

Abstract
Objectives
Low‐dose interleukin‐2 (IL‐2) has shown promising clinical benefits in the treatment of systemic lupus erythematosus (SLE), but how this therapy alleviates pathogenic humoral immunity remains not well understood. The dilemma is that IL‐2 can suppress both follicular helper and regulatory T (Tfh and Tfr) cells, which counteract each other in regulating autoantibody production.
Methods
Female NZB/W F1 mice received recombinant human IL‐2 (3 × 104 IU/dose) in three treatment regimens: (1) short, daily for 7 days; (2) medium, daily for 14 days, and (3) long, every second day for 28 days. Tfh (Foxp3−CXCR5+Bcl6+), Tfr (Foxp3+CXCR5+Bcl6+), germinal centre (GC, B220+GL‐7+Fas+) and antibody‐secreting cell (ASC, B220−CD138+TACI+) were analysed by flow cytometry. Serum anti‐dsDNA level was determined by ELISA. Kidney pathology was evaluated by H&E and immunofluorescence staining. Circulating Tfh and Tfr cells in SLE patients treated with low‐dose IL‐2 from a previous clinical trial (NCT02084238) was analysed.
Results
Low‐dose IL‐2 treatment consistently increased Tfr/Tfh ratio in mice and SLE patients, because of a stronger suppression on Tfh cells than Tfr cells. Three treatment regimens revealed distinct immunological features. Tfh suppression was observed in all regimens, but Tfr suppression and GC reduction were recorded in just medium and long regimens. Only the long treatment regimen resulted in inhibited ASC differentiation in spleen, which was accompanied by reduced anti‐dsDNA titres and ameliorated kidney pathology.
Conclusion
Low‐dose IL‐2 therapy increases the Tfr/Tfh ratio, and a less frequent and prolonged treatment can alleviate pathogenic humoral immunity and improve renal function.
Keywords: B cell, humoral immunity, interleukin‐2, systemic lupus erythematosus, Tfr/Tfh ratio
We designed and examined three treatment regimens of low‐dose interleukin‐2 (IL‐2) therapy in lupus‐prone NZB/W F1 mice and characterised the immune responses in detail. Low‐dose IL‐2 therapy increases the Tfr/Tfh ratio and a less frequent and prolonged treatment can alleviate pathogenic humoral immunity and improve renal function, which provide necessary rationale and new insights in optimising low‐dose IL‐2 therapy.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease that is characterised by the production of autoantibodies and the manifestations in multiple tissues. 1 , 2 , 3 Studies on the complex immunological pathogenesis of SLE have revealed the critical participation of both T and B cells in the development and exacerbation of the disease. 4 , 5 The aberrant activation of B cells is a common feature of SLE. 6 , 7 Not only do B cells serve as antigen‐presenting cells to promote the activation and differentiation of T cells, 8 but they also produce autoantibodies, such as anti‐double‐strand DNA (anti‐dsDNA) and antinuclear antibodies (ANA) to mediate organ damages. 9 , 10 For example, autoantigen–autoantibody complexes and complement components (particularly C3) deposit at the glomerular basement membrane of kidneys, which attract robust immune cell infiltration and exacerbate local inflammation and tissue damage. 11 , 12 Belimumab (anti‐BAFF) alone or in combination with rituximab (anti‐CD20) sequentially depleting or reducing circulating B cells shows objective efficacies in treating patients with active refractory lupus and reduces cumulative dose of corticosteroids, stressing the significance of pathogenic B cells in SLE. 13 , 14 , 15 , 16
The activation of B cells and their differentiation into antibody‐secreting cells (ASCs) are critically dependent on follicular helper T (Tfh) cells via co‐stimulation and helper cytokines. 17 , 18 , 19 We and others demonstrated that excessive Tfh function drove aberrant germinal centre (GC) formation and autoantibody production in murine models and detected a positive correlation between Tfh activities and autoantibody profiles in patients with SLE. 4 , 20 , 21 Tfh cell‐mediated B‐cell response is counteracted by follicular regulatory T (Tfr) cells, also localising to B follicles. The genetic ablation of Tfr cells in mice led to enhanced GC formation and autoantibody production. 22 , 23 , 24 It is now widely accepted that balanced actions between Tfh and Tfr cells are required to maintain normal humoral immunity. 25 , 26 On the flip side, a tilt to reduced Tfr/Tfh ratios has been often reported in many rheumatic diseases including SLE. 27 , 28 Therapies in SLE aim to reinstate the Tfr/Tfh balance in order to achieve the induction of immune tolerance.
Tfr/Tfh balance in order to achieve the induction of immune tolerance. Past decade observed a renaissance of interleukin‐2 (IL‐2) therapy, represented by the application of low‐dose IL‐2 in managing inflammatory and autoimmune disorders. 29 , 30 , 31 , 32 , 33 , 34 Following pioneer clinical trials of low‐dose IL‐2 therapy in graft‐versus‐host disease (GvHD) and vasculitis, we conducted the first open‐labelled and a subsequent randomised clinical trial of low‐dose IL‐2 therapy in SLE patients, showing acceptable safety and efficacy. We recorded the reduction of Tfh cell differentiation in SLE patients after low‐dose IL‐2 therapy, 32 which agreed with the reported inhibition of Tfh cell differentiation by IL‐2 in mouse models. 35
An emerging question is how low‐dose IL‐2 therapy regulates Tfr cells. Notably, in the mouse model of infection by H1N1 influenza virus, IL‐2 treatment reduced Tfr cells and led to the enhanced generation of histone‐specific ASCs and the production of ANA. 36 Another study suggested that IL‐2 drove the conversion of Tfh cells to Tfr cells in culture. 28 If low‐dose IL‐2 therapy suppresses both Tfh and Tfr cells, which counteract each other in regulating humoral immunity, what is the overall effect of this new therapy to pathogenic humoral immunity, particularly autoantibody production in autoimmune diseases? As this is a difficult question to be directly addressed in human clinical trials, we decided to go back using mouse models to dissect the effect of low‐dose IL‐2 therapy in the regulation of Tfh vs Tfr cells.
In this study, we carefully examined the dynamic impact of low‐dose IL‐2 therapy in female NZB/W F1 mice as a murine lupus model. IL‐2 treatment was found consistently increasing Tfr/Tfh ratio, because of a stronger suppression on Tfh cells than Tfr cells. Prolonged treatment of IL‐2 inhibited GC formation and ASC differentiation in spleen, which was accompanied by reduced anti‐dsDNA titres and ameliorated renal pathology. The improvement of Tfr/Tfh ratio by low‐dose IL‐2 therapy was also validated in patients with SLE.
Results
Short treatment regimen of low‐dose IL‐2 therapy reduced Tfh but not Tfr cells in NZB/W F1 mice
The NZB/W F1 mouse is a widely used murine model to study immunopathogenesis of lupus. Female NZB/W F1 mice show aberrant B‐ and T‐cell activation and produce autoantibodies as early as 12 weeks of age, with the spontaneous renal disease at 5 months. 37 , 38 Female NZB/W F1 mice at 20 weeks of age were intraperitoneally injected of PBS or recombinant human IL‐2 (3 × 104 IU). Human IL‐2 stimulates mouse T cells at similar concentrations of mouse IL‐2 and has been widely used in mouse models. 32 , 35 , 39 This dose was reported as a low‐dose therapy in murine models. 35 We first tested a short therapy regimen of daily treatment for 7 days.
As expected, 7‐day treatment of low‐dose IL‐2 expanded Treg cells (B220−CD4+Foxp3+) in spleens, showing an increase in frequency by 1.7‐fold (Figure 1a and b). The frequency of effector T cell (Teff, CD4+Foxp3−CD44+) was reduced by ~ 20% in IL‐2‐treated mice than in PBS controls (Figure 1a and c). Notably, IL‐2 treatment led to a significant reduction by ~ 2‐fold for the frequency of Tfh cells (Bcl6+CXCR5+) in CD44+ Teff cells or total CD4+ T cell (Figure 1a, d and e) and a significant reduction of Tfh cell number (Figure 1f) suggesting a prominent inhibition of Tfh cells by IL‐2. The frequency of Tfr cells (Bcl6+CXCR5+) in CD4+Foxp3+ Treg cells was reduced after IL‐2 treatment (Figure 1a and g), which was due to the expansion of total Treg cells rather than the net reduction of Tfr frequency in total CD4+ T cells or the number of Tfr cells (Figure 1h and i). Overall, IL‐2 treatment not only increased the Tfr/Tfh ratio changes (by 2‐fold), but also Tfr/Tfh cell number changes (by 1.75‐fold) (Figure 1j and k).
Figure 1.
