

Abstract
Background
Isocitrate dehydrogenase (IDH) wildtype (wt) grade II gliomas are a rare and heterogeneous entity. Survival and prognostic factors are poorly defined.
Methods
We searched retrospectively all patients diagnosed with diffuse World Health Organization (WHO) grades II and III gliomas at our center (1989–2020).
Results
Out of 517 grade II gliomas, 47 were “diffuse astrocytomas, IDHwt.” Tumors frequently had fronto-temporo-insular location (28/47, 60%) and infiltrative behavior. We found telomerase reverse transcriptase (TERT) promoter mutations (23/45, 51%), whole chromosome 7 gains (10/37, 27%), whole chromosome 10 losses (10/41, 24%), and EGFR amplifications (4/43, 9%), but no TP53 mutations (0/22, 0%). Median overall survival (OS) was 59 months (vs 19 mo for IDHwt grade III gliomas) (P < 0.0001). Twenty-nine patients (29/43, 67%) met the definition of molecular glioblastoma according to cIMPACT-NOW update 3. Median OS in this subset was 42 months, which was shorter compared with patients with IDHwt grade II gliomas not meeting this definition (median OS: 57 mo), but substantially longer compared with IDHwt grade III gliomas meeting the definition for molecular glioblastoma (median OS: 17 mo, P < 0.0001). Most patients with IDHwt grade II gliomas met cIMPACT criteria because of isolated TERT promoter mutations (16/26, 62%), which were not predictive of poor outcome (median OS: 88 mo). Actionable targets, including 5 gene fusions involving FGFR3, were found in 7 patients (24%).
Conclusions
Our findings highlight the importance of histological grading and molecular profiling for the prognostic stratification of IDHwt gliomas and suggest some caution when assimilating IDHwt grade II gliomas to molecular glioblastomas, especially those with isolated TERT promoter mutation.
Keywords: diffuse low grade gliomas, FGFR3, gene fusion, IDH-wildtype, molecular markers
Key Points.
IDH-wildtype diffuse grade II gliomas should be distinguished from grade III because of a lower burden of genetic alterations (including EGFR amplifications, whole chromosome 7 gain/whole chromosome 10 loss, TERT promoter mutations, TP53 mutations, deletions of cyclin-dependent kinase inhibitor 2A, and chromosome 9p loss) and a much better outcome.
With a median overall survival of 88 months, IDH-wildtype grade II gliomas with isolated TERT promoter mutations should not be assimilated to molecular glioblastomas.
Importance of the Study.
The cIMPACT-NOW update 3 has recently established that IDHwt histological grade II and III diffuse gliomas with EGFR amplifications, and/or combined whole chromosome 7 gain and whole chromosome 10 loss, and/or TERT promoter mutations should be considered as bona fide glioblastomas. Our data suggest that, while true for histological grade III gliomas, these considerations do not fit a subset of grade II gliomas, and namely those with isolated TERT promoter mutations (median overall survival: 88 mo). These findings highlight the importance of histological grade, in parallel to molecular profile, for the prognostic stratification of IDHwt lower-grade gliomas and suggest that IDHwt gliomas with grade II histology (<2 mitosis per 10 high power fields) and isolated TERT promoter mutations should not be assimilated to molecular glioblastomas.
Lower grade (ie, World Health Organization [WHO] grades II and III) diffuse gliomas form a heterogeneous group of tumors including entities characterized by different malignant behavior. The IDH mutation and the chromosome 1p/19q codeletion represent the main diagnostic and prognostic markers in this group.1,2 The IDH mutation is an independent predictor of prolonged survival and its prevalence is inversely correlated with tumor grade.2
Isocitrate dehydrogenase wildtype (IDHwt) grade II diffuse gliomas correspond to a rare subgroup of low-grade tumors associated with dismal prognosis and poor response to treatments.3 Due to the lack of large prospective studies, there is still conflicting evidence regarding the clinical and molecular profile associated with these tumors, and survival estimates widely range from 4.7 to 8.4 years across studies.3–6 Given the rarity of IDHwt grade II diffuse gliomas, most of the studies analyzed grade II and III gliomas altogether7,8 to generate more solid data. However, evidence suggests that IDHwt grade II and grade III tumors significantly differ in terms of prognosis and biological behavior: while IDHwt grade III gliomas strikingly resemble primary glioblastomas,7,9IDHwt grade II neoplasms display less malignant features.4,6
A recent consensus from the cIMPACT-NOW consortium has proposed that grades II and III IDHwt astrocytomas harboring epidermal growth factor receptor (EGFR) amplification, and/or combined whole chromosome 7 gain and whole chromosome 10 loss (+7/−10), and/or telomerase reverse transcriptase (TERT) promoter mutation should be considered as bona fide glioblastomas, given their poor outcome,10,11 though these recommendations are not yet part of the WHO classification.
The aim of this study was to better define the outcome of IDHwt grade II diffuse gliomas compared with IDH-mutant (IDHmut) grade II and IDHwt grade III diffuse gliomas, highlighting the main prognostic factors in this cohort.
Materials and Methods
We performed retrospective research in the OncoNeuroTek database for all patients with diagnoses of WHO grades II and III diffuse gliomas between January 1989 and February 2020. After dividing WHO grades II and III gliomas in molecular subgroups based on their IDH1/2 and chromosome 1p/19q codeletion status,7,12 we focused on the subgroup of patients with IDHwt grade II gliomas. The clinical, radiological, histological, and molecular features of the patients in this subgroup were thoroughly reviewed to ensure an accurate patient selection. All available histological specimens were independently reviewed by 2 expert neuropathologists (K.M., F.B.), who assigned an integrated diagnosis according to the 2016 WHO Classification of Tumors of the Central Nervous System.1 Discordant assessments were resolved after collective discussion and additional immunohistochemical and molecular studies. Immunohistochemical staining for IDH1 R132H, which was performed systematically in all cases, allowed identification and exclusion of IDH1-mutant patients who were falsely negative on Sanger sequencing because of massive contamination of the tumor specimen with normal tissue. Immunostainings for alpha thalassemia/mental retardation syndrome X-linked (ATRX), p53, H3K27M, FGFR3, EGFR, CD34, and neurofilaments allowed to better characterize and classify the tumor and to exclude circumscribed gliomas. Grading was assigned using widely accepted criteria13 that have recently been embraced by the cIMPACT consortium for lower-grade IDHmut diffuse astrocytomas14: tumors with high cellularity, marked nuclear atypia, and ≥2 mitoses (per 10 high power fields for biopsy specimens and per 30 high power fields Ki67 hotspots for resection specimens) were attributed grades III–IV histology.1,14 Patients with midline tumors were excluded from the study if positive for the H3F3A K27M mutation on immmunohistochemistry and/or DNA sequencing, as they are assigned grade IV according to the 2016 WHO classification. MRI scans acquired at diagnosis were systematically reviewed to verify that imaging features were compatible with diffuse glioma and to exclude the presence of gross nodules of contrast enhancement that would suggest that it was sampled the periphery of a higher-grade neoplasm. Patients who, upon the revision of histological, molecular, and MRI features, had a confirmed diagnosis of “diffuse astrocytoma, IDH-wildtype (grade II)” were included in subsequent analyses.
