

PURPOSE
Rhabdomyosarcoma (RMS) is the most common pediatric soft-tissue sarcoma and accounts for 3% of all pediatric cancer. In this study, we investigated germline sequence and structural variation in a broad set of genes in two large, independent RMS cohorts.
MATERIALS AND METHODS
Genome sequencing of the discovery cohort (n = 273) and exome sequencing of the secondary cohort (n = 121) were conducted on germline DNA. Analyses were performed on 130 cancer susceptibility genes (CSG). Pathogenic or likely pathogenic (P/LP) variants were predicted using the American College of Medical Genetics and Genomics (ACMG) criteria. Structural variation and survival analyses were performed on the discovery cohort.
RESULTS
We found that 6.6%-7.7% of patients with RMS harbored P/LP variants in dominant-acting CSG. An additional approximately 1% have structural variants (ATM, CDKN1C) in CSGs. CSG variants did not influence survival, although there was a significant correlation with an earlier age of tumor onset. There was a nonsignificant excess of P/LP variants in dominant inheritance genes in the patients with FOXO1 fusion–negative RMS patients versus the patients with FOXO1 fusion–positive RMS. We identified pathogenic germline variants in CSGs previously (TP53, NF1, DICER1, mismatch repair genes), rarely (BRCA2, CBL, CHEK2, SMARCA4), or never (FGFR4) reported in RMS. Numerous genes (TP53, BRCA2, mismatch repair) were on the ACMG Secondary Findings 2.0 list.
CONCLUSION
In two cohorts of patients with RMS, we identified pathogenic germline variants for which gene-specific therapies and surveillance guidelines may be beneficial. In families with a proband with an RMS-risk P/LP variant, genetic counseling and cascade testing should be considered, especially for ACMG Secondary Findings genes and/or with gene-specific surveillance guidelines.
INTRODUCTION
Rhabdomyosarcoma (RMS) accounts for 3% of all pediatric cancer and is the most common soft-tissue tumor in childhood and adolescence.1 The WHO classification scheme divides RMS into four subtypes: alveolar (ARMS), embryonal (ERMS), pleomorphic, and sclerosing or spindle cell.2 ERMS accounts for 70% of all RMS and is associated with heterozygous loss of 11p15.5.3 ERMS typically presents in children under 10 years of age, most commonly in the head and neck region and genitourinary system. ARMS is most commonly driven by the PAX3-FOXO1 or PAX7-FOXO1 gene fusions, termed fusion-positive RMS. The incidence of ARMS is similar across age groups; however, compared with ERMS, the median age at diagnosis is higher.4,5 The most common site for ARMS is in the deep tissues of the extremities.6 Five-year event-free survival (EFS) for RMS is around 53%-67% for children younger than 15 years and 30%-51% for children older than 15 years.7 A multimodal approach to treatment is used across risk groups (low, intermediate, high), which includes surgery, radiotherapy, and chemotherapy, typically vincristine, dactinomycin, and cyclophosphamide.8 Risk groups are determined by the TNM stage, clinical group, and fusion status; approximately 50% of the patients are classified as intermediate-risk. 9 RMS survivors are at risk for long-term morbidities, including treatment-related secondary malignancies.10
CONTEXT
Key Objective
To determine the prevalence of germline pathogenic or likely pathogenic (P/LP) variants in known cancer susceptibility genes (CSG) in children and young adults with rhabdomyosarcoma (RMS).
Knowledge Generated
In patients with RMS, approximately 7% harbor germline P/LP variants and approximately 1% harbor structural variants in dominant-acting CSG, including TP53, NF1, DICER1, mismatch repair genes, BRCA2, CBL, CHEK2, and SMARCA4. RMS patients with P/LP variants in CSG had an earlier age of tumor onset compared with patients without P/LP variants, although these variants did not influence overall survival. Numerous genes were on the American College of Medical Genetics and Genomics (ACMG) Secondary Findings 2.0 list.
Relevance
Children with RMS should have germline sequencing. Those who have a germline P/LP variant in a CSG should have genetic counseling and cascade testing for other family members, especially for ACMG Secondary Findings genes and/or those with gene-specific surveillance guidelines.
Pathogenic germline variants are observed in 10%-12% of children with cancer.11-13 One study found that approximately 4% of patients with advanced adult cancers harbored pathogenic germline variants that suggested targets for therapy.14 RMS is associated with numerous hereditary cancer disorders, including Li-Fraumeni, Lynch, neurofibromatosis type 1, Noonan, Costello, DICER1, and Beckwith-Wiedemann syndromes. In addition, several studies have identified RMS-associated somatic variation, including NRAS, KRAS, HRAS, FGFR4, PIK3CA, CTNNB1, FBXW7, and BCOR with Tyr kinase/RAS/PIK3CA axis mutations, and MYOD1 mutations.3,15
The characterization of RMS-associated germline genetic variants may influence the selection of therapeutic agents (eg, PARP [poly(ADP-ribose) polymerase] inhibitors for DNA repair defects), enable cascade genetic testing of at-risk family members, facilitate tumor surveillance for those at risk, and increase understanding of RMS etiology. To identify germline variants, comprehensive analysis of large numbers of individuals with clinical- and molecular-annotated RMS is required. Thus, we investigated germline sequence and structural variation in a broad set of genes in two large and independent RMS cohorts: (1) the discovery cohort, derived from a clinical trial open to patients with intermediate-risk RMS; and (2) the secondary cohort, derived from an independent group of patients with mixed clinical risk and treatment.
MATERIALS AND METHODS
Patients and Ethics Statement
For the discovery cohort, patient DNA samples were collected from the Children's Oncology Group (COG) clinical therapeutic trial ARST0531 investigating intermediate-risk RMS.8 Consent for banking and future use of germline material was obtained through the linked COG study D9902 (NCT00919269), a companion biobanking protocol to COG ARST0531. For the secondary cohort, patient DNA samples were collected from the Cooperative Human Tissue Network (n = 31), COG (n = 69, D9902, and D9602; NCT00002995), Children's Hospital at Westmead (n = 19), and NCI clinical trial 10-C-0086 (n = 2). Histologic diagnosis and clinical information were compiled by each source, and histology was reviewed when available. Written consent was obtained from all participants (or their parent or guardian). All protocols underwent institutional review board review and approval.
Sequencing, Filtering, and Annotation
Exome (ES) and genome sequencing (GS) was processed and mapped using previously published methods.16 ANNOVAR17 was used to annotate gene, population allele frequencies, and in silico pathogenicity. ClinVar classification was annotated using clinical laboratories meeting minimum requirements for data sharing to support quality assurance (badged lab) (2018-06-04 version).18 InterVar classification was annotated using python version 0.1.7 20180118.19 All variants were filtered using publicly available databases (Exome Sequencing Project, 1000 Genomes Project, Exome Aggregation Database without The Cancer Genome Atlas [TCGA], Genome Aggregation Database without TCGA) where any population allele frequency <1%. Details are in the Appendix Supplementary Methods.