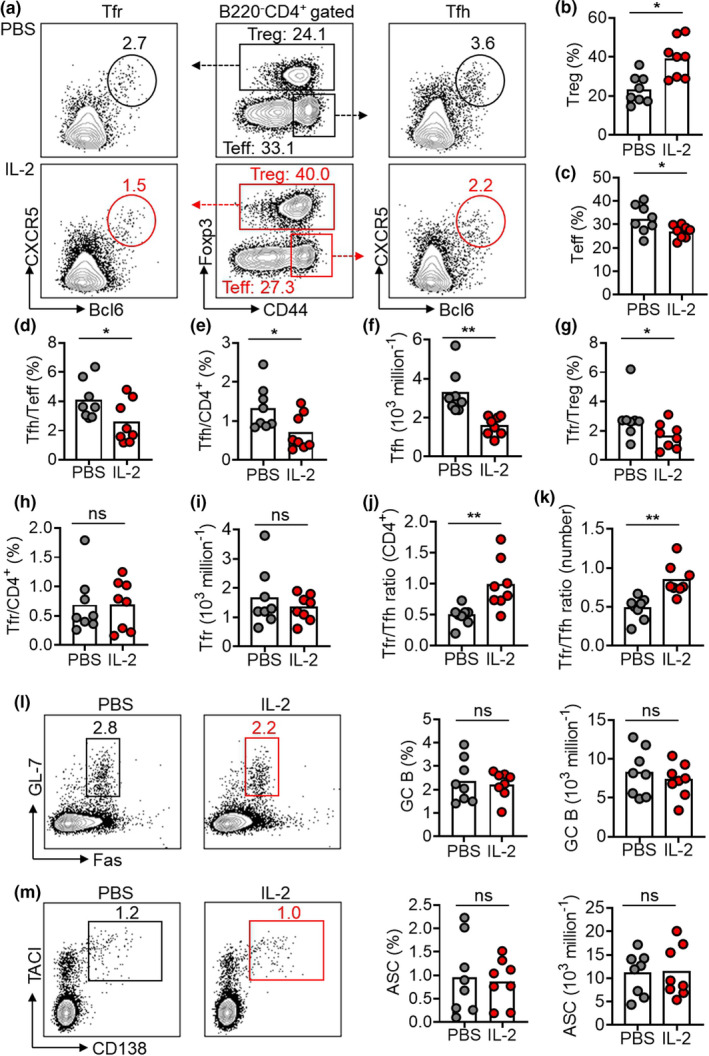
7‐day low‐dose IL‐2 therapy suppressed Tfh but not Tfr cells in NZB/W F1 mice. (a) Representative FACS plots showing Treg cells (B220−CD4+Foxp3+), Teff cells (CD4+Foxp3−CD44+), Tfh cells (CD4+Foxp3−CD44+CXCR5+Bcl6+) and Tfr cells (CD4+Foxp3+CXCR5+Bcl6+) in spleens of female NZB/W F1 mice with short treatment regimen of low‐dose IL‐2 or PBS for 7 days in spleens of female NZB/W F1 mice with IL‐2 or PBS treatment. (b–e) Frequency statistics of Treg (b), Teff (c), Tfh in Teff (d), Tfh in CD4+ T cell (e) and the statistics of Tfh cell number (f) in spleens of female NZB/W F1 mice with IL‐2 or PBS treatment. (g–i) Frequency statistics of Tfr cells in Treg cells (g), Tfr cells in CD4+ T cell (h) and the statistics of Tfh cell numbers (i) in spleens of female NZB/W F1 mice with IL‐2 or PBS treatment. (j, k) The ratio of Tfr cell frequency in CD4+ T cells to Tfh cell frequency in CD4+ T cells [Tfr/Tfh ratio (CD4+)] (j) and the ratio of Tfr cell number to Tfh cell numbers (Tfr/Tfh ratio (number) (k) in spleens of female NZB/W F1 mice with IL‐2 or PBS treatment. (l, m) Representative FACS plots and statistics showing the frequencies and the absolute numbers of GC B cells (B220+GL‐7+Fas+) (l) and ASC (B220− CD138+TACI+) (m) in spleens of female NZB/W F1 mice with IL‐2 or PBS treatment. Data are shown for individual (dots, n = 8) and mean (bars) values. Data are representative of three independent experiments and analysed by the Mann–Whitney U‐test. *P < 0.05 and **P < 0.01. ASC, antibody‐secreting cell; GC, germinal centre; NZB/W F1, F1 hybrid between the New Zealand Black (NZB) and New Zealand White (NZW) strains; Teff, effector T cell; Tfh, T follicular helper cell; Tfr, T follicular regulatory cell; Treg, regulatory T cell.
The development of spontaneous GCs in NZB/W F1 mice has been known for over 30 years ago, 37 and was reported to rely on Tfh cells. 40 Despite the increase of Tfr/Tfh ratio, there was no significant change of the frequencies and the absolute number for GC B cells (B220+GL‐7+Fas+) or ASCs (B220−CD138+TACI+) in spleens of IL‐2‐treated mice (Figure 1l and m). Anti‐dsDNA titres remained unaltered by the 7‐day IL‐2 treatment (Supplementary figure 1). These results revealed that a short period (7 days) of low‐dose IL‐2 treatment in NZB/W F1 mice could change the balance of Tfh and Tfr cells by increasing the Tfr/Tfh ratio, which primarily results from a stronger inhibition of IL‐2 to Tfh cells rather than Tfr cells. However, such short period of intervention was insufficient to correct the spontaneous GC response in this model.
Medium treatment regimen of low‐dose IL‐2 therapy suppressed spontaneous GC responses in NZB/W F1 mice
Next, we doubled the treatment duration of low‐dose IL‐2 therapy for 14 days as a medium treatment regimen. Consistent with results in the short treatment regimen, 14‐day IL‐2 treatment also increased Treg cells and reduced Teff cells (Supplementary figure 2a and b). The frequencies of Tfh cells in Teff cells and total CD4+ T cells decreased by 1.5‐ and 2.5‐fold, respectively, in IL‐2‐treated mice than in control mice (Figure 2a–c). Consistently, IL‐2 treatment also decreased the absolute number of Tfh cells (Figure 2d). Notably, the frequency of Tfr cells declined not only in Treg cells (by 2.4‐fold) but also in CD4+ T cells (by 1.7‐fold) (Figure 2a, e and f). Furthermore, the absolute number of Tfr decreased by 1.9‐fold (Figure 2g). This suggested a longer IL‐2 treatment led to an objective reduction of Tfr cells (Figure 2f and g), which was otherwise not observed in short treatment regimen (Figure 1h and i). Both Tfr/Tfh ratio (CD4+) and Tfr/Tfh ratio (number) were upregulated by 14‐day IL‐2 treatment (Figure 2h and i). Accordingly, although the medium treatment regimen again increased the Tfr/Tfh ratio (CD4+), the incremental level was smaller in the medium treatment regimen (by 1.4‐fold; Figure 2h) than that in the short treatment regimen (by 2‐fold; Figure 1j), because of the reduction of both Tfh and Tfr cells by the medium treatment regimen.
Figure 2.