The clinical and paraclinical characteristics in patients with IDHwt grade II gliomas were analyzed and compared with patients with IDHwt grade III gliomas. We separately assessed how many patients in the group of IDHwt grade II and IDHwt III gliomas met the definition of “diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, grade IV” according to cIMPACT-NOW update 310 and compared their clinicomolecular features to patients with IDHwt tumors of the same grade not meeting this definition.
All tumor samples and clinical data were collected upon written informed consent in accordance with the tenets of the Declaration of Helsinki. The study was approved by the ethical committee CPP “Ile-de-France VI.”
Molecular Analyses
Tumor DNA was extracted from formalin-fixed paraffin-embedded (FFPE) and/or from snap frozen tumor samples (−80°C). DNA was extracted using commercial kits (GeneJET FFPE DNA purification kit, Thermo Scientific [FFPE tissue]; QIAamp DNA mini kit, Qiagen [frozen tissue]) or by automated DNA extraction (Maxwell, Promega). The mutational status of IDH1 (codon 132), IDH2 (codon 172), H3F3A (codon 27 and 34), FGFR1 (codons 546 and 656), BRAF (codon 600), and TERT promoter (-250 and -228) was obtained by Sanger sequencing following standard PCR amplification, using previously reported primers15–17 or from next-generation sequencing (NGS). Information on copy number was acquired by comparative genomic hybridization (CGH-array)18 or by copy number analysis from NGS data. FGFR3-TACC3 fusions were assessed by real-time (RT)-PCR amplification followed by Sanger sequencing from RNA extracted from snap-frozen tumor tissue.19 A subset of patients who tested negative for FGFR3-TACC3 fusions on RT-PCR but had suggestive cell morphology underwent a wider research for fusion genes using Illumina NGS panels: the Archer Comprehensive Thyroid Lung Fusion Plex (MiniSeq), which identifies rearrangements in ALK, AKT1, BRAF, CALCA, CCND1, CTNNB1, DDR2, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, FOXL4, GNAS, HRAS, IDH1, IDH2, KRAS, KRT20, KRT7, MAP2K1, MET, NRAS, NRG1, NTRK1, NTKR2, NTRK2, PIK3CA, PPARG, PTH, RAF1, RET, ROS1, SLC5A5, THADA, TTF, or the AmpliSeq FOCUS (Miseq), which identifies rearrangements in ABL1, ALK, AKT3, AXL, BRAF EGFR, ERBB2, ERG, ETV1, ETV4, ETV5, FGFR1, FGFR2, FGFR3, MET, NTRK1, NTRK2, NTRK3, PDGFRA, PPARG, RAF1, RET, and ROS1.
Statistical Analyses
Categorical variables were compared using the chi-square or the Fisher’s exact test. Quantitative variables were compared using Student’s t-test or the Mann–Whitney test. Overall survival (OS) was estimated by the Kaplan–Meier method and survival curves were compared using the log-rank test. The Cox model was used for continuous variables survival analyses. Hierarchical clustering and multidimensional association tables were used to explore associations between variables. The Pearson or the Spearman correlation test was used to assess statistically significant correlations between variables. For all analyses, the established threshold for statistical significance was P = 0.05. All statistical analyses were performed using “R” software packages.
Results
IDHwt Grade II Gliomas
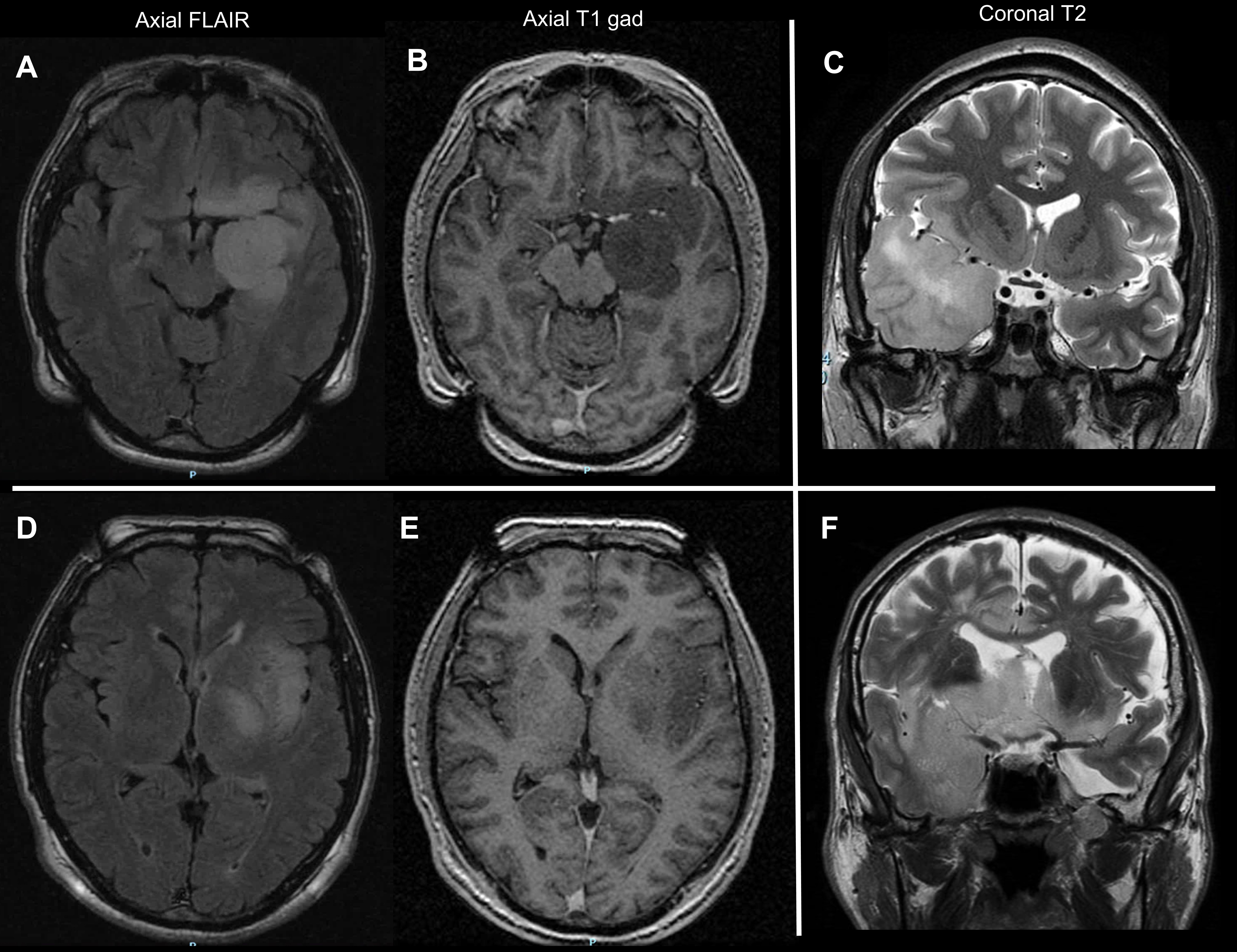
The process of case selection is illustrated in Fig. 1. The clinical and molecular characteristics of the 47 patients who were assigned a diagnosis of “diffuse astrocytoma, IDH-wildtype (grade II)” upon centralized histological review are summarized in Table 1. The median age at diagnosis was 55.0 years (range, 19.6–82.1). Thirty-6 patients were male (36/47, 77%). Median preoperative Karnofsky performance status (KPS) was 90 (range, 70–100). Tumors commonly had their epicenter in the temporal lobe, extending to the fronto-basal lobe and the insula (28/47, 60%) (Supplementary Figure 1, panel A–C). Infiltration was commonly extensive, involving the ipsilateral deep gray matter (10/32, 57%) (Supplementary Figure 1, panel D–E), the cortex of adjacent lobes (5/32, 16%) (Supplementary Figure 