguingly, we found that the 13C-NMR signal of [2-13C] glycine and [2-13C] serine was significantly higher when MOLM13 and K562 cells were cultured in the media with [2-13C] fructose as compared to [2-13C] glucose (Figure 1D; Figures S1B and S1C); we did not find this metabolic difference in the culture media of THP1 and KASUMI1 cells. We quantitatively compared the ratio of [2-13C] glycine to [2-13C] lactate across the four cell lines, as these metabolites were generated from [2-13C] glucose or fructose via different pathways and this approach could reveal the differential contribution normalized to the uptake of the nutrient (Figure S1D); [2-13C] glycine is the end product of the SSP and [2-13C] lactate is the end product of glycolysis. The ratio analysis of [2-13C] glycine to [2-13C] lactate clearly demonstrated that MOLM13 and K562 cells metabolized fructose through the SSP more significantly than THP1 and KASUMI1 cells (Figure S1E). Liquid chromatography-mass spectrometry (LC-MS) analysis of intracellular metabolites in MOLM13 cells incubated with [U-13C6] glucose or fructose consistently showed that the total pool size of serine and glycine as well as their fractional enrichment were much higher in the fructose-rich condition compared to the glucose-rich condition (Figure 1E). The intracellular total pool size and enrichment of lactate were significantly lower in the fructose-rich condition, which was also consistent with the 13C-NMR media analysis (Figure 1D). Considering that the pool size of unlabeled metabolites was similar between the two conditions, the difference in the total pool size of metabolites was mainly due to the contribution from fully labeled metabolites (Figure S1F). To confirm whether the flux through the SSP was elevated in MOLM13 cells, we conducted a time course analysis of extracellular metabolites of these cells; the level of extracellular [2-13C] glycine increased with time significantly faster in the [2-13C] fructose media compared to the [2-13C] glucose media (3.35 vs. 1.39 μM/hr), while the level of [2-13C] lactate increased significantly slower (0.03 vs. 0.52 mM/hr) (Figures 2A and 2B). The time course analysis of intracellular metabolites of MOLM13 cells incubated with [U-13C6] fructose confirmed that the pool size and fractional enrichment of serine (m+3) and glycine (m+2) continuously increased with time (Figures 2C and 2D).
Figure 1. Leukemic cells exhibit distinct metabolism of fructose.

(A) Growth curves of MOLM13 and K562 cells in the culture media with 10 mM glucose or fructose (mean ± S.D., n = 4 biological replicates).
(B) Comparison of growth rates of four leukemic cell lines. The growth rate in the fructose-rich condition was normalized to that in the glucose-rich condition (The box-whisker plot shows the center line as median, the box limits as upper and lower quartiles, and the whiskers to minimum and maximum values).
(C) Changes in metabolites in the culture media of the four leukemic cell lines (mean ± S.D., n = 3 biological replicates).
(D) Comparison of 13C-NMR spectra of the media after MOLM13 cells were cultured with 10 mM [2-13C] glucose or fructose for 2 days. The NMR peak corresponding to each metabolite was compared based on the peak area, and the fold difference was calculated with the NMR spectra from the fructose-rich condition normalized to that from the glucose-rich condition. Each dot indicates the mean value of the fold difference from three biological replicates.
(D) Comparison of the intracellular level of serine, glycine, and lactate and their enrichment in MOLM13 cells cultured with 10 mM [U-13C] glucose or fructose for 2 days. Signal intensity for the metabolites from the fructose-rich condition was normalized to that from the glucose-rich condition (mean ± S.D., n = 3 biological replicates).
All statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 2. Metabolic flux through the serine synthesis pathway is higher in the fructose-rich condition.

(A) Representative 13C-NMR spectra of the media after MOLM13 cells were cultured for 1, 3, and 7 hrs with 10 mM [2-13C] glucose or fructose. LAC, lactate; GLY, glycine.
(B) Quantitative comparison of the 13C-NMR peak area in the spectra from (A) (mean ± S.D., n = 3 biological replicates).
(C) Changes in the fractional enrichment of intracellular metabolites in MOLM13 cells cultured in the media with 10 mM [U-13C6] fructose for 0, 0.5, 1, 3, and 7 hrs (mean ± S.D., n = 3 biological replicates). 3-PG, 3-phosphoglycerate.
(D) Changes in the absolute amount of fully-labeled intracellular metabolites in MOLM13 cells from (C) (mean ± S.D., n = 3 biological replicates).
Statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
GLUT5 level is a key factor that determines the difference in fructose metabolism.
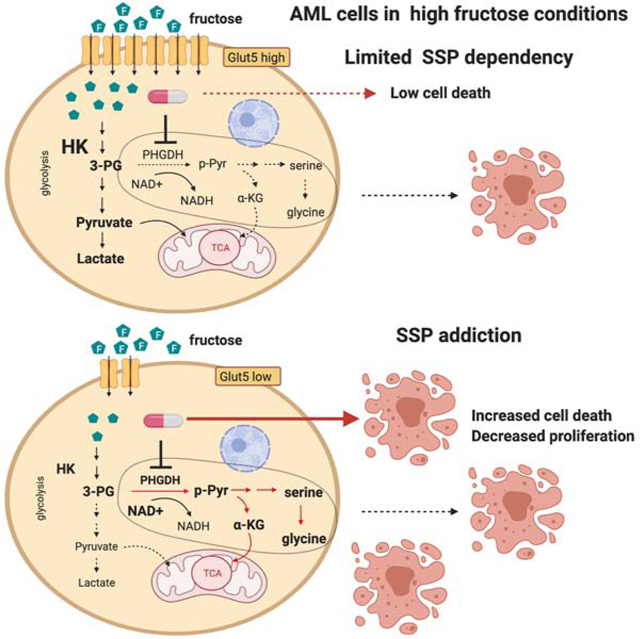
To identify putative differences that drive fructose metabolism in these two groups (MOLM13 and K562 vs. THP1 and KASUMI1), we surveyed the expression level of metabolic enzymes and the predominant fructose transporter, GLUT5 (Figure 3A; Figure S2A), as it was reported that GLUT5 expression was higher in patients with AML compared to healthy controls and the patients with a higher level of GLUT5 showed a poor prognosis (Figures S2B and S2C). We found that GLUT5 level was dramatically different between the groups; MOLM13 and K562 cells had a markedly lower abundance of GLUT5, compared to THP1 and KASUMI1 cells. Using flow cytometry analysis, we confirmed that the difference in the total amount of GLUT5 was readily observed on a per cell basis and similar to the difference in its abundance on the plasma membrane (Figure 3B). We next compared GLUT5 abundance across seven additional AML cell lines (KG1, TF1, NOMO1, MV411, U937, OCIAML2, and OCIAML3) as well as their fructose metabolism (Figure 3C). The cells with a low level of GLUT5 (median fluorescence intensity of less than 350) exhibited a more than 5-fold higher ratio of [2-13C] glycine to [2-13C] lactate in the [2-13C] fructose media compared to the [2-13C] glucose media, indicating that the relative contribution of carbons through the SSP was higher in the presence of high fructose (Figure 3D). To further confirm our hypothesis that GLUT5 expression determines the fate of fructose metabolism, we genetically modulated the GLUT5 level: overexpression of GLUT5 in K562 cells (GLUT5-low) and knockdown of GLUT5 in THP1 and OCIAML3 cells (GLUT5-high). The GLUT5 overexpression in K562 cells led to not only higher consumption of fructose and generation of lactate, but also lower generation of glycine, which made their fructose metabolism similar to that of GLUT5-high cells (Figures 3E and 3F; Figure S2D); the ratio of [2-13C] glycine to [2-13C] lactate was substantially lower with GLUT5 overexpression in the fructose-rich condition. As the counterpart, the GLUT5 knockdown in THP1 and OCIAML3 cells significantly decreased the fructose consumption and increased the ratio of [2-13C] glycine to [2-13C] lactate in the [2-13C] fructose media compared to the [2-13C] glucose media (Figures 3G and 3H; Figures S2E-S2G). Therefore, the GLUT5 expression level determines the level of fructose uptake, which in turn regulates the SSP flux. We also analyzed the GLUT5 expression level in mononuclear cells collected from eight patients with AML, one of which was concurrently analyzed with MOLM13 (GLUT5-low) and THP1 (GLUT5-high) cells to test whether the patient samples could be functionally classified as GLUT5-low or -high. Intriguingly, they predominantly exhibited GLUT5 abundance similar to that of MOLM13 cells, even with their different genetic backgrounds (Figure 3I; Table S1). These results imply that most AML cells will utilize the SSP in fructose-rich environments.
Figure 3. Lower level of GLUT5 leads to higher flux through the SSP in fructose-rich conditions.

(A and B) Immunoblot analysis of metabolic enzymes (A) and flow cytometry analysis of GLUT5 (B) in the four leukemic cell lines (mean ± S.D., n = 3 biological replicates). MFI, median fluorescence intensity.
(C) Flow cytometry analysis of GLUT5 in seven additional AML cell lines. MOLM13 and THP1 cell lines were analyzed together as a reference for GLUT5-low and GLUT5-high cells.
(D) Ratio of 13C-NMR peak area of [2-13C] glycine to that of [2-13C] lactate in the NMR spectrum of the media after the AML cells were cultured with 10 mM [2-13C] glucose or fructose for 2 days (mean ± S.D., n = 3 biological replicates).
(E) Flow cytometry analysis of GLUT5 in K562 cells with GLUT5 overexpression (GLUT5-OV) or empty vector overexpression (Ctrl-OV).
(F) Changes in the level of extracellular metabolites after K562 cells with GLUT5-OV and Ctrl-OV were cultured with 10 mM [2-13C] fructose for 2 days (mean ± S.D., n = 3 biological replicates).