Cancer Susceptibility Genes
Analysis focused on germline variants in 130 cancer susceptibility genes (CSG) from the study of Rahman20 and published RMS-associated genes21 (Data Supplement, online only). These candidate genes include those with autosomal-dominant (n = 76), autosomal-recessive (n = 28), both autosomal-dominant and autosomal-recessive inheritance (n = 16), X-linked (n = 5), Y-linked (n = 1), and unknown (n = 4) modes of inheritance.
Automated and Manual Review of Variation
The Appendix Figure A1 shows the scheme by which variant pathogenicity was determined. Panel sequencing was performed on a subset of genes. Details are given in the Appendix Supplementary Methods.
Structural Variant Analysis
Germline structural variant (SV) analysis was performed on the discovery cohort using DELLY v0.7.722 and Manta v1.0.323 for the discovery of deletions, duplications, and inversions, and MELT v2.0.524 for the discovery of mobile element insertions (MEIs) in 130 CSG (Appendix Supplementary Methods).
PCR Validation of Structural Variation
A series of polymerase chain reactions (PCR) were performed to validate breakpoints identified by bioinformatic means; the PCR primers used and experimental details are listed in the Appendix Supplementary Methods.
Statistical Analysis
A total of two predictor variables (pathogenic or likely pathogenic [P/LP] variants in 130 CSG with autosomal dominant (AD) and autosomal dominant and autosomal recessive (AD/AR) genes [102 genes]) were analyzed for association in the 256 RMS patients with confirmed intermediate-risk features and for which EFS and overall survival (OS) data were available in the discovery cohort. Each predictor variable was tested for univariate association with EFS and OS using log-rank tests and for association within multivariate Cox proportional hazard regressions. The P- value presented for each of the univariate models is the P-value from the log-rank test. Details are in the Appendix Supplementary Methods.
RESULTS
Patient Demographics
Table 1 lists the demographic and clinical data for the discovery (n = 273 germline GS) and secondary (n = 121 germline ES) cohorts. The distributions of sex, histology, and fusion status in the discovery cohort are similar to what has been reported in the literature for intermediate RMS. The distribution of histology and fusion status in the secondary cohort reflects the heterogeneity of risk in that group.
TABLE 1.
Demographics of Participants Enrolled in the Discovery and Secondary Rhabdomyosarcoma Cohorts

Frequency of P/LP Variants in Dominant and Dominant or Recessive CSG was 7.7% in the Discovery Cohort and 6.6% in the Secondary Cohort
In 43 patients in the discovery cohort, there were 44 unique P/LP variants (allele count = 58) in 23 genes; in 16 patients in the secondary cohort, there were 17 unique variants (allele count = 21) in 13 genes (Table 2). Using genes with dominant and dominant or recessive inheritance (102 CSGs), the frequency of P/LP variants was 7.7% in the discovery cohort and 6.6% in the secondary cohort (Table 3). Among FOXO1 fusion–negative tumors, the frequency of P/LP variants (by-person) in all 130 CSG was 15.5% in the discovery cohort and 12.5% in the secondary cohort (Table 2); using the 102 genes with only dominant and dominant or recessive inheritance, the frequency of P/LP variants (by-person) was 9.8% in the discovery cohort and 8.3% in the secondary cohort (Table 3).
TABLE 2.
Frequency of Pathogenic or Likely Pathogenic Germline Variants in All 130 Cancer Susceptibility Genes

TABLE 3.
Frequency of Pathogenic or Likely Pathogenic Germline Variants in 102 Dominant and Dominant or Recessive Cancer Susceptibility Genes

Germline P/LP Variants Observed in Known and Novel RMS-Associated Genes
At the variant-specific level, there were seven heterozygous variants (Table 4) observed in both cohorts in genes with dominant (TP53) and recessive (MUTYH, GBA, SBDS, ERCC2) patterns of inheritance. Biallelic ERCC2 variants (p.A717G and p.L461V) were observed in one person in both the discovery and secondary cohorts; phase could not be determined but is likely in cis.25-27 At the gene-specific level, P/LP variants were observed in eight genes in both cohorts, including DICER1, TP53 (dominant inheritance), ATM, COL7A1 (dominant and recessive inheritance), ERCC2, GBA, MUTYH, and SBDS (recessive inheritance). At the pathway-specific level, variation in Lynch syndrome genes (MSH2 and MSH6) was found in both cohorts.
TABLE 4.
Pathogenic or Likely Pathogenic Variants Found in Both the Discovery and Secondary Cohorts

In addition to observing P/LP variants in established, dominant RMS-associated genes (TP53, DICER1, NF1, MSH6, MSH2), we observed P/LP variants in dominant-acting genes in which pathogenic germline variation has never (or rarely) been reported in RMS, including BRCA2, CBL, CHEK2, FH, RET, and SMARCA4 (Fig 1, Data Supplement). For BRCA2, we observed one P/LP allele in a single individual and two P/LP alleles in a second individual in the discovery cohort. Phase could not be determined for the biallelic BRCA2 variants because of genomic distance. A missense variant (p.L318P) was observed in FGFR4, a gene somatically mutated in RMS,21 but to date without recognized germline phenotypic consequence (predicted LP by InterVar). A custom-built multigene panel using germline DNA from patients in the discovery cohort verified 100% (9/9) of the tested P/LP variants.
FIG 1.

Oncoprint of pathogenic and likely pathogenic variants in 130 cancer susceptibility genes in the (A) discovery cohort (B) secondary cohort, sorted by histology type.
P/LP Germline Variants in Dominant Genes in Participants with FOXO1 Fusion–Negative Tumors
In the discovery cohort, 67/273 (25%) of the patients had a confirmed fusion-positive tumor; of those, 2/67 (3.0%) harbored a P/LP variant in genes with a dominant mode of inheritance, whereas 17/174 (9.8%) patients with a fusion-negative tumor had a dominant P/LP variant. This difference trended toward significance (Fisher's exact test, P = .08). In the secondary cohort, 38/121 (31%) of the patients had a confirmed fusion-positive tumor; of those, 2/38 (5.3%) harbored P/LP variants in genes with a dominant mode of inheritance, whereas 6/72 (8.3%) patients with a fusion-negative tumor had a dominant P/LP variant (Fisher's exact test, P = .56).