1, panel C), the brainstem (3/32, 9%), and the contralateral hemisphere (5/32, 16%) (Supplementary Figure 1, panel F). As a consequence of tumor location, size, and highly infiltrative behavior, surgery commonly consisted of biopsy (27/44, 61%). Strikingly, in this population of IDHwt, H3K27 and G34-wildtype gliomas, immunohistochemical studies showed loss of ATRX nuclear expression in 6 cases (6/38, 16%). Seventeen patients were studied by NGS and 30 by Sanger sequencing plus CGH-array. The most common molecular alterations included TERT promoter mutations (23/45, 51%), whole chromosome 7 gain (10/37, 27%), and whole chromosome 10 loss (10/41, 24%). Cyclin-dependent kinase inhibitor 2A (CDKN2A) deletions (5/43, 12%), EGFR amplifications (4/43, 9%), and chromosome 9p loss (3/42, 7%) were far less common. One patient with a temporal tumor harbored a FGFR1 mutation (1/26, 4%), and one patient with a bithalamic tumor harbored a BRAF V600G mutation (1/35, 3%). None of the tumors harbored TP53 mutations (0/22), PTEN deletions (0/18), PDGFRα amplifications (0/17), or the chromosome 1p/19q codeletion (0/42). Twenty-nine patients underwent the research for fusion genes, with the detection of gene fusions in 5 (5/29, 17%), including 4 fusions FGFR3 (exon 17 or 18)‒TACC3 (exon 5, 8, 11 or 23) and one fusion FGFR3 (exon 17)‒MYH14 (exon 23).
Fig. 1.

Algorithm for patient selection.
Table 1.
Clinical and molecular features in the whole cohort of IDHwt grade II gliomas (n = 47), in the subgroup of patients meeting the definition of “diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma (grade IV)” (molecular GBM) (n = 29) and in the subgroup of patients not meeting this definition (n = 14)
| IDHwt Grade II Gliomas, Whole Cohort (n = 47) | IDHwt Grade II Gliomas Meeting the Definition of Molecular GBM (n = 29) | IDHwt Grade II Gliomas NOT Meeting the Definition of Molecular GBM (n = 14) | P-value* | |
|---|---|---|---|---|
| Age, y, median (range) | 55.0 (19.6–82.1) | 58.6 (20.8–82.1) | 34.5 (19.6–65.4) | 0.00057 |
| Male | 36/47 (77%) | 24/29 (83%) | 10/14 (71%) | 0.44 |
| Preoperative KPS, median (range) | 90 (70–100) | 90 (70–100) | 90 (70–100) | 0.83 |
| Tumor location | ||||
| Fronto-temporo-insular | 28/47 (60%) | 19/29 (66%) | 5/14 (36%) | 0.0055 |
| Fronto-callosal or parieto- callosal | 4/47 (9%) | 4/29 (14%) | 0/14 (0%) | |
| Other | 11/47 (23%) | 6/29 (21%) | 5/14 (36%) | |
| Thalamo-mesencephalic | 4/47 (9%) | 0/29 (0%) | 4/14 (29%) | |
| Extent of resection | ||||
| Biopsy | 27/44 (61%) | 19/28 (68%) | 7/12 (58%) | 0.88 |
| Partial resection | 9/44 (20%) | 5/28 (18%) | 2/12 (17%) | |
| Gross total resection | 8/44 (18%) | 4/28 (14%) | 3/12 (25%) | |
| Initial treatment | ||||
| Chemotherapy alone | 13/38 (34%) | 10/25 (40%) | 3/9 (33%) | 0.74 |
| Sequential radiochemotherapy | 7/38 (18%) | 5/25 (20%) | 1/9 (11%) | |
| Stupp protocol | 12/38 (32%) | 7/25 (28%) | 3/9 (33%) | |
| Radiotherapy alone | 1/38 (3%) | 0/25 (0%) | 0/9 (0%) | |
| Surveillance | 5/38 (13%) | 3/25 (12%) | 2/9 (22%) | |
| Radiological progression | ||||
| Infiltrative | 13/18 (72%) | 11/15 (73%) | 2/3 (67%) | 0.45 |
| Nodular enhancing | 5/18 (28%) | 4/15 (27%) | 1/3 (33%) | |
| Molecular profile | ||||
| TERT promoter mutation | 23/45 (51%) | 23/28 (82%) | 0/14 (0%) | <0.0001 |
| EGFR amplification | 4/43 (9%) | 4/26 (15%) | 0/14 (0%) | 0.28 |
| 7+ | 10/37 (27%) | 10/26 (38%) | 0/10 (0%) | 0.016 |
| 10- | 10/41 (24%) | 10/26 (38%) | 0/14 (0%) | 0.011 |
| 7+/−10 | 7/41 (17%) | 7/26 (27%) | 0/14 (0%) | 0.075 |
| 9p loss | 3/42 (7%) | 3/26 (12%) | 0/14 (0%) | 0.54 |
| CDKN2A deletion | 5/43 (12%) | 3/26 (12%) | 1/14 (7%) | 1 |
| Median OS, mo | 59.1 | 42.2 | 56.7 | 0.2 |
7+ = whole chromosome 7 gain; 9p = chromosome 9p; −10 = whole chromosome 10 loss; 7+/−10 = whole chromosome 7 gain and whole chromosome 10 loss; FU = follow-up. *P-values refer to the comparison between patients meeting the definition for molecular GBM and patients not meeting this definition; in bold statistically significant results.