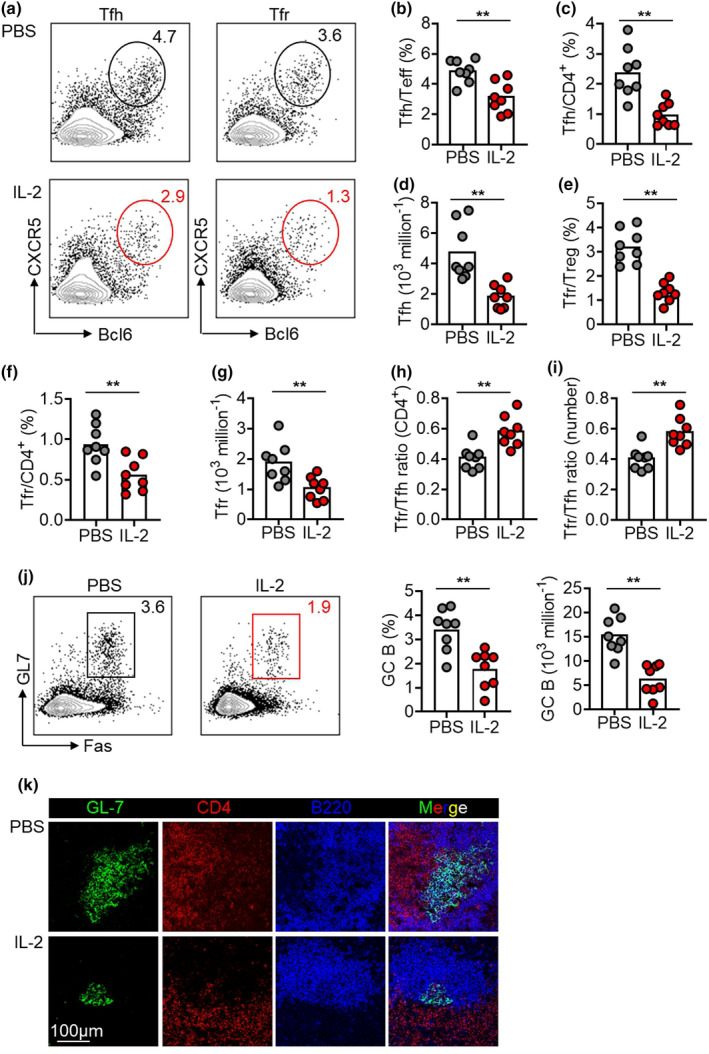
14‐day low‐dose IL‐2 therapy mitigated spontaneous GC responses in NZB/W F1 mice. (a–g) Representative FACS plots and statistics showing Tfh cells in Teff cells (b), Tfh in CD4+ T cells (c), Tfh cell numbers (d), Tfr cells in Treg cells (e), Tfr in CD4+ T cells (f), Tfr cell numbers (g) in spleens of NZB/W F1 mice with PBS or IL‐2 treatment. (h, i) The ratio of Tfr cell frequency in CD4+ T cells to Tfh cell frequency in CD4+ T cells [Tfr/Tfh ratio (CD4+)] (h) and the ratio of Tfr cell number to Tfh cell numbers [Tfr/Tfh ratio (number)] (i) in spleens of NZB/W F1 mice with medium treatment regimen of low‐dose IL‐2 or PBS for 14 days. (j) Representative FACS plots and statistics showing the frequencies and the absolute numbers of GC B cells (B220+GL‐7+Fas+) in spleens of NZB/W F1 mice with medium treatment regimen of low‐dose IL‐2 or PBS for 14 days. (k) Immunofluorescence staining of GL‐7 (green), CD4 (red) and B220 (blue) GC B cells in spleens of NZB/W F1 mice with PBS or IL‐2 treatment. Scale bar: 100 μm. Data are shown for individual (dots, n = 8) and mean (bars) values. Data are representative of three independent experiments and analysed by the Student's unpaired t‐test or the Mann–Whitney U‐test. **P < 0.01. GC, germinal centre; NZB/W F1, F1 hybrid between the New Zealand Black (NZB) and New Zealand White (NZW) strains; Teff, effector T cell; Tfh, T follicular helper cell; Tfr, T follicular regulatory cell; Treg, regulatory T cell.
The other major difference was also recorded between short vs medium treatment regimens, with GC B cells only falling in mice treated with IL‐2 for 14 days (Figure 2j) but not 7 days (Figure 1l). The suppression of spontaneous formation of GC B cells by the medium treatment regimen of low‐dose IL‐2 was verified by the immunofluorescence staining of splenic tissue sections, showing a remarkable reduction of GC areas positive for the marker GL‐7 after IL‐2 treatment (Figure 2k). However, even a longer treatment of IL‐2 for 14 days was unable to significantly inhibit the differentiation of ASCs or the production of anti‐dsDNA antibodies (Supplementary figure 2c and d). Moreover, the deposits immune complex and C3 in kidneys were comparable between IL‐2‐ vs PBS‐treated mice under the medium regimen (data not shown). Together, these results suggest although a medium treatment regimen of 14 days reduced spontaneous GC formation in NZB/W F1 mice, the pathogenic humoral immune responses were largely unaffected.
Long treatment regimen of low‐dose IL‐2 therapy alleviated pathogenic humoral immunity
In the clinical trials of low‐dose IL‐2 therapy, IL‐2 was administrated daily or every second day in different regimens, but the rationale to guide the choice remains elusive. 33 , 41 , 42 In order to understand how the frequency and duration of low‐dose IL‐2 therapy regulate humoral immunity in autoimmune diseases, we designed a third long treatment regimen whereby IL‐2 was administered every second day for 28 days. The long treatment regimen (28 days) and medium treatment regimen (14 days) delivered IL‐2 with a same dose in total but by a reduced frequency.
In the comparison of mice treated with IL‐2 every second day and PBS as controls, we recorded a 2‐fold reduction of Teff cells (Figure 3a and b) and a trend of increase of Treg cells but, unexpectedly, not reaching the statistical significance (19.9% vs 24.6%, P‐value = 0.32; Figure 3a and c). In the long treatment regimen, the frequencies of Tfh cells in Teff cells and total CD4+ T cell decreased by 2.1‐ and 5.1‐fold, respectively (Figure 3d and e), while the Tfr frequencies in Treg cell and total CD4+ T cell decline by 2.3‐ and 2.1‐fold, respectively (Figure 3g and h). Furthermore, 28‐day IL‐2 treatment decreased the number of Tfh (by 4.6‐fold) and Tfr cells (by 2.3‐fold) (Figure 3f and i). The net change of the Tfr/Tfh ratio was an upregulation by 2‐fold in 28‐day IL‐2 treatment (Figure 3j), consistent with elevated Tfr/Tfh ratios by short and medium treatment regimens.
Figure 3.
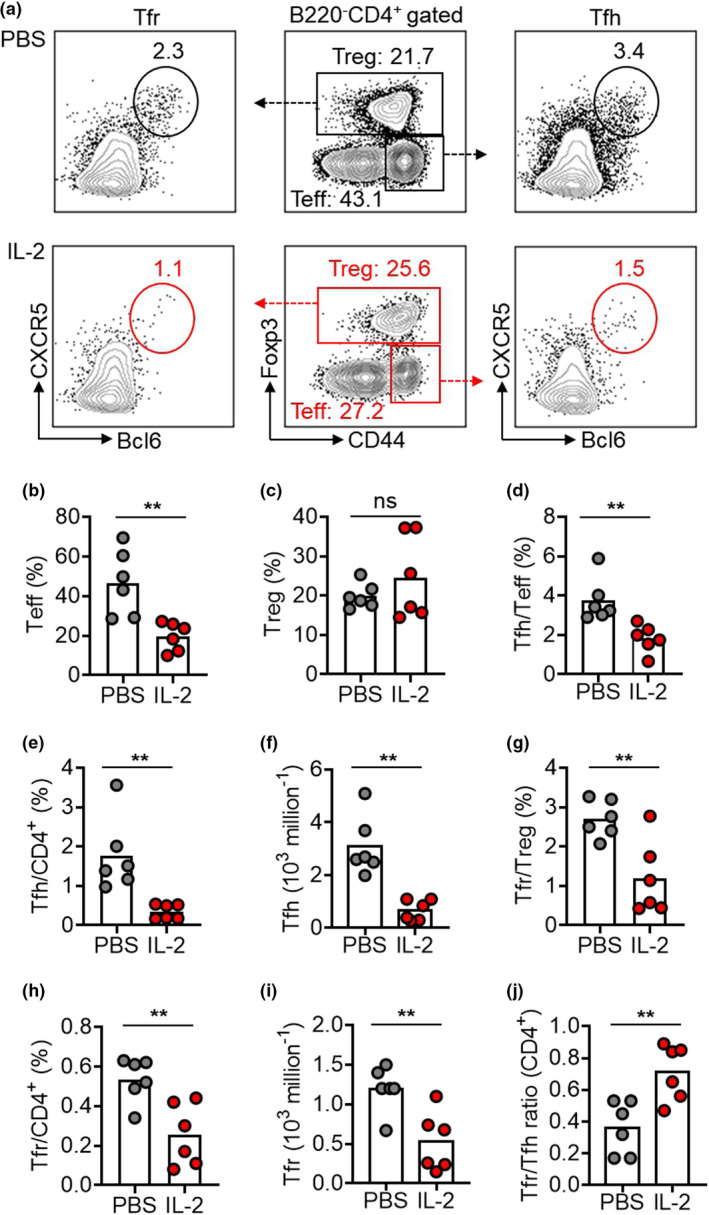
28‐day low‐dose IL‐2 therapy inhibited Tfr cells but upregulated the Tfr/Tfh ratio in NZB/W F1 mice. (a–j) Representative FACS plots (a) and statistics showing Teff cells (b), Treg (c), Tfh cells in Teff cells (d), Tfh cells in CD4+ T cells (e), the numbers of Tfh cells (f), Tfr cells in Treg cells (g), Tfr cells in CD4+ T cells (h), the numbers of Tfr cells (i) and the ratio of Tfr/Tfh cells (CD4+) (j) in spleens of NZB/W F1 mice with long treatment regimen of low‐dose IL‐2 or PBS with every second day for 28 days. Data are shown for individual (dots, n = 6) and mean (bars) values. Data are representative of three independent experiments and analysed by the Student's unpaired t‐test or the Mann–Whitney U‐test. **P < 0.01. NZB/W F1, F1 hybrid between the New Zealand Black (NZB) and New Zealand White (NZW) strains; Teff, effector T cell; Tfh, T follicular helper cell; Tfr, T follicular regulatory cell; Treg, regulatory T cell.