(G) Flow cytometry analysis of GLUT5 in THP1 cells with GLUT5 knockdown (sh-GLUT5) or scramble knockdown (sh-Ctrl).
(H) Ratio of 13C-NMR peak area of [2-13C] glycine to that of [2-13C] lactate in the NMR spectrum of the media after THP1 cells with sh-GLUT5 and sh-Ctrl were cultured with 10 mM [2-13C] fructose for 2 days (mean ± S.D., n = 3 biological replicates). GLC, glucose; FRC, fructose.
(I) Flow cytometry analysis of GLUT5 in mononuclear cells collected from eight patients with AML. The Patient 6 sample was concurrently analyzed with MOLM13 and THP1 cell lines as a reference for GLUT5-low and GLUT5-high cells (right).
All statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001.
Normal hematopoietic stem/progenitor cells rarely increase the SSP flux in fructose-rich conditions.
To test if normal hematopoietic cells exhibit this metabolic program, we investigated GLUT5 abundance and fructose metabolism in normal hematopoietic stem and progenitor cells (CD34+ population collected from cord blood). While their GLUT5 surface abundance was heterogeneous, CD34+ cells exhibited a similar amount of GLUT5 as MOLM13 cells (Figure 4A). However, their glucose and fructose metabolism were similar to those of GLUT5-high cells (THP1 and KASUMI1), and they did not exhibit a higher flux through the SSP in the fructose-rich condition (Figures 4B–4D). Future studies investigating the role of fructose metabolism in normal hematopoietic stem and progenitor cells would be necessary, but these results imply that the high SSP flux may confer metabolic advantages to cancer cells and targeting this pathway has a potential therapeutic advantage.
Figure 4. Normal CD34+ cells express a low level of GLUT5, but their SSP flux is not higher in fructose-rich conditions compared to glucose-rich conditions.

(A) Flow cytometry analysis of GLUT5 in MOLM13 and CD34+ cells (mean ± S.D., n = 3 biological replicates). MFI, median fluorescence intensity.
(B) Growth curves of CD34+ cells in 10 mM glucose or fructose media (mean ± S.D., n = 3 biological replicates).
(C) Changes in metabolites in the media from the experiment (B) (mean ± S.D., n = 3 biological replicates).
(D) Comparison of the intracellular level of serine and glycine and their enrichment in CD34+ cells cultured with 10 mM [U-13C] glucose or fructose for 2 days (mean ± S.D., n = 3 biological replicates).
All statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Leukemic cells metabolize fructose through HK, rather than KHK.
Fructose is predominantly metabolized through ketohexokinase (KHK), rather than hexokinase (HK), in the small intestine and liver, the canonical organs of fructose metabolism (Andres-Hernando et al., 2020; Diggle et al., 2009; Goncalves et al., 2019; Jang et al., 2018; Lanaspa et al., 2018), but it is unknown whether leukemic cells are dependent on the same pathway for fructose metabolism. In order to address this question, we genetically inhibited KHK in both MOLM13 and THP1 cells (Figure S3A). Intriguingly, the growth rate of MOLM13 and THP1 cells were unchanged with KHK knockdown in fructose-rich conditions, and the level of [2-13C] glycine was not significantly different when KHK-knockdown MOLM13 cells were incubated in the media with [2-13C] fructose (Figures S3B and S3C). Additionally, incubation of MOLM13 cells with a selective KHK inhibitor (Maryanoff et al., 2011) showed no difference in growth when comparing glucose-rich and fructose-rich conditions (Figure S3D). It has been widely recognized that fructose phosphorylation by KHK leads to the depletion of intracellular ATP, followed by the activation of phosphofructokinase (PFK), a rate-limiting enzyme in glycolysis (van den Berghe et al., 1977; Lanaspa et al., 2018; Liu et al., 2020; Mäenpää et al., 1968). Our analysis of the intracellular level of ATP and AMP in MOLM13 cells cultured in glucose-rich or fructose-rich conditions showed that the ratio of ATP to AMP was similar between both conditions (Figure S3E), further supporting that KHK plays a minimal role in AML fructose metabolism. This could be due to a lower abundance of KHK in the AML cells compared to liver cells as demonstrated by assessing HepG2 cells, a liver cancer cell line (Figure S3F). As indicated by the ATP to AMP ratio, MOLM13 cells did not increase their glucose consumption when fructose was added in the same media (Figure S3G); even with 10 mM [2-13C] fructose supplementation, the [2-13C] lactate generation was not significantly changed, which implied that i) 13C-NMR signal of [2-13C] lactate was mostly from glucose-derived lactate and ii) the overall glycolytic rate was not changed. Intriguingly, [2-13C] glycine peak was nearly undetectable when both glucose and [2-13C] fructose were provided, while there was a distinct peak when [2-13C] fructose was provided without glucose (Figure S3H).
These findings led to our next hypothesis that fructose is metabolized through HK2, rather than KHK, in AML cells, as HK2 has been shown to be upregulated in various types of cancers, including AML, and be associated with a higher glycolytic flux in the malignant cells (Ju et al., 2017; Patra et al., 2013; Wang et al., 2014). To test this hypothesis, we evaluated the fructose metabolism in AML cells with HK2 knockdown, which clearly decreased [2-13C] lactate generation from [2-13C] fructose both in MOLM13 (GLUT5-low) and THP1 (GLUT5-high) cells (Figures S4A and S4B). In addition, we observed that [2-13C] glycine generation was decreased less significantly than [2-13C] lactate in MOLM13 cells and the generation was actually increased in THP1 cells with HK2 knockdown. This is further evidence that fructose is predominantly metabolized through HK2; the HK2 knockdown led to a significant decrease in the carbon flux from fructose, which made the GLUT5-high cells’ fructose metabolism similar to that of the GLUT5-low cells.
Alternatively, we tested the inhibition of fructose-directed SSP flux with the supplementation of 2-deoxy-D-glucose (2-DG), as 2-DG is metabolized by HK and its product (2-DG-6-phosphate) inhibits HK activity. In the presence of 2-DG, MOLM13 cells produced significantly less [2-13C] lactate and glycine in the media with [2-13C] fructose (Figure S4C). Lastly, we measured glucose and fructose consumption in the media of MOLM13 cells in the presence of both sugars. We found that fructose levels decreased only when glucose became limited (less than 1 mM) (Figure S4D). Overall, these results clearly demonstrate that the AML cells metabolize fructose through HK2, rather than KHK, which is in contrast to other organs and cellular contexts.
GLUT5-low leukemic cells are more dependent on the SSP in fructose-rich conditions.
We hypothesized that GLUT5-low leukemic cells are more dependent on the SSP in fructose-rich conditions (Figure 5A). To test this hypothesis, we modulated the SSP flux by targeting phosphoglycerate dehydrogenase (PHGDH) (Locasale et al., 2011; Possemato et al., 2011), the rate-limiting enzyme in the SSP. Our analyses on public database showed that PHGDH level was significantly elevated in patients with AML as compared to healthy controls (GSE9476, National Center for Biotechnology Information) (Figure S5A) and the patients with a higher level of PHGDH expression showed a poor prognosis (Cancer Genome Atlas Research Network et al., 2013; Tyner et al., 2018) (Figure S5B). This is consistent with the previous finding that PHGDH was among four of the genes of which elevated expression could predict a poor prognosis in patients with AML (Nguyen et al., 2019). Importantly, the knockdown of PHGDH resulted in a lower level of extracellular [2-13C] glycine derived from [2-13C] fructose, and it significantly hampered the cell growth in the fructose-rich condition (Figures 5B and 5C). Using two PHGDH inhibitors, NCT-503 (Pacold et al., 2016) and CBR-5884 (Mullarky et al., 2016), we found that the proliferation of GLUT5-low leukemic cells was inhibited more dramatically in the fructose-rich condition compared to the glucose-rich condition, whereas the proliferation of GLUT5-high cells was unchanged in the two conditions (Figure 5D; Figure S5C). Furthermore, GLUT5-overexpression was able to mitigate the effect of PHGDH inhibition and rescue the proliferation rate of GLUT5-low cells in the fructose-rich condition (Figure S5D).
Figure 5. GLUT5-low leukemic cells are dependent on SSP in the fructose-rich condition.