Histology and Demographics of Patients With Germline P/LP Variants in CSG
We used OncoPrint to visualize P/LP variants and clinical data, including age at enrollment, stage, histology, and fusion status (Fig 1, Data Supplement). We have very limited demographic data for the secondary cohort compared with discovery cohort (Data Supplement). Therefore, only sex was tested for the secondary cohort. The age at enrollment for patients harboring any CSG P/LP variants (mean = 6.8 years) was younger than those without any CSG P/LP variants (mean = 7.6 years) but was not statistically significant (Mann-Whitney U test, P = .21). However, if restricted to genes with dominant or dominant or recessive inheritance, patients with P/LP variants were significantly younger (mean = 4.4 years) compared with patients without P/LP variants (mean = 7.7 years; Mann-Whitney U test, P = .002; Fig 2). There were no significant differences in the sex of the patients in regard to the risk of P/LP in both cohorts (Fisher's exact test, P = .87 and .59).
FIG 2.

Box-and-whisker plot comparing age at enrollment in dominant and dominant or recessive CSG in P/LP variant carriers v no P/LP variant carriers in discovery RMS patients. CSG, cancer susceptibility genes; P/LP, pathogenic or likely pathogenic; RMS, rhabdomyosarcoma.
Heterozygous Deletions in CDKN1C and ATM Identified in Two Patients
Analysis of structural variation identified two individuals with PCR-confirmed deletions in ATM and CDKN1C (Appendix Fig A2) that were absent from noncancer cohorts (gnomAD and 1000 Genomes Project). One subject with an ERMS harbored a deletion spanning exon 1 of CDKN1C, a tumor-suppressor gene associated with Beckwith-Wiedemann syndrome, a congenital overgrowth disorder characterized by an increased susceptibility to pediatric cancer, including RMS. A second individual with ERMS harbored a deletion in the kinase domain of the DNA-damage response tumor suppressor, ATM, thus likely altering stability and impairing protein kinase activity. Neither individual harbored concomitant sequence P/LP variants in any of the 130 analyzed CSG. No pathogenic duplications, inversions, or MEIs were discovered in CSGs in any of the patients. Overall, we observed structural variation affecting the exons of CSGs in 2/273 (0.7%) of children with RMS.
No Significant Differences in OS or EFS in Participants With P/LP Variants
There was no significant difference in OS or EFS between P/LP carriers versus non-P/LP carriers (Appendix Fig A3) for either univariate or multivariate analysis except univariate analysis with age at enrollment for 102 AD, AD/AR genes (P-value = .01).
DISCUSSION
We used a conservative, clinically based approach to classify germline variation in a set of predefined CSG in discovery (n = 273, predominately intermediate-risk) and secondary (n = 121, unselected risk group) cohorts of children and young adults with RMS. Our study is notable for the large size of two independent and fully sequenced cohorts, a comprehensive analysis of genome-wide SVs of the discovery cohort, and survival analyses of patients harboring germline pathogenic variants. We note that in a separate, unselected cohort of 615 RMS pediatric patients, 7.3% carried a cancer predisposition variant in a known CSG, in comparison to 1.4% in population-based controls.28 We identified pathogenic germline variants in numerous CSG that have been previously (TP53, NF1, DICER1, mismatch repair genes), rarely (BRCA2, CBL, CHEK2, SMARCA4), or never (FGFR4) reported in children and young adults with RMS. The frequency and type of P/LP variants we found are comparable to another recent, large exome sequencing study of germline risk in RMS.28 We identified pathogenic structural variation (ATM, CDKN1C) in two individuals and estimate the frequency of such variation in the RMS population. Our work found an excess of dominant-acting variants in individuals with fusion-negative tumors, which has implications for the design of future germline discovery studies. Our work can inform clinical care: In two cohorts of patients with RMS, we identified pathogenic variants for which gene-specific therapies and surveillance guidelines may be beneficial. In addition, in families with P/LP variants in RMS-risk genes, genetic counseling and cascade testing should be considered for all members, regardless of age.
Using the American College of Medical Genetics and Genomics (ACMG) or Association for Molecular Pathology criteria, the most common pathogenic germline variants we found were in TP53, NF1, DICER1, and the mismatch repair pathway genes. These variants were observed exclusively in individuals with nonalveolar histology. In both the discovery and secondary cohorts, we observed a nonsignificant excess of P/LP alleles in genes with dominant inheritance in individuals with FOXO1 fusion–negative tumors. These two complementary observations highlight the potency of FOXO1 fusion genes and suggest that the effect of germline variation in FOXO1 fusion–positive tumors in the pathogenesis of RMS may be of secondary importance. These observations have consequences in the design of studies to identify germline variation that increases the risk for RMS. To be adequately powered, studies investigating the effects of dominant-acting variation should emphasize the recruitment of individuals with FOXO1 fusion–negative tumors. The role of germline variation in the pathogenesis of FOXO1 fusion–positive tumors (if any) also would require adequately powered exome studies with appropriate controls.
This study uncovered pathogenic variants in genes not previously linked to RMS susceptibility. In both the discovery and secondary cohorts, a single individual with biallelic ERCC2 variants was observed. Previously published work shows that the two alleles, p.A717G and p.L461V, are found together in cis and create a complex allele that is associated with decreased DNA repair function.26 Overall, for recessive genes, heterozygous alleles in ERCC2, GBA, MUTYH, SBDS were found in both cohorts; however, given the lack of matched controls, the meaning of these observations is uncertain. In the discovery cohort, we identified P/LP variants in CBL (associated with a Noonan-like disorder) and SMARCA4 (associated with risk for rhabdoid tumors, a histologically distinct entity from RMS). In the secondary cohort, we found P/LP variants in RET (multiple endocrine neoplasia type 2), and FH (hereditary leiomyomatosis and renal cell cancer). With the exception of CBL-associated Noonan,29 these other genes have not been previously linked to RMS. Our findings, if replicated, expand the phenotypic consequences of P/LP variants in ERCC2, CBL, SMARC4, RET, and FH and should inform revision of genetic counseling and clinical care for individuals harboring these variants. A rare variant in FGFR4 (somatically mutated in RMS21) was classified as LP. However, since there is no known human, phenotypic consequence arising from FGFR4 germline variants and since this was an InterVar-based classification, this finding should be interpreted with caution but merits follow-up.
Pediatric cancer, including RMS, has been observed in families with germline pathogenic BRCA1 and BRCA2 variants.30,31 In pediatric pan-cancer studies, pathogenic variants in BRCA2 were observed in individuals with RMS in two different reports (1/43 [2.3%]12; 1/21 [4.8%]13). In our data, we observed P/LP variants in BRCA2 in 1% of FOXO1 fusion–negative RMS in the discovery cohort, a rate comparable to other similar-sized studies.28 We observed P/LP variants in other adult cancer genes, including sequence (in both cohorts) and structural variation in ATM. Lastly, we observed a single participant with a P/LP variant in CHEK2, a gene with wide pleiotropy and recognized risk for a variety of adult-onset cancers, including sarcomas.32 Our observations of RMS susceptibility from germline pathogenic variants in ATM, BRCA2, CHEK2, MSH2, and MSH6 blur the historic distinction in risk between adult and pediatric cancer genes, especially given the statistically significant earlier age of enrollment of children with RMS and dominant-acting P/LP variants. Some of these genes (TP53, BRCA2, mismatch repair) are on the ACMG Secondary Findings 2.0 list.33 Larger studies are needed to quantify the risk of RMS from germline pathogenic variants. For now, in families with a proband with an RMS-risk P/LP variant, genetic counseling and cascade testing should be considered, especially for genes with specific surveillance guidelines (eg, National Comprehensive Care Network [NCCN], DICER134) or on the ACMG Secondary Findings list.