Initial treatment included concomitant radiochemotherapy with temozolomide followed by adjuvant temozolomide according to the Stupp protocol (12/38, 32%), sequential radiochemotherapy with temozolomide or procarbazine/lomustine/vincristine (PCV) (7/38, 18%), chemotherapy alone (13/38, 34%), or radiotherapy alone (1/38, 3%). Tumor progression occurred either through an infiltrative pattern (Fig. 2, panel C-D) or through the appearance of gross nodules of contrast enhancement (Fig. 2, panel G-H). Patients with nodular progression showed poorer outcomes compared with patients evolving through an infiltrative pattern (median OS: 22 vs 88 mo, P = 0.03) (Supplementary Figure 2, panel A). Three patients underwent surgery at progression: in 1 patient, a TERT promoter mutation appeared 12 years after initial surgery, while an EGFR amplification appeared 3 and 5 years after initial surgery in 2 patients with known TERT promoter mutations.
Fig. 2.

Radiological patterns of progression in IDHwt grade II gliomas. Panel A-D: example of an infiltrative pattern of progression. Compared with images at diagnosis (panel A, B), images at progression (panel C, D) show a substantial extension of tumor infiltration along the right temporal and insular lobe and the ipsilateral thalamus (panel C), without the appearance of enhancing abnormalities (panel D). Panel E-H: example of a nodular pattern of progression. Compared with images at diagnosis (panel E, F), images at progression (panel G, H) show the appearance of a gross nodule of contrast enhancement in the right insula (panel H), surrounded by extensive perilesional edema (panel G, H).
The median OS for IDHwt grade II gliomas was 59 months (vs 101 mo for IDHmut 1p/19q non-codeleted and 176 mo for IDHmut 1p/19q codeleted gliomas, P < 0.0001) (Fig. 3, panel A). Higher preoperative KPS (P = 0.04) was associated with improved OS. Whole chromosome 10 loss (median OS: 88 vs 33 mo, P = 0.03), the +7/−10 signature (median OS: 88 vs 33 mo, P = 0.02), and chromosome 9p loss (median OS: 88 vs 19 mo, P < 0.0001) were associated with poorer OS (Supplementary Figure 2, panel B-D). A similar trend was observed for whole chromosome 7 gain (median OS: 88 vs 41 mo, P = 0.2), TERT promoter mutations (median OS: 88 vs 41 mo, P = 0.2), and CDKN2A deletions (median OS: 59 vs 42 mo, P = 0.3). Association matrix showed that whole chromosome 10 loss was associated with whole chromosome 7 gain (P < 0.001), EGFR amplifications (P < 0.01), chromosome 9p loss (P < 0.01), and CDKN2A deletions (P < 0.01) (Supplementary Figure 3, panel A-B).
Fig. 3.

Overall survival in IDHwt grade II gliomas compared with IDHmut grade II (panel A) and to IDHwt grade III (panel B) gliomas; overall survival in IDHwt grade II and III gliomas meeting the cIMPACT-NOW definition for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma (grade IV)” (panel C-F). Panel A: survival curves for IDHmut 1p/19q codeleted (black line) vs IDHmut 1p/19q non-codeleted (dark gray line) vs IDHwt (light gray line) grade II gliomas (median OS: 176 vs 101 vs 59 mo, P < 0.0001). Panel B: survival curves for IDHwt grade II (gray line) vs IDHwt grade III (black line) gliomas (median OS: 59 vs 19 months, P < 0.0001). Panel C: survival curves for patients with IDHwt grade III gliomas meeting (black line) and not meeting (gray line) the cIMPACT definition for molecular glioblastoma (median OS: 17 vs 23 months, P = 0.07). Panel D: survival curves for patients with IDHwt grade II (gray line) and IDHwt grade III (black line) gliomas meeting the cIMPACT definition of molecular glioblastoma (median OS: 42 vs 17 months, P < 0.0001). Panel E: survival curves for patients with IDHwt grade II (gray line) and grade III (black line) gliomas meeting the cIMPACT definition for molecular glioblastoma because of isolated TERT promoter mutations (median OS: 88 vs 22 mo, P = 0.002). Panel F: survival curves for patients with IDHwt grade II (gray line) and grade III (black line) meeting the cIMPACT definition for molecular glioblastoma because of EGFR amplifications and/or the +7/−10 signature (median OS: 37 vs 18 mo, P = 0.02).
IDHwt Grade III Gliomas
Table 2 compares the clinical and molecular features of IDHwt grade II (n = 47) and grade III (n = 255) gliomas. The median age at diagnosis for IDHwt grade III gliomas was 56.1 years-old (vs 55.0 years, P = 0.26). One-hundred-fifty-four patients (154/255, 60%) were male (vs 36/47 (77%), P = 0.048). Median preoperative KPS was 80 (vs 90, P = 0.0025). Surgery commonly consisted of biopsy (115/237, 49%) (P = 0.31). Initial treatment was represented by concomitant or sequential radiochemotherapy (129/220, 59%) or, less frequently, by chemotherapy (53/220, 24%) or radiotherapy (38/220, 17%) alone (P < 0.001).