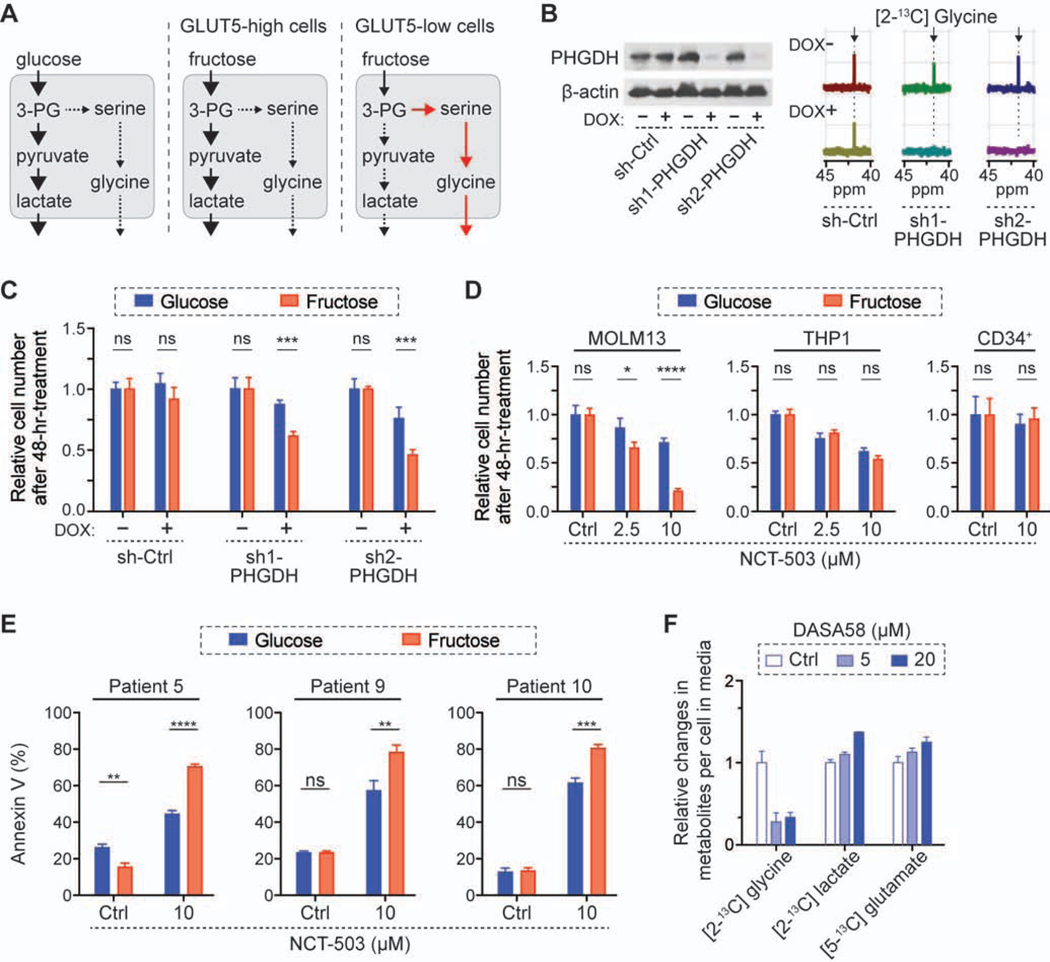
(A) Schematics of metabolic changes in the fructose condition. 3-PG, 3-phosphoglycerate.
(B) Effect of PHGDH knockdown on the SSP activity. After 3-day culture with doxycycline (2 μg/mL), MOLM13 cells were cultured in 10 mM [2-13C] fructose for another 2 days, followed by 13C-NMR analysis of the culture media. For control (sh-Ctrl), shRNA against Renilla luciferase was used.
(C) Comparison of growth rates of MOLM13 cells in 10 mM glucose or fructose media after genetic knockdown of PHGDH (mean ± S.D., n = 3 biological replicates).
(D) Comparison of growth rates of MOLM13, THP1, CD34+ cells in 10 mM glucose or fructose media with treatment of PHGDH inhibitor, NCT-503.
(E) Comparison of apoptosis in mononuclear cells from patients with AML with NCT-503 treatment. The cells were treated in 10 mM glucose or fructose media for 72 hrs (Patient 5) or 48 hrs (Patients 9 and 10).
(F) Relative changes in the metabolites in the media after 2-day culture of MOLM13 cells in 10 mM [2-13C] fructose with different doses of PKM2 activator, DASA58. The metabolite levels were normalized to the level with control treatment (DMSO, 0.1%).
All bar graphs show mean ± S.D. (n = 3 biological replicates), and all statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
PHGDH inhibitor treatment for 48 hours substantially hindered the proliferation of AML cell lines but did not affect the proliferation of their normal counterpart, CD34+ cells (Figure 5D). Moreover, the inhibitor induced significant apoptosis in primary patient AML cells, which was further increased in fructose versus glucose conditions (Figure 5E). Additionally, GLUT5 abundance in these primary AML cells across various subtypes and genetic backgrounds were also similar to that of MOLM13 cells (Figure S5E; Table S1). These differential responses of PHGDH inhibition imply that the SSP pathway dependence is a unique feature of leukemic cells and targeting the SSP could be more effective under certain conditions.
Recent studies have shown that the low kinase activity of the M2 isoform of pyruvate kinase (PKM2) leads to accumulation of glycolytic intermediates, which can provide substrates for the SSP, and PKM2 activation reduces the SSP flux (Kung et al., 2012; Oh et al., 2018; Ye et al., 2012). To confirm whether SSP inhibition, not PHGDH inhibition itself, affected the proliferation of leukemic cells, we activated PKM2 in both culture conditions. Consistent with previous studies, we observed that a substantially lower amount of labeled glycine was generated with the treatment of PKM2 activator, DASA58 (Anastasiou et al., 2012), and a higher amount of labeled lactate was generated in a dose dependent manner (Figure 5F). A higher amount of labeled glutamate with treatment of DASA58 could be attributed to a greater flux of pyruvate into the mitochondria (Park et al., 2016) and inhibition of conversion from glutamate to alpha-ketoglutarate through the SSP (Possemato et al., 2011). PKM2 activation, which reduced the SSP flux, hampered leukemic cell proliferation in the fructose-rich condition more significantly than the glucose-rich condition (Figure S5F). This was reconfirmed using treatment with another PKM2 activator, TEPP46 (Anastasiou et al., 2012), verifying the role of the SSP on fructose metabolism in AML cells (Figure S5G).
The ratio of NAD+/NADH determines the SSP flux in leukemic cells.
To investigate the underlying mechanism driving a higher flux through the SSP, we compared the expression level of the metabolic enzymes downstream of the metabolite 3-phosphoglycerate (3-PG) in glucose-rich and fructose-rich conditions, as the SSP is a metabolic branch of glycolysis at the level of 3-PG (Figure S1D). Surprisingly, there were no substantial changes in the expression and phosphorylation levels of the enzymes downstream of 3-PG, PKM2, and lactate dehydrogenase A (LDHA), after 2-day incubation in each condition (Figure S6A). Additionally, the activity of PKM in both conditions was not significantly different and the level of PKCζ, an upstream regulator of PHGDH (Ma et al., 2013), was also not significantly changed (Figure S6B) [AU: where is that data shown?] [RE: Figure S6B. We edited accordingly]. Instead, we found that the level of redox cofactors, NAD+ and NADH, was different between the two culture conditions (Figure 6A); in particular, the NADH level was substantially lower in the fructose-rich condition. This difference can explain the higher oxygen consumption rate in the fructose-rich condition (Figure 6B), mediated by a higher glutamine consumption (Figure S1A).
Figure 6. The higher ratio of NAD+/NADH in MOLM13 cells cultured in the fructose-rich condition leads to the higher SSP flux, beneficial for providing the anaplerotic precursor, aKG.

(A) The signal intensity of intracellular redox cofactors, NAD+ and NADH, in the cells cultured in the media with glucose or fructose.
(B) Oxygen consumption rate (OCR) of the cells cultured in the media with glucose or fructose. The graphs show mean ± S.D. (n = 8 biological replicates).
(C) Changes in the ratio NAD+/NADH in the cells and the [2-13C] glycine level in the [2-13C] fructose media with supplementation of sodium lactate (20 mM) for 24 hours. The glycine level was normalized to the level without lactate supplementation.
(D) Changes in the ratio NAD+/NADH in the cells and the [2-13C] glycine level in the [2-13C] glucose media with supplementation of sodium pyruvate (4 mM) for 24 hours. The glycine level was normalized to the level without pyruvate supplementation.
(E) Changes in the [2-13C] glycine level in the [2-13C] fructose media with supplementation of N-acetyl-L-Cysteine (NAC), nucleosides (thymidine, uridine, adenosine, cytidine, and inosine; 100 mM of each), or dimethyl-alpha ketoglutarate (DM-aKG, 2.5 mM).
(F) Comparison of the proliferation rate of MOLM13 cells in the fructose media with DM-aKG (2.5 mM).
(G) Comparison of the contribution of glutamine to alpha-ketoglutarate (aKG) in the cells cultured in glucose or fructose media. Total signal intensity and relative intensity of aKG (m+5)/(m+0) were analyzed with LC-MS after the cells were cultured with 4 mM [U-13C5] glutamine for 24 hrs. GLC, glucose; FRC, fructose.
(H) Schematics displaying the metabolic changes in leukemic cells between the glucose-rich and fructose-rich conditions. All graphs show mean ± S.D. (n = 3 biological replicates), unless otherwise specified. and all statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The enzymatic reaction of PHGDH requires NAD+ to convert 3-PG to 3-phosphohydroxypyruvate (p-Pyr), and the NAD+ availability has been shown to constrain the SSP activity in PHGDH-high cancer cells (Diehl et al., 2019; Murphy et al., 2018). We hypothesized that a higher ratio of NAD+/NADH resulted in a higher flux through the SSP in fructose-rich conditions, and we tested it by modulating the ratio with supplementation of lactate or pyruvate in the media. As expected, lactate supplementation in the fructose-rich condition led to a lower ratio of NAD+/NADH and in turn less generation of glycine (Figure 6C). In contrast, pyruvate supplementation in the glucose-rich condition led to a higher NAD+/NADH ratio and more flux through the SSP, resulting in more generation of glycine (Figure 6D). We also tested whether GLUT5 overexpression would change the NAD+/NADH ratio in fructose-rich conditions. In K562 cells with GLUT5 overexpression (Figure 3E), the ratio was substantially lower only in the fructose-rich condition, not in the glucose-rich condition (Figure S6C). The similar ratio of NAD+/NADH between glucose-rich and fructose-rich conditions with GLUT5 overexpression could explain the similar metabolic rate of glucose and fructose (Figures 1C and 3F). These data further support our hypothesis that the SSP flux is driven by the higher ratio of NAD+/NADH in fructose-rich conditions, compared to glucose-rich conditions.
A higher flux through the SSP supports TCA anaplerosis from glutamine in fructose-rich conditions.