Strengths of our study include its large size, independent cohorts broadly representative of North American pediatric cancer populations, clinically based classification of pathogenicity, and availability of structural variation analysis in the discovery cohort. Limitations include a lack of family history data and, in the secondary cohort, more limited clinical information. This study of predominantly intermediate-risk and unselected RMS may not be generalizable to all RMS-risk groups.
In this study, we found that 8%-10% of children and young adults with predominately intermediate-risk, fusion-negative RMS harbored P/LP variants in dominant-acting CSG. An additional approximately 1% of patients with RMS have structural germline variants in CSGs. There are multiple clinical and research consequences arising from our findings. To be adequately powered, studies investigating the effects of variants in dominant-acting CSGs should emphasize the recruitment of individuals with FOXO1 fusion–negative tumors. Clinically, the identification of such germline variation for the individual could be critically important to fulfill eligibility criteria for clinical trials to guide targeted treatments (eg, use of MEK [for RAS pathway variants] or PARP inhibitors for DNA repair variants) and permits gene-specific imaging and/or laboratory and imaging surveillance available now for many CSG. The identification of germline variation in patients with RMS is critically important as it would prompt genetic counseling and cascade genetic testing to identify other family members, whose risk of RMS (or numerous other cancers, as with DICER1, MSH2, MSH6, NF1, RET, TP53) may then be prospectively managed with NCCN or other published guidelines.34 This is especially true for genes on the ACMG Secondary Findings list.33 In children and young adults with RMS without a pathogenic germline P/LP variant, detection of germline structural variation in RMS-risk genes should be considered. More speculatively, we identified individuals with RMS and pathogenic germline variants in ERCC2, FGFR4, FH, RET, and SMARCA4 which, if replicated, may contribute to further understanding of RMS etiology and opportunities for intervention.
APPENDIX. APPENDIX. Supplementary Methods
Exome Sequencing (ES)
DNA preparation.
For each sample, 200-ng genomic DNA was purified using Agencourt AMPure XP Reagent (Beckman Coulter Inc, Brea, CA) according to the manufacturer’s protocol. An adapter-ligated library was prepared with the KAPA HyperPlus Kit (KAPA Biosystems, Wilmington, MA) using Bioo Scientific NEXTflex DNA Barcoded Adapters (Bioo Scientific, Austin, TX) according to the KAPA-provided protocol.
Prehybridization Ligation Mediated (LM)-Polymerase Chain Reaction (PCR).
Genomic DNA sample libraries were amplified prehybridization by ligation-mediated PCR consisting of one reaction containing 20 μL library DNA, 25 μL 2× KAPA HiFi HotStart ReadyMix, and 5 μL 10× Library Amplification Primer Mix (which includes two primers whose sequences are 5′-AATGATACGGCGACCACCGA-3′ and 5′-CAAGCAGAAGACGGCATACGA-3′). PCR cycling conditions were as follows: 98°C for 45 seconds followed by five cycles of 98°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. The last step was an extension at 72°C for 1 minute. The reaction was kept at 4°C until further processing. The amplified material was cleaned with Agencourt AMPure XP Reagent (Beckman Coulter Inc, Brea, CA) according to the KAPA-provided protocol. Amplified sample libraries were quantified using Quant-iT PicoGreen dsDNA Reagent (Life Technologies, Carlsbad, CA).
Liquid-Phase Sequence Capture.
Before hybridization, amplified sample libraries with unique barcoded adapters were combined in equal amounts into 1.1-μg pools for multiplex sequence capture. Exome sequence capture was performed with NimbleGen’s SeqCap EZ Human Exome Library, v3.0, with 64 Mb of exonic sequence targeted (Roche NimbleGen, Inc., Madison, WI). Before hybridization, the following components were added to the 1.1-μg pooled sample library, 4 μL of NEXTflex HE Universal Oligo 1,250 μM (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′), 40-μL total 25 μM NEXTflex INV-HE blocking oligos, equal volumes of each blocking oligo complementary to the barcodes in the pool (5′-CAAGCAGAAGACGGCATACGAGATXGTGACT GGAGTTCAGACGTGTGCTCTTCCGATCT/C3 Spacer/-3′, where X is 8 bases of sequence specific to adapter barcode used for library construction), and 5 μL of 1 mg/mL COT-1 DNA (Invitrogen, Inc, Carlsbad, CA). Samples were dried down by puncturing a hole in the plate seal and processing in an Eppendorf 5301 Vacuum Concentrator (Eppendorf, Hauppauge, NY) set to 60°C for approximately 1 hour. To each dried pool, 7.5 μL of NimbleGen Hybridization Buffer and 3.0 μL of NimbleGen Hybridization Component A were added and placed in a heating block for 10 minutes at 95°C. The mixture was then transferred to 4.5 μL of EZ Exome Probe Library and hybridized at 47°C for 64-72 hours. Washing and recovery of captured DNA were performed as described in the NimbleGen SeqCap EZ Library SR Protocol.
Posthybridization LM-PCR.
Pools of captured DNA were amplified by ligation-mediated PCR consisting of one reaction for each pool containing 20 μL captured library DNA, 25 μL 2× KAPA HiFi HotStart ReadyMix, and 5 μL 10× Library Amplification Primer Mix (which includes two primers whose sequences are 5′-AATGATACGGCGACCACCGA-3′ and 5′-CAAGCAGAAGACGGCATACGA-3′). PCR cycling conditions were as follows: 98°C for 45 seconds followed by 8 cycles of 98°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. The last step was an extension at 72°C for 1 minute. The reaction was kept at 4°C until further processing. The amplified material was cleaned with Agencourt AMPure XP Reagent (Beckman Coulter Inc, Brea, CA) according to the NimbleGen SeqCap EZ Library SR Protocol. Pools of amplified captured DNA were then quantified via Kapa’s Library Quantification Kit for Illumina (Kapa Biosystems, Woburn, MA) on the LightCycler 480 (Roche, Indianapolis, IN).
Exome sequencing.