Table 2.
Clinical and molecular features in IDHwt grade II (n = 47) and IDHwt grade III (n = 255) gliomas
| IDHwt Grade II And III Gliomas, Whole Cohort (n = 302) | IDHwt Grade II Gliomas (n = 47) | IDHwt Grade III Gliomas (n = 255) | P-value* | |
|---|---|---|---|---|
| Age, y, median (range) | 56.1 (8.5–84.1) | 55.0 (19.6–82.1) | 56.1 (8.5–84.1) | 0.26 |
| Male | 190/302 (63%) | 36/47 (77%) | 154/255 (60%) | 0.048 |
| Preoperative KPS, median (range) | 90 (20–100) | 90 (70–100) | 80 (20–100) | 0.0025 |
| Extent of resection | ||||
| Biopsy | 142/270 (53%) | 27/44 (61%) | 115/237 (49%) | 0.31 |
| Partial resection | 60/270 (22%) | 9/44 (20%) | 51/237 (22%) | |
| Gross total resection | 68/270 (25%) | 8/44 (18%) | 60/237 (25%) | |
| Initial treatment | ||||
| Chemotherapy alone | 67/258 (26%) | 14/38 (37%) | 53/220 (24%) | <0.001 |
| Concomitant or sequential RT-CHT | 147/258 (57%) | 18/38 (47%) | 129/220 (59%) | |
| Radiotherapy alone | 39/258 (15%) | 1/38 (3%) | 38/220 (17%) | |
| Surveillance | 5/258 (2%) | 5/38 (13%) | 0/220 (0%) | |
| Molecular profile | ||||
| TERT promoter mutation | 176/274 (64%) | 23/45 (51%) | 151/230 (66%) | 0.0092 |
| EGFR amplification | 77/275 (28%) | 4/43 (9%) | 73/235 (31%) | 0.00088 |
| 7+ | 102/206 (50%) | 10/37 (27%) | 91/173 (53%) | 0.0062 |
| 10- | 119/259 (46%) | 10/41 (24%) | 111/222 (50%) | 0.0010 |
| 7+/−10 | 85/236 (36%) | 7/41 (17%) | 77/199 (39%) | 0.018 |
| 9p loss | 59/260 (23%) | 3/42 (7%) | 57/222 (26%) | 0.0082 |
| CDKN2A deletion | 76/274 (27%) | 5/43 (12%) | 72/235 (31%) | 0.0033 |
| TP53 mutation | 22/123 (18%) | 0/22 (0%) | 22/104 (21%) | <0.0001 |
| Median OS, mo | 21.9 | 59.1 | 19.1 | < 0.0001 |
7+ = whole chromosome 7 gain; 9p = chromosome 9p; 10- = whole chromosome 10 loss; 7+/−10 = whole chromosome 7 gain and whole chromosome 10 loss; RT-CHT = radiochemotherapy. *P-values refer to the comparison between IDHwt grades II and III gliomas; in bold statistically significant results.
Compared with IDHwt grade II, IDHwt grade III gliomas had a higher prevalence of TERT promoter mutations: 151/230 (66%) vs 23/45 (51%), P = 0.0092; EGFR amplifications; 73/235 (31%) vs 4/43 (9%), P = 0.00088; whole chromosome 7 gain: 91/173 (53%) vs 10/37 (27%), P = 0.0062; whole chromosome 10 loss: 111/222 (50%) vs 10/41 (24%), P = 0.0010; chromosome 9p loss: 57/222 (26%) vs 3/42 (7%), P = 0.0082; CDKN2A deletions: 72/235 (31%) vs 5/43 (12%), P = 0.0033; and TP53 mutations (22/104 (21%) vs 0/22 (0%), P < 0.0001.
The median OS for IDHwt grade III gliomas was 19 months (vs 59 mo for IDHwt grade II gliomas, P < 0.0001) (Fig. 3, panel B). Younger age at diagnosis (P = 0.0019) and higher preoperative KPS (P = 0.0030) were associated with prolonged OS. EGFR amplifications (median OS: 22 vs 16 mo, P = 0.04) and whole chromosome 10 loss (median OS: 23 vs 18 mo, P = 0.03) were associated with poorer OS, and a similar trend was observed for TERT promoter mutations (median OS: 22 vs 17 mo, P = 0.05) and CDKN2A deletions (median OS: 20 vs 16 mo, P = 0.05) (Supplementary Figure 2, panel E-H).
Association matrix showed that whole chromosome 10 loss associated with whole chromosome 7 gain (P < 0.001), EGFR amplifications (P < 0.01), chromosome 9p loss (P < 0.001) and CDKN2A deletions (P < 0.01). TERT promoter mutations associated with whole chromosome 7 gain (P < 0.001), EGFR amplifications (P = 0.04), and older age at diagnosis (P = 0.02) (Supplementary Figure 3, panel C-D).
IDHwt Grades II and III Gliomas Meeting cIMPACT-NOW Criteria for “Diffuse Astrocytic Glioma, IDH-Wildtype, with Molecular Features of Glioblastoma (Grade IV)”
Twenty-nine patients in the group of IDHwt grade II gliomas (29/43, 67%) and 166 patients in the group of IDHwt grade III gliomas (166/224, 74%) met cIMPACT-NOW criteria for the definition of “diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma (WHO grade IV).” 10
Patients with IDHwt grade II gliomas and molecular features of glioblastoma were older (median age at diagnosis: 58.6 vs 34.5, P = 0.00057) and more frequently had fronto-temporo-insular tumors (19/29 (66%) vs 5/14 (36%), P = 0.0055) compared with patients with IDHwt grade II gliomas lacking defining molecular alterations (Table 1). Extent of resection (P = 0.88) and treatment schemes (P = 0.74) did not substantially differ between the 2 groups, as most patients had been treated before the publication of cIMPACT criteria. The median OS in patients with IDHwt grade II gliomas and molecular features of glioblastoma was 42 months (vs 57 months in patients with IDHwt grade II gliomas lacking defining features, P = 0.2). Neither age (P = 0.31) nor molecular features of GBM (P = 0.17) were associated with survival on the Cox model.
