

Abstract
Aims/Introduction
Emerging evidence shows that epigenetic modifications occurring during fetal development in response to intrauterine exposures could be one of the mechanisms involved in the early determinants of adult metabolic disorders. This study aimed to investigate whether the placental maternally expressed gene 3 (MEG3) deoxyribonucleic acid (DNA) methylation profile is associated with maternal gestational diabetes mellitus status and newborn birthweight.
Materials and Methods
Samples for measurement were collected from 23 women with gestational diabetes mellitus and 23 healthy controls. MEG3 gene expression and DNA methylation levels were assessed using quantitative real‐time polymerase chain reaction and MethylTargetTM, respectively. Pearson correlation analyses were used to examine associations between placental DNA methylation levels and clinical variables of interest. The associated results were adjusted by multivariate linear regression for maternal age, body mass index, height, gestational age and newborn sex as confounders.
Results
We found that the DNA methylation levels in the MEG3 differentially methylated region were significantly different between the gestational diabetes mellitus and control groups on the maternal side of the placenta (40.64 ± 2.15 vs 38.33 ± 2.92; P = 0.004). Furthermore, the mean MEG3 DNA methylation levels were correlated positively with maternal fasting glucose concentrations (R = 0.603, P < 0.001) and newborn birthweight (R = 0.568, P < 0.001).
Conclusions
The placental DNA methylation status in the MEG3 differentially methylated region was correlated with maternal glucose concentrations and newborn birthweight. These epigenetic adaptations might contribute to late‐onset obesity, underlining the adverse intrauterine environment.
Keywords: Epigenetics, Gestational diabetes mellitus, Maternally expressed gene 3 differentially methylated region
The placental deoxyribonucleic acid methylation status in the maternally expressed gene 3 differentially methylated region was correlated with maternal fasting glucose concentrations and the newborn birthweight. These epigenetic adaptations might contribute to late‐onset obesity, underlining the adverse intrauterine environment.

Introduction
Gestational diabetes mellitus (GDM) is a common metabolic disorder, defined as any degree of glucose intolerance with onset or first recognition during pregnancy 1 . The prevalence of GDM is estimated to range from 6.5 to 25% 2 , 3 . Accumulating studies have shown that GDM could cause short‐ and long‐term complications in both mothers and offspring. Maternal adverse outcomes associated with GDM include pre‐eclampsia, postpartum hemorrhage and susceptibility to future metabolic diseases 4 , 5 . Fetuses exposed to the maternal hyperglycemic environment are at increased risk of stillbirth, premature birth, macrosomia, shoulder dystocia and neonatal respiratory distress syndrome 1 , 6 . In addition, the offspring of women with GDM are at elevated risk of developing diabetes, cardiovascular disease, overweight and obesity, with potential transgenerational effects 7 .
The underlying mechanisms involved in developmental programming are still obscure. One potential molecular mechanism that has been proposed to explain the long‐term effect of exposure to a deleterious fetal environment is epigenetics. Epigenetics refers to regulating gene expression without altering the nucleotide sequence. Deoxyribonucleic acid (DNA) methylation is the most stable and widely studied epigenetic mechanism, which occurs as the addition of a methyl group to the 5‐carbon position of cytosine residues within CpG islands (genomic regions rich in CpG dinucleotides). Abnormal DNA methylation mediated by DNA methyltransferase enzymes might result in inappropriate gene silencing, thus regulating gene expression and subsequent cellular functions.
The placenta is critical for regulating fetal development, producing hormones and responding to environmental signals. Adverse conditions in utero might alter placental gene regulation and subsequent placental physiology, consequently, fetal development. A hyperglycemic environment during pregnancy (GDM and pregestational diabetes) disturbs endothelial cell cycle pathways 8 , changes inflammatory patterns 9 , induces decidual vasculopathy 10 and impacts on vascular permeability of the placenta 11 . Epigenetic adaptations in the placenta makes it specifically susceptible to the environment 12 . Epigenetic modifications in the placenta, mainly carried out by genomic imprinting and DNA methylation, contribute to these short‐ and long‐term outcomes. Previous studies reported dysregulation in placental lipoprotein lipase and IGF2/H19 DNA methylation exposed to the intrauterine hyperglycemic environment 13 , 14 .
Maternally expressed gene 3 (MEG3) is an imprinted gene located on human chromosome 14q32.3 within the DLK1‐MEG3 locus 15 . MEG3 encodes a long non‐coding ribonucleic acid (RNA), and has been reported to participate in various types of human diseases 16 , 17 , 18 , 19 . The expression of MEG3 is regulated by two differentially methylated regions (DMRs) – the intergenic differentially methylated region (IG‐DMR) and the MEG3‐DMR 20 , 21 . Previous studies have shown that epigenetic modification of the DLK1‐MEG3 domain is altered in type 1 and type 2 diabetes 19 , 22 . Hypermethylation of MEG3‐DMR in mice with diabetes results in decreased MEG3 messenger RNA (mRNA) levels 23 . A genome imprinting study showed that the Dlk1‐Dio3 locus is responsible for fetal growth in mice 24 . Hence, the MEG3 DNA methylation profile appears to have a potential role in GDM‐related developmental programming. We supposed that maternal hyperglycemia could alter MEG3 DNA methylation status, possibly contributing to fetal development. The objectives of the present study were to investigate the association between maternal glycemic status with fetal growth and to examine the role of MEG3 DNA methylation profile in this association.