The human reference genome and the known gene transcript annotation were downloaded from the UCSC database (http://genome.ucsc.edu/), version hg19 (corresponding to the Genome Reference Consortium assembly GRCh37). Sequencing reads were first trimmed using the Trimmomatic program (v0.32),1 which marks all low-quality stretches (average quality score < Q15 in a 4-bp sliding window) and reports the longest high-quality stretch of each read. Only read pairs with both ends no shorter than 36 bp were used. Reads were then aligned to the hg19 reference genome using Novoalign software (v3.00.05) (http://www.novocraft.com). Duplicate reads because of either optical or PCR artifacts were removed from further analysis using the MarkDuplicates module of the Picard software (v1.126) (http://picard.sourceforge.net/). Additionally, our analysis used only properly aligned read pairs, in the sense that the two ends of each pair must be mapped to the reference genome in complementary directions and must reflect a reasonable fragment length (300 ± 100 bp). These high-quality alignments for each individual were further refined according to a local realignment strategy around known and novel sites of insertion and deletion polymorphisms using the RealignerTargetCreator and IndelRealigner modules from the Genome Analysis Toolkit2 (GATK v3.1). Bam file level recalibration was also performed using the BaseRecalibrator module from GATK.
Variant discovery and genotype calling of multiallelic substitutions, insertions and deletions were performed on all individuals globally using the UnifiedGenotyper and HaplotypeCaller modules from Genome Analysis Toolkit (GATK v3.1) as well as the FreeBayes variant caller (v9.9.2). The Ensemble variant calling pipeline (v0.2.2; http://bcb.io/2013/02/06/an-automated-ensemble-method-for-combining-and-evaluating-genomic-variants-from-multiple-callers/) was then implemented to integrate analysis results from the above three callers. Then, the Ensemble variant calling pipeline applies a machine learning algorithm called support vector machine to identify an optimal decision boundary on the basis of the variant calling results out of multiple variant callers, with an aim to improve the caller’s receiver operating characteristic—in other words, a more balanced decision between false positives and true positives.
In addition, insertions and deletions were left-aligned at both postalignment (BAM) and postvariant calling (VCF) levels using GATK’s LeftAlignIndels and LeftAlignVariants modules, respectively.
We imported Northern and Western European ancestry (CEU), African ancestry (YRI), and Asian ancestry (ASI) (Han Chinese and Japanese) patients from 1000 Genomes (1 KG) for the admixture analysis. For each of the ethnic groups, we imported 419,035 SNPs overlapping to the rhabdomyosarcoma (RMS) secondary cohort and filtered out monomorphic SNPs in any of the ethnic groups and ended up with 35,502 SNPs. We used these SNPs to estimate the ethnic components of the RMS secondary cohort. The estimation was made with an in-house imputation of the algorithm described in the work of Pritchard et al.3 We considered CEU > 0.8 as White, YRI > 0.8 as Black or African American, ASI > 0.8 as Asian, and the rest as admix.
Genome Sequencing
Five hundred nanogram to 1 μg of genomic DNA was submitted to The Centre for Applied Genomics for genomic library preparation and genome sequencing. DNA samples were quantified using the Qubit High Sensitivity Assay, and purity was assessed using the Nanodrop OD260/280 ratio. Approximately 500-700 ng of DNA was used as input material for library preparation using the Illumina TruSeq PCR-free DNA Library Prep Kit following the manufacturer’s recommended protocol. In brief, DNA was fragmented to 400 bp on average using sonication on a Covaris LE220 instrument. Fragmented DNA was end-repaired; A-tailed and indexed TruSeq Illumina adapters with overhang-T will be added to the DNA. Libraries were then validated on a Bioanalyzer DNA High Sensitivity chip to check for size and absence of primer dimers and quantified by quantitative PCR (qPCR) using Kapa Library Quantification Illumina/ABI Prism Kit protocol (KAPA Biosystems). Validated libraries were pooled in equimolar quantities and paired-end sequenced on an Illumina HiSeq X platform following Illumina’s recommended protocol to generate paired-end reads of 150-bases in length. SNV and indel detection was performed using HaplotypeCaller in a gVCF model of GATK v.4.0.2.1.4 All detected variants were filtered to remove false positive using the following requirements: read position 10-90, strandedness 1%-99%, distance to 3′ > 20, homopolymer < 5, map quality difference < 30, read length difference < 25, and mismatch quality sum (MMQS) difference < 100. Variants with less than five alternate reads detected using bam-readcount were removed.
Automated and Manual Review of Variation
Any variants that had a read depth < 10 or allelic balance of heterozygous calls < 0.2 or > 0.8 were excluded from the analysis. Only variants with refGene nonsynonymous exonic and splicing were included in the analysis. All rare variants (minor allele frequency [MAF] < 1%) from the candidate gene list were classified into pathogenic (P), likely pathogenic (LP), variant of uncertain significance, likely benign, and benign first from ClinVar calls, made based on clinical laboratories meeting minimum requirements for data sharing to support quality assurance5 (badged lab) by ClinGen (https://www.clinicalgenome.org/lablist/) using an archive database downloaded on May 20, 2018 (nonbadged lab calls were disregarded). At least one call from ClinVar badged lab was needed to classify variants and a majority rule applied. If no ClinVar calls were available, Human Gene Mutation Database (HGMD) (2019.1)6 disease-causing mutation variants were subjected to manual review according to American College of Medical Genetics (ACMG)-Association of Molecular Pathology (AMP) guidelines.7 Next, variation was interpreted by InterVar.8 Final calls were prioritized in the following order: ClinVar, HGMD manual review, and InterVar (Appendix Fig 1). All the P/LP variants in BAM files were reviewed.
FIG A1.

Flow diagram of unique variant classification. (A) Discovery cohort, (B) secondary cohort. B, benign; DM, disease causing; HGMD, human gene mutation database; LB, likely benign; LP, likely pathogenic; P, pathogenic; VUS, variant of unknown significance.
Multigene Panel Sequencing Analysis
Next-generation sequencing library preparation and custom-targeted capture.
Blood-derived genomic DNAs (100 ng) from 276 intermediate-risk RMS germline samples were sheared using a Covaris S2 instrument (Covaris, Woburn, MA). Libraries were prepared using the Ovation Ultralow Library System (NUGEN #0329) according to the manufacturer’s instruction. Library enrichment for a 59-gene custom panel was done with the Roche SeqCap EZ Choice Library (cat #06266339001) and the SeqCap EZ Reagent Kit Plus v2 (NimbleGen #06-953-247-001) using the manufacturer’s protocol. Individual libraries were combined into pools of 12 before hybridization. Captured libraries were sequenced on the Illumina HiSeq2000 channel using the HiSeq 101 Cycle Paired-End sequencing protocol. The 59-gene custom capture was selected based on their known contribution to hereditary cancer syndromes (59 genes include ALK, APC, ATM, AXIN2, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, CTNNB1, DICER1, FLCN, MAX, MEN1, MLH1, MLH3, MRE11A, MSH2, MSH6, MUTYH, NBN, NF1, PALB2, PHOX2B, PKHD1, PMS1, PMS2, POLD1, POLE, POT1, PRSS1, PTCH1, PTEN, RAD50, RAD51B, RAD51C, RET, RINT1, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMARCB1, STK11, SUFU, TCF7L2, TGFBR2, TMEM127, TP53, VHL, WT1, XRCC2, XRCC3). Generated reads were aligned to the hg19 reference genome with Burrows-Wheeler Alignment Tool.9 Base quality score recalibration, indel realignment, duplicate removal, and SNP/INDEL calling and other processing to variant calls were completed via the GATK.10 Samples that failed library preparation were excluded from the analysis (n = 18) as well as samples with less than an average of 10× coverage over the panel (n = 7) as determined by bedtools.11,17
Structural Variant Analysis
Filtering and annotation of deletions, duplications, and inversions.