Patients with IDHwt grade III gliomas and molecular features of glioblastoma were older (median age at diagnosis: 58.8 vs 43.9 years, P < 0.0001) and more frequently received biopsy (80/149 (54%) vs 19/47 (40%), P = 0.063) compared with patients with IDHwt grade III gliomas lacking defining molecular alterations (Supplementary Table 1). The median OS patients with IDHwt grade III gliomas and molecular features of glioblastoma was 17 months (vs 23 mo for IDHwt grade III gliomas lacking defining features, P = 0.07) (Fig. 3, panel C). Age (P = 0.0019) was associated with survival on Cox model, while molecular features of GBM did not reach significance (P = 0.058).
Therefore, IDHwt grade II and IDHwt grade III gliomas meeting the definition for molecular glioblastoma clearly had different OS (42 vs 17 mo, P < 0.0001) (Fig. 3, panel D). Table 3 compares clinical and molecular features in the 2 groups. Most patients in the group of IDHwt grade II gliomas met the definition of molecular glioblastoma because of a single criterion, most frequently isolated TERT promoter mutations (16/26, 62%). Conversely, patients with IDHwt grade III gliomas generally met the definition of molecular glioblastoma because of multiple criteria as, besides TERT promoter mutations, most of them had additional defining alterations such as EGFR amplifications or the +7/−10 signature (93/131, 71%).
Table 3.
Comparison between IDHwt grade II (n = 29) and IDHwt grade III (n = 166) gliomas meeting cIMPACT-NOW criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma (grade IV)” (molecular GBM)
| IDHwt Grade II and III Gliomas Meeting the definition of molecular GBM (n = 195) | IDHwt Grade II Gliomas Meeting the Definition of Molecular GBM (n = 29) | IDHwt Grade III Gliomas Meeting the Definition of Molecular GBM (n = 166) | P-value* | |
|---|---|---|---|---|
| Age, y, median (range) | 58.7 (20.8–83.0) | 58.6 (20.8–82.1) | 58.8 (22.2–83.0) | 0.86 |
| Male | 124/195 (64%) | 24/29 (83%) | 100/166 (60%) | 0.022 |
| Preoperative KPS, median (range) | 90 (60–100) | 90 (70–100) | 80 (60–100) | 0.026 |
| Extent of resection | ||||
| Biopsy | 99/177 (56%) | 19/28 (68%) | 80/149 (54%) | 0.33 |
| Partial resection | 31/177 (18%) | 5/28 (18%) | 26/149 (17%) | |
| Gross total resection | 47/177 (27%) | 4/28 (14%) | 43/149 (29%) | |
| Treatment | ||||
| Chemotherapy alone | 41/169 (24%) | 10/25 (40%) | 30/144 (21%) | <0.0001 |
| Concomitant or sequential RT-CHT | 98/169 (58%) | 12/25 (48%) | 87/144 (60%) | |
| Radiotherapy alone | 27/169 (16%) | 0/25 (0%) | 27/144 (19%) | |
| Surveillance | 3/169 (2%) | 3/25 (12%) | 0/144 (0%) | |
| Molecular profile | ||||
| TERT promoter mutation | 171/187 (91%) | 23/28 (82%) | 148/159 (93%) | 0.12 |
| EGFR amplification | 77/180 (43%) | 4/26 (15%) | 73/155 (47%) | 0.0037 |
| 7+ | 96/141 (68%) | 10/26 (38%) | 86/116 (74%) | 0.00089 |
| 10- | 114/170 (67%) | 10/26 (36%) | 105/145 (72%) | 0.00045 |
| 7+/−10 | 83/151 (55%) | 7/26 (27%) | 76/126 (60%) | 0.00194 |
| 9p loss | 47/170 (28%) | 3/26 (12%) | 44/145 (30%) | 0.057 |
| CDKN2A deletion | 57/180 (32%) | 3/26 (12%) | 54/155 (35%) | 0.023 |
| Number of c-IMPACT criteria met | ||||
| One | 52/154 (34%) | 20/26 (77%) | 33/129 (26%) | <0.0001 |
| Two or 3 | 102/154 (66%) | 6/26 (23%) | 96/129 (74%) | |
| Reason for meeting c-IMPACT criteria: | ||||
| TERT promoter mutation without EGFR amplification or +7/−10 | 43/156 (28%) | 16/26 (62%) | 28/131 (21%) | <0.0001 |
| EGFR amplification and/ or +7/−10 without TERT promoter mutation | 15/156 (10%) | 5/26 (19%) | 10/131 (8%) | |
| TERT promoter mutation plus EGFR amplification and/or +7/−10 | 98/156 (63%) | 5/26 (19%) | 93/131 (71%) | |
| Median OS, mo | 19.2 | 42.2 | 17.2 | <0.0001 |
RT-CHT = radiochemotherapy; +7 = whole chromosome 7 gain; −10 = whole chromosome 10 loss. *P values refer to the comparison between the 2 subgroups identified by initial WHO grade; in bold statistically significant results.
We then evaluated the ability of the different criteria to capture tumor malignant behavior. Isolated TERT promoter mutations, without EGFR amplifications or the +7/−10 signature, were associated with a median OS of 88 months in IDHwt grade II and 22 months in IDHwt grade III gliomas (P = 0.002) (Fig. 3, panel E). Conversely, the presence of EGFR amplifications and/or of the +7/−10 signature, regardless of the presence of TERT promoter mutations, was associated with a median OS of 37 months in IDHwt grade II and 18 months in IDHwt grade III gliomas (P = 0.02) (Fig. 3, panel F).
Discussion
From a large cohort of 1360 “lower grade” (ie, grades II and III) gliomas, we extracted 101 IDHwt grade II gliomas, whose radiological, molecular, and histological features were thoroughly revised to guarantee a rigorous patient selection. We ensured to exclude IDH1-mutant gliomas falsely negative on Sanger sequencing because of contamination with normal tissue, peripheral samples of high-grade IDHwt gliomas, and H3K27M-mutant midline gliomas. The analyses were then restricted to 47 patients who had a confirmed diagnosis of “diffuse astrocytoma, IDH-wildtype (grade II)” following centralized histological review, which was independently conducted by 2 expert neuropathologists through comprehensive morphological and immunohistochemical studies.
Representing less than 15% of low-grade diffuse gliomas,4,8,20IDHwt grade II gliomas are uncommon, especially in women, and are associated with older age at diagnosis, fronto-temporo-insular location and a highly invasive behavior,3,6,7 with frequent infiltration of adjacent cortex and deep gray matter.21 This highly infiltrative pattern accounts for the prevalence of biopsy over resection and the scarce tissue availability for translational studies. With no TP53 mutations and, as expected,12 no 1p/19q codeletions, our IDHwt grade II gliomas correspond to the previously defined “triple negative” grade II gliomas3: usually large and highly infiltrative fronto-temporal-insular tumors, which could be merely defined as IDHwt grade II diffuse gliomas.