Methods
Participants
GDM was diagnosed using a 75‐g oral glucose tolerance testing (OGTT) carried out at 24–28 weeks'’ gestation, according to the International Association of Diabetes in Pregnancy Study Group criteria 25 . Women were diagnosed with GDM if at least one value exceeded the following thresholds: fasting plasma glucose ≥5.1 mmol/L, 1‐h plasm glucose ≥10.0 mmol/L, 2‑h plasma glucose ≥8.5 mmol/L 26 and recommended with dietary treatment. The glycated hemoglobin levels were also measured at the time of OGTT. We excluded women with pre‐GDM, multifetation, hyperthyroidism, cardiovascular disease and pre‐eclampsia. The gestational age‐matched women with normal glucose tolerance were in the control group. Body mass index (BMI) was calculated according to the standard formula (kg/m2) in the first trimester and before delivery.
The study was approved by the ethics committee of the Women’s Hospital, School of Medicine, Zhejiang University (IRB‐20200194‐R), and followed the Declaration of Helsinki. Written informed consent was obtained from all participants.
Placental tissue sampling
Placenta samples were collected in the minutes after delivery (<30 min postpartum) by well‐trained research staff, and kept in RNALater (Qiagen, Valencia, CA, USA) at −80°C until nucleic acid extraction. We selected tissues from both sides of the placenta, the maternal side consisted of the intervillous tissues and chorionic villi, and the fetal side consisted mainly of fetal villous tissue. Analyses were carried out on both sides independently.
RNA extraction and real‐time polymerase chain reaction analyses
Total RNA was extracted from collected placenta tissues using the Trizol reagent (Invitrogen, Carlsbad, CA, USA), and then reverse‐transcribed to complementary DNA with the primeScript™ RT reagent Kit (TaKaRa, Otsu, Japan) according to the manufacturer’s instructions, respectively. The quantitative real‐time polymerase chain reaction (PCR) was carried out with SYBR® Premix Ex Taq™ (TaKaRa) in an Applied Biosystems 7900HT (ABI, Foster City, CA, USA). Glyceraldehyde 3‐phosphate dehydrogenase was chosen as the endogenous control, and the relative expression of MEG3 was processed with the method 27 . All the samples were run in triplicate. The primer sequences used are shown in Table S1.
DNA extraction and methylation‐specific PCR
Genomic DNA was purified from placenta samples with the Genomic DNA Purification Kit (Invitrogen), and bisulfite conversion was carried out with the EpiTect Bisulfite Kit (Qiagen) according to the manufacturer’s protocols. Regions of interest were carried out for multiplex and index PCR reaction, as described in a previous study 28 . The PCR amplicons were purified by the QIAquick Gel Extraction Kit (Qiagen) and loaded onto Illumina NextSeq 500 (Illumina, San Diego, CA, USA) according to the manufacturer’s protocols. Specific primers for PCR amplification are presented in Table S2.
Statistical analysis
Statistical analyses were carried out using SPSS 25.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 8.0 (GraphPad Software Inc., La Jolla, CA, USA), and results were presented as the mean ± standard deviation. Differences in continuous variables among groups were carried out by the independent two‐sample Student’s t‐test. Categorical variables are reported as numbers (percentages) and were compared by Fisher’s exact test. Pearson correlation analysis was carried out to examine associations between placental MEG3 methylation profile and clinical variables of interest: maternal glucose levels of OGTT and neonatal birthweight. The associated results were adjusted by multivariate linear regression using the enter method for maternal age, BMI (during the first and third trimester), height, gestational age and newborn sex. Differences were assigned statistically significant at P < 0.05, P < 0.01 or P < 0.001. The sample size was estimated to obtain a significant Pearson correlation coefficient of at least 0.4 between methylation levels and clinical variables. The total estimated sample size was 46 by accepting an alpha risk of 0.05 and a beta risk of 0.2.
Results
Study participants
A total of 46 women with normal glucose tolerance (n = 23) or GDM (n = 23) participated in the present study. Descriptive statistics of mothers and newborns are presented in Table 1. The maternal characteristics, such as maternal age, height, BMI, gestational weeks and weight gain, were similar in the two groups. Meanwhile, fasting plasma glucose (4.31 ± 0.31 vs 4.91 ± 0.57 mmol/L, P < 0.001), 1‐h plasma glucose (7.36 ± 1.32 vs 10.60 ± 1.84 mmol/L, P < 0.001), 2‐h plasma glucose (6.65 ± 1.07 vs 9.37 ± 1.41 mmol/L, P < 0.001) and glycated hemoglobin (5.05 ± 0.26 vs 5.34 ± 0.43 (%), P = 0.009) concentrations were higher in the GDM group compared with the control group. Furthermore, the newborn birthweight of the GDM group was higher than the control group (3,178.91 ± 331.72 vs 3,541.30 ± 504.97 g, P = 0.006).