An ensemble approach was taken, retaining only the deletions, duplications, and inversions called by both DELLY and Manta, based on the following size-specific constraints for breakpoint consensus between the two tools: breakpoints within 100 bp for structural variation (SV) ≤ 10 kbp, breakpoints within 1 kbp for SVs ≥ 10 kbp and ≤ 50 kbp, and breakpoints within 10 kbp for SVs ≥ 50 kbp. A high-quality SV set was obtained by applying additional filtering criteria. A panel of normal controls was created to remove technical false positives using 72 germline genomes sequenced on the same HiSeqX platform (150bp paired end sequencing, minimum of 30× depth coverage). SVs present in ≥ 3 panel of normal genomes were removed. Annotation was performed with AnnotSV and subsequently filtered to remove SVs with an AnnotSV score < 3 and present in publicly available normal databases: 1000 Genomes Project and gnomAD. The remaining exonic SVs were subject to manual review in Integrative Genomics Viewer.
PCR validation of structural variation.
In all instances, amplification was performed in a 50-µL reaction volume, and PCR products were examined by agarose gel electrophoresis. The reaction consisted of 25 µL of a standard Taq master mix, 2.5 µL each of both the forward and reverse primers (10 µM), and 100 ng of the relevant template DNA (patient DNA and control human genomic DNA), with the remaining volume being nuclease-free water. Primer sequences for ATM are 5′- TGGGTAATAATTTCCAAGTGAAACTCCCCA-3′ and 5′- GAGTTGCAGGGGGATAATAGTGATGATGTG-3′, and CDKN1C are 5′- TCAGCTTTGTTTACGTCGCCGCGCAATGTG-3′ and 5′-AGGCGCCGGAGCAGCTGCCTAGT-3′. An initial denaturation of 30 seconds was performed at 98°C followed by 35 cycles of amplification, which consisted of a 10-second denaturation step performed at 98°C, a 10-second annealing step performed at 60°C, and an extension step of 30 seconds or 3 minutes, for CDKN1C and ATM, respectively (Appendix Figs 2A and A3), performed at 72°C. A final extension of 5 minutes was performed at 72°C.
FIG A2.

Structural variant PCR validation using 1% agarose gel in tris-acetate-EDTA buffer: (A) CDKN1C and (B) ATM. Columns: A, patient DNA; B, control genomic DNA; C, no template.
FIG A3.
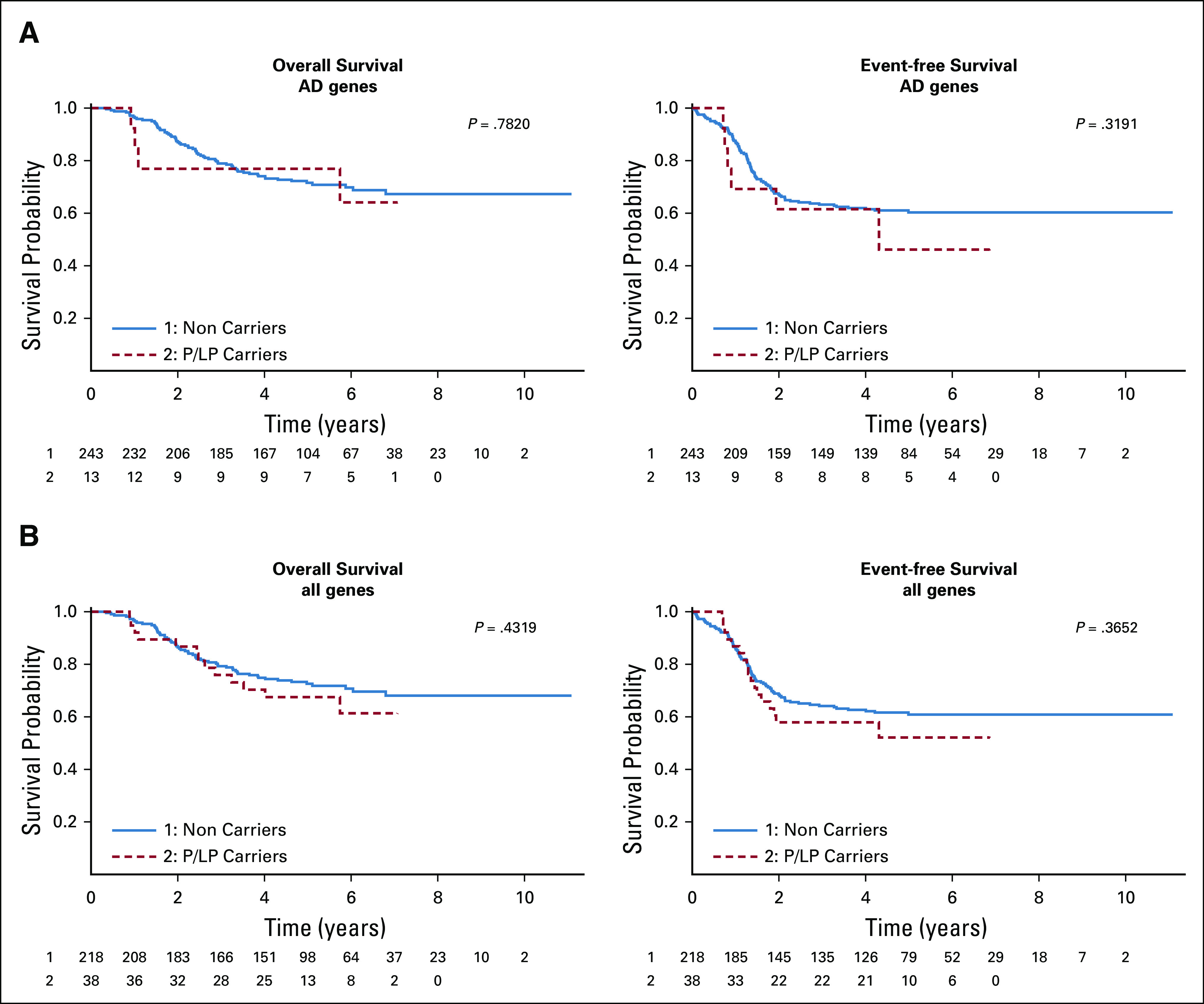
(A) Overall survival analysis and event-free survival analysis in AD genes only and (B) all genes in Discovery cohort.