There was a remarkable reduction of GC frequencies (by 2.5‐fold) and absolute cell numbers (by 3.1‐fold) in mice treated with IL‐2 in comparison with mice treated with PBS (Figure 4a). Strikingly, in contrast to short or medium treatment regimens, we, for the first time, observed IL‐2 treatment significantly inhibited the differentiation of ASCs, shown as a 2.9‐fold reduction in frequency and a 2.3‐fold reduction in absolute cell numbers (Figure 4b). In parallel, serum anti‐dsDNA titres declined by 3.9‐fold in IL‐2‐treated mice than in control mice (Figure 4c). IgG and C3 deposits in the glomerular basement membranes lead to sustained inflammation and renal damage. We observed IL‐2 treatment reduced C3 and total IgG deposits in renal glomeruli (Figure 4d). Indeed, IgG1, IgG2a, IgG2b and IgG3 deposits were all reduced by IL‐2 treatment (Figure 4e). These data demonstrate that the treatment of IL‐2 for 28 days, despite a reduced frequency of every second day, efficiently mitigated pathogenic humoral immunity in NZB/W F1 mice.
Figure 4.
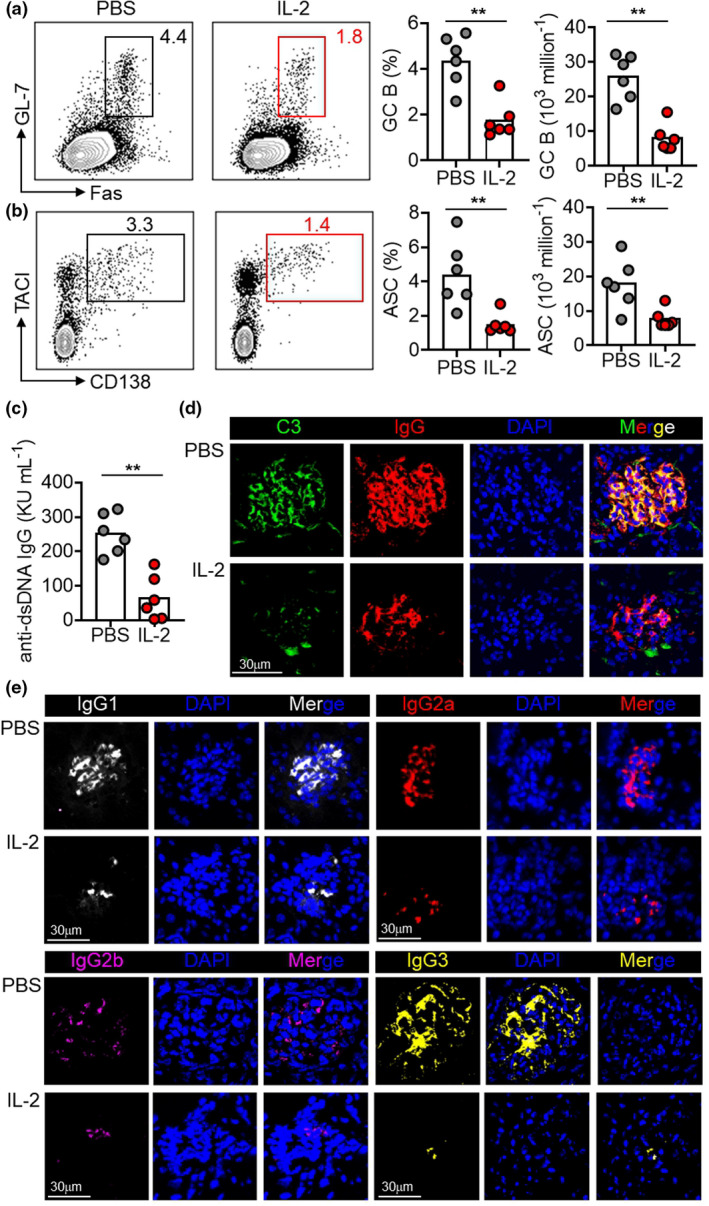
28‐day low‐dose IL‐2 therapy attenuated pathogenic humoral immunity. (a, b) Representative FACS plots and statistics showing the ratio and cell numbers of GC B cells (a) and ASCs (b) in spleens of NZB/W F1 mice with long treatment regimen of low‐dose IL‐2 or PBS with every second day for 28 days. (c) ELISA measurement of anti‐dsDNA IgG titre in the serum of NZB/W F1 mice with PBS or IL‐2 treatment. (d, e) Immunofluorescence staining of deposits C3 (green), IgG (red) (d), IgG1 (white), IgG2a (red), IgG2b (pink) and IgG3 (yellow) (e) in kidneys of NZB/W F1 mice with PBS or IL‐2 treatment. Nucleus were stained with DAPI (blue). Scale bar: 30 μm. Data are shown for individual (dots, n = 6) and mean (bars) values. Data are representative of three independent experiments and analysed by the Student's unpaired t‐test or the Mann–Whitney U‐test. **P < 0.01. ASC, antibody‐secreting cell; C3, complement component 3; GC, germinal centre; and NZB/W F1, F1 hybrid between the New Zealand Black (NZB) and New Zealand White (NZW) strains.
Long treatment regimen of low‐dose IL‐2 therapy alleviated renal histopathology and improved renal function
As a major complication of SLE, lupus nephritis is driven by the production of autoantibodies, especially ANA, and the formation of glomerular immune deposits. 11 , 12 Lupus nephritis is also featured in lupus‐prone mice, including NZB/W F1 mice. After observing the suppression of the pathogenic humoral immunity in NZB/W F1 mice, we next examined how low‐dose IL‐2 therapy with the long treatment regimen influenced the renal histopathology and function. Compared with C57BL/6 mice, NZB/W F1 mice with PBS treatment developed severe renal damages, shown by the expansion and sclerosis of glomeruli, crescent formation, tubular dilation and intratubular casts, and massive cell infiltration around blood vessels in the kidney (Figure 5a). IL‐2 treatment significantly alleviated renal histopathology, shown by the reductions in glomerular, tubular and perivascular histological scores (Figure 5a). Moreover, IL‐2 treatment also markedly reduced mesangial matrix expansion, measured by periodic acid–Schiff (PAS) staining (Figure 5b). Proteinuria represents a key measure to assess glomerulonephritis, and the method of spot urinary albumin/creatinine ratio was developed. 12 IL‐2 treatment reduced urinary albumin by 4.3‐fold without changing urinary creatinine (Figure 5c and d). The resulted drop of the albumin/creatinine ratio by 5.6‐fold (Figure 5e) indicated that the every second day treatment of IL‐2 for 28 days significantly ameliorated renal histopathology and improved renal function in NZB/W F1 mice.
Figure 5.