Table 1.
Characteristics of the mothers and newborns
| Characteristics | Control (n = 23) | GDM (n = 23) | P‐value |
|---|---|---|---|
| Maternal age (years) | 31.87 ± 4.67 | 33.87 ± 4.85 | 0.161 |
| Height (cm) | 160.35 ± 6.51 | 158.48 ± 4.41 | 0.261 |
| Gestational age at birth (weeks) | 38.22 ± 1.17 | 38.04 ± 1.19 | 0.619 |
| First trimester BMI (kg/m2) | 22.12 ± 3.32 | 23.20 ± 2.75 | 0.235 |
| Third trimester BMI (kg/m2) | 27.25 ± 3.11 | 27.69 ± 2.78 | 0.618 |
| Weight gain (%) † | 24.02 ± 8.76 | 19.77 ± 7.46 | 0.083 |
| Fasting glucose levels (mmol/L) | 4.31 ± 0.31 | 4.91 ± 0.57 | <0.001 * |
| 1‐h OGTT glucose levels (mmol/L) | 7.36 ± 1.32 | 10.60 ± 1.84 | <0.001 * |
| 2‐h OGTT glucose levels (mmol/L) | 6.65 ± 1.07 | 9.37 ± 1.41 | <0.001 * |
| HbA1c (%) | 5.05 ± 0.26 | 5.34 ± 0.43 | 0.009 * |
| Birthweight (g) | 3,178.91 ± 331.72 | 3,541.30 ± 504.97 | 0.006 * |
| Newborn sex, male (%) | 12 (52.2%) | 14 (60.9%) | 0.767 |
Data are presented as Mean ± SD and number (%).
Weight gain between the first trimester and third trimester (% of initial body weight).
P < 0.05 (in bold).
mRNA expression
To investigate the possible role of intrauterine hyperglycemia in the alteration of MEG3 expression, we first assessed the mRNA expression of MEG3 in the placenta using quantitative real‐time PCR. As shown in Figure 1a, the mRNA expressions of MEG3 on the maternal placental side were significantly reduced in the GDM patients compared with the healthy controls. Interestingly, this downregulation did not occur on the fetal side of the placenta (Figure 1b).
Figure 1.
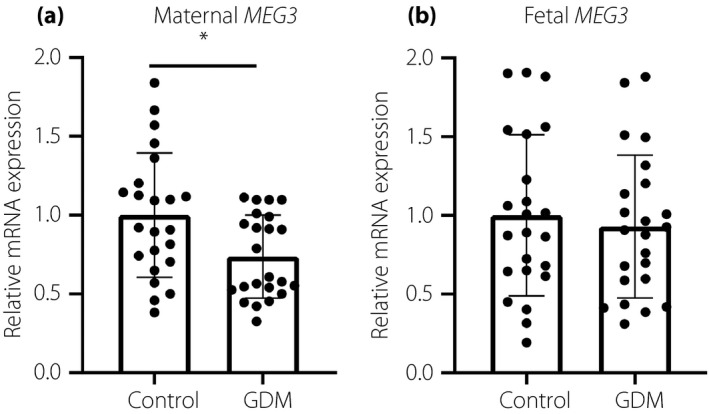
Analysis of relative maternally expressed gene 3 (MEG3) messenger ribonucleic acid (mRNA) levels in gestational diabetes mellitus (GDM; n = 23) and control (n = 23) placentas. (a) On the maternal side of the placenta, MEG3 expression was significantly downregulated in GDM patients compared with controls. (b) On the fetal side, there was no significant difference between GDM and control groups. Data are presented as the mean ± standard error; *P < 0.05.
Methylation status of MEG3‐DMR in placental tissues
Then, we analyzed the DNA methylation levels of the MEG3‐DMR in both maternal and fetal sides of placental tissues. Overall, 35 CpG dinucleotides in the MEG3‐DMR were the epigenotype. The individual CpG analyses are presented in Table S3. On the maternal side of the placenta, the DNA methylation levels at seven CpG islands’ loci were significantly higher in the GDM group compared with the control group (Figure 2a); whereas on the fetal side, only single CpG site showed significant hypermethylation in the GDM group (Figure 2c). On average, the mean MEG3 methylation levels on the maternal placental side were significantly different between the two groups (40.64 ± 2.15 vs 38.33 ± 2.92; P = 0.004; Figure 2b). However, there were no significant differences in mean MEG3 methylation between women with or without GDM on the fetal side of the placenta. (39.87 ± 4.97 vs 38.36 ± 4.62; P = 0.294; Figure 2d).
Figure 2.