To identify potential metabolic benefits of the higher SSP flux, we investigated the three most important functions of the SSP in cellular metabolism: oxidative stress management (Samanta et al., 2016; Ye et al., 2014), nucleotides synthesis (Zhang et al., 2017), and alpha-ketoglutarate (aKG) generation (Possemato et al., 2011). First, we incubated MOLM13 cells with the antioxidant, 10 mM N-Acetyl-L-Cysteine (NAC), to test whether the higher SSP flux is critical for the management of oxidative stress. Although leukemic cells exhibited a higher oxygen consumption in the fructose-rich condition (Figure 6B), the level of reactive oxygen species was not significantly different (Figure S6D) and further antioxidant supplementation did not alter the level of [2-13C] glycine in the media with [2-13C] fructose (Figure 6E). We then explored the incubation of cells with nucleosides (thymidine, uridine, adenosine, cytidine, and inosine) or dimethyl-aKG (cell-permeable form of aKG) to test whether the higher SSP flux was critical for nucleotides synthesis or TCA anaplerosis. In both cases, a less amount of glycine was generated, but the effect was more significant in the case of DM-aKG supplementation (Figure 6E). As cell growth was not significantly affected by DM-aKG supplementation (Figure 6F), this data suggests that the higher SSP flux is critically beneficial for generating the anaplerotic precursor in the fructose-rich condition. Furthermore, by tracing [U-13C5] glutamine metabolism, we confirmed that the contribution of glutamine to the pool of aKG and its downstream TCA metabolites was much higher in the fructose-rich condition than the glucose-rich condition (Figure 6G; Figure S6E). While there are multiple pathways that convert glutamate to aKG, the significantly higher SSP flux along with more glutamine consumption indicates that the SSP plays an essential role in supporting TCA anaplerosis from glutamine in the fructose-rich condition (Figure 6H).
Leukemic cells are more dependent on the SSP in vivo in the presence of high fructose.
To assess the relevance of this metabolism in vivo, we utilized a xenograft model of MOLM13 cells. First, we modeled a fructose-rich environment in bone marrow of leukemic mice similar to that in patients with leukemia, where fructose concentration was more than 1 mM (Chen et al., 2016); an intraperitoneal (IP) injection of fructose with a dose of 4 g/kg resulted in 5.72 ± 1.76 mM (mean ± S.D.) of fructose and 3.35 ± 1.38 mM of glucose in the bone marrow of leukemic mice (30 min. post-injection) (Figure 7A). Interestingly, the same injection resulted in a much lower level of fructose (2.99 ± 1.14 mM) and a much higher level of glucose (8.87 ± 4.54) in the bone marrow of normal NSG mice. This model recapitulates a higher level of fructose previously observed in patients with AML (Chen et al., 2016) and provides another evidence of limited glucose in the tumor microenvironment (Ho et al., 2015; Sullivan et al., 2019b; Urasaki et al., 2012). Utilizing this model system, we measured isotopically enriched metabolites in MOLM13 cells engrafted in bone marrow 5 hr post-IP injection of [U-13C6] glucose, [U-13C6] fructose, or [U-13C6] fructose with the PHGDH inhibitor, NCT-503 (Figure 7B). Consistent with our in vitro experiments, the enrichment of glycine (m+2) was significantly higher with the injection of [U-13C6] fructose compared to [U-13C6] glucose, and PHGDH inhibition resulted in lower enrichment of glycine (m+2), making the fructose injection condition analogous to the glucose condition (Figure 7C). Given the GLUT5-low leukemic cells’ sensitivity to SSP inhibition in fructose-rich conditions compared to glucose-rich conditions in vitro (Figures 5C and 5D), we created a xenograft model of MOLM13 cells with doxycycline-inducible PHGDH knockdown accompanied by serial dosing of glucose or fructose (4 g/kg, IP injection three times a week) (Figure 7D). In all the conditions, PHGDH knockdown led to markedly lower engraftment of human leukemic cells (hCD45+) in the bone marrow, and importantly, the effect on the engraftment was more pronounced in the mice with fructose injection than with glucose or no sugar injection (Figure 7E; Figures S7A and S7B). Intriguingly, we realized that the level of PHGDH expression in hCD45+ cells at the endpoint of the survival experiment was comparable among all the conditions, indicating a selection against PHGDH knockdown in vivo (Figures S7C and S7D); while the less engraftment with PHGDH knockdown led to significantly extended survival, the effect of serial injection of fructose was precluded likely due to the outgrowth of poorly knockdown cells.
Figure 7. In vivo experiments with xenograft AML mouse models demonstrate that leukemic cells are dependent on SSP in fructose-rich conditions.

(A) Glucose and fructose concentration in bone marrow of xenograft leukemia mice and healthy mice measured at 30 min. after IP injection of fructose (4 g/kg).
(B) Schematic of the in vivo experiment with infusion of [U-13C6] glucose or fructose. NSG mice transplanted with MOLM13 cells were grouped into three different experimental conditions; one group for glucose infusion, another for fructose infusion, and the other for fructose infusion with PHGDH inhibitor (NCT-503) treatment. GLC, glucose; FRC, fructose; PHGDHi, PHGDH inhibitor. 5 hours after injection, all the mice were sacrificed and human leukemic cells were isolated from mouse bone marrow based on hCD45 marker.
(C) Enrichment of glycine (m+2) in the cells collected in the experiment (B). n = 9 for the [U-13C6] glucose infusion group, n = 8 for the [U-13C6] fructose infusion group, n = 10 for the [U-13C6] fructose infusion with PHGDH inhibitor treatment group.
(D) Schematic of the in vivo experiment with doxycycline-inducible PHGDH knockdown. NSG mice transplanted with MOLM13 cells with sh-Ctrl or sh2-PHGDH (Figure 5B) were grouped into three different experimental conditions; one group for serial IP injection of glucose (4 g/kg), another for fructose (4 g/kg), and the other for PBS three times a week. After four weeks of injection, all the mice were sacrificed and the engraftment of human leukemic cells was analyzed based on hCD45 marker.
(E) Engraftment level of hCD45+ cells in bone marrow from the mice in the experiment (D).
(F) Schematic of the in vivo experiment with a patient-derived AML mouse model (cells from Patient 5 in Figure 3I and Table S1). One group was for fructose (4 g/kg) along with vehicle treatment, and the other group was for fructose along with NCT-503 (40 mg/kg) treatment. After 6 weeks of daily injection, all the mice were sacrificed and the engraftment of human leukemic cells was analyzed based on hCD45 marker.
(G) Engraftment level of hCD45+ cells in bone marrow from the mice in the experiment (F). Statistical analyses were conducted with unpaired two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. GLC, glucose; FRC, fructose.
Lastly, we sought to demonstrate the SSP-mediated fructose metabolism in patient-derived xenograft (PDX) models. In the first model (Patient 5 in Table S1), we treated the mice with daily IP injection of NCT-503 along with fructose (Figure 7F). Consistent with the PHGDH-knockdown in vivo model, NCT-503 treatment led to substantially lower engraftment of hCD45+ cells in bone marrow (Figure 7G). Our isotopic tracing analysis with an IP injection of [U-13C6] fructose 5 hours before sacrifice confirmed that fructose-derived carbons contributed to the SSP metabolism in the PDX model (Figure S7E), as it did in the xenograft model with MOLM13 cells (Figure 7C). Variations in enrichment could be due to the low engraftment rate (less than 30%) in this PDX model (Figure 7G). In another PDX model with higher engraftment (up to 90%, Patient 9 in Table S1), we also detected labeled glycine (Figure S7F). Although the enrichment was not significantly higher with the injection of [U-13C6] fructose compared to [U-13C6] glucose (P-value of 0.061) (Figure S7G), the overall trend was consistent with the xenograft model with MOLM13 cells (Figure 7C). Taken together, these data suggest that leukemic cells become more dependent on the SSP under fructose-rich conditions and that the SSP inhibition could provide a novel therapeutic option for patients with leukemia, especially with a high level of fructose in their bone marrow.
DISCUSSION
Metabolic plasticity of cancer cells allows them to adapt and proliferate in the tumor microenvironment, where nutrients are severely limited. For example, a metabolic imbalance between the high demand of glucose by cancer cells and its poor supply by dysfunctional vasculature often leads to a significantly lower level of glucose in the tumor microenvironment than that in circulation or in normal tissues (Ho et al., 2015; Sullivan et al., 2019b; Urasaki et al., 2012). Multiple studies have uncovered diverse metabolic strategies of cancer cells to compensate for the limited supply of glucose, many of which are related to increased dependence on mitochondrial oxidative phosphorylation (OXPHOS) in low-glucose environments (Birsoy et al., 2014). This metabolic adaptation often comes with an increased consumption of glutamine, but it still requires a carbon flux through glycolysis, which is consistent with our experimental data. Interestingly, fructose level in cancer patients has been reported to be higher than that of a normal control group, and fructose has been suggested as an alternative carbon source to run glycolysis, at least at certain steps, when glucose is limited (Chen et al., 2016; Fan et al., 2017; Hui et al., 2009; Liu et al., 2010; Weng et al., 2018). As dietary fructose consumption has markedly increased over the past decades due to widespread use of high-fructose corn syrup, a deeper understanding of how cancer cells utilize fructose for their malignant growth is critically required for the development of more effective prevention and treatment strategies. In this report, we demonstrate that leukemic cells proliferate well in both glucose-rich and fructose-rich conditions, but they are more dependent on the SSP in fructose-rich conditions. This metabolic reprogramming is mediated by a change in the level of NAD+ and NADH, which are cofactors of PHGDH, the rate-limiting enzyme in the SSP. The SSP has been reported to play a critical role in multiple cancers (Locasale et al., 2011; Possemato et al., 2011; Reid et al., 2018; Wei et al., 2019; Weng et al., 2018), but the importance of the redox cofactors, NAD+ ad NADH, on regulating the SSP is beginning to be appreciated (Diehl et al., 2019; Murphy et al., 2018). We have revealed that a higher SSP flux, driven by a higher ratio of NAD+/NADH in fructose-rich conditions, contributes to the generation of aKG from glutamine and facilitates TCA anaplerosis, consistent with a previous study in breast cancer (Possemato et al., 2011).