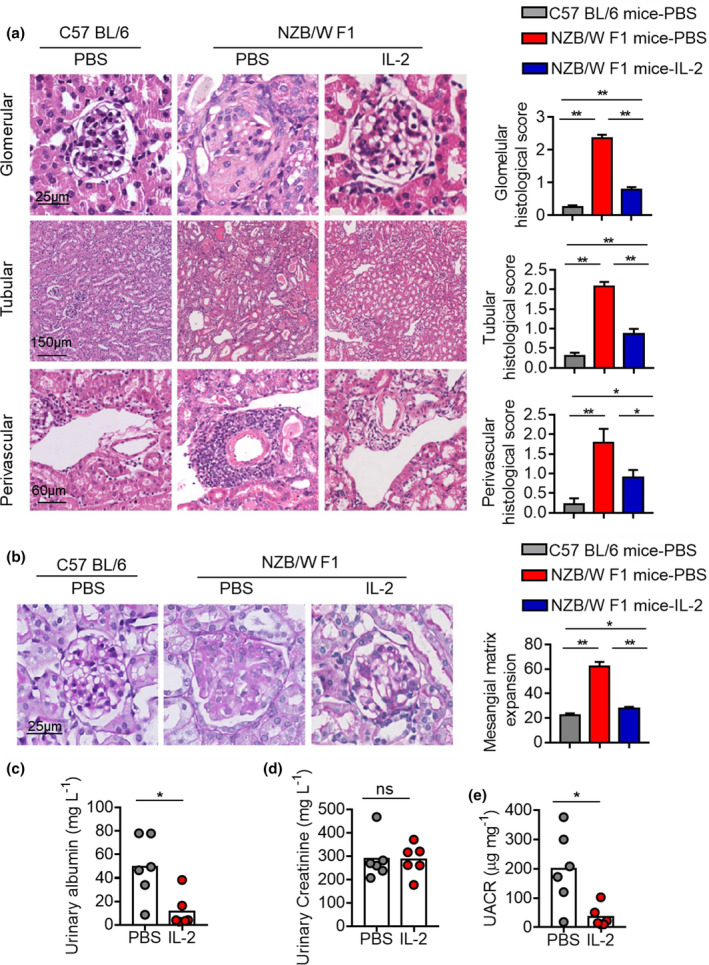
28‐day low‐dose IL‐2 therapy ameliorated renal pathology and improved renal function. (a) H&E staining and histological scores of renal tissues from C57BL/6 mice and NZB/W F1 mice with long treatment regimen of low‐dose IL‐2 or PBS with every second day for 28 days. Scale bar: glomerular 25 μm, tubular 150 μm and perivascular 60 μm. Data are expressed as means ± SE. (b) Representative images of PAS staining of renal sections and quantification of mesangial matrix index of glomerulus in C57BL/6 mice with PBS treatment and NZB/W F1 mice with long treatment regimen of low‐dose IL‐2 therapy with every second day for 28 days. Scale bar: 25 μm. Data are expressed as means ± SE. (c–e) Urinary albumin (c), urinary creatinine (d) and urinary albumin‐to‐creatinine ratio (UACR) (e) in NZB/W F1 mice with PBS or IL‐2 treatment. Data are shown for individual (dots, n = 6) and mean (bars) values. Data are representative of three independent experiments. The Student's unpaired t‐test was employed for comparisons between two groups; one‐way ANOVA followed by the post hoc Tukey test for multiple comparisons was used for groups of three or more. *P < 0.05 and **P < 0.01. NZB/W F1, F1 hybrid between the New Zealand Black (NZB) and New Zealand White (NZW) strains and PAS, periodic acid–Schiff.
Low‐dose IL‐2 treatment increased the Tfr/Tfh ratio in SLE patients
In a previous open‐labelled clinical trial of low‐dose IL‐2 therapy in SLE (three cycles, 1 million IU rhIL‐2 subcutaneously injected every second day for 2 weeks plus a 2‐week break), we found low‐dose IL‐2 suppressed the active Tfh generation. 32 We next asked whether low‐dose IL‐2 regulates Tfr cells and improves the Tfr/Tfh ratio in SLE patients by exploring the same data set. After three cycles of low‐dose IL‐2 therapy, the frequency of CXCR5+ cells in CD4+ T cells was significantly reduced (P‐value = 0.0001; Figure 6a and b), and Tfr cells (CXCR5+CD25highCD127low) in total CD4+ T cells slightly declined but did not reach a statistical significance (2.01% vs 1.41%; P‐value = 0.09) at 12 weeks (Figure 6a and c). Despite this, the Tfr/Tfh ratios were significantly improved by 1.4‐fold (P‐value = 0.0043; Figure 6d). Altogether, low‐dose IL‐2 treatment elevated the Tfr/Tfh ratio in SLE patients, in agreement with the observation in NZB/W F1 mice.
Figure 6.
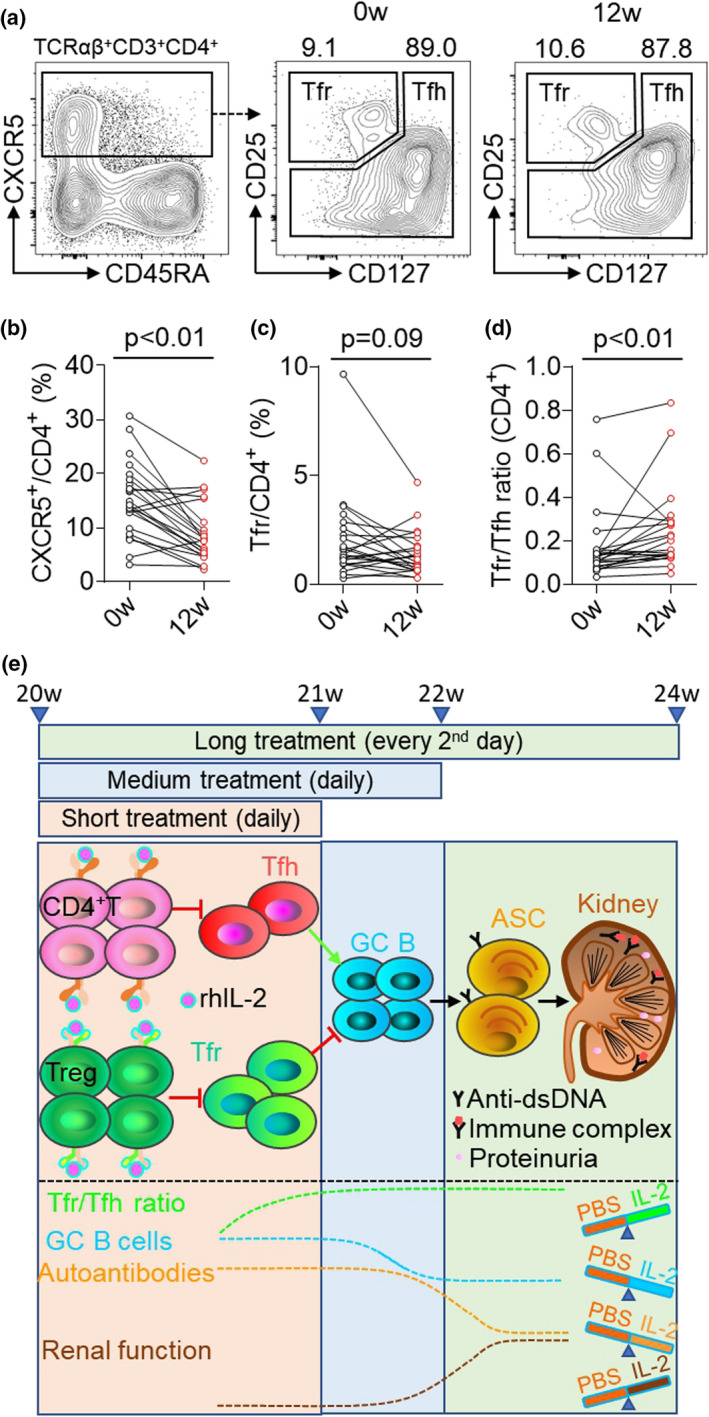
Low‐dose IL‐2 therapy elevated the Tfr/Tfh ratio in SLE patients. (a) Representative FACS plots showing circulating TCRαβ+CD3+CD4+CXCR5+CD25+CD127− Tfr cells in the PBMC of SLE patients at baseline (0w) and after low‐dose IL‐2 therapy (12w). (b–d) Statistics of CXCR5+ cells in total CD4+ T cells (b), Tfr cells in total CD4+ T cells (c) and the Tfr/Tfh ratio (d) at baseline (0w) and after IL‐2 therapy (12w). (e) Graphic model of the proposed low‐dose IL‐2 therapy mitigates pathogenic humoral immunity in NZB/W F1 mice via a step‐by‐step process. Short treatment regimen of low‐dose IL‐2 therapy inhibits Tfh cells and increases Tfr/Tfh ratio, medium treatment regimen of low‐dose IL‐2 therapy reduces Tfr cells and GC B cells, and long treatment regimen of low‐dose IL‐2 therapy mitigates pathogenic humoral immunity, alleviates renal histopathology and improved renal function. Data are shown for individual (dots, n = 23) and mean (bars) values and analysed by the Student's paired t‐test. PBMC, peripheral blood mononuclear cell and SLE, systemic lupus erythematosus.
The expression of CD25 and CD122 on Tfr is higher than Tfh cells
To understand whether the differential expression of IL‐2 receptors CD25 and CD122 may explain a better sensitivity of Tfh than Tfr cells to IL‐2 therapy, we measured the expressions of IL‐2 receptors on Tnaive, Treg, Tfh and Tfr cells in NZB/W F1 mice and SLE patients by flow cytometry. In spleens of NZB/W F1 mice, CD25 was highly expressed on CD4+Foxp3+ Treg cells. Both CXCR5+BCL6+Foxp3− Tfh and CXCR5+BCL6+Foxp3+ Tfr cells expressed very low levels of CD25, with even lower on Tfh cells (Figure 7a). CD122 expression was comparable between Treg and Tfr cells, which were significantly higher than that on Tfh cells (Figure 7b). In PBMC from SLE patients, we also detected similar results showing higher expression of CD25 and CD122 on Tfr cells than those on Tfh cells (Figure 7c–g). Therefore, the phenomenon that Tfh cells were more sensitive than Tfr cells to low‐dose IL‐2 therapy in NZB/W F1 mice and SLE patients is likely caused by the distinct signal pathways between Tfh and Tfr differentiation rather than the differential expression of IL‐2 receptors on established Tfh and Tfr cells leading to different IL‐2 responsiveness.