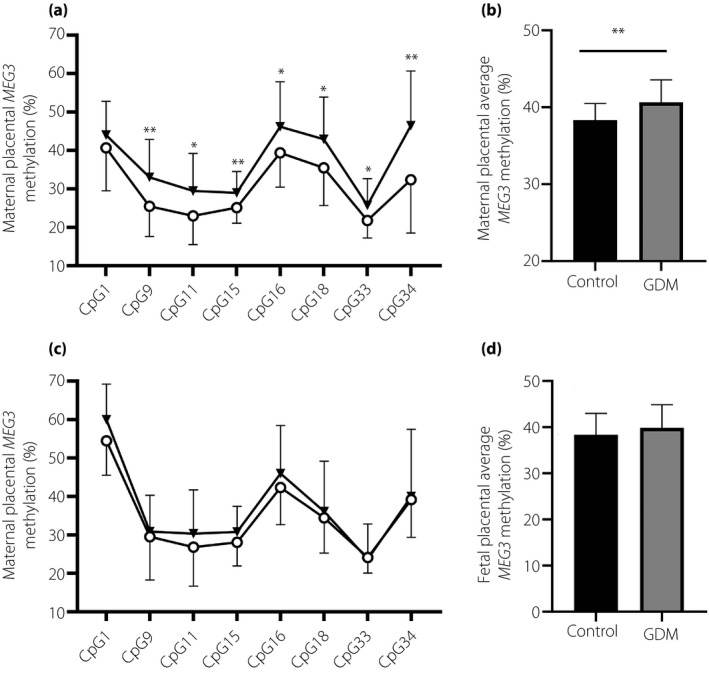
Comparison of maternally expressed gene 3 (MEG3) deoxyribonucleic acid methylation levels in gestational diabetes mellitus (GDM; n = 23) and control (n = 23) placentas. (a,b) Methylation status of MEG3 differentially methylated region (DMR) on the maternal side, including the (a) mean MEG3 methylation level and (b) specific individual CpG sites. (c,d) MEG3‐DMR methylation status on the fetal side, including the (c) mean MEG3 methylation level and (d) specific individual CpG sites. GDM and the control group are indicated by triangles (▼) and circles (○), respectively. Data are presented as the mean ± standard error; *P < 0.05; **P < 0.01.
Correlations between placental MEG3 methylation levels, maternal hyperglycemia and fetal growth
Furthermore, we investigated whether the placental methylation status of the MEG3‐DMR might be associated with maternal and newborn characteristics. On the maternal side of the placenta, we found that the increasing level of maternal fasting glucose was positively correlated with methylation levels at five MEG3 CpG islands (CpG9[chr14:101,292,225], CpG15[chr14:101,292,460], CpG16[chr14:101,292,463], CpG18[chr14:101,292,483] and CpG19[chr14:101,292,493]; data not shown). Furthermore, correlations were also observed between newborn birthweight and placental DNA methylation levels at five MEG3 CpG islands (CpG9[chr14:101,292,225], CpG11[chr14:101,292,392], CpG12[chr14:101,292,398], CpG15[chr14:101,292,460] and CpG16[chr14:101,292,463]; data not shown). Of note, the levels of MEG3 DNA methylation on the maternal side at three specific CpG islands (CpG9, CpG15 and CpG16) were correlated with both maternal glucose status (R = 0.447, P = 0.002; R = 0.461, P = 0.001; R = 0.579, P < 0.001, respectively) and newborn birthweight (R = 0.495, P < 0.001; R = 0.450, P = 0.001; R = 0.471, P = 0.001, respectively; Figure 3). The correlations remained significant after adjusting for maternal age, height, BMI, gestational age and newborn sex as confounders by carrying out multivariable linear regression (Table 2). Furthermore, the higher mean methylation levels of the MEG3‐DMR on the maternal side of the placenta also showed a significant correlation with higher maternal fasting glucose concentration (R = 0.603, P < 0.001) and newborn birthweight (R = 0.568, P < 0.001; Figure 4). These relationships substantially improved after adjusting for confounding variables (Table 2). It suggests that hypermethylation in the MEG3‐DMR, especially at CpG9, CpG15 and CpG16, is related to intrauterine hyperglycemia and conceptus growth. However, there was no correlation between DNA methylation levels of the MEG3‐DMR and the maternal glucose status or newborn birthweight on the fetal side of the placenta (data not shown).
Figure 3.