Compared with IDHwt grade III gliomas, IDHwt grade II gliomas had a lower burden of molecular alterations, including EGFR amplifications (9% vs 31%), whole chromosome 7 gain (27% vs 53%), whole chromosome 10 loss (24% vs 50%), TERT promoter mutations (51% vs 66%), TP53 mutations (0% vs 21%), chromosome 9p loss (7% vs 26%), and CDKN2A deletions (12% vs 31%). Their median OS was 59 months that, while much shorter than IDHmut grade II gliomas, was 3 times the median OS of IDHwt grade III tumors (19 months, P < 0.0001). These findings reveal deep differences between IDHwt grade II and IDHwt grade III gliomas in terms of both genetic profile and outcome.
The distinction between histological grades II and III gliomas primarily relies on proliferative activity expressed as mitotic index, which is known to have a heavier prognostic impact in IDHwt than in IDHmut gliomas.4,22 The 2016 WHO classification does not provide clear thresholds for the evaluation of mitotic count. The use of different thresholds for grading assessment margin could explain the inconsistent results obtained in different studies on prognosis and molecular profile of IDHwt grade II and III gliomas and, in our view, represents an urgent issue to address in the forthcoming WHO classification.23 The grading criteria used here have been widely used in the past and have been adopted by the cIMPACT to separate grade II and III IDHmut diffuse astrocytomas14: in the same way, they allow here a prognostic stratification of IDHwt gliomas, suggesting that the same mitotic threshold is associated with different outcome in both IDHmut and IDHwt gliomas.
Among the tumors meeting the cIMPACT-NOW definition of “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV,” 10 only those with grade III histology had a survival similar to IDHwt glioblastomas, while tumors with grade II histology had a survival almost 3 times longer.7,9 In fact, more than half of the patients with IDHwt grade II gliomas in our series met cIMPACT-NOW criteria because of isolated TERT promoter mutations and this was not predictive of poor outcome. TERT promoter mutations are detected in IDHmut 1p/19q codeleted diffuse gliomas a well as in several other primary brain tumors and are associated with different prognostic significance depending on tumor histology and IDH status.24,25 These considerations suggest some caution when assimilating IDHwt grade II gliomas to molecular glioblastomas, especially if the sole criterion met is an isolated TERT promoter mutation. In addition, our findings suggest that other molecular markers, besides the ones identified by the cIMPACT-NOW consortium, could be helpful for the prognostic stratification of IDHwt lower grade diffuse gliomas, such as chromosome 9p loss that was as a strong predictor of poor outcome in our cohort.
The uncertainties on the malignant behavior of IDHwt grade II gliomas, together with their rarity, have prevented to reach a consensus on the standard of treatment for these tumors. As clinical trials conducted in low-grade gliomas failed to indicate a clear treatment strategy for this small population,26–28 the guidelines of the European Association of Neuro-Oncology leave an ample discretional margin on the choice of individual treatment schemes, suggesting that this choice should rely on age, KPS, and MGMT promoter methylation status,29 which are recognized prognostic indicators in IDHwt diffuse gliomas. The absence of clinical standard advocates a careful multidisciplinary approach based on close clinical surveillance and case-by-case decisions. DNA methylation profiles identified 3 subgroups of IDHwt grade II gliomas, one of them with molecular similarities to pilocytic astrocytomas and more favorable outcome.30 However, as most of our patients had a simple biopsy due to highly infiltrative pattern, the scarce available material did not allow such analysis. In our study, we found actionable molecular targets in 7 patients (7/29, 24%), including 1 BRAF V600 mutation, 1 activating FGFR1 mutation, and 5 FGFR3 fusions. FGFR3-TACC3 fusions have been reported in only 3% of IDHwt grades II–IV gliomas but they are of high interest for clinicians as they can be targeted by specific inhibitors.19 Moreover FGFR3-TACC3 fusions characterize a subgroup of IDHwt gliomas with a specific molecular and metabolic profile.31,32 While grade II IDHwt represent less than 10% of IDHwt gliomas, we found here a clear overrepresentation of FGFR3 fusions compared with what is expected for the whole population of IDHwt gliomas, with 4 FGFR3-TACC fusions and one FGFR3-MYH14 fusion out of 29 patients tested (17%; P < 0.01). The systematic screening for fusion genes is therefore of special interest to better classify this population and to provide novel therapeutic targets.
In conclusion, our results highlight the importance of histological grade for the prognostic stratification of IDHwt lower-grade gliomas and warns on the importance of carefully integrating molecular features and histology for diagnostic, prognostic, and theranostic purposes. There is a clear need for further molecular characterization, search for actionable targets, and new clinical trials in this population.
Funding
This work was in part supported by the grant INCa-DGOS-Inserm_12560 of the SiRIC CURAMUS and by funding from the program “investissements d’avenir” ANR-10-IAIHU-06. MS was supported by a grant from the Ligue Nationale contre le Cancer (LNCC; équipe labellisée).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Muriel Brandel, Armelle Rametti, Yseult Cardona, Julie Lerond, and Amithys Rahimian for database management and technical support.
Conflict of interest statement. The authors report no disclosures relevant to the manuscript.
Authorship statement. Study conception and design: GB, ALDS, MS. Molecular analysis: GB, EG, MG, YS, YM. Histological analysis: SR, FB, CV, KM. Statistical analysis and manuscript drafting: GB, MS. All authors contributed to case recruitment and data acquisition. All authors provided a critical review of the manuscript for intellectual content and approved the final version of the manuscript.
Contributor Information
Giulia Berzero, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France; Department of Brain and Behavioral Sciences, University of Pavia, Italy.
Anna Luisa Di Stefano, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France; Department of Neurology, Foch Hospital, Suresnes, France.
Susanna Ronchi, University Hospitals of Pitié Salpêtrière, Charles Foix, R Escourolle Laboratory, Paris, France.
Franck Bielle, University Hospitals of Pitié Salpêtrière, Charles Foix, R Escourolle Laboratory, Paris, France.
Chiara Villa, Department of Pathology, Foch Hospital, Suresnes, France.
Erell Guillerm, University Hospitals of La Pitié Salpêtrière, Charles Foix, Functional Unit of Oncogenetics and Molecular Angiogenetics, Department of Genetics, Paris, France.