A higher dependency on the SSP implies that PHGDH inhibition has therapeutic potential in leukemia, possibly more so in the presence of high fructose. Recently, targeting PHGDH has been suggested as a promising therapeutic strategy in certain types of cancers, because not only does this rate-limiting enzyme in the SSP support proliferation of cancer cells, but also it leads to the development of drug resistance; for example, liver cancer cells as well as multiple myeloma cells have been shown to increase the PHGDH expression as a resistance mechanism to oxidative stress induced by drug treatment (Wei et al., 2019; Zaal et al., 2017). Therefore, there has been an increasing effort to develop PHGDH inhibitors (Mullarky et al., 2016, 2019; Pacold et al., 2016). Our in vitro and in vivo experiments clearly showed that leukemic cells are more sensitive to genetic and pharmacological inhibition of PHGDH in fructose-rich conditions compared to glucose-rich conditions; especially, in our in vivo experiments with fructose IP injection, we recapitulated a low level of glucose and a high level of fructose in the tumor microenvironment in patients with leukemia (Chen et al., 2016), and demonstrated that not only PHGDH inhibition reduced disease burden but also PHGDH was required for leukemogenesis. We then elucidated a novel mechanism of how GLUT5 level determines the metabolic rate of fructose-derived carbons through the SSP, uncovering the link between the SSP and fructose metabolism in AML cells. As shown in our flow cytometry analyses, the majority of samples from patients with AML expressed the GLUT5 level similar to MOLM13 cells (GLUT5-low), and thus we expect that the dependence on the SSP of malignant cells could be widely found in individuals with AML, especially with a high level of fructose.
In summary, our data reveals a critical role of the SSP in the metabolic program of leukemic cells, which allows them to utilize fructose as an alternative fuel when glucose is deprived. This study also leads to important biological questions, including why PHGDH expression is elevated in leukemic cells and what determines the GLUT5 expression in hematopoiesis and leukemogenesis. Considering that the tumor microenvironment is glucose-limited and leukemic cells are more dependent on the SSP than their normal counterparts, targeting PHGDH could be exploited as a promising strategy for AML treatment.
Limitations of Study
While our study provides evidence that AML cells proliferate well in fructose-rich conditions and their GLUT5 level plays a critical role in their fructose metabolism, our validation in primary AML cells from patients is limited. A correlation study evaluating the fructose level in bone marrow of patients and the SSP activity in their cells from bone marrow would further validate our findings. Future investigation is also needed to understand the role of PHGDH and the SSP in normal hematopoietic stem and progenitor cells as well as their metabolic reprogramming in fructose-rich conditions, which is critically important for the translation of these findings to patients. Lastly, our in vivo model with IP injections of fructose recapitulated a fructose-rich tumor microenvironment in patients with AML, demonstrating a therapeutic potential of PHGDH inhibition for patients, especially with a high level of fructose in their bone marrow. [AU: huh? The meaning of the phrase “, utilizing fructose as therapeutic” is quite unclear. Please rephrase] [RE: We have rephrased to make it clear]. A long-term in vivo study with high-fructose diet conditions would allow us to further support these metabolic findings in a pathophysiological setting.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Kayvan R. Keshari (rahimikk@mskcc.org)
Materials availability
There are no restrictions to the availability of all materials mentioned in the manuscript.
Data and Code Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Flow cytometry data were analyzed using FlowJo (v.10). NMR spectra were analyzed using Chenomx NMR Suite (v.8) and MNova (Mestrelab, v.10). No custom software/code was used.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture.
All cells were grown at 37C in a 5% CO2 humid atmosphere. MOLM13, K562, THP1, U937, TF1, KG1, NOMO1, MV411, OCIAML2, and OCIAML3 cell lines were cultured in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin streptomycin (P/S). KASUMI1 cell line was cultured in RPMI-1640 medium, supplemented with 20% FBS and 1% PS. The cell lines were tested for mycoplasma and inter-species cross-contamination by isoenzyme and short tandem repeat analyses at the Integrated Genomics Operation Core Facility at Memorial Sloan Kettering Cancer Center during the research.
Patient samples.
Human bone marrow mononuclear cells and peripheral blood mononuclear cells from patients with AML were obtained with informed consent under protocols approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center. Briefly, frozen AML patient cells were thawed in RPMI-1640 medium (with 20% FBS and 1% P/S), washed, stained with antibodies (Glut5-Alexa647, R&D Systems; CD45-PerCP/Cy5.5, Thermo Fisher), and analyzed on the BD Fortessa Instrument.
Patient-derived xenograft (PDX) in vitro culture.
PDXs (sex isolated from the bone marrow of mice showing more than 90% human engraftment were cultured in glucose-deprived IMDM + 4 mM glutamine + 10 mM glucose or fructose, 10% dialyzed FBS, 1% P/S and a cocktail of cytokines (100 nM SCF, 10 nM IL3, 10 nM FLT3-ligand, 10 nM GM-CSF, and 10 nM TPO) + 2-mercaptoethanol for 48 hours in presence of 10 μM NCT-503 or DMSO.
CD34+ isolation and culture.
Mononucleated cells were isolated from a pool of 10 fresh human cord blood (sex:F) using a Ficoll gradient. CD34+ cells were enriched using human CD34 MicroBeads (Miltenyi Biotec) and Auto MACS pro separator. CD34+ enriched cells were cultured in glucose-deprived IMDM + 4 mM glutamine + 10 mM [U-13C6] glucose or fructose, 10% dialyzed FBS, 1% Pen/strep and a cocktail of cytokines (100 nM SCF, 10 nM IL6, 10 nM FLT3-ligand, 10 nM GM-CSF, and 10 nM TPO) for 48 hours. The supernatant and the cell pellet were collected for further NMR and LC-MS analyses, respectively.
In vivo experiments.
Animal housing and all the experimental procedures were authorized by the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center (protocol 11–10-025). NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG, The Jackson Laboratory), were housed 5 per cage in a temperature- and humidity-controlled colony room (temperature of 22–24 °C and humidity of 30–70%), maintained on a 12-h light/dark cycle (06:00 to 18:00 light on), with standard or Doxycycline food (ENVIGO, Dox Diet TD 07573) and water provided ad libitum and environmental enrichments. For tracing, PHGDH Knock Down and drug experiments, we used 8–12 weeks old females per group and we repeated the experiment twice. We performed survival experiment once. Mice were randomly assigned to experimental groups. During all the experiments, mice were monitored daily controlling behavior, weight and furrow. Their health was constantly monitored, and they were euthanized at first sign of physical pain. The total number of mice for each experiment is detailed in the figure legends. All the mice used for the experiments were imported to the facility and used after necessary acclimatization.
METHOD DETAILS
Assessment of cell proliferation rate and viability.
Cell number and viability were measured using Cellometer® Mini cell counter (Nexcelom Bioscience); the cell suspension was mixed at 1:1 ratio with Trypan Blue Solution (Sigma-Aldrich) before the measurement. To monitor the proliferation rate for 10 days, cells were split and resuspended into fresh media every three days with a final concentration of 400k cells/mL.
Western blot:
Cells were collected, washed once with PBS and lysed in 1X Laemmli sample buffer (BioRad). Standard procedures were used for immunoblotting: 10 μg of total protein lysate was loaded on a 4–15% SDS-PAGE gel, transferred to a nitrocellulose membrane and incubated overnight with primary antibodies (dilution 1:1000) at 4 °C. Proteins were detected using ECL Western Blotting Substrate (Promega).
Analysis of extracellular metabolites using high-field NMR.
The cell culture media was filtered through Amicon® Ultra-0.5 Centrifugal Filter (Millipore) by centrifugation (14000 rpm for 30 min). The filtered media was mixed with the internal standard solution with a ratio of 9 to 1; the internal standard solution was prepared in D2O with 5 mM 2,2-Dimethyl-2-silapentane-5-sulfonate sodium salt (DSS), 100 mM imidazole, 0.2 mM NaN3, and 10 mM 13C-Urea. 600 μL of the mixed sample was loaded into a NMR tube (Wilmad®) and 1H/13C-NMR spectra were acquired in a 600MHz NMR system (AVANCE III, Bruker). The acquired spectra were analyzed with a commercial software (Chenomx for 1H-NMR spectra and MestReNova for 13C-NMR spectra) to quantify the metabolites level.
Analysis of intracellular metabolites using LC-MS.
The cells were cultured with [U-13C6] glucose or fructose for a certain period of time, washed twice with ice-cold PBS, and lysed with 80% methanol (in water). Targeted LC-MS analyses were conducted on a Q Exactive™ Orbitrap™ Mass Spectrometer (Thermo Scientific) coupled to a Vanquish UPLC system (Thermo Scientific), and the Mass Spectrometer was operated in polarity-switching mode. A SeQuant® ZIC®-HILIC column (2.1 mm i.d. × 150 mm, Merck) was used for separation of metabolites, and the flow rate was 150 μL/min. Buffers consisted of 100% acetonitrile for A, and 0.1% ammonium hydroxide / 20 mM ammonium acetate in water for B. Gradient ran from 85 to 30% A for 20 min, followed by a wash with 30% A and re-equilibration at 85% A. Metabolites and their 13C-isotopologues were identified on the basis of standard retention times and exact mass within 5 ppm. The relative quantification was performed based on the metabolite peak area. All data analyses were performed using in-house written scripts.