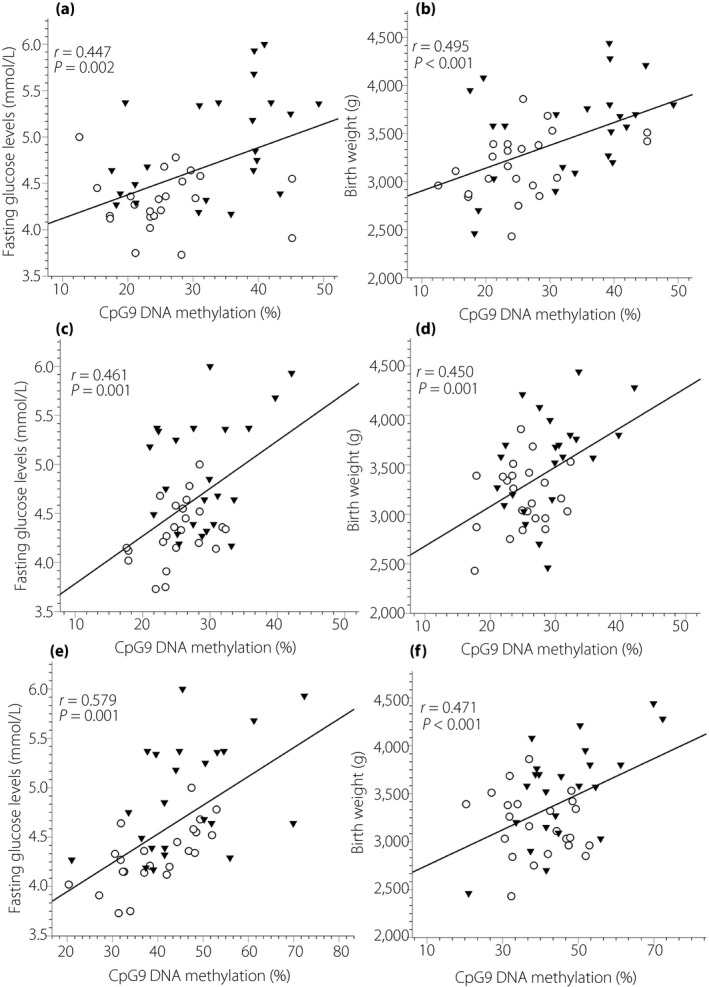
Pearson correlation for the association between placental deoxyribonucleic acid (DNA) methylation levels at specific individual CpG sites in the maternally expressed gene 3 (MEG3) differentially methylated region (DMR) and maternal glucose levels and newborn birthweight. (a,b) The correlation between the MEG3 methylation and (a) maternal fasting glucose levels and (b) newborn birthweight at CpG9, (c,d) at CpG15, and (e,f) at CpG16. GDM (n = 23) and the control (n = 23) group are indicated by triangles (▼) and circles (○), respectively.
Table 2.
Multivariate linear models analyzing the correlation of maternally expressed gene 3 deoxyribonucleic acid methylation with selected clinical variables
| β | P | R 2 | |
|---|---|---|---|
| Mean methylation levels of MEG3‐DMR (%) | |||
| Fasting plasma glucose | 0.491 | 0.001 | 0.488 |
| Birthweight | 0.547 | 0.002 | 0.478 |
| Methylation level at CpG9 | |||
| Fasting plasma glucose | 0.463 | 0.004 | 0.376 |
| Birthweight | 0.483 | 0.015 | 0.330 |
| Methylation level at CpG15 | |||
| Fasting plasma glucose | 0.482 | 0.005 | 0.274 |
| Birthweight | 0.612 | 0.003 | 0.285 |
| Methylation level at CpG16 | |||
| Fasting plasma glucose | 0.665 | 0.000 | 0.445 |
| Birthweight | 0.547 | 0.009 | 0.258 |
Models are adjusted for maternal age, height, body mass index, gestational age at delivery and newborn gender.
MEG3‐DMR, maternally expressed gene 3 differentially methylated region.
Figure 4.

Pearson correlation for the association between the mean placental deoxyribonucleic acid (DNA) methylation of maternally expressed gene 3 differentially methylated region (MEG3‐DMR) and maternal fasting glucose levels and newborn birthweight. (a,b) Correlation between the (a) meanMEG3‐DMR DNA methylation and fasting glucose levels and (b) newborn birthweight.
Discussion
The developmental origins hypothesis posits that prenatal environmental variations could disturb fetal programming, and lead to a lifetime risk of obesity and chronic diseases 29 , 30 . In addition, growing clinical and epidemiological evidence recognizes that exposure to GDM is associated with large for gestational age and obesity in later life. To date, the exact molecular mechanism of developmental programming remains unknown, while epigenetic modification might elucidate the phenomenon. In the present study, we investigated maternal fasting glucose levels in the second trimester regarding neonatal birthweight, and evaluated the role of DNA methylation differences within the MEG3‐DMR in this association. For the first time, we showed that the hypermethylation of MEG3‐DMR on the maternal side of the placenta was associated with maternal hyperglycemia and neonatal birthweight. In a total of 35 CpG islands analyzed, we found hypermethylation of three specific CpG sites in the MEG3‐DMR as the most potential candidate gene sites for further research. Interestingly, these phenomena were not found on the fetal side. These results might indicate that DNA methylation of the MEG3‐DMR plays an essential role in response to intrauterine hyperglycemia, and this epigenetic alteration is a link to offspring development.