Statistical Analysis
Survival/multivariate analysis.
Event-free survival (EFS) was defined as time from enrollment on a Children's Oncology Group study until event (relapse, SMN, or death) or until last contact. Patients who did not experience an EFS event were considered censored at the time of last contact. Overall survival was defined as time from enrollment on a COG study until death or until last contact. Patients who were alive at last contact were considered censored at the time of last contact.
Each multivariate model was produced using stepwise selection, starting with a model containing only the predictor variable being tested (which was forced to be in the model) and allowing for the addition of the following demographic and clinical variables: age at enrollment (as a categorical variable), sex, race, ethnicity, RMS type, RMS stage, N-stage, and T-stage. The final model included the predictor variable being tested (which was forced to be in the model) and any demographic variables found to be significant. The P-value presented for each of the multivariate models is the type 3 P value from the Cox proportional hazard regression for the predictor variable given that any significant demographic and diagnostic variables are also in the model. Each predictor variable was a 0/1 categorical variable with 1 indicating the subject being a P/LP carrier in 130 CSGs or 102 AD, AD/AR genes, and 0 indicating the subject not being a P/LP carrier.
Age at enrollment was defined as fractional years from birth until enrollment on a COG trial. It was treated as a continuous variable. Age category at enrollment was defined as years from birth until enrollment on a COG trial. It was treated as a categorical variable with the following categories: < 1 year old, 1-9 years old, and ≥ 10 years old. Patient sex was treated as a categorical variable with two categories (male and female). Patient race was treated as a categorical variable with four categories (White, Black, Others, and Unknown). Patient ethnicity was treated as a categorical variable with three categories (Hispanic, Non-Hispanic, and Unknown). RMS type was treated as a categorical variable with three categories (Alveolar, Embryonal, and Not Otherwise Specified). RMS stage was treated as a categorical variable with three categories (stage I, stage II, and stage III). N-stage (nodal involvement) was treated as a categorical variable with 3 categories (No/N0, Yes/N1, and Not Evaluated/Unknown). T-stage was treated as a categorical variable with 2 categories (T1 and T2).
Multigene panel comparison.
Germline results were compared with the results of a subset of patients from the same COG discovery cohort, which were run earlier on a custom-made multigene panel from the University of Utah (see Supplemental Methods).
REFERENCES
- 1.Lohse M, Bolger AM, Nagel A, et al. : RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res 40:W622-W6272012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DePristo MA, Banks E, Poplin R, et al. : A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491-4982011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics 155:945-9592000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna A, Hanna M, Banks E, et al. : The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297-13032010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landrum MJ, Lee JM, Benson M, et al. : ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46:D1062-D10672018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenson PD, Ball EV, Mort M, et al. : Human gene mutation database (HGMD®): 2003 update. Hum Mutat 21:577-5812003 [DOI] [PubMed] [Google Scholar]
- 7.Richards S, Aziz N, Bale S, et al. : Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405-4242015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Wang K: InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet 100:267-2802017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754-17602009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DePristo MA, Banks E, Poplin R, et al. : A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491-4982011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quinlan AR, Hall IM: BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26:841-8422010 [DOI] [PMC free article] [PubMed] [Google Scholar]
EQUAL CONTRIBUTION
J.K., N.L., V.S., E.L.Y., and T.W.-O. contributed equally to this work. J.D.S., J.K., D.M., and D.R.S. contributed equally to this work.
SUPPORT
Supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics of the National Cancer Institute, Bethesda, MD, a Foundation scheme grant (143234) from the Canadian Institutes for Health Research and a Terry Fox New Frontiers Program Project Grant (1081) (D.M.) and NCTN Operations Center Grant U10 CA180886, Human Specimen Banking Grants U24 CA114766 and 1U24-CA196173, and NCTN Statistics & Data Center Grants U10 CA 180899 and U10 CA098413 of the Children's Oncology Group and Children's Oncology Group Chair's Grant U10 CA098543 from the National Cancer Institute of the National Institutes of Health. Additional research support was provided by a grant from the WWWW (QuadW) Foundation, Inc (www.QuadW.org) to the Children's Oncology Group. COG ARST0531 was also supported by St. Baldrick's Foundation. Support was also provided in part through the Cancer Prevention and Research Institute of Texas grant RP170071 (P.L.). JDS is supported by Hyundai Hope on Wheels, Soccer for Hope Foundation, Li-Fraumeni Syndrome Association, Kneaders Bakery & Café Hope Campaign, 5 For The Fight (Qualtrics), and the Elephant p53 (EP53) Program funded through Huntsman Cancer Institute by the State of Utah. N.L. was supported in part from the University of Toronto MD/PhD program, the McLaughlin Center, a Ruggles Family Innovation Award, and a CIHR MD/PhD Studentship; V.S. was supported in part from the Ontario Graduate Scholarship, MBP Excellence Scholarship, and a Vector Institute Research Grant; T.W.-O. was supported in part by CONACYT. This work used the computational resources of the NIH High Performance Computing Biowulf cluster.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Talia Wegman-Ostrosky
Travel, Accommodations, Expenses: Pfizer
Donald A. Barkauskas
Employment: Genentech
Stock and Other Ownership Interests: Genentech
Patents, Royalties, Other Intellectual Property: U.S. patent based on Ph.D. research in glioblastoma
Luke D. Maese
Honoraria: Jazz Pharmaceuticals
Consulting or Advisory Role: Jazz Pharmaceuticals
Sean V. Tavtigian
Patents, Royalties, Other Intellectual Property: I hold patents on BRCA1, BRCA2, and PTEN. However, the claims in these patents related to genetic testing have been overturned by the US Supreme Court. Continuing claims are irrelevant to the paper. My royalty stream ended more than 2 years ago.
Anna Goldenberg
Research Funding: 4UandMe
Douglas S. Hawkins
Research Funding: Loxo, Bristol-Myers Squibb, Merck Sharp Dohme, Bayer, Lilly, Eisai, Amgen, Seattle Genetics, Incyte, Jazz Pharmaceuticals
Travel, Accommodations, Expenses: Loxo, Bayer, Celgene, AstraZeneca
Joshua D. Schiffman
Employment: PEEL Therapeutics, Inc.
Leadership: PEEL Therapeutics, Inc.
Stock and Other Ownership Interests: ItRunsInMyFamily.com, PEEL Therapeutics, Inc.
Honoraria: Affymetrix
Consulting or Advisory Role: N-of-One, Fabric Genomics
Javed Khan
Research Funding: Lentigen
Patents, Royalties, Other Intellectual Property: Monoclonal antibody-based therapeutics targeting fibroblast growth factor receptor 4 (FGFR4) for potential treatment of human cancers expressing FGFR4
David Malkin
Consulting or Advisory Role: Bayer, Canada
Douglas R. Stewart
Employment: Genome Medical, LLC
No other potential conflicts of interest were reported.