Laurent Capelle, University Hospitals of La Pitié Salpêtrière, Charles Foix, Department of Neurology 2, Paris, France.
Bertrand Mathon, University Hospitals of La Pitié Salpêtrière, Charles Foix, Department of Neurology 2, Paris, France.
Alice Laurenge, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France.
Marine Giry, Sorbonne University, Brain and Spinal Cord Institute, Paris, France.
Yohann Schmitt, Sorbonne University, Brain and Spinal Cord Institute, Paris, France.
Yannick Marie, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; Onconeurotek Tumor Bank, University Hospitals of Pitié Salpêtrière, Charles Foix, Paris, France.
Ahmed Idbaih, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France.
Khe Hoang-Xuan, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France.
Jean-Yves Delattre, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France; Onconeurotek Tumor Bank, University Hospitals of Pitié Salpêtrière, Charles Foix, Paris, France.
Karima Mokhtari, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, R Escourolle Laboratory, Paris, France; Onconeurotek Tumor Bank, University Hospitals of Pitié Salpêtrière, Charles Foix, Paris, France.
Marc Sanson, Sorbonne University, Brain and Spinal Cord Institute, Paris, France; University Hospitals of Pitié Salpêtrière, Charles Foix, Department of Neurology 2 Mazarin, Paris, France; Onconeurotek Tumor Bank, University Hospitals of Pitié Salpêtrière, Charles Foix, Paris, France.
References
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 2. Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27(25):4150–4154. [DOI] [PubMed] [Google Scholar]
- 3. Metellus P, Coulibaly B, Colin C, et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010;120(6):719–729. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5): 458–468. [DOI] [PubMed] [Google Scholar]
- 5. Chan AK, Yao Y, Zhang Z, et al. Combination genetic signature stratifies lower-grade gliomas better than histological grade. Oncotarget. 2015;6(25):20885–20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aibaidula A, Chan AK, Shi Z, et al. Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro Oncol. 2017;19(10):1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wijnenga MMJ, Dubbink HJ, French PJ, et al. Molecular and clinical heterogeneity of adult diffuse low-grade IDH wild-type gliomas: assessment of TERT promoter mutation and chromosome 7 and 10 copy number status allows superior prognostic stratification. Acta Neuropathol. 2017;134(6):957–959. [DOI] [PubMed] [Google Scholar]
- 9. Tabouret E, Nguyen AT, Dehais C, et al. ; for POLA Network . Prognostic impact of the 2016 WHO classification of diffuse gliomas in the French POLA cohort. Acta Neuropathol. 2016;132(4):625–634. [DOI] [PubMed] [Google Scholar]
- 10. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018;136(5):805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tesileanu CMS, Dirven L, Wijnenga MMJ, et al. Survival of diffuse astrocytic glioma, IDH1/2 wildtype, with molecular features of glioblastoma, WHO grade IV: a confirmation of the cIMPACT-NOW criteria. Neuro Oncol. 2020;22(4):515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Labussière M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74(23): 1886–1890. [DOI] [PubMed] [Google Scholar]
- 13. Perry A. Pathology of low-grade gliomas: an update of emerging concepts. Neuro Oncol. 2003;5(3):168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brat DJ, Aldape K, Colman H, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 2020;139(3):603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Labussière M, Boisselier B, Mokhtari K, et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology. 2014;83(13):1200–1206. [DOI] [PubMed] [Google Scholar]
- 16. Reyes-Botero G, Giry M, Mokhtari K, et al. Molecular analysis of diffuse intrinsic brainstem gliomas in adults. J Neurooncol. 2014;116(2):405–411. [DOI] [PubMed] [Google Scholar]
- 17. Picca A, Berzero G, Bielle F, et al. FGFR1 actionable mutations, molecular specificities, and outcome of adult midline gliomas. Neurology. 2018;90(23):e2086–e2094. [DOI] [PubMed] [Google Scholar]
- 18. Labussière M, Rahimian A, Giry M, et al. Chromosome 17p homodisomy is associated with better outcome in 1p19q non-codeleted and IDH-mutated gliomas. Oncologist. 2016;21(9):1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Stefano AL, Fucci A, Frattini V, et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin Cancer Res. 2015;21(14):3307–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasselblatt M, Jaber M, Reuss D, et al. Diffuse astrocytoma, IDH-wildtype: a dissolving diagnosis. J Neuropathol Exp Neurol. 2018;77(6):422–425. [DOI] [PubMed] [Google Scholar]
- 21. Izquierdo C, Barritault M, Poncet D, et al. Radiological characteristics and natural history of adult IDH-wildtype astrocytomas with TERT promoter mutations. Neurosurgery. 2019;85(3):E448–E456. [DOI] [PubMed] [Google Scholar]
- 22. Olar A, Wani KM, Alfaro-Munoz KD, et al. IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol. 2015;129(4):585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komori T. Updating the grading criteria for adult diffuse gliomas: beyond the WHO2016CNS classification. Brain Tumor Pathol. 2020;37(1):1–4. [DOI] [PubMed] [Google Scholar]
- 24. Labussière M, Di Stefano AL, Gleize V, et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br J Cancer. 2014;111(10):2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stichel D, Ebrahimi A, Reuss D, et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018;136(5):793–803. [DOI] [PubMed] [Google Scholar]
- 26. Ceccarelli M, Barthel FP, Malta TM, et al. ; TCGA Research Network . Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buckner JC, Shaw EG, Pugh SL, et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med. 2016;374(14):1344–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baumert BG, Hegi ME, van den Bent MJ, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bell EH, Zhang P, Shaw EG, et al. Comprehensive genomic analysis in NRG oncology/RTOG 9802: a phase III trial of radiation versus radiation plus procarbazine, lomustine (CCNU), and vincristine in high-risk low-grade glioma. J Clin Oncol. 2020; JCO1902983. 10.1200/JCO.19.02983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weller M, van den Bent M, Tonn JC, et al. ; European Association for Neuro-Oncology (EANO) Task Force on Gliomas . European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):e315–e329. [DOI] [PubMed] [Google Scholar]
- 31. Di Stefano AL, Picca A, Saragoussi E, et al. Clinical, molecular and radiomic profile of gliomas with FGFR3-TACC3 fusions. Neuro Oncol. 2020;22(11):1614–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frattini V, Pagnotta SM, Tala, et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. 2018;553(7687):222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.