As a mediator of environmental and maternal inputs throughout pregnancy, the placenta is central to coordinating fetal development. The epigenetic alterations of the placenta responding to maternal and fetal signals can contribute to determining fetal growth potential and influence health outcomes throughout life. Studies in both humans and animal models have shown that the DNA methylation profile of the placenta is one of the critical mechanisms. A previous animal study found that dysregulation of methylation patterns can lead to adverse placenta morphology and birth outcome 31 . A wide array of environmental and maternal signals might alter gene methylation in the placenta, such as maternal depressed mood 32 , maternal hyperglycemia 33 , poor nutritional 34 or over‐nutrition status 35 and pre‐eclampsia 36 . There is compelling evidence that DNA methylation dysregulation underlies the associations between adult diseases and intrauterine hyperglycemia. Genome‐wide DNA methylation variation in the GDM‐exposed placenta compared controls identified 1,708 methylation variable positions achieving significant difference 37 . Another placental epigenome‐wide association study identified methylation at fiftee15n CpG islands that were associated with newborn birthweight 38 . Thus far, the focus regarding placental epigenetic marking progress in GDM was set on specific DNA methylation loci. Bouchard et al. 39 reported a significant correlation between maternal glycemic status and DNA methylation levels at the adiponectin gene in the placenta. This relationship could increase the risk of developing diabetes and obesity in the offspring. Another relevant study was carried out by Houde et al. 40 , who found that the 2‐h post‐OGTT results are correlated with adenosine triphosphate‐binding cassette transporter A1 DNA methylation levels of the placenta. Then, the same authors found the association between maternal glycemia and placental lipoprotein lipase DNA methylation levels 13 . In addition, we added a novel finding that the hypermethylation status of the MEG3‐DMR on the maternal side of the placenta is a link between maternal hyperglycemia and large neonatal birthweight. The variability of MEG3‐DMR hypermethylation was only observed on the maternal placental side in the GDM group, and is perhaps a result of the intrauterine hyperglycemic environment.
Genomic imprinting is an epigenetic modification that causes unequal gene expression depending on their parental origin. It is one of the candidate mechanisms that contribute to regulating fetal growth in mammals. The animal model study by our team showed that intrauterine hyperglycemia could alter the expression of the imprinted genes of the placenta, 35 upregulated and 10 downregulated 41 . The study of the placenta exposed to intrauterine hyperglycemia suggested that altered methylation percentages at the multiple imprint regulatory regions, including the MEST and IGF2/H19, are associated with large for gestational age or macrosomia (birthweight >4 kg) 35 , 42 . Similar results were also identified in the children of obese mothers 43 . The present findings, together with the previous studies, support the idea that epigenetic modifications at some imprinted loci have been crucial regulators of both neonatal and long‐term health outcomes.
MEG3 is a maternally expressed imprinting gene in humans, and produces a long non‐coding RNA transcript 15 . The MEG3 promoter region is rich in CpG islands and there are two DMRs – the IG‐DMR the MEG3‐DMR 20 . Previous studies showed that the methylation status of the MEG3‐DMR plays an essential role in regulating the MEG3 expression, which mediated cell proliferation, migration and apoptosis in pituitary tumor 34 , retinoblastoma 11 , cervical cancer 44 , acute myeloid leukemia 45 , and type 1 and 2 diabetes 19 , 46 . Mounting studies have reported that MEG3 is involved in the progression of diabetes‐related diseases. For example, Qiu et al. 47 found MEG3 knockdown aggravated diabetes‐related retinal microvascular dysfunction in mice by the inactivation of PI3k/Akt signaling. Additionally, MEG3 protected against podocyte injury in diabetic nephropathy by regulating Wnt/β‐catenin signaling 48 . Studies of patients with diabetes also suggested that increased methylation at the MEG3‐DMR is sufficient to repress MEG3 expression, and impacts the sensitivity of β‐cells to cytokine‐mediated oxidative stress 23 . However, the role of MEG3 in fetal development and responding to the intrauterine hyperglycemic environment remains unknown. In the present study, we endeavored to show increased methylation levels of the MEG3‐DMR in the GDM‐exposed placenta, and whether the hypermethylation status is correlated with fetal growth. We showed that methylation levels in the MEG3‐DMR of the placenta are positively correlated with newborn birthweight. Interestingly, a recent study carried out by Prats‐Puig et al. 49 reported that placental MEG3‐DMR methylation correlated negatively with length and weight gain during the first postnatal year. Similarly, a follow‐up study also showed that children exposed to the diabetic environment in utero had a lower bodyweight at the age of 5 years than non‐exposed children 50 . One possible explanation for this variation in rates of weight gain in the early postnatal is catch‐up growth 51 . Another potential explanation is that GDM exposure might impact childhood growth; whether this relationship is mediated by epigenetic modification needs to be investigated.
The present study had several strengths. We analyzed the placental DNA methylation profile on the maternal and fetal side, independently. The hypermethylation of MEG3‐DMR on the maternal side is strongly associated with maternal fasting glucose levels and newborn birthweight. Intriguingly, these relationships were not found on the fetal placental side. This supports the idea that maternal lifestyle is vital for the optimal development of the fetus 52 . Second, the individual CpG island of the MEG3‐DMR was independently analyzed for the association between maternal glucose levels and birthweight. We found three specific sites to determine the relationship. Although our findings were generated confined to GDM patients, it is essential to realize that epigenetic modification of imprinting gene MEG3 could be a hopeful mechanism involved in fetal programming in general ways. This altered DNA methylation markers might allow the early approach to assessing the prenatal or postnatal growth of offspring. Nevertheless, the present study had certain limitations. The number of participants might not be adequate, limiting the power of our study. In addition, a postnatal follow‐up study should be carried out to show the impact of these epigenetic alterations on later‐life health.
In conclusion, this is the first report showing that DNA methylation perturbations within the imprinted gene, MEG3‐DMR, of the placenta are correlated with maternal glucose concentrations and newborn birthweight. Our investigations provide supporting evidence involved in fetal programming of obesity predisposition in adult life, underlining the consequences of adverse intrauterine exposures.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 | Primer list for quantitative real‐time polymerase chain reaction.