Analysis of the intracellular level of NAD+ and NADH.
We used a commercially available assay kit, NAD/NADH-Glo™ Assay (Promega). Briefly, 10,000 cells of interest were collected, washed with PBS, resuspended in 50 μL PBS, and loaded into a well in a 96-well plate. 50 μL of the lysis solution, 0.2N NaOH + 1% (v/v) dodecyltrimethylammonium bromide (DTAB), was added into the same well. After 5 min. of incubation at room temperature, the lysed cell sample (100 μL) was divided into two wells; 50 μL for NAD+ and another 50 μL for NADH. 25 μL of 0.4N HCl solution was added into the well for NAD+ measurement (not into the well for NADH measurement), followed by incubation in an oven at 60ºC for 15 min. After the plate was cooled to room temperature for 10 min., add 25 μL of 0.5M Trizma base solution into the well for NAD+ measurement and add 50 μL of a mixture of 0.4N HCl/ 0.5M Trimza base solution (1:1) into the well for NADH measurement, which made a final volume of 100 μL in both wells. 100 μL of the detection reagent of NAD/NADH-Glo™ Assay was added into each well and gently mixed. After 30 min. incubation at room temperature, the luminescence level was measured with a plate reader.
Analysis of oxygen consumption rate in glucose- or fructose-rich conditions.
MOLM13 cells incubated in 10 mM glucose or fructose media for 2 days were resuspended in custom-designed glucose or fructose assay medium, respectively, with a concentration of 500k cells/mL; the assay medium was prepared by supplementing Seahorse XF Base Medium (Agilent) with 1 mM pyruvate, 4 mM glutamine, and 10 mM glucose or fructose. 100 μL of the cell suspension was loaded into a well in a Seahorse XF Cell Culture Microplate (96-well plate) coated with Cell-Tak solution (Corning). After the cells were adhered to the bottom of each well, a Seahorse XF Cell Mito Stress Test assay was run in a XFe96 Extracellular Flux Analyzer (Agilent) as described in the assay manual.
Supplementation of N-acetyl-L-Cysteine (NAC) or dimethyl-aKG (DM-aKG) into cell culture media.
10 mM NAC (Sigma-Aldrich) or 2.5 mM DM-aKG (Sigma-Aldrich) was dissolved into the media and sterilized with 0.22-μm-pore filter (Millipore). The prepared media was used within an hour.
Supplementation of nucleosides into cell culture media.
Thymidine, uridine, adenosine, cytidine, and inosine (Sigma-Aldrich) were dissolved into the media with a final concentration of 100 mM of each and sterilized with 0.22-μm-pore filter (Millipore: SCGP00525). The prepared media was used within a day.
PHGDH knockdown using short hairpin RNAs (shRNAs).
For PHGDH knockdown, we used lentiviral particles containing doxycycline-inducible shRNA vectors (LT3GEPIR) targeting PHGDH or Renilla luciferase (control). The lentiviral particles were generated from the Gene Editing & Screening Core Facility at Memorial Sloan Kettering Cancer Center (MSK). MOLM13 cells were incubated with the lentiviral particles in the presence of 4 μg/ml polybrene for 24 hr, washed with PBS, and suspended in fresh media for another 24 hr. The infected cells were selected with 2 μg/ml puromycin (3-day treatment). To induce shRNA expression, the selected cells were treated with 2 μg/ml doxycycline. We confirmed the efficient knockdown of PHGDH using two independent shRNAs.
KHK knockdown using shRNAs.
For KHK knockdown, we used lentiviral particles containing shRNA vectors (SGEP) targeting KHK or Renilla luciferase (control). The lentiviral particles were generated from the Gene Editing & Screening Core Facility at MSK. MOLM13 and THP1 cells were incubated with the lentiviral particles in the presence of 4 μg/ml polybrene for 24 hr, washed with PBS, and suspended in fresh media for another 24 hr. The infected cells were selected with 2 μg/ml puromycin (3-day treatment). We confirmed the efficient knockdown of KHK using two independent shRNAs.
KHK inhibitor.
N⁸-(Cyclopropylmethyl)-N⁴-(2-(methylthio)phenyl)-2-(1-piperazinyl)-pyrimido[5,4-d]pyrimidine-4,8-diamine (Sigma-Aldrich) was dissolved in DMSO and added into cell culture media.
PHGDH inhibitor.
NCT-503 (Cayman Chemical) or CBR-5884, generously provided from Dr. Edouard Mullarky in Dr. Lewis C. Cantley group, was dissolved in DMSO and added into cell culture media.
Analysis of the intracellular levels of PHGDH.
To perform PHGDH intracellular staining, MOLM13 cells transduced with shRNA targeting PHGDH or Renilla (control) were fixed with 1.6% paraformaldehyde (PFA) for 10 min at room temperature, then centrifuged (@1500 rpm for 4 min) and resuspended in 90% methanol for permeabilization and incubated overnight. The following day the methanol was removed, and the cells were stained with the primary rabbit polyclonal anti-human PHGDH antibody (dilution 1:50) followed by Alexa Fluor 488 goat anti-rabbit IgG (dilution 1:200) and analyzed on the BD Fortessa instrument. For the ex vivo analysis of intracellular PHGDH, bone marrow cells were collected at the time of sacrifice and stained with anti-human CD45 antibody (CD45-PerCP/Cy5.5, dilution 1:100) and anti-mouse CD45 (APC or V540, dilution 1:200), then fixed and permeabilized as described above. Human CD45 staining was repeated after permeabilization together with PHGDH staining, followed by secondary. The data were analyzed on the BD Fortessa instrument. For the survival experiment, bone marrow cells were collected at the time of death, frozen and analyzed all together at the final endpoint, as described above.
Patient-derived xenograft (PDX) in vitro viability assay.
PDXs isolated from the bone marrow of mice showing more than 90% human engraftment were cultured for 48 hours in presence of 10 μM NCT-503 or DMSO. Cells were stained with anti-human CD45-PercP/Cy 5.5, anti-mouse CD45-V450, and Annexin V-PE/DAPI and analyzed using a BD Fortessa instrument.
In vivo experiments.
For the in vivo tracing experiments, 8–12 weeks old females NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG, The Jackson Laboratory) were transplanted intravenously with 1×106 MOLM13 cells. At clear signs of diseases (hunchback, weight loss, appearance of extra-medullary masses) the mice were injected intraperitoneally (IP) with 4 g/kg of [2-13C] glucose or fructose dissolved in PBS and sacrificed 5 hours later. The group of mice treated with NCT-503 was injected IP with the drug at 40 mg/kg 30 minutes prior to the sugar injection. A second drug treatment was administered after 3 hours from the sugar injection in order to match the shelf-life of NCT503 and keep the enzyme inhibited. At sacrifice, bone marrow was harvested and processed by crashing the bones in cold PBS. Human cells were positively selected using human CD45 MicroBeads (Miltenyi Biotec) following the manufacturer instruction. Auto MACS pro separator was used for the procedure. After separation, positive enriched human CD45 were counted and a dry pellet was snap frozen and stored at −80°. The same procedure was applied to mice engrafted with Patient 5-derived leukemic cells, the day of sacrifice. Mice engrafted with Patient 9-derived leukemic cells were subjected to the same treatment except the human engraftment was over 95% and enrichment with CD45 beads was not needed (experimental workflow is shown in Figure S7F).
Effect of PHGDH inhibition on tumor burden and survival.
8–12 weeks old females NSG mice (The Jackson Laboratory) were transplanted intravenously with 1×106 MOLM13 cells transduced with doxycycline-inducible shRNA vectors (LT3GEPIR) targeting PHGDH or Renilla luciferase (control) (n=5 mice /group). Doxycycline was administered in the chow 2 days after the transplantation, and fructose was IP inj ected 3 times a week at 4 g/kg. The mice were sacrificed when the controls showed evident signs of disease and their bone marrow was collected and processed as described above. Tumor load was assessed by calculating the percentage of human CD45+ cells by FACS. To investigate overall survival, mice transplanted with shRNA targeting PHGDH or control were randomized into 2 groups each (n=7–8 mice /group) and IP injected with PBS or fructose (4 g/kg), 3 times a week. Long-rank tests were used for statistical significance.
Patient-derived xenograft.
The primary cells from Patient 5 (Table S1) were first transplanted in irradiated (200 rad) NSG mice (The Jackson Laboratory) at a dose of 3×106 cells/mouse in order to expand them. Secondary transplant was performed from fresh when the primary mouse was sacrificed with a dose of 1×106 cells/mouse. Mice were treated with NCT-503 (40 mg/kg) or vehicle (5% ethanol, 35% PEG 300, 60% of aqueous solution of 2-hydroxypropyl-beta cyclodextrin) and a solution of 4 g/kg fructose dissolved in PBS. The treatment started 1 week after the transplant and the drug and fructose were administered daily for 5 weeks. At sacrifice, the bone marrow was collected, and the extracted cells were analyzed using FACS to check for human engraftment using the following antibodies: anti-human CD45-PercP/Cy 5.5 (dilution 1:100) and anti-mouse CD45-APC or V450 (dilution 1:200).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data representation and statistical analysis
In vitro data are displayed as bar plots of three independent biological replicates, composed of 3–4 technical replicates each. At least triplicates for the in vitro data were considered as the minimum number necessary to have standard deviation. This way we ensured that the techniques provide comparable results between similar experiments. In vivo data are displayed as dot plots of at least two independent experiments. Plots include mean and S.D. Statistical significance was calculated using unpaired two-tailed Student’s t-test with PRISM software. Results are considered significant when P-value < 0.05. The power was estimated according to the criteria described in doi:10.4103/0976-500X.119726 and pilot experiments performed in our laboratory. We estimated that 5–7 mice/group would be necessary to achieve 80% power and a P-value of 0.05 for both xenograft models of PHGDH KD and drug administration. Animals were randomly assigned to control or experimental groups, while ensuring equal age and sex matching in the experimental groups and blinding was performed during data collection. No animals were excluded from the study. For flow cytometry, no formal blinding was performed but control and experimental samples were processed and analyzed using standardized procedures.