Figure 7.
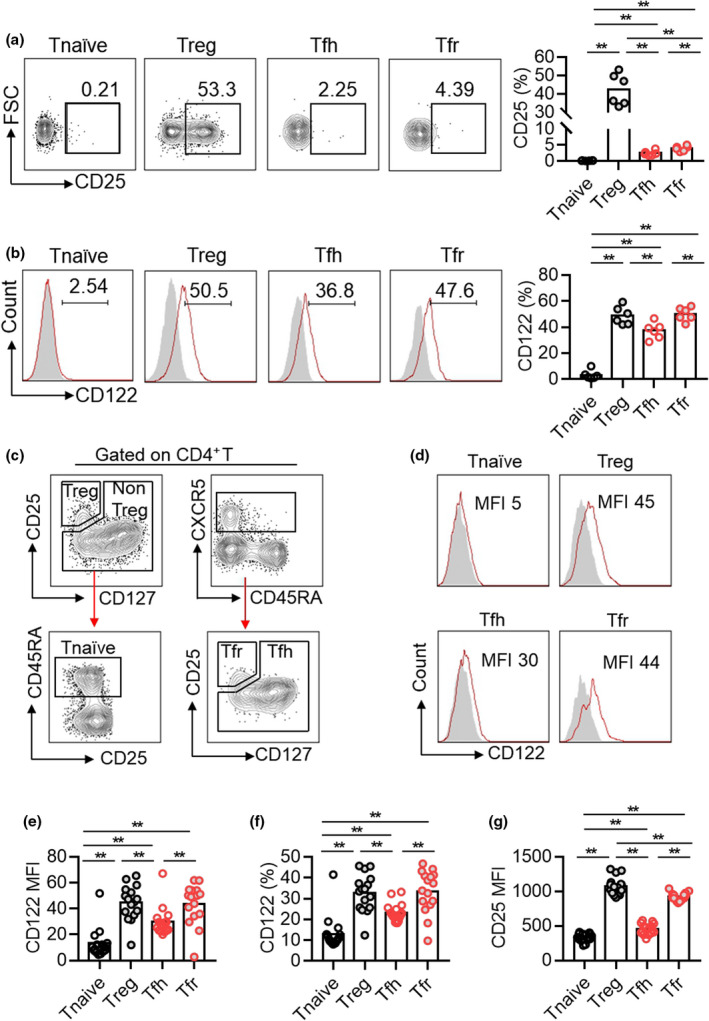
Flow cytometric analysis of CD25 and CD122 expressions in Tnaive, Treg, Tfh and Tfr cells in spleens of NZB/W F1 mice and in PBMC of SLE patients. (a, b) Representative dot plots and percentage statistics of CD25 (a) and CD122 (b) expressions in Tnaive (B220−CD4+CD44−CD62L+), Treg (B220−CD4+Foxp3+), Tfh (B220−CD4+CD44+CXCR5+BCL6+) and Tfr (B220−CD4+Foxp3+CXCR5+BCL6+) cells in spleens of 5‐month‐old NZB/W F1 mice. Data are shown for individual (dots, n = 6). Data are representative of three independent experiments. (c–g) Representative dot plots and the statistics of the median fluorescence index (MFI) of CD122 (e), the frequency of CD122 positive portion (f) and the median fluorescence index (MFI) of CD25 (g) among Tnaive (CD3+CD4+CD25−CD45RA+), Treg (CD3+CD4+CD25+CD127−), Tfh (CD3+CD4+CXCR5+CD25−) and Tfr (CD3+CD4+CXCR5+CD25+CD127−) cells in PBMC from SLE patients. Data are shown for individual (dots, n = 16) and mean (bars) values and analysed by one‐way ANOVA followed by the post hoc Tukey test. **P < 0.01.
Discussion
Here, we designed and examined three treatment regimens of low‐dose IL‐2 therapy in lupus‐prone NZB/W F1 mice and characterised the immune responses in detail. We found that low‐dose IL‐2 treatment for a period of 7, 14 or 28 days consistently upregulated the Tfr/Tfh ratio. In addition, the improvement of the Tfr/Tfh ratio by low‐dose IL‐2 therapy in SLE patients was verified using the data from a published clinical trial. Notably, short treatment of IL‐2 for 7 days in NZB/W F1 mice was able to reduce only Tfh cells but not Tfr cells. In SLE patients, low‐dose IL‐2 therapy significantly suppressed Tfh generation and only modestly but non‐significantly reduced Tfr cells. Therefore, it appeared that in both mice and humans, low‐dose IL‐2 therapy demonstrated a more pronounced inhibition on Tfh cells than Tfr cells.
Tfr and Tfh cells essentially control the formation of GC B cells and ASCs, as well as autoantibody production in many autoimmune diseases including SLE. 22 , 23 , 25 The imbalanced Tfr/Tfh ratios were reported in patients with SLE, which were shown to correlate with excessive B cell responses and autoantibody production. 43 Our study demonstrated that low‐dose IL‐2 therapy in NZB/W F1 mice improved the Tfr/Tfh ratio and gradually alleviated spontaneous GC formation, excessive ASC differentiation and autoantibody production. The mitigation of such pathogenic humoral immunity in NZB/W F1 mice by low‐dose IL‐2 appeared to be a step‐by‐step process (Figure 6e). Although the increased of the Tfr/Tfh ratio by IL‐2 treatment was detected as early as 7‐day treatment, a therapeutic period of 14 and 28 days was necessary to reduce GC formation, while the suppression of ASC differentiation and autoantibody production only happened after the long treatment regimen of 28 days. With the inhibition of autoantibody production by the long treatment regimen, immunocomplex and C3 deposits in the kidney were profoundly reduced, accompanied by ameliorated renal histopathology and improved renal function.
In NZB/W F1 mice and SLE patients, Tfr cells showed a weaker sensitivity to IL‐2 than that of Tfh cells. However, in a mouse model of influenza infection, the treatment of 15 000 IU IL‐2 from day 20 to day 30 postinfection was shown to deplete Tfr cells without obvious changes on Tfh cells and GC B cells, suggesting Tfr cells were more sensitive than Tfh cells to IL‐2 treatment. 36 It is alarming that the reduced Tfr/Tfh ratio by IL‐2 treatment in this infection model led to the aberrant generation of self‐reactive ASCs and the production of autoantibodies. 36 This is unlikely the case in autoimmune diseases since IL‐2 treatment was found to reduce autoantibody production in clinical trials in inflammatory and autoimmune diseases. 31 , 32 In both NZB/W F1 mice and SLE patients, the expressions of CD25 and CD122 were higher on Tfr cells than those on Tfh cells, which indicate a likely better sensitivity of established Tfr cells to IL‐2 than Tfh cells. Indeed, a recent study reported that Tfh cells in GCs poorly respond to IL‐2. 44 IL‐2 potently suppressed Tfh cells primarily through inhibiting BCL6 expression required for primed CD4+ T cells to commit Tfh differentiation. 35 Thus, IL‐2 therapy likely inhibited Tfh early differentiation rather than deleting established Tfh cells. For Tfr cells, IL‐2 therapy expands Treg cells but simultaneously exerts the inhibition of Treg to Tfr differentiation. Both positive and negative effects of IL‐2 therapy on Tfr generation might offset each other which reduces the overall responsiveness of Tfr cells to IL‐2 therapy. Tfh differentiation and Tfr differentiation are regulated not only by IL‐2 but also by other pathways, in particular antigen stimulation. Therefore, the discrepancy of IL‐2 therapy observed between infection vs autoimmune disease models could lie in the distinct activation of other regulatory pathways for Tfh and Tfr differentiation which thus differentially modulates the sensitivity to IL‐2 in Tfh and Tfr cells.
As a major limitation of our study, the characterisation of IL‐2 treatment effect largely focused on the frequencies of Tfh and Tfr cells as well as the bulk antibody responses, while the fine‐tuning of Tfh and Tfr function after IL‐2 therapy has not been examined. The low numbers of Tfh and Tfr cells impede the feasibility to evaluate their function by in vitro culture assays. Future studies are required to deploy cutting‐edge technologies such as single‐cell RNA sequencing to delineate the heterogeneity and functionality of Tfh and Tfr cells regulated by low‐dose IL‐2 therapy. In future, the regulatory effects of human IL‐2 on human Tfh and Tfr cells could also been validated in humanised mouse models, such as by engrafting human T cells in NOD scid gamma (NSG) mice.