Table S2 | Primer list for MethylTarget sequencing.
Table S3 | Methylation levels of individual CpG island in the maternally expressed gene 3 differentially methylated region of the placenta.
Acknowledgment
The study was supported by Nature Science Foundation of Zhejiang Province (grants: LY20H040009, LQ20H040008), and Medical Health Science and Technology Project of Zhejiang Province (grant: 2018244300).
J Diabetes Investig 2021; 12: 1074–1082
References
- 1. Metzger BE, Lowe LP, Dyer AR, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med 2008; 358: 1991–2002. [DOI] [PubMed] [Google Scholar]
- 2. Berger H, Crane J, Farine D, et al. Screening for gestational diabetes mellitus. J Obstet Gynaecol Can 2002; 24: 894–912. [DOI] [PubMed] [Google Scholar]
- 3. Ad A. Standards of medical care in diabetes‐2017: summary of revisions. Diabetes Care 2017; 40: S4–S5. [DOI] [PubMed] [Google Scholar]
- 4. Mack LR, Tomich PG. Gestational diabetes: diagnosis, classification, and clinical care. Obstet Gynecol Clin North Am 2017; 44: 207–217. [DOI] [PubMed] [Google Scholar]
- 5. Farrar D, Simmonds M, Bryant M, et al. Hyperglycaemia and risk of adverse perinatal outcomes: systematic review and meta‐analysis. BMJ 2016; 354: i4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mitanchez D, Yzydorczyk C, Siddeek B, et al. The offspring of the diabetic mother–short‐ and long‐term implications. Best Pract Res Clin Obstet Gynaecol 2015; 29: 256–269. [DOI] [PubMed] [Google Scholar]
- 7. Mitanchez D, Burguet A, Simeoni U. Infants born to mothers with gestational diabetes mellitus: mild neonatal effects, a long‐term threat to global health. J Pediatr 2014; 164: 445–450. [DOI] [PubMed] [Google Scholar]
- 8. Acar N, Korgun ET, Cayli S, et al. Is there a relationship between PCNA expression and diabetic placental development during pregnancy? Acta Histochem 2008; 110: 408–417. [DOI] [PubMed] [Google Scholar]
- 9. Corrêa‐Silva S, Alencar AP, Moreli JB, et al. Hyperglycemia induces inflammatory mediators in the human chorionic villous. Cytokine 2018; 111: 41–48. [DOI] [PubMed] [Google Scholar]
- 10. Huynh J, Yamada J, Beauharnais C, et al. Type 1, type 2 and gestational diabetes mellitus differentially impact placental pathologic characteristics of uteroplacental malperfusion. Placenta 2015; 36: 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leach L, Taylor A, Sciota F. Vascular dysfunction in the diabetic placenta: causes and consequences. J Anat 2009; 215: 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Novakovic B, Yuen RK, Gordon L, et al. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genom 2011; 12: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Houde AA, St‐Pierre J, Hivert MF, et al. Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles. J Dev Orig Health Dis 2014; 5: 132–141. [DOI] [PubMed] [Google Scholar]
- 14. Ge ZJ, Liang QX, Luo SM, et al. Diabetic uterus environment may play a key role in alterations of DNA methylation of several imprinted genes at mid‐gestation in mice. Reprod Biol Endocrinol 2013; 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyoshi N, Wagatsuma H, Wakana S, et al. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000; 5: 211–220. [DOI] [PubMed] [Google Scholar]
- 16. Al‐Rugeebah A, Alanazi M, Parine NR. MEG3: an oncogenic long non‐coding RNA in different cancers. Pathol Oncol Res 2019; 25: 859–874. [DOI] [PubMed] [Google Scholar]
- 17. Dong Z, Zhang A, Liu S, et al. Aberrant methylation‐mediated silencing of lncRNA MEG3 functions as a ceRNA in esophageal cancer. Mol Cancer Res 2017; 15: 800–810. [DOI] [PubMed] [Google Scholar]
- 18. Gao Y, Huang P, Zhang J. Hypermethylation of MEG3 promoter correlates with inactivation of MEG3 and poor prognosis in patients with retinoblastoma. J Transl Med 2017; 15: 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kameswaran V, Bramswig NC, McKenna LB, et al. Epigenetic regulation of the DLK1‐MEG3 microRNA cluster in human type 2 diabetic islets. Cell Metab 2014; 19: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. da Rocha ST, Edwards CA, Ito M, et al. Genomic imprinting at the mammalian Dlk1‐Dio3 domain. Trends Genet 2008; 24: 306–316. [DOI] [PubMed] [Google Scholar]
- 21. Lin SP, Youngson N, Takada S, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1‐Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 2003; 35: 97–102. [DOI] [PubMed] [Google Scholar]
- 22. Wallace C, Smyth DJ, Maisuria‐Armer M, et al. The imprinted DLK1‐MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet 2010; 42: 68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kameswaran V, Golson ML, Ramos‐Rodríguez M, et al. The dysregulation of the DLK1‐MEG3 locus in islets from patients with type 2 diabetes is mimicked by targeted epimutation of its promoter with TALE‐DNMT constructs. Diabetes 2018; 67: 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin SP, Coan P, da Rocha ST, et al. Differential regulation of imprinting in the murine embryo and placenta by the Dlk1‐Dio3 imprinting control region. Development 2007; 134: 417–426. [DOI] [PubMed] [Google Scholar]