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies for immunoblot | ||
| HK2 (C64G5), rabbit mAb (1:1000) | Cell Signaling Technology (CST) | # 2867 |
| Aldolase A (D73H4), rabbit mAb (1:1000) | CST | # 8060 |
| Aldolase B, rabbit pAb (1:1000) | Abcam | # ab153828 |
| LDHA, rabbit pAb (1:1000) | CST | # 2012 |
| Phospho-LDHA (Tyr10), rabbit pAb (1:1000) | CST | # 8176 |
| PHGDH, rabbit pAb (1:1000) | Sigma-Aldrich | # HPA021241 |
| Actin (D6A8) HRP-conjugated, rabbit mAb (1:10000) | CST | # 12620 |
| KHK, rabbit pAb (1:1000) | Abcam | # ab154405 |
| PKM1 (D30G6), rabbit mAb (1:1000) | CST | # 7067 |
| PKM2 (D78A4), rabbit mAb (1:1000) | CST | # 4053 |
| Phospho-PKM2 (Tyr105), rabbit pAb (1:1000) | CST | # 3827 |
| GCK, mouse mAb (1:1000) | R&D Systems | # MAB7840 |
| PKCζ (C24E6), rabbit mAb (1:1000) | CST | # 9368 |
| Phospho-PKCζ (190D10), rabbit mAb (1:1000) | CST | # 2060 |
| Glut5 (E-2), mouse mAb (1:1000) | Santa Cruz Biotechnology | # sc-271055 |
| Antibodies for flow cytometry | ||
| Human CD45 PerCP Cy5.5 clone 2D1 (1:100) | Thermo Fisher Scientific | # 45-9459-42 |
| Mouse CD45.1 V450 clone A20 (1:200) | Thermo Fisher Scientific | # 48-0453-80 |
| Mouse CD45 APC clone 30-F11 (1:200) | BD Biosciences | # 559864 |
| PHGDH, rabbit pAb (1:50) | Thermo Fisher Scientific | # PA5-24633 |
| Rabbit IgG (H+L) Alexa Fluor 488 (1:200) | Thermo Fisher Scientific | # A-11008 |
| Human Glut5 Alexa Fluor 647 (1:50) | R&D Systems | # FAB1349R |
| Biological Samples | ||
| Human acute myeloid leukemia specimens | MSK The Hematologic Oncology Tissue Bank (HOTB) | N/A |
| Commercial Assays | ||
| CD45 MicroBeads, human | Miltenyi Biotec | # 130-045-801 |
| NAD/NADH-Glo Assay kit | Promega | # G9071 |
| EnzyChrom Glucose Assay kit | BioAssay Systems | # EBGL-100 |
| EnzyChrom Fructose Assay kit | BioAssay Systems | # EFRU-100 |
| CellROX Deep Red Reagent | Thermo Fisher Scientific | # C10422 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | # 103015–100 |
| Chemicals, peptides, and recombinant proteins | ||
| Ketohexokinase (KHK) Inhibitor - Calbiochem | Sigma-Aldrich | # 420640 |
| NCT-503 | Cayman Chemical | # 19718 |
| CBR-5884 | Dr. Lewis C. Cantley group | # 19236 |
| DASA58 | Cayman Chemical | # 13941 |
| TEPP46 (ML-265) | Cayman Chemical | # 13942 |
| Puromycin dihydrochloride | Sigma-Aldrich | # P9620 |
| Doxycycline hyclate | Sigma-Aldrich | # D9891 |
| Cell-Tak Cell and Tissue Adhesive | Corning | # 354240 |
| N-acetyl-L-Cysteine | Sigma-Aldrich | # A7250 |
| Dimethyl alpha-ketoglutarate | Sigma-Aldrich | # 349631 |
| Recombinant Human SCF | PeproTech | # 300–07 |
| Recombinant Human IL6 | PeproTech | # 200–06 |
| Recombinant Human IL3 | PeproTech | # 200–03 |
| Recombinant Human Flt3-Ligand | PeproTech | # 300–19 |
| Recombinant Human GM-CSF | PeproTech | # 300–03 |
| Recombinant Human TPO | PeproTech | # 300–18 |
| 16% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences | # 15710-S |
| Annexin V-PE | BioLegend | # 640947 |
| DAPI | Thermo Fisher Scientific | # D1306 |
| Trypan Blue | Sigma-Aldrich | # T8154 |
| 2xLaemmli Buffer | Bio-Rad | #1610747 |
| ECL Western Blotting Substrate | Thermo Scientific | #1859698 #1859701 |
| 4–15% SDS-PAGE gel | Bio-Rad | #4561084 |
| Metabolites and enzymes | ||
| Phospho(enol)pyruvic acid monosodium salt hydrate (PEP) | Sigma-Aldrich | # P0564 |
| NADH (sodium salt hydrate) | Cayman Chemical | # 16078 |
| Adenosine 5′-diphosphate sodium salt (ADP) | Sigma-Aldrich | # A2754 |
| Sodium pyruvate | Thermo Fisher Scientific | # 11360070 |
| Sodium lactate | Sigma-Aldrich | # L7022 |
| [U-13C] Glucose | Sigma-Aldrich | # 389374 |
| [U-13C] Fructose | Sigma-Aldrich | # 587621 |
| [U-13C] Glutamine | Sigma-Aldrich | # 605166 |
| [2-13C] Glucose | Sigma-Aldrich | # 310794 |
| [2-13C] Fructose | Sigma-Aldrich | # 492140 |
| Thymidine | Sigma-Aldrich | # T1895 |
| Uridine | Sigma-Aldrich | # U3003 |
| Adenosine | Sigma-Aldrich | # A4036 |
| Cytidine | Sigma-Aldrich | # C4654 |
| Inosine | Sigma-Aldrich | # I4125 |
| Lactate dehydrogenase (LDHA) | Sigma-Aldrich | # SAE0049 |
| Pyruvate kinase (PK) | Sigma-Aldrich | # P9136 |
| Experimental Models: Cell Lines | ||
| KG1 | MSK | N/A |
| MOLM13 | MSK | N/A |
| TF1 | ATCC | # CRL-2003 |
| NOMO1 | MSK | N/A |
| MV411 | MSK | N/A |
| U937 | MSK | N/A |
| OCIAML2 | MSK | N/A |
| OCIAML3 | MSK | N/A |
| THP1 | MSK | N/A |
| KASUMI1 | MSK | N/A |
| K562 | MSK | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Jackson Laboratory | #005557 |
| Software and Algorithms | ||
| Graphpad Prism | Graphpad | https://www.graphpad.com/ |
| Chenomx | Chenomx | https://www.chenomx.com |
| MestReNova | Mesterlab Research | https://mestrelab.com |
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| FACSDiva | BD Biosciences | https://www.bdbiosciences.com |
Research Highlights.
AML cells proliferate in glucose-deprived environments with fructose supplementation
Fructose is mainly metabolized through hexokinase, not ketohexokinase, in AML cells
AML cells upregulate the serine synthesis pathway in fructose-rich environments
PHGDH inhibition in high fructose conditions markedly reduces leukemia progression
ACKNOWLEDGMENTS
The authors thank Dr. Moonjung Jung, Dr. Sui-Seng Tee, Dr. Ralph Garippa, and Daniel Zakheim for providing insightful comments on designing experiments. All the LC-MS analyses on intracellular metabolites were conducted by the Proteomics and Metabolomics Core Facility at the Weill Cornell Medicine Meyer Cancer Center. This work was supported in part by US NIH grants R21CA212958 (K.R.K.), R01CA237466 (K.R.K.), R01CA252037 (K.R.K.), Cancer Center Support Grant P30CA008748 (K.R.K. and M.G.K.), K99CA226357 (S.J.), R35CA197588 (L.C.C.), R01DK101989 (M.G.K.), R01CA193842 (M.G.K.), R01HL135564 (M.G.K.), and R01CA225231 (M.G.K.), and by grants from the Center for Molecular Imaging and Nanotechnology at Memorial Sloan Kettering Cancer Center (K.R.K.), Kimmel Scholar Award (M.G.K.), V-Scholar Award (M.G.K.), Geoffrey Beene Award (M.G.K.), Leukemia Lymphoma Society Career Development Award (M.G.K.), Starr Cancer Consortium (M.G.K.), Alex’s Lemonade Stand A Award (M.G.K.), Lauri Strauss Leukemia Foundation and AIL (Associazione Italiana contro Leucemie, Linfomi e Mielomi) through SIES (Societa’ Italiana Ematologia Sperimentale) (A.M.S.).