One unique design of our study was to compare the medium treatment regimen (daily treatment for 14 days) and the long treatment regimen (every second day treatment for 28 days) which delivered the same accumulative dose of IL‐2. Despite comparable pharmacokinetics of IL‐2 treatment given daily or every second day (Supplementary figure 3a and b), the long treatment regimen showed a better pharmacodynamics by reducing ASC differentiation, anti‐dsDNA titres and immunocomplex deposits in the kidney. The suppression of pathogenic humoral immunity supports the choice of a less frequent but prolonged treatment regimen of low‐dose IL‐2 therapy, which could be further tested in clinical trials.
Altogether, our study, for the first time, demonstrates that low‐dose IL‐2 therapy elevates the Tfr/Tfh ratio and alleviates pathogenic humoral immunity in lupus‐prone mice and SLE patients, which provide necessary rationale and new insights in optimising low‐dose IL‐2 therapy.
Methods
Human subjects
A total of 16 female SLE patients from Renji hospital Affiliated to Shanghai Jiao Tong University School of Medicine were recruited. SLE patients fulfilled the 1997 American College of Rheumatology classification criteria for SLE. 45 This study was approved by the Ethics Committee of Renji Hospital and was conducted in accordance with good clinical practice. Detailed demographic and clinical characteristics of the subjects are listed in Supplementary table 3.
Mice
NZW and NZB mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA), and C57BL/6 mice were purchased from Shanghai Jihui Laboratory Animal Care Co., Ltd (Shanghai, China). All mice were maintained under specific pathogen‐free conditions in the animal facility of Renji Hospital, Shanghai Jiao Tong University School of Medicine. Female NZB/W F1 mice (F1 hybrid between the NZB and NZW strains) with 20 weeks of age were used for experiments. All mice experiments were performed in accordance with the animal welfare guidelines under approved protocols of Renji Hospital, School of Medicine, Shanghai Jiao Tong University.
IL‐2 treatment
Female NZB/W F1 mice (20 weeks of age) were randomly divided into two groups based on body weight, proteinuria and anti‐dsDNA titres. One group of mice were intraperitoneally administrated with low‐dose recombinant human rhIL‐2 [recombinant human IL‐2Ser125, Beijing SL Pharma, 3 × 104 international unit (IU); Beijing, China] following three treatment regimens: (1) short, daily for 7 days; (2) medium, daily for 14 days, and (3) long, every second day for 28 days. The other group of control mice were intraperitoneally administrated with equal volume of PBS.
Flow cytometry
Single‐cell suspension of spleens from IL‐2‐ or PBS‐treated female NZW/B F1 mice was stained with the fluorochrome‐conjugated antibodies: CD4 (RM4‐5), CD44 (IM7), Foxp3 (FJK‐16s), CD138 (281‐2), GL‐7 (GL7), Bcl6 (K112‐91), B220 (RA3‐6B2), TACI (8F10), CD25 (PC61), CD122 (TM‐β1), CD62L (MEL‐14) and CXCR5 (L138D7; purchased from BD Biosciences, San Jose, CA, USA, or BioLegend, San Diego, CA, USA). PBMCs were separated from the peripheral blood of SLE patients by density gradient centrifugation and were stained with fluorochrome‐conjugated antibodies: CD3 (UCHT1), CD19 (SJ25C1), CD4 (SK3), CD25 (M‐A251), CD122 (TU27), CD127 (A019D5), CD45RA (HI100) and CXCR5 (J252D4). Live/dead cells were distinguished using the Zombie Aqua Fixable Viability kit (BioLegend). Data were collected by a flow cytometer (Fortessa X20, BD) and analysed by FlowJo software (BD).
ELISA
Anti‐dsDNA IgG titre in the serum was measured using the anti‐dsDNA IgG ELISA kit (Alpha Diagnostic International, San Antonio, TX, USA) following the manufacturer's protocol.
Urinary albumin and urinary creatinine
Twenty‐four‐hour urine samples were collected and stored at −80°C. Urinary albumin was measured by the Roche urine trace albumin kit (Roche, Basel, Switzerland), and urinary creatinine was measured by the Urinary Creatinine kit (Maccura Biotechnology, Sichuan, China) following the manufacturers' protocols.
Immunofluorescence staining
Frozen sections of spleens (4 μm) were incubated with PE‐conjugated anti‐CD4 (RM4‐5, BioLegend), Alexa Fluor 488‐conjugated anti‐GL7 (GL7, BD) and Alexa Fluor 647‐conjugated anti‐B220 (RA3‐6B2, BD). Frozen sections of kidneys (4 μm) were stained with goat anti‐mouse IgG (Sigma‐Aldrich, Darmstadt, Germany), FITC rat anti‐mouse C3 (Santa Cruz Biotechnology, Dallas, TX, USA), donkey anti‐goat CY3 (Proteintech, Wuhan, China), Alexa Fluor 647‐conjugated IgG1, PE‐conjugated IgG2a (BioLegend), biotin anti‐IgG2b (BioLegend), biotin anti‐IgG3 (BioLegend) and PE streptavidin. Nuclei were identified by DAPI staining. Images were obtained with a laser scanning confocal microscope (Zeiss, Oberkochen, Germany) and analysed by Photoshop CS4 software (Adobe, San Jose, CA, USA).
Renal pathology
Renal pathology was evaluated according to the previously described protocol 46 in a blinded manner. Glomerular pathology was evaluated by assessing 20 glomerular cross sections (gcs) per kidney and scored each glomerulus on a semiquantitative scale: 0 = normal [35–40 cells per glomerular cross sections (gcs)]; 1 = mild [glomeruli with few lesions showing slight proliferative changes, mild hypercellularity (41–50 cells per gcs)]; 2 = moderate (glomeruli with moderate hypercellularity (51–60 cells per gcs), including segmental and/or diffuse proliferative changes, hyalinosis); and 3 = severe [glomeruli with segmental or global sclerosis and/or severe hypercellularity (> 60 cells per gcs), necrosis and crescent formation]. Interstitial/tubular pathology was assessed in 10 randomly selected high‐power fields (hpf) based on the number of infiltrates and damaged tubules. 0 = normal, 1 = mild, 2 = moderate and 3 = severe. Perivascular cell accumulation was determined by scoring the number of cell layers surrounding the majority of vessel walls (score: 0 = none, 1 = ≤ 5 cell layers, 2 = 5–10 cell layers and 3 = ≥ 10 cell layers). PAS staining was applied to quantify the glomerular mesangial matrix expansion and analysed by ImageJ (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data were analysed with GraphPad Prism (Version 8.0, Insightful Science, San Diego, CA, USA). The significance was determined by the Student's t‐test, the Mann–Whitney U‐test or one‐way ANOVA followed by the post hoc Tukey test. Data are shown as mean ± SEM, and P < 0.05 was considered statistically significant.
Study approval
All the experiments were performed with the use of protocols approved by the Institutional Animal Care and Use Committee at Renji Hospital and the Ethics Committee of Renji Hospital, Shanghai Jiao Tong University School of Medicine.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Kaili Liang: Data curation; Formal analysis; Methodology; Software; Supervision; Writing‐original draft. Jing He: Data curation; Methodology. Yunbo Wei: Conceptualization; Data curation. Qunxiong Zeng: Formal analysis; Software. Dongcheng Gong: Data curation; Methodology. Jiahuan Qin: Data curation; Methodology. Huihua Ding: Data curation; Formal analysis. Zhian Chen: Data curation; Formal analysis; Methodology. Ping Zhou: Data curation; Investigation. Peng Niu: Data curation; Methodology. Qian Chen: Data curation; Methodology. Chenguang Ding: Data curation; Investigation. Liangjing Lu: Data curation; Investigation. Xiaoxiang Chen: Data curation; Methodology. Zhanguo Li: Data curation; Project administration; Writing‐review & editing. Nan Shen: Conceptualization; Funding acquisition. Di Yu: Conceptualization; Funding acquisition; Investigation; Project administration; Writing‐review & editing. Jun Deng: Conceptualization; Data curation; Funding acquisition; Investigation; Project administration; Writing‐original draft.
Supporting information
Acknowledgments
We acknowledge Ms Jufang Yao from the animal facility of Renji Hospital, Shanghai Jiao Tong University School of Medicine for technical assistance. This project was supported by the National Key Research and Development Program of China (2017YFC0909003 to DY), the National Natural Science Foundation of China (82071816 to JD, 82071792 to DY and 31630021, 81421001 and 31930037 to NS), Health Commission of Hubei Province Scientific Research Project (2019WJ2019H136 to DY) and Innovative Research Team of High‐level Local Universities in Shanghai led by NS.