- 25. Metzger BE, Gabbe SG, Persson B, et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010; 33: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 1998; 15: 539–553. [DOI] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 28. Zhao BH, Jiang Y, Zhu H, et al. Placental delta‐like 1 gene DNA methylation levels are related to mothers’ blood glucose concentration. J Diabetes Res 2019; 2019: 9521510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barker DJP. The origins of the developmental origins theory. J Intern Med 2007; 261: 412. [DOI] [PubMed] [Google Scholar]
- 30. Bateson P, Barker D, Clutton‐Brock T, et al. Developmental plasticity and human health. Nature 2004; 430: 419–421. [DOI] [PubMed] [Google Scholar]
- 31. Serman L, Vlahović M, Sijan M, et al. The impact of 5‐azacytidine on placental weight, glycoprotein pattern and proliferating cell nuclear antigen expression in rat placenta. Placenta 2007; 28: 803–811. [DOI] [PubMed] [Google Scholar]
- 32. Liu Y, Murphy SK, Murtha AP, et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics 2012; 7: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bouchard L, Thibault S, Guay SP, et al. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010; 33: 2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 2008; 105: 17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. El Hajj N, Pliushch G, Schneider E, et al. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes 2013; 62: 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chelbi ST, Mondon F, Jammes H, et al. Expressional and epigenetic alterations of placental serine protease inhibitors: SERPINA3 is a potential marker of preeclampsia. Hypertension 2007; 49: 76–83. [DOI] [PubMed] [Google Scholar]
- 37. Finer S, Mathews C, Lowe R, et al. Maternal gestational diabetes is associated with genome‐wide DNA methylation variation in placenta and cord blood of exposed offspring. Human Mol Genet 2015; 24: 3021–3029. [DOI] [PubMed] [Google Scholar]
- 38. Tekola‐Ayele F, Zeng X, Ouidir M, et al. DNA methylation loci in placenta associated with birthweight and expression of genes relevant for early development and adult diseases. Clin Epigenet 2020; 12: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bouchard L, Hivert MF, Guay SP, et al. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes 2012; 61: 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Houde AA, Guay SP, Desgagné V, et al. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics 2013; 8: 1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang Y, Yu YC, Ding GL, et al. Intrauterine hyperglycemia induces intergenerational Dlk1‐Gtl2 methylation changes in mouse placenta. Oncotarget 2018; 9: 22398–22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Su R, Wang C, Feng H, et al. Alteration in expression and methylation of IGF2/H19 in placenta and umbilical cord blood are associated with macrosomia exposed to intrauterine hyperglycemia. PLoS One 2016; 11: e0148399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soubry A, Murphy SK, Wang F, et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. J Int J Obes 2015; 39: 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J, Lin Z, Gao Y, et al. Downregulation of long noncoding RNA MEG3 is associated with poor prognosis and promoter hypermethylation in cervical cancer. J Exp Clin Cancer Res 2017; 36: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sellers ZP, Bolkun L, Kloczko J, et al. Increased methylation upstream of the MEG3 promotor is observed in acute myeloid leukemia patients with better overall survival. Clin Epigenet 2019; 11: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. You L, Wang N, Yin D, et al. Downregulation of long noncoding RNA Meg3 affects insulin synthesis and secretion in mouse pancreatic beta cells. J Cell Physiol 2016; 231: 852–862. [DOI] [PubMed] [Google Scholar]
- 47. Qiu GZ, Tian W, Fu HT, et al. Long noncoding RNA‐MEG3 is involved in diabetes mellitus‐related microvascular dysfunction. Biochem Biophys Res Commun 2016; 471: 135–141. [DOI] [PubMed] [Google Scholar]
- 48. Che X, Deng X, Xie K, et al. Long noncoding RNA MEG3 suppresses podocyte injury in diabetic nephropathy by inactivating Wnt/β‐catenin signaling. PeerJ 2019; 7: e8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prats‐Puig A, Carreras‐Badosa G, Bassols J, et al. The placental imprinted DLK1‐DIO3 domain: a new link to prenatal and postnatal growth in humans. Am J Obstet Gynecol 2017; 217: 350.e1–350.e13. [DOI] [PubMed] [Google Scholar]
- 50. Gagné‐Ouellet V, Houde AA, Guay SP, et al. Placental lipoprotein lipase DNA methylation alterations are associated with gestational diabetes and body composition at 5 years of age. Epigenetics 2017; 12: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kk O, ML A, Pm E, et al. Association between postnatal catch‐up growth and obesity in childhood: prospective cohort study. BMJ 2000; 320: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol 2011; 31: 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Primer list for quantitative real‐time polymerase chain reaction.
Table S2 | Primer list for MethylTarget sequencing.
Table S3 | Methylation levels of individual CpG island in the maternally expressed gene 3 differentially methylated region of the placenta.