AUTHOR CONTRIBUTIONS
Conception and design: Jung Kim, Nicholas Light, Philip J. Lupo, Sean V. Tavtigian, Adam Shlien, Anna Goldenberg, Douglas S. Hawkins, Joshua D. Schiffman, Javed Khan, David Malkin, Douglas R. Stewart
Financial support: Anna Goldenberg, Joshua D. Schiffman, Javed Khan, David Malkin, Douglas R. Stewart
Provision of study materials or patients: Mingyi Wang, Daniel Catchpoole, Javed Khan
Collection and assembly of data: Nicholas Light, Vallijah Subasri, Erin L. Young, Donald A. Barkauskas, David Hall, Philip J. Lupo, Kristine Jones, Sean V. Tavtigian, Frank Telfer, Jun S. Wei, Xinyu Wen, Daniel Catchpoole, Douglas S. Hawkins, Joshua D. Schiffman, Javed Khan
Data analysis and interpretation: Jung Kim, Nicholas Light, Vallijah Subasri, Erin L. Young, Talia Wegman-Ostrosky, Donald A. Barkauskas, David Hall, Philip J. Lupo, Rajesh Patidar, Luke D. Maese, Mingyi Wang, Dongjing Wu, Frank Telfer, Anna Goldenberg, Stephen X. Skapek, Jun S. Wei, Xinyu Wen, Douglas S. Hawkins, Joshua D. Schiffman, Javed Khan, David Malkin
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
REFERENCES
- 1.Egas-Bejar D, Huh WW: Rhabdomyosarcoma in adolescent and young adult patients: Current perspectives. Adolesc Health Med Ther 5:115-1252014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jo VY, Fletcher CD: WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 46:95-1042014 [DOI] [PubMed] [Google Scholar]
- 3.Shern JF, Yohe ME, Khan J: Pediatric rhabdomyosarcoma. Crit Rev Oncog 20:227-2432015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ognjanovic S, Linabery AM, Charbonneau B, et al. : Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer 115:4218-42262009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skapek SX, Ferrari A, Gupta AA, et al. : Rhabdomyosarcoma. Nat Rev Dis Primers 5:1.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kashi VP, Hatley ME, Galindo RL: Probing for a deeper understanding of rhabdomyosarcoma: Insights from complementary model systems. Nat Rev Cancer 15:426-4392015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith MA, Altekruse SF, Adamson PC, et al. : Declining childhood and adolescent cancer mortality. Cancer 120:2497-25062014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawkins DS, Chi YY, Anderson JR, et al. : Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: A report from the Children's Oncology Group. J Clin Oncol 36:2770-27772018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breitfeld PP, Meyer WH: Rhabdomyosarcoma: New windows of opportunity. Oncologist 10:518-5272005 [DOI] [PubMed] [Google Scholar]
- 10.Zong X, Pole JD, Grundy PE, et al. : Second malignant neoplasms after childhood non-central nervous system embryonal tumours in North America: A population-based study. Eur J Cancer 84:173-1832017 [DOI] [PubMed] [Google Scholar]
- 11.Parsons DW, Roy A, Yang Y, et al. : Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2:616-6242016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Walsh MF, Wu G, et al. : Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373:2336-23462015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grobner SN, Worst BC, Weischenfeldt J, et al. : The landscape of genomic alterations across childhood cancers. Nature 555:321-3272018 [DOI] [PubMed] [Google Scholar]
- 14.Mandelker D, Zhang L, Kemel Y, et al. : Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA 318:825-8352017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Stewart E, Shelat AA, et al. : Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 24:710-7242013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Luo W, Wang M, et al. : Prevalence of pathogenic/likely pathogenic variants in the 24 cancer genes of the ACMG secondary findings v2.0 list in a large cancer cohort and ethnicity-matched controls. Genome Med 10:99.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K, Li M, Hakonarson H: ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landrum MJ, Lee JM, Benson M, et al. : ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46:D1062-D10672018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Wang K: InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet 100:267-2802017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahman N: Realizing the promise of cancer predisposition genes. Nature 505:302-3082014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shern JF, Chen L, Chmielecki J, et al. : Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4:216-2312014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rausch T, Zichner T, Schlattl A, et al. : DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28:i333-i3392012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Schulz-Trieglaff O, Shaw R, et al. : Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32:1220-12222016 [DOI] [PubMed] [Google Scholar]
- 24.Gardner EJ, Lam VK, Harris DN, et al. : The mobile element locator tool (MELT): Population-scale mobile element discovery and biology. Genome Res 27:1916-19292017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takayama K, Salazar EP, Lehmann A, et al. : Defects in the DNA repair and transcription gene ERCC2 in the cancer-prone disorder xeroderma pigmentosum group D. Cancer Res 55:5656-56631995 [PubMed] [Google Scholar]
- 26.Zhou X, Khan SG, Tamura D, et al. : Abnormal XPD-induced nuclear receptor transactivation in DNA repair disorders: Trichothiodystrophy and xeroderma pigmentosum. Eur J Hum Genet 21:831-8372013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orioli D, Compe E, Nardo T, et al. : XPD mutations in trichothiodystrophy hamper collagen VI expression and reveal a role of TFIIH in transcription derepression. Hum Mol Genet 22:1061-10732013 [DOI] [PubMed] [Google Scholar]
- 28.Li H Sisoudlya SD. Khayat MM et al. : Prevalence of germline cancer-predisposition variants in pediatric rhabdomyosarcoma: a report from the Children's Oncology Group. JNCI 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji J, Navid F, Hiemenz MC, et al. : Embryonal rhabdomyosarcoma in a patient with a germline CBL pathogenic variant. Cancer Genet 231-232:62-662019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks GA, Stopfer JE, Erlichman J, et al. : Childhood cancer in families with and without BRCA1 or BRCA2 mutations ascertained at a high-risk breast cancer clinic. Cancer Biol Ther 5:1098-11022006 [DOI] [PubMed] [Google Scholar]
- 31.Magnusson S, Borg A, Kristoffersson U, et al. : Higher occurrence of childhood cancer in families with germline mutations in BRCA2, MMR and CDKN2A genes. Fam Cancer;7:331-3372008 [DOI] [PubMed] [Google Scholar]
- 32.Naslund-Koch C, Nordestgaard BG, Bojesen SE: Increased risk for other cancers in addition to breast cancer for CHEK2*1100delC heterozygotes estimated from the copenhagen general population study. J Clin Oncol 34:1208-12162016 [DOI] [PubMed] [Google Scholar]
- 33.Kalia SS, Adelman K, Bale SJ, et al. : Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med 19:249-2552017 [DOI] [PubMed] [Google Scholar]
- 34.Schultz KAP, Williams GM, Kamihara J, et al. : DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 24:2251-22612018 [DOI] [PMC free article] [PubMed] [Google Scholar]