Footnotes
DECLARATION OF INTERESTS
L.C.C. is a founder and member of the SAB of Agios Pharmaceuticals and of Petra Pharmaceuticals. These companies are developing novel therapies for cancer. L.C.C.’s laboratory also receives some financial support from Petra Pharmaceuticals. K.R.K. serves on the SAB of NVision Imaging Technologies. M.G.K. is a consultant for Accent Therapeutics and M.G.K.’s laboratory receives some financial support from 28–7. E.M. is currently an employee of Clarion Healthcare. All other authors do not have competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- Anastasiou D, Yu Y, Israelsen WJ, Jiang J, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. (2012). Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, and Lanaspa MA (2020). Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aune D, Chan DSM, Vieira AR, Navarro Rosenblatt DA, Vieira R, Greenwood DC, Cade JE, Burley VJ, and Norat T. (2012). Dietary fructose, carbohydrates, glycemic indices and pancreatic cancer risk: A systematic review and meta-analysis of cohort studies. Ann. Oncol 23, 2536–2546. [DOI] [PubMed] [Google Scholar]
- van den Berghe G, Bronfman M, Vanneste R, and Hers HG (1977). The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J 162, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, and Sabatini DM (2014). Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu P, Chen K, Xiang K, Johnson C, Crown SB, Rakhilin N, Ai Y, Wang L, Xi R, Astapova I, et al. (2018). Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 27, 1249–1262.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG, Hoadley K, Triche TJ, Laird PW, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreño D, Corro N, Torres-Estay V, Véliz LP, Jaimovich R, Cisternas P, San Francisco IF, Sotomayor PC, Tanasova M, Inestrosa NC, et al. (2019). Fructose and prostate cancer: toward an integrated view of cancer cell metabolism. Prostate Cancer Prostatic Dis. 22, 49–58. [DOI] [PubMed] [Google Scholar]
- Chen WL, Wang YY, Zhao A, Xia L, Xie G, Su M, Zhao L, Liu J, Qu C, Wei R, et al. (2016). Enhanced Fructose Utilization Mediated by SLC2A5 Is a Unique Metabolic Feature of Acute Myeloid Leukemia with Therapeutic Potential. Cancer Cell 30, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, Meigs JB, D’Agostino RB, Gaziano JM, and Vasan RS (2007). Soft Drink Consumption and Risk of Developing Cardiometabolic Risk Factors and the Metabolic Syndrome in Middle-Aged Adults in the Community. Circulation 116, 480–488. [DOI] [PubMed] [Google Scholar]
- Diehl FF, Lewis CA, Fiske BP, and Heiden M.G. Vander (2019). Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, Hayward BE, Asipu A, and Bonthron DT (2009). Ketohexokinase: Expression and Localization of the Principal Fructose-metabolizing Enzyme. J. Histochem. Cytochem 57, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, and Rabinowitz JD (2017). One-Carbon Metabolism in Health and Disease. Cell Metab. 25, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Liu H, Liu M, Wang Y, Qiu L, and Cui Y. (2017). Increased utilization of fructose has a positive effect on the development of breast cancer. PeerJ 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang S-K, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K, et al. (2019). High-fructose corn syrup enhances intestinal tumor growth in mice. Science (80-. ). 363, 1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, MacIntyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui H, Huang D, McArthur D, Nissen N, Boros LG, and Heaney AP (2009). Direct spectrophotometric determination of serum fructose in pancreatic cancer patients. Pancreas 38, 706–712. [DOI] [PubMed] [Google Scholar]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, and Rabinowitz JD (2018). The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 27, 351–361.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S, Eskandari R, Park SM, Alvarez J, Tee SS, Weissleder R, Kharas MG, Lee H, and Keshari KR (2017). Real-time quantitative analysis of metabolic flux in live cells using a hyperpolarized micromagnetic resonance spectrometer. Sci. Adv 3, e1700341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang D-H, Gersch MS, Benner S, and Sánchez-Lozada LG (2007). Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr 86, 899–906. [DOI] [PubMed] [Google Scholar]
- Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang J, Lu WH, Xu RH, Li J, Li Y, et al. (2017). ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 31, 2143–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, Delabarre B, Cianchetta G, Sethumadhavan S, Wang X, et al. (2012). Small molecule activation of pkm2 in cancer cells induces serine auxotrophy. Chem. Biol 19, 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, Milagres T, Kuwabara M, Wempe MF, Rabinowitz JD, et al. (2018). Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Invest 128, 2226–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Huang D, McArthur DL, Boros LG, Nissen N, and Heaney AP (2010). Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 70, 6368–6376. [DOI] [PubMed] [Google Scholar]
- Liu L, Li T, Liao Y, Wang Y, Gao Y, Hu H, Huang H, Wu F, Chen Y, Xu S, et al. (2020). Triose Kinase Controls the Lipogenic Potential of Fructose and Dietary Tolerance. Cell Metab. 32, 1–14. [DOI] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, et al. (2013). Control of nutrient stress-induced metabolic reprogramming by PKC in tumorigenesis. Cell 152, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäenpää PH, Raivio KO, and Kekomäki MP (1968). Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science (80-. ). 161, 1253–1254. [DOI] [PubMed] [Google Scholar]
- Marriott BP, Cole N, and Lee E. (2009). National Estimates of Dietary Fructose Intake Increased from 1977 to 2004 in the United States. J. Nutr 139, 1228S–1235S. [DOI] [PubMed] [Google Scholar]
- Maryanoff BE, O’Neill JC, McComsey DF, Yabut SC, Luci DK, Jordan AD, Masucci JA, Jones WJ, Abad MC, Gibbs AC, et al. (2011). Inhibitors of ketohexokinase: Discovery of pyrimidinopyrimidines with specific substitution that complements the ATP-binding site. ACS Med. Chem. Lett 2, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzarella L, Botteri E, Matthews A, Gatti E, Di Salvatore D, Bagnardi V, Breccia M, Montesinos P, Bernal T, Gil C, et al. (2020). Obesity is a risk factor for acute promyelocytic leukemia: Evidence from population and cross-sectional studies and correlation with FLT3 mutations and polyunsaturated fatty acid metabolism. Haematologica 105, 1559–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, Wong JCY, Christen S, Takahashi H, Singh PK, et al. (2016). Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. U.S.A 113, 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E, Xu J, Robin AD, Huggins DJ, Jennings A, Noguchi N, Olland A, Lakshminarasimhan D, Miller M, Tomita D, et al. (2019). Inhibition of 3-phosphoglycerate dehydrogenase (PHGDH) by indole amides abrogates de novo serine synthesis in cancer cells. Bioorganic Med. Chem. Lett 29, 2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JP, Giacomantonio MA, Paulo JA, Everley RA, Kennedy BE, Pathak GP, Clements DR, Kim Y, Dai C, Sharif T, et al. (2018). The NAD+ Salvage Pathway Supports PHGDH-Driven Serine Biosynthesis. Cell Rep. 24, 2381–2391.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen CH, Glüxam T, Schlerka A, Bauer K, Grandits AM, Hackl H, Dovey O, Zöchbauer-Müller S, Cooper JL, Vassiliou GS, et al. (2019). SOCS2 is part of a highly prognostic 4-gene signature in AML and promotes disease aggressiveness. Sci. Rep 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M, Song J, Yang Y, Kong M, Abeywardana T, Yang Y, and Jiang L. (2018). CARM1 suppresses de novo serine synthesis by promoting PKM2 activity. J. Biol. Chem 293, 15290–15303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJYM, Possemato R, Chen WW, Sullivan LB, Fiske BP, et al. (2016). A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol 12, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Josan S, Jang T, Merchant M, Watkins R, Hurd RE, Recht LD, Mayer D, and Spielman DM (2016). Volumetric spiral chemical shift imaging of hyperpolarized [2–13c]pyruvate in a rat c6 glioma model. Magn. Reson. Med 75, 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al. (2013). Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poynter JN, Richardson M, Blair CK, Roesler MA, Hirsch BA, Nguyen P, Cioc A, Warlick E, Cerhan JR, and Ross JA (2016). Obesity over the life course and risk of acute myeloid leukemia and myelodysplastic syndromes. Cancer Epidemiol. 40, 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X, Dai Z, Gao X, Wang Q, Liu Y, et al. (2018). Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, and Semenza GL (2016). PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 76, 4430–4442. [DOI] [PubMed] [Google Scholar]
- Strober JW, and Brady MJ (2019). Dietary fructose consumption and triple-negative breast cancer incidence. Front. Endocrinol 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, Meeth K, Muir A, Darnell AM, Bosenberg MW, et al. (2019a). Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Heiden MGV, and Muir A. (2019b). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. (2018). Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urasaki Y, Heath L, and Xu CW (2012). Coupling of glucose deprivation with impaired histone H2B monoubiquitination in tumors. PLoS One 7, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, et al. (2014). Hexokinase 2-Mediated Warburg Effect Is Required for PTEN- and p53-Deficiency-Driven Prostate Cancer Growth. Cell Rep. 8, 1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Lee D, Law CT, Zhang MS, Shen J, Chin DWC, Zhang A, Tsang FHC, Wong CLS, Ng IOL, et al. (2019). Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y, Fan X, Bai Y, Wang S, Huang H, Yang H, Zhu J, and Zhang F. (2018). SLC2A5 promotes lung adenocarcinoma cell growth and metastasis by enhancing fructose utilization. Cell Death Discov. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Adane B, Khan N, Alexeev E, Nusbacher N, Minhajuddin M, Stevens BM, Winters AC, Lin X, Ashton JM, et al. (2018). Subversion of Systemic Glucose Metabolism as a Mechanism to Support the Growth of Leukemia Cells. Cancer Cell 34, 659–673.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, and Thompson CB (2012). Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. U.S.A 109, 6904–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, Finley LWS, Lu C, Lindsten T, Cross JR, et al. (2014). Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal EA, Wu W, Jansen G, Zweegman S, Cloos J, and Berkers CR (2017). Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Zheng A, Hydbring P, Ambroise G, Ouchida AT, Goiny M, Vakifahmetoglu-Norberg H, and Norberg E. (2017). PHGDH Defines a Metabolic Subtype in Lung Adenocarcinomas with Poor Prognosis. Cell Rep. 19, 2289–2303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files). Flow cytometry data were analyzed using FlowJo (v.10). NMR spectra were analyzed using Chenomx NMR Suite (v.8) and MNova (Mestrelab, v.10). No custom software/code was used.