Contributor Information
Zhanguo Li, Email: li99@bjmu.edu.cn.
Nan Shen, Email: nanshensibs@gmail.com.
Di Yu, Email: di.yu@uq.edu.au.
Jun Deng, Email: jun.deng@aliyun.com.
References
- 1. Kaul A, Gordon C, Crow MK et al. Systemic lupus erythematosus. Nat Rev Dis Primers 2016; 2: 16039. [DOI] [PubMed] [Google Scholar]
- 2. Tsokos GC. Systemic lupus erythematosus. N Engl J Med 2011; 365: 2110–2121. [DOI] [PubMed] [Google Scholar]
- 3. Aringer M, Costenbader K, Daikh D et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis 2019; 78: 1151–1159. [DOI] [PubMed] [Google Scholar]
- 4. He J, Tsai LM, Leong YA et al. Circulating precursor CCR7loPD‐1hi CXCR5+CD4 + T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013; 39: 770–781. [DOI] [PubMed] [Google Scholar]
- 5. Scharer CD, Blalock EL, Mi T et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat Immunol 2019; 20: 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yurasov S, Wardemann H, Hammersen J et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med 2005; 201: 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luzina IG, Atamas SP, Storrer CE et al. Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol 2001; 70: 578–584. [PubMed] [Google Scholar]
- 8. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat Rev Immunol 2010; 10: 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma K, Li J, Wang X et al. TLR4+CXCR4+ plasma cells drive nephritis development in systemic lupus erythematosus. Ann Rheum Dis 2018; 77: 1498–1506. [DOI] [PubMed] [Google Scholar]
- 10. Cambridge G, Leandro MJ, Teodorescu M et al. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum 2006; 54: 3612–3622. [DOI] [PubMed] [Google Scholar]
- 11. Scott DG, Rowell NR. Immunohistological studies of the kidney in systemic lupus erythematosus and systemic sclerosis using antisera to IgG, C3, fibrin, and human renal glomeruli. Ann Rheum Dis 1974; 33: 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arbuckle MR, McClain MT, Rubertone MV et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 2003; 349: 1526–1533. [DOI] [PubMed] [Google Scholar]
- 13. Gualtierotti R, Borghi MO, Gerosa M et al. Successful sequential therapy with rituximab and belimumab in patients with active systemic lupus erythematosus: a case series. Clin Exp Rheumatol 2018; 36: 643–647. [PubMed] [Google Scholar]
- 14. Kraaij T, Kamerling SWA, de Rooij ENM et al. The NET‐effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J Autoimmun 2018; 91: 45–54. [DOI] [PubMed] [Google Scholar]
- 15. Kraaij T, Arends EJ, van Dam LS et al. Long‐term effects of combined B‐cell immunomodulation with rituximab and belimumab in severe, refractory systemic lupus erythematosus: 2‐year results. Nephrol Dial Transplant 2020; gfaa117: 1–10. 10.1093/ndt/gfaa117. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doria A, Stohl W, Schwarting A et al. Efficacy and safety of subcutaneous belimumab in anti‐double‐stranded DNA‐positive, hypocomplementemic patients with systemic lupus erythematosus. Arthritis Rheumatol 2018; 70: 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol 2016; 34: 335–368. [DOI] [PubMed] [Google Scholar]
- 18. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol 2015; 16: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014; 41: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang W, Zhang H, Liu S et al. Excessive CD11c+Tbet+ B cells promote aberrant TFH differentiation and affinity‐based germinal center selection in lupus. Proc Natl Acad Sci USA 2019; 116: 18550–18560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choi JY, Ho JH, Pasoto SG et al. Circulating follicular helper‐like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol 2015; 67: 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Linterman MA, Pierson W, Lee SK et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med 2011; 17: 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chung Y, Tanaka S, Chu F et al. Follicular regulatory T cells expressing Foxp3 and Bcl‐6 suppress germinal center reactions. Nat Med 2011; 17: 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clement RL, Daccache J, Mohammed MT et al. Follicular regulatory T cells control humoral and allergic immunity by restraining early B cell responses. Nat Immunol 2019; 20: 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deng J, Wei Y, Fonseca VR, Graca L, Yu D. T follicular helper cells and T follicular regulatory cells in rheumatic diseases. Nat Rev Rheumatol 2019; 15: 475–490. [DOI] [PubMed] [Google Scholar]
- 26. Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol 2015; 36: 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu B, Wang S, Zhou M et al. The ratio of circulating follicular T helper cell to follicular T regulatory cell is correlated with disease activity in systemic lupus erythematosus. Clin Immunol 2017; 183: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hao H, Nakayamada S, Yamagata K et al. Conversion of T Follicular Helper Cells to T Follicular Regulatory Cells by Interleukin‐2 Through Transcriptional Regulation in Systemic Lupus Erythematosus. Arthritis Rheumatol 2021; 73: 132–142. [DOI] [PubMed] [Google Scholar]
- 29. Spolski R, Li P, Leonard WJ. Biology and regulation of IL‐2: from molecular mechanisms to human therapy. Nat Rev Immunol 2018; 18: 648–659. [DOI] [PubMed] [Google Scholar]
- 30. Koreth J, Matsuoka K, Kim HT et al. Interleukin‐2 and regulatory T cells in graft‐versus‐host disease. N Engl J Med 2011; 365: 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saadoun D, Rosenzwajg M, Joly F et al. Regulatory T‐cell responses to low‐dose interleukin‐2 in HCV‐induced vasculitis. N Engl J Med 2011; 365: 2067–2077. [DOI] [PubMed] [Google Scholar]
- 32. He J, Zhang X, Wei Y et al. Low‐dose interleukin‐2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med 2016; 22: 991–993. [DOI] [PubMed] [Google Scholar]
- 33. von Spee‐Mayer C, Siegert E, Abdirama D et al. Low‐dose interleukin‐2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis 2016; 75: 1407–1415. [DOI] [PubMed] [Google Scholar]
- 34. Yan JJ, Lee JG, Jang JY, Koo TY, Ahn C, Yang J. IL‐2/anti‐IL‐2 complexes ameliorate lupus nephritis by expansion of CD4+CD25+Foxp3+ regulatory T cells. Kidney Int 2017; 91: 603–615. [DOI] [PubMed] [Google Scholar]
- 35. Ballesteros‐Tato A, Leon B, Graf BA et al. Interleukin‐2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 2012; 36: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Botta D, Fuller MJ, Marquez‐Lago TT et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat Immunol 2017; 18: 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol 1985; 37: 269–390. [DOI] [PubMed] [Google Scholar]
- 38. Andrews BS, Eisenberg RA, Theofilopoulos AN et al. Spontaneous murine lupus‐like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med 1978; 148: 1198–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mosmann TR, Yokota T, Kastelein R, Zurawski SM, Arai N, Takebe Y. Species‐specificity of T cell stimulating activities of IL 2 and BSF‐1 (IL 4): comparison of normal and recombinant, mouse and human IL 2 and BSF‐1 (IL 4). J Immunol 1987; 138: 1813–1816. [PubMed] [Google Scholar]
- 40. Hu YL, Metz DP, Chung J, Siu G, Zhang M. B7RP‐1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J Immunol 2009; 182: 1421–1428. [DOI] [PubMed] [Google Scholar]
- 41. He J, Zhang R, Shao M et al. Efficacy and safety of low‐dose IL‐2 in the treatment of systemic lupus erythematosus: a randomised, double‐blind, placebo‐controlled trial. Ann Rheum Dis 2020; 79: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Humrich JY, von Spee‐Mayer C, Siegert E et al. Rapid induction of clinical remission by low‐dose interleukin‐2 in a patient with refractory SLE. Ann Rheum Dis 2015; 74: 791–792. [DOI] [PubMed] [Google Scholar]
- 43. Liu C, Wang D, Song Y, Lu S, Zhao J, Wang H. Increased circulating CD4+CXCR5+FoxP3+ follicular regulatory T cells correlated with severity of systemic lupus erythematosus patients. Int Immunopharmacol 2018; 56: 261–268. [DOI] [PubMed] [Google Scholar]
- 44. Papillion A, Powell MD, Chisolm DA et al. Inhibition of IL‐2 responsiveness by IL‐6 is required for the generation of GC‐TFH cells. Sci Immunol 2019; 4: eaaw7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40: 1725. [DOI] [PubMed] [Google Scholar]
- 46. Kikawada E, Lenda DM, Kelley VR. IL‐12 deficiency in MRL‐Fas lpr mice delays nephritis and intrarenal IFN‐gamma expression, and diminishes systemic pathology. J Immunol 2003; 170: 3915–3925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials