

Abstract
The prevalence of cardiovascular and metabolic disease coupled with kidney dysfunction is increasing worldwide. This triad of disorders is associated with considerable morbidity and mortality as well as a substantial economic burden. Further understanding of the underlying pathophysiological mechanisms is important to develop novel preventive or therapeutic approaches. Among the proposed mechanisms, compromised nitric oxide (NO) bioactivity associated with oxidative stress is considered to be important. NO is a short-lived diatomic signalling molecule that exerts numerous effects on the kidneys, heart and vasculature as well as on peripheral metabolically active organs. The enzymatic l-arginine-dependent NO synthase (NOS) pathway is classically viewed as the main source of endogenous NO formation. However, the function of the NOS system is often compromised in various pathologies including kidney, cardiovascular and metabolic diseases. An alternative pathway, the nitrate–nitrite–NO pathway, enables endogenous or dietary-derived inorganic nitrate and nitrite to be recycled via serial reduction to form bioactive nitrogen species, including NO, independent of the NOS system. Signalling via these nitrogen species is linked with cGMP-dependent and independent mechanisms. Novel approaches to restoring NO homeostasis during NOS deficiency and oxidative stress have potential therapeutic applications in kidney, cardiovascular and metabolic disorders.
Subject terms: End-stage renal disease, Renovascular hypertension, Type 2 diabetes, Glomerulus, Nephrons
Nitric oxide (NO) has important roles in the regulation of kidney, cardiovascular and metabolic functions. This Review discusses the physiological roles of NO and its effects on kidney function, as well as its association with cardiometabolic complications and novel approaches to restoring NO homeostasis.
Key points
Nitric oxide and other bioactive nitrogen species have pivotal roles in multiple physiological functions, including modulation of the kidney, cardiovascular and metabolic systems; in the kidney, nitric oxide has a crucial role in autoregulation and modulation of tubular transport.
Nitric oxide is classically derived from l-arginine-dependent nitric oxide synthases, but can also be formed via serial reduction of inorganic nitrate and nitrite, that is, the nitrate–nitrite–nitric oxide pathway.
The nitrate–nitrite–nitric oxide pathway can be boosted via the diet and is of particular importance in conditions where the activity of the nitric oxide synthase system is reduced, such as hypoxia, ischaemia or low pH.
Signalling via bioactive nitrogen species is linked with both cGMP-dependent and independent mechanisms.
Reduced nitric oxide bioactivity has been associated with ageing and kidney, cardiovascular and metabolic disorders, which are often coupled with oxidative stress.
Novel pharmacological and nutritional strategies that increase nitric oxide bioactivity and reduce oxidative stress could be potential therapies for preventing and treating kidney disease and associated cardiometabolic complications.
Introduction
The prevalence of cardiovascular disorders, including hypertension, and metabolic disorders such as type 2 diabetes mellitus (T2DM), is increasing worldwide. These disorders are closely coupled with the development and progression of kidney disease, which significantly increases patient morbidity and mortality1,2. The resulting societal economic burden is immense and further understanding of the underlying pathophysiological mechanisms is urgently needed to enable the development of novel preventive and therapeutic nutritional and pharmacological strategies2. The kidney, cardiovascular and metabolic phenotypes (that is, kidney disease, cardiovascular disease and T2DM) are interrelated, suggesting that this triad of disorders share common underlying pathological mechanisms. The exact causes of these disorders, the interactions between organ systems and the complex pathophysiological mechanism(s) that underlie the initiation, maintenance and progression of disease are complex and not fully understood.
Potential mechanisms that might contribute to the development of kidney disease, cardiovascular disease and T2DM include hyperglycaemia, altered lipid metabolism, low-grade inflammation, overactivity of the renin–angiotensin–aldosterone system (RAAS), increased sympathetic nerve activity and altered microbiota3–6. In addition, several studies have suggested a substantial contribution of increased generation of NADPH oxidase-derived and mitochondria-derived reactive oxygen species (ROS) and oxidative stress coupled with reduced nitric oxide (NO) bioactivity and endothelial dysfunction7–10. NO is a short-lived diatomic signalling molecule that exerts multiple effects on kidney, cardiovascular and metabolic functions, including modulation of renal autoregulation, tubular fluid and electrolyte transport, vascular tone, blood pressure, platelet aggregation, immune cell activation, insulin-glucose homeostasis and mitochondrial function. The classical view is that nitric oxide synthase (NOS) systems are the main source of endogenous NO formation. However, an alternative pathway exists whereby the supposedly inert oxidation products of NO, that is, inorganic nitrate and nitrite, undergo serial reductions to form NO and other closely related bioactive nitrogen oxide species11–13.
The important role of NO in the regulation of kidney, cardiovascular and metabolic functions in health and disease has led to substantial interest in the identification of methods to therapeutically modulate NO bioactivity. In this Review, I discuss the physiological roles of NO, the direct and indirect effects by which this molecule influences kidney function and its association with cardiometabolic complications. I also highlight novel approaches to restoring NO homeostasis during NOS deficiency with a focus on the alternative nitrate–nitrite–NO pathway, which can be boosted via dietary intake, particularly by eating green leafy vegetables.
The classical NO synthase systems
NO is endogenously generated by numerous cells throughout the body via three different NOS systems (Fig. 1)14–16. Neuronal NOS (nNOS; also known as NOS1) and endothelial NOS (eNOS; also known as NOS3) are constitutively expressed, whereas inducible NOS (iNOS; also known as NOS2) is mainly associated with inflammatory conditions16,17. l-Arginine, molecular oxygen, NADPH and tetrahydrobiopterin (BH4) are equally important substrates or co-factors that lead to equimolar generation of NO and l-citrulline18,19. In the endothelium, eNOS-derived NO has a central role in the regulation of blood flow and maintenance of endothelial integrity. The activity of eNOS and nNOS is regulated by intracellular calcium, which activates calmodulin. In turn, calmodulin binds and increases NOS enzyme activity. This process results in NO-mediated activation of soluble guanylate cyclase (sGC) and increased formation of cyclic GMP (cGMP), which activates cGMP-dependent protein kinases. This NO–sGC–cGMP signalling pathway mediates many of the effects of NO bioactivity on cardiovascular, kidney and metabolic functions20. However, other bioactive nitrogen oxide species formed by reactions of NO can induce other important physiological signalling pathways, including post-translational modifications of proteins, independent of cGMP signalling21–23 (Fig. 1). These bioactive nitrogen oxide species include mobile nitrosyl−heme (heme-NO)24, dinitrosyl–iron complexes25, S-nitrosothiols26, nitrogen dioxide (NO2)27, dinitrogen trioxide27, nitrosopersulfides28, nitroxyl29 and peroxynitrite30. Oxidation of NO to form the more stable anions nitrite and nitrate also provides more distant NO-like bioactivity.
Fig. 1. The NOS pathway and potential effects of NO on cardiovascular, renal and metabolic functions.

Nitric oxide (NO) is endogenously formed by three different nitric oxide synthase (NOS) isoforms: neuronal NOS (nNOS), inducible (iNOS) and endothelial NOS (eNOS). The activity of these enzymes is oxygen dependent and requires l-arginine and several co-factors (calmodulin, nicotinamide adenine dinucleotide phosphate (NADPH), tetrahydrobiopterin (BH4), flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN)). NO binds to the reduced heme site (Fe2+) of soluble guanylyl cyclase (sGC), which activates this enzyme, leading to the formation of the second messenger cyclic GMP (cGMP) from GTP. NO is a short-lived molecule that is oxidized in blood and tissues to form nitrite (NO3−), nitrate (NO2−) and other bioactive nitrogen species. NO bioactivity has been associated with numerous favourable effects in cardiovascular, renal and metabolic systems, mainly via cGMP-dependent mechanisms, although cGMP-independent mechanisms have also been reported. These mechanisms are multifactorial and involve modulation of protein function and immune cells, reductions in angiotensin II (Ang II) signalling, oxidative stress and sympathetic nerve activity and modulation of mitochondrial function. GFR, glomerular filtration rate.
NOS are also modulated via complex post-translational modifications, including acylation, nitrosylation, phosphorylation, acetylation, glycosylation and glutathionylation at various sites, as well as via protein–protein interactions and regulation of subcellular localization, which can increase or decrease their enzymatic activity31–33. Acute changes in nNOS and eNOS activity in the vasculature and the kidney are mainly modulated via post-translational mechanisms, whereas chronic changes in NO synthesis are regulated by altered eNOS or nNOS transcription and translation18,34,35. Activation of iNOS is associated with inflammatory processes and leads to significantly higher levels of NO than those that are produced by constitutive activation of other NOS isoforms. The resulting acute increases in NO have beneficial antimicrobial effects against bacteria, viruses and fungi. However, induction of iNOS is also associated with chronic low-grade inflammation in cardiovascular, metabolic and kidney disorders36.
The constitutive NOS systems are generally thought to be the main source of endogenous NO production and signalling under normal, healthy conditions, but are often dysfunctional in pathological conditions including cardiovascular disorders9 and chronic kidney disease (CKD)37,38. This dysfunction is associated with reduced NO bioactivity. The mechanisms that contribute to reduced NO formation and compromised signalling are multifactorial and include reduced NOS expression, limited substrate availability, uncoupling of NOS, elevated levels of endogenous NOS inhibitors such as asymmetric dimethylarginine and compromised signalling in states of oxidative stress due to direct scavenging by ROS or oxidation of the heme group in sGC (Fe2+ to Fe3+), which renders it insensitive to activation by NO9,39.
Kidney expression of NOS isoforms
Expression of all three NOS isoforms and sGC has been reported in the kidney, although some discrepancies exist regarding the expression levels along the nephron when comparing data from human and animal studies. Moreover, splice variants of nNOS and iNOS40,41 can influence their activity and function. In the macula densa, splice variants (α, β and γ isoforms) have been reported to have a substantial impact on the function of nNOS42, thus modulating renin secretion and autoregulatory mechanisms.
Expression of nNOS in the macula densa and eNOS in the kidney vasculature has been consistently documented; some but not all studies have also reported eNOS expression in tubular epithelial cells. In normal, healthy human kidney tissue samples, nNOS protein and mRNA expression was detected in most segments of the nephron, including the macula densa, proximal tubule, thick ascending limb (TAL) of the loop of Henle, distal tubule and collecting duct43. eNOS was only expressed in the endothelium and iNOS was not detected in any of the tested segments. The expression of NOS, which was confirmed using enzyme activity studies, was generally found to be higher in the cortex than in the medulla. Data from the Human Protein Atlas support these findings, indicating that nNOS is expressed in cortical tubules but not in glomeruli and that eNOS is expressed in glomeruli but not in tubules. Moreover, iNOS is expressed at low levels in tubules but not in glomeruli. Whether or not constitutive iNOS expression has a functional role in the healthy kidney is controversial, but a substantial body of evidence demonstrates increased iNOS expression and activity during pathological conditions associated with inflammation, such as ischaemia–reperfusion injury (IRI)44, ureteral obstruction45, lipopolysaccharide-induced endotoxemia or sepsis46 and CKD47,48.
The nitrate–nitrite–NO pathway
Redox reactions with other radicals and transition metals, such as those in heme proteins, rapidly metabolize NO (t1/2 ~ 0.05–1 s)49 to form other more stable nitrogen oxide species, including nitrite and nitrate50,51. As these anions are mainly excreted by the kidneys, the sum of their total urinary excretion (termed NOx) during a 24-h period has often been used to estimate whole-body NOS activity. However, circulating nitrate and nitrite can also be converted back to bioactive NO species via endogenous serial reduction, that is, the nitrate–nitrite–NO pathway11–13 (Fig. 2).
Fig. 2. The generation of bioactive NO in mammals.

Nitric oxide (NO) is classically viewed to be formed via the NO synthase (NOS) pathway but can also be generated via a fundamentally different mechanism, the nitrate (NO3−)–nitrite (NO2−)–NO pathway. During conditions of normal oxygen tension and pH, NO and other bioactive nitrogen species are oxidized to form inorganic nitrite and nitrate in the blood and tissues. Circulating NO3− and NO2− can be reduced back to NO and other bioactive nitrogen species via non-enzymatic and enzymatic systems. This alternative pathway of NO generation is of particular importance during low oxygen tension (that is, ischaemia and hypoxia) and acidic conditions. In addition to NOS-derived NO3−, which is formed following oxidation of NO, dietary inorganic nitrate is a major contributor to the pool of this anion in the body. In particular, green leafy vegetables and beetroot contain high levels of inorganic nitrate. Commensal oral bacteria are crucial for the reduction of NO3− to NO2−, whereas conversion of NO2− to NO occurs in the acidic milieu of the stomach and in the circulation as a result of non-enzymatic and enzymatic systems (for example, deoxyhaemoglobin (deoxy-Hb), deoxymyoglobin (deoxy-Mb), xanthine oxidoreductase (XOR) and mitochondrial complexes). eNOS, epithelial NOS; iNOS, inducible NOD; nNOS, neuronal NOS.
Moreover, dietary intake contributes substantially to the body pool of nitrate and nitrite52,53. Ingested nitrate that enters the circulation is actively taken up by the salivary glands and then concentrated and excreted in the saliva (this process is known as enterosalivary circulation of nitrate)54,55. Accumulating evidence shows that commensal bacteria in the oral cavity have a crucial role in the first step of the reduction of nitrate to nitrite56. In the acidic gastric milieu, swallowed nitrite is rapidly protonated and non-enzymatically forms NO and other nitrogen species with nitrosating and nitrating properties57. However, most of the swallowed nitrate/nitrite is rapidly and efficiently reabsorbed in the gastrointestinal system and enters the circulation53 where several non-enzymatic (deoxyhaemoglobin, deoxymyoglobin) and enzymatic systems (xanthine oxidoreductase (XOR), mitochondrial complexes and liver cytochromes) further reduce nitrite to NO58,59. Nitrate and nitrite can signal not only via the classical NO–sGC–cGMP pathway but also via nitration and nitros(yl)ation mechanisms that are mediated via other bioactive nitrogen species independently of sGC-cGMP signalling (Fig. 3). These bioactive nitrogen species can influence various cellular functions via modification of proteins, lipids, nucleosides, metals and transnitrosation/transnitrosylation.
Fig. 3. cGMP-independent signalling via bioactive nitrogen species.
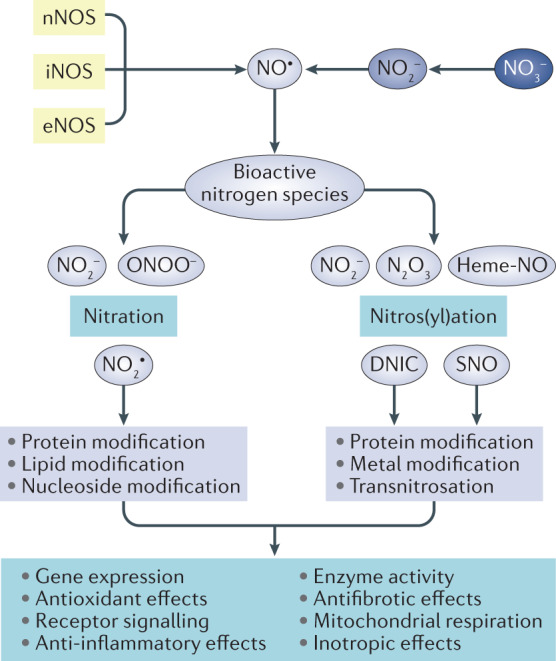
The nitric oxide synthase (NOS) systems and serial reductions of nitrate (NO3−) and nitrite (NO2−) lead to the formation of nitric oxide (NO•) and other bioactive nitrogen species. These species can undergo nitration or nitrosation/nitrosylation reactions independent of cyclic GMP (cGMP) signalling and modify proteins, lipids, nucleosides and metals as well as induce transnitration, which can alter gene expression, receptor signalling, enzyme activity and mitochondrial function and elicit antioxidant, anti-inflammatory, antifibrotic and inotropic effects. DNIC, dinitrosyl−iron complexe; eNOS, epithelial NOS; heme-NO, nitrosyl-heme; iNOS, inducible NOS; N2O3, dinitrogen trioxide; nNOS, neuronal NOS; NO2•, nitrogen dioxide; ONOO−, peroxynitrite; SNO, S-nitrosothiols.
In contrast to NOS-dependent NO generation, the nitrate–nitrite–NO pathway is oxygen independent and potentiates during conditions of low oxygen tension (that is, hypoxia and ischaemia) and low pH60–62. This effect can be explained by more efficient non-enzymatic reduction of nitrite by protonation under more acidic conditions63. During hypoxic conditions, enhanced activity of enzymes such as XOR and the formation of deoxyhaemoglobin also contribute to increased bioactivity of NO by facilitating the reduction of nitrite, and potentially also nitrate, to NO63,64. However, signalling by nitrate- and nitrite-derived bioactive species also occurs during normoxia in humans, as evidenced by blood pressure reduction and vasodilatation following treatment with nitrate and nitrite, respectively (discussed further below).
Role of NO in renal autoregulation
Renal autoregulatory mechanisms operate together to maintain relatively constant blood flow and glomerular filtration rate (GFR) despite variations in renal perfusion pressure over a wide range (80–180 mmHg). These mechanisms are crucial to prevent barotrauma65.
Myogenic response and tubuloglomerular feedback
Autoregulation is largely mediated by the myogenic response, macula densa-derived tubuloglomerular feedback (TGF) and their interactions65. Both mechanisms regulate pre-glomerular tone primarily via changes in the afferent arteriolar diameter, which is the effector site. Moreover, the tone and contractility of the afferent arterioles are modulated by the concentration and interaction of several endogenous vasoactive substances, including NO, angiotensin II (Ang II) and adenosine, within the juxtaglomerular apparatus, as well as by the activity of the sympathetic nervous system65–67.
The mechanisms that contribute to myogenic and TGF responses and their complex interactions in health, hypertension, kidney disease and diabetes, involve changes in NO and ROS signalling65. The myogenic response and TGF as well as their interaction are modulated by NOS-derived NO. The effects of non-selective and selective NOS inhibitors on renal autoregulation, mediated by the myogenic and TGF responses, have been assessed in various experimental models. In rat kidneys in vivo, the initial increase in renal vascular resistance during the first 5 s after an increase in perfusion pressure, which corresponds to the myogenic response, was greatly exaggerated in the setting of non-selective NOS inhibition68. However, no major effect of NOS inhibition was observed in the later phase (5–25 s) after an increase in perfusion pressure, corresponding to the TGF response. Another study in rats in vivo demonstrated that NOS inhibition reduced vascular conductance and augmented the myogenic response, as evidenced by a more abrupt reduction in vascular admittance gain (in the region corresponding to the myogenic response) and a steeper regression of admittance on frequency69. Moreover, selective inhibition of nNOS in the macula densa did not induce substantial vasoconstriction but did potentiate the myogenic response, suggesting interaction between the two autoregulatory responses. In rat hydronephrotic kidney preparations, which lack functional TGF, NOS inhibition had no effect on pressure-induced changes in afferent arteriole diameter (that is, the myogenic response)69. Ex vivo experiments using isolated and perfused single arterioles, showed no differences in arteriolar responses following increased perfusion pressure (that is, myogenic response) between vessels from eNOS knockout mice and wild-type controls70. Another study using in vitro blood-perfused juxtamedullary nephron preparations showed that inhibition of nNOS increased the arteriolar autoregulatory response to increased perfusion pressure71.
These findings clearly indicate an important role of NOS-derived NO in renal autoregulation. The contribution of eNOS versus nNOS in modulating myogenic responses is debated owing to differing findings depending on the experimental setting. However, the available data support a predominant role of macula densa nNOS-derived NO in dampening the speed and the strength of the myogenic response65. The exact cellular events by which NO attenuates afferent arteriolar vascular smooth muscle cell contraction during myogenic responses are incompletely understood65. NO, cGMP or its target protein kinase G (PKG; also known as PRKG1) and cyclic adenosine monophosphate or protein kinase A could dampen Ca2+ signalling or sensitivity, and thereby moderate arteriolar tone65,72, via multiple mechanisms, for example, by inhibiting voltage-operated calcium channels or transient receptor potential cation channels, by activating large-conductance calcium-activated potassium channels, by suppressing ADP-ribosyl cyclase activity and thus leading to reduced ryanodine receptor-mediated Ca2+ mobilization or by NO-mediated interaction and/or scavenging of ROS.
TGF mechanisms are largely activated by increased tubular sodium-chloride load at the macula densa, which increases the activity of the apical Na+-K+-2Cl− cotransporter (NKCC2; also known as SLC12A1) and in turn other tubular transporters, leading to ATP generation and/or metabolism and the formation of adenosine. The resulting activation of adenosine A1 (refs73,74) and/or purinergic P2 (ref.75) receptors on adjacent vascular smooth muscle cells stimulates calcium-dependent signalling and contraction of the afferent arteriole76 (Fig. 4). The available evidence suggests that nNOS is largely expressed in macula densa cells and has a functional role in the regulation of TGF and in at least the short-term regulation of volume homeostasis77. Early in vivo micropuncture studies in rats showed that local pharmacological inhibition of NOS in the macula densa was associated with decreased glomerular capillary pressure, indicating a sensitized and exaggerated TGF response78. This reduction in glomerular capillary pressure following NOS inhibition was abolished by simultaneous tubular administration of the NKCC2 blocker furosemide. Subsequent studies using different approaches (for example, ex vivo double microperfused JGA preparations79 and transgenic nNOS knockout mice80) provided further evidence that nNOS dampens TGF responses. Compromised nNOS function in the macula densa has been implicated in hypertension, kidney disease and diabetes81. Early experimental studies showed that spontaneously hypertensive rats and the Milan hypertensive strain of rats have abnormal nNOS function82,83 and that chronic inhibition of nNOS increased TGF sensitivity, reduced GFR and salt and water excretion and subsequently led to hypertension84. Although nNOS is expressed in the human kidney43, its functional role during renal autoregulation in health and disease is still a largely unexplored field.
Fig. 4. Effects of NO on sodium transporters in the nephron.

Nitric oxide (NO) is generally considered to inhibit tubular sodium reabsorption along the nephron. However, differing results have been obtained in acute and chronic conditions, in different experimental settings (in vivo versus ex vivo or in vitro) and in different species. Moreover, the effects of NO on tubular sodium (Na+) handling seem to be dependent on hormonal activity, particularly via interaction with the renin–angiotensin–aldosterone system. In the proximal tubule, neuronal NO synthase (nNOS) and endothelial NOS (eNOS)-derived NO has been reported to inhibit the basolateral sodium–potassium pump (Na+/K+-ATPase) and the apical sodium/hydrogen exchanger 3 (NHE3), as well as to modulate the activity of the basolateral Na+/HCO3− cotransporter. In the thick ascending limb (TAL) of the loop of Henle, eNOS-derived NO inhibits NHE3 and may also inhibit the apical Na+-K+-2Cl− cotransporter (NKCC2). eNOS-derived NO also inhibits NKCC2 in macula densa cells. Activation of nNOS in the macula densa can inhibit paracrine signalling mediated via adenosine triphosphate (ATP) and adenosine (ADO), which forms part of the tubuloglomerular feedback mechanism following activation of purinergic P2 and/or adenosine A1 receptors located on vascular smooth muscle cells in the afferent arteriole. nNOS expression has been demonstrated in the distal tubule but the potential effects of NO on specific transporters in this segment of the nephron (for example, the Na+/Cl− cotransporter) are currently not clear. Finally, in collecting duct cells, nNOS-derived NO can inhibit the epithelial sodium channel (ENaC).
Overall, the physiological significance of interactions between the vascular and tubular mechanisms that mediate autoregulation in the kidney remains elusive. These interactions are influenced by the balance of positive and negative modulators of vasomotor tone of afferent arterioles, which can be generated by macula densa and tubular cells. NO influences renal myogenic response and TGF as well as their interactions, but the primary source of NO generation is still debated.
Medullary blood flow and pressure natriuresis
The kidney medulla is perfused from cortical arterioles and the vasa recta capillary system of juxtamedullary nephrons. Measurement of medullary blood flow is considerably more complex than measurement of cortical blood flow, which might partly explain the variable results regarding the efficiency of medullary autoregulation in different studies and species. The descending vasa recta are surrounded by contractile pericytes that can generate a myogenic response85. Different autoregulatory responses have been described in human outer medullary descending vasa recta of different diameters. In large diameter segments, contractions were observed in response to increased luminal pressure, whereas no significant change was observed in those with a small diameter86. The same study showed concentration-dependent constriction of descending vasa recta in response to Ang II. NOS inhibition has also been shown to induce constriction of isolated rat descending vasa recta; this vasoconstriction could be reversed by an NO donor or by pharmacological inhibition of oxidative stress using a NOX inhibitor or a superoxide dismutase mimetic87.
Paracrine agents including NO, prostaglandins and ATP have been proposed to modulate medullary autoregulatory and pressure natriuretic responses. In rat juxtamedullary nephron preparations, inhibition of macula densa nNOS led to significant increases in afferent arteriolar myogenic contraction in response to increased perfusion pressure71,88,89. By contrast, stimulation of NO production in these nephron preparations dampened pressure-induced contraction of cortical radial artery and afferent arterioles by reducing autoregulatory responses.
Two main hypotheses exist regarding the interactions between renal autoregulation and pressure natriuresis in response to increased renal perfusion pressure with excellent autoregulation of cortical blood flow in the presence or absence of efficient autoregulation of medullary blood flow. The major difference between these hypotheses concerns the mediating factor(s) and the relative importance of a primary change in renal cortical NO generation versus a primary change in medullary blood flow65. In general, the slope of the natriuretic response to increased renal perfusion pressure is attenuated by inhibition of NOS. Moreover, increased RAAS activity, sympathetic nerve activity and excessive formation of ROS, especially in the kidney medulla, may inhibit pressure natriuresis.
Abnormal NO homeostasis coupled with increases in Ang II and ROS and anomalous renal autoregulation (either increased activity contributing to hypertension or decreased activity in the chronic state) have been demonstrated in experimental models of hypertension (for example, spontaneously hypertensive rat, Milan hypertensive strain of rat, Dahl salt-sensitive rat, Goldblatt renovascular hypertension, Ang II-induced hypertension, DOCA-salt hypertension, brown Norway rat), CKD (for example, reduced renal mass models) and T2DM (for example, obese Zucker diabetic rat and chronic high fat diet)65. Together, these studies suggest that augmented renal autoregulation (in particular TGF) may contribute to the development of hypertension, whereas decreased renal autoregulation can lead to both hypertension-induced and diabetes-induced nephropathies.
Modulation of sodium transport by NO
Sodium and water homeostasis is mainly regulated via the actions of hormones (that is, aldosterone and vasopressin) in the kidney as well as Ang II and endothelin signalling. However, other endogenous compounds that do not circulate at high levels, such as NO, contribute substantially to the renal handling of sodium and water via different mechanisms. In general, NO inhibits tubular sodium reabsorption along the nephron90,91; however, the acute and chronic actions of NO in specific tubular segments during health and disease warrant further investigation. In particular, the effects of NO on sodium and fluid reabsorption in proximal tubules are debated as interactions with Ang II and biphasic effects have been reported. These differing effects may be explained by different experimental settings, models and species differences92.
Given the short half-life of NO in vivo, its actions are mainly thought to be mediated via autocrine or paracrine signalling. However, NO might also act as an endocrine hormone93, potentially via heme-NO signalling24. As discussed above, NO in the kidney originates not only from eNOS in the vasculature but also from tubular epithelial nNOS and potentially iNOS during pathological conditions associated with inflammation. Early studies that used pharmacological approaches to investigate the specific role of NOS-derived NO on tubular function were sometimes difficult to interpret because non-selective inhibition of systemic NO generation increased blood pressure, reduced renal perfusion and impacted on renal autoregulatory mechanisms. However, subsequent studies using more selective pharmacological inhibitors or genetic knockout approaches demonstrated that NOS inhibition can reduce sodium and fluid excretion without inducing substantial haemodynamic changes65.
Along the nephron, approximately 67% of the filtered sodium load is reabsorbed in the proximal convoluted tubules; 25% in the TAL of the loop of Henle; 5% in the distal convoluted tubule, connecting tubule and initial collecting tubule; and 3% in the inner medullary collecting duct94. nNOS and/or eNOS-derived NO have been reported to inhibit the basolateral sodium-potassium-pump (Na+/K+-ATPase) in the proximal tubule, apical sodium/hydrogen exchanger 3 (NHE3; also known as SLC9A3) in the proximal tubule and TAL of the loop of Henle, apical NKCC2 in the TAL of the loop of Henle and apical epithelial sodium channel (ENaC) in the cortical collecting duct91,95 (Fig. 4). Although nNOS is expressed in the distal tubule, its potential role in modulating transporters in this part of the nephron (for example, the Na+/Cl− cotransporter (NCC; also known as SLC12A3)) is not clear.
Early studies that used systemic administration of NOS inhibitors support an inhibitory effect of NO on proximal tubular sodium reabsorption96,97. However, a 2014 study using isolated human proximal tubules demonstrated that Ang II dose-dependently stimulated proximal tubular sodium transport (as demonstrated by increased activity of NHE3 and the basolateral Na+–HCO3− cotransporter) via NO-CGMP-mediated phosphorylation of ERK98. This study also showed that treatment with the NO donor sodium nitroprusside reduced sodium transporter activity in mouse and rat proximal tubules, but had the opposite effect in human proximal tubules. Further investigation is needed to understand the reason for this discrepancy and to clarify if similar phenomena exist for other transporters and in other segments of the nephron. The effect of NO on tubular reabsorption could potentially be concentration dependent and involve interaction with regulatory hormonal systems such as the RAAS. Although the effects of NO on proximal tubular reabsorption is debated, NO clearly has an important role in kidney physiology and compromised NO bioactivity is associated with kidney disease and associated cardiovascular and metabolic disorders7,38,39.
Tubular handling of NO metabolites
As mentioned above, NO is rapidly metabolized to form nitrite and nitrate, which are mainly excreted by the kidneys. Although urinary excretion of NOx is often reported, this measurement mainly reflects nitrate, which is found in plasma at almost 1,000-fold higher concentrations and has a substantially longer half-life than nitrite (t1/2 ~6 h versus t1/2 ~30 min); thus, it is much more stable in urine. As diet is a major contributor to the pool of circulating nitrate and nitrite in the body, accumulated excretion of NOx can only be used to estimate NOS function during strict dietary restrictions. Moreover, kidney NOS activity might substantially influence the total excretion of NOx, at least during high salt intake. Among mice fed a high salt diet, those with deletion of nNOS in the collecting duct had approximately 50% lower NOx excretion than controls and developed hypertension99.
Early studies in young, healthy volunteers showed that only 60% of orally administered 15N-nitrate was excreted in the urine as nitrate within 48 h, with minimal amounts (0.1%) excreted in the faeces100,101. A small amount of 15N labelled nitrate was also excreted as ammonia or urea in the urine (2–4%) and faeces (0.2%), but the handling and/or removal of the remaining dose (36–38%) is still not clear. Some nitrate is likely distributed to the muscle pool and elimination from the body via exhalation of nitrogen gas is also possible.
A small clinical study showed that healthy volunteers with normal kidney function (eGFR >60 ml/min/1.73 m2) had significantly higher fractional excretion of nitrate (median 16.3%; 95% CI 8.7–22.8) than patients with CKD and eGFR ≤30 ml/min/1.73 m2 (median 10.3%, 95% CI 96.9–4.4)102. In patients with CKD, renal nitrate clearance correlated positively with kidney function. Reduced fractional excretion of nitrate in patients with reduced eGFR was associated with increased plasma nitrate levels. These findings might be explained by altered glomerular filtration and tubular handling of nitrate during kidney disease, but could also be related to reduced NOS-derived bioactivity in patients with CKD, leading to reduced production of oxidized NO metabolites in the circulation to which the kidneys might adapt by reabsorbing more or secreting less nitrate.
A randomized controlled trial that investigated sex differences in renal nitrate handling in adults (n = 231) with elevated blood pressure reported that during dietary nitrate restriction, urinary nitrate concentration, amount of nitrate excreted, renal nitrate clearance and fractional excretion of nitrate were significantly lower in women than in men103. However, no association was observed between plasma nitrate concentration or fractional excretion of nitrate and GFR in either sex. Following high dietary nitrate intake for 5 weeks, fractional excretion of nitrate markedly increased and no sex differences in renal handling of nitrate were observed. This study suggests that tubular nitrate reabsorption might be higher in women than in men, but the underlying mechanisms warrant further investigation.
In the absence of intrarenal generation, the fractional excretion of nitrate correlates linearly with plasma levels and has been calculated to be approximately 3−10% in anesthetized dogs and rats, with major reabsorption taking place in the proximal tubules104,105. In healthy volunteers, inhibition of carbonic anhydrase using acetazolamide lowered proximal tubular reabsorption of nitrite and nitrate and increased their content in the urine, suggesting a role of carbonic anhydrase-dependent mechanisms in this reabsorption106. Evidence suggests that nitrate reabsorption also takes place in later segments of the nephron; clearance and stop-flow studies in dogs showed that inhibition of NKCC2 with furosemide reduced the tubular reabsorption of nitrate from 97% to 87% during inhibition of intrarenal NOS and from 90% to 84% without NOS inhibition107.
Another possible candidate for nitrate reabsorption is the chloride–bicarbonate exchanger pendrin (also known as SLC26A4), which is expressed in intercalated cells in the distal convoluted tubule, the connecting tubule and the cortical collecting duct108. In vitro studies have shown that pendrin expression is reduced in mouse cortical collecting ducts and connecting tubules in the presence of NO donors, and upregulated during inhibition of NOS109. Sialin (also known as SLC17A5) transports nitrate from the plasma into the salivary glands54. High apical expression of sialin has been reported in distal tubule cells110, suggesting that this transporter might also contribute to renal reabsorption of nitrate.
Most of the current knowledge regarding the tubular handling of nitrate and nitrite is based on the excretion of nitrate. Both of these anions are freely filtered in the glomeruli but whether similar tubular transport mechanisms exist for nitrate and nitrite along the nephron is unknown. Further studies are needed to identify nitrate and nitrite transporters in the human kidney. Such studies would not only advance understanding of the nitrate–nitrite–NO pathway in health and disease, but could potentially lead to novel therapeutic strategies.
Approaches to restoring NO bioactivity
Despite several decades of research focused on understanding NO biology and developing novel tools to increase the bioactivity of this signalling molecule in various disorders, particularly in the cardiovascular system9,39, the number of approved clinical applications is limited. Four main approaches could potentially increase NO bioactivity (Fig. 5). First, increasing or restoring endogenous NOS activity, for example, by supplementation with l-arginine, l-citrulline or BH4; inhibiting arginase activity; lowering endogenous levels of NOS inhibitors; stimulating hydrogen sulfide (H2S) formation; or using drugs such as statins, ACE2 activators, type-2 angiotensin II receptor (AT2) agonists and Mas receptor agonists111 that might dampen oxidative stress and facilitate eNOS activation. Second, giving substances that directly increase NO generation independently of the NOS system, for example, inhaled NO gas or inorganic nitrite, hybrid drugs that attach a NO-releasing moiety to an existing pharmacological agent, increasing H2S signalling, treatment with organic nitrates or supplementation with inorganic nitrate or nitrite. Third, limiting NO metabolism, for example, by dampening oxidative stress and thereby preventing scavenging of NO, and fourth, facilitating downstream signalling pathways, for example, using phosphodiesterase inhibitors, sGC stimulators or sGC activators9,39. Some of the existing and promising future approaches to increasing NO generation and signalling are discussed below.
Fig. 5. Strategies to restore NO bioactivity.

Endogenous nitric oxide (NO) bioactivity originates from the classical NO synthase (NOS) pathway and the alternative nitrate (NO3−)–nitrite (NO2−)–NO pathway. Signalling via NO and other bioactive nitrogen species involves soluble guanylate cyclase (sGC)-dependent formation of cyclic GMP (cGMP) as well as cGMP-independent effects. Kidney, cardiovascular and metabolic disorders are associated with reduced NO production and/or signalling, which might result from increased production of reactive oxygen species (ROS) from NADPH oxidase (NOX), mitochondria or uncoupled eNOS owing to substrate or co-factor deficiency. Several approaches might increase NO bioactivity. First, increasing or restoring endogenous NOS activity by supplementing with l-arginine, l-citrulline or tetrahydrobiopterin (BH4), inhibiting arginase or blocking the endogenous NOS inhibitor asymmetric dimethylarginine (ADMA). Statins, novel modulators of the renin–angiotensin–aldosterone system (RAAS) and compounds that increase hydrogen sulfide (H2S) formation or signalling might also restore NOS activity. Second, inhalation of NO or inorganic NO2−, treatment with organic nitrates that directly or indirectly increase NO generation independent of the NOS system or dietary supplementation with inorganic nitrate. Compared with inhalation of NO and organic nitrates, the dietary approach, using inorganic nitrate, has a more favourable pharmacokinetic and pharmacodynamic profile and is associated with lower risk of tolerance and adverse effects. Third, limiting NO metabolism, for example, by reducing the generation of ROS and thereby preventing scavenging of NO, for example, using novel antioxidants and NOX inhibitors. In addition, angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor (AT1) antagonists, which block angiotensin II (Ang II) formation and signalling, respectively, might reduce mitochondrial and NOX-derived ROS generation. Another novel approach is to oppose classical Ang II signalling by targeting the ACE2−neutral endopeptidase (NEP)−Ang(1−7)−AT2−Mas receptor pathway using an ACE2 stimulator, AT2 receptor agonist or Mas receptor agonist. Activation of this pathway has been associated with increased eNOS function. Finally, NO signalling can be facilitated by modulating downstream signalling pathways using phosphodiesterase 5 (PDE5) inhibitors, which inhibit the breakdown of cGMP, sGC modulators (both stimulators and activators) or compounds that increase H2S signalling. R-ONO2, nitrate ester.
Inhaled NO gas
Since the FDA approval of inhaled NO for the treatment of persistent pulmonary hypertension in neonates in 1999, this approach has been used off-label in various clinical settings112. Concerns exist regarding chronic use of inhaled NO, especially in patients with multiple-organ failure, owing to the risks of methaemoglobin formation (due to binding of NO to haemoglobin, which reduces its oxygen-carrying capacity) and development of kidney dysfunction. A systematic review and meta-analysis of randomized trials showed that NO inhalation therapy increased the risk of acute kidney injury (AKI) in patients with acute respiratory distress syndrome (ARDS) but not in non-ARDS populations113. The underlying mechanisms likely involve modulation of pre- and post-glomerular arteriolar resistance and altered tubular handling of salt and water, which is supported by previous animal and human studies113. Kidney function and markers of AKI should therefore be closely monitored in patients who require inhaled NO therapy.
Organic nitrates
Nitroglycerin (also known as glyceryl trinitrate) dilates venous capacitance vessels, aorta, medium-to-large coronary arteries and collaterals. This organic nitrate and structurally similar compounds were used to treat angina, acute myocardial infarction and severe hypertension even before the discovery of the role of NO in physiology114. Chronic use of organic nitrates has been associated with tolerance and risk of adverse effects, including hypotension and endothelial dysfunction114, which limit their therapeutic applications.
Arginase inhibition
The NOS isoforms compete for L‑arginine with two other enzymes, arginase and arginine methyltransferase, which convert L‑arginine into urea and L‑ornithine or asymmetric dimethylarginine (ADMA), respectively. ADMA in turn inhibits NOS activity by directly competing with L‑arginine for binding to NOS, leading to NOS uncoupling115. Two isozymes of arginase exist; arginase 1 is primarily located in the cytoplasm of hepatocytes and red blood cells116, whereas arginase 2 is located in the mitochondria of several tissues in the body, with high abundance in the kidney (Human Protein Atlas). Increased arginase activity and elevated ADMA levels, together with reduced NO synthesis, have been associated with endothelial dysfunction and increased cardiovascular risk in patients with CKD38,117,118. Moreover, arginase inhibition has been shown to improve microvascular endothelial function in patients with coronary artery disease and T2DM119,120.
Experimental studies have shown that dietary inorganic nitrate can decrease arginase expression and activity, which may contribute to the salutary effects of nitrate in cardiovascular and metabolic disease121,122. Increased arginase 2 expression and activity have been associated with kidney failure, diabetic kidney disease (DKD) and hypertensive nephropathy, and favourable effects of arginase inhibition have been demonstrated in kidney disease models38,118. Further studies are required to investigate the potential clinical benefits of attenuating arginase function in patients with kidney disease.
H2S formation and signalling
The signalling molecule H2S has many similarities with NO and affects a wide range of physiological functions, including modulation of cardiovascular, renal and metabolic systems123–125. H2S is formed endogenously in most organs, including the kidney, via enzymatic and non-enzymatic reactions124. Stimulation of H2S production might enhance the NO–sGC–cGMP–PKG pathway by increasing NO production and its downstream signalling. H2S can also increase eNOS activation via mechanisms that involve mobilization of intracellular Ca2+ and promotion of phosphorylation126,127. In addition, H2S might increase NO production independent of NOS via stimulation of XOR-dependent reduction of nitrite to NO128. H2S has also been shown to activate sGC and/or directly increase cGMP levels via inhibition of phosphodiesterase129. The interactions and crosstalk that occur between the NO and H2S signalling systems are complex and involve formation of S/N-hybrid species130. Treatment with slow-releasing H2S donors is associated with protective effects in animal models of cardiovascular, kidney and metabolic diseases123–125, but these results await further clinical translation.
Phosphodiesterase inhibition
cGMP is hydrolysed to guanosine monophosphate (GMP) by phosphodiesterase. To date, phosphodiesterase 5 (PDE5), which is expressed in various tissues including the cardiovascular and renal systems, has been the main focus of research, but other phosphodiesterase isozymes have also been suggested to modulate NO-mediated cGMP-dependent and independent signalling.
PDE5 inhibitors block cGMP breakdown and thereby lead to increased or prolonged NO signalling. These compounds have been proven to lower blood pressure in preclinical and clinical studies and to exert kidney and cardiovascular protective effects in various experimental models of IRI, heart failure131, CKD and DKD132. PDE5 is highly expressed in the kidney (in the glomeruli, mesangial cells, cortical tubules and inner medullary collecting duct) and the kidney-protective effects of PDE5 inhibitors are thought to extend far beyond their antihypertensive effect132. In 5/6 nephrectomized rats, 8 weeks of treatment with a PDE5 inhibitor initiated immediately after nephrectomy prevented the development of hypertension and ameliorated kidney injury and proteinuria133. However, this profound kidney protection was lost if PDE5 inhibition was initiated at a later stage (that is, 4 weeks after nephrectomy) when proteinuria was already evident.
PDE5 inhibitors are currently clinically approved for the treatment of pulmonary hypertension, erectile dysfunction and lower urinary tract symptoms134. However, promising preclinical and early clinical findings suggest that additional therapeutic indications could be possible in the future. For example, a phase II trial demonstrated that once daily treatment with a long-acting PDE5 inhibitor for 12 weeks decreased albuminuria in 256 patients with T2DM and overt nephropathy135. Importantly, this kidney-protective effect was observed despite simultaneous treatment with RAAS blockers and independent of any changes in blood pressure or GFR.
Modulation of sGC
Small compounds that target sGC are currently used to treat patients with pulmonary hypertension, but novel sGC stimulators and activators might have therapeutic value in other cardiovascular and kidney disorders136,137. For example, clinical trials have demonstrated favourable effects of these agents in patients with heart failure with preserved ejection fraction138 or heart failure with reduced ejection fraction139. In various preclinical models of kidney disease, including hypertensive and diabetic nephropathy, unilateral ureteral obstruction and acute glomerular nephritis, treatment with sGC stimulators and activators has consistently been associated with kidney-protective effects, as evidenced by improved creatinine clearance, reduced proteinuria and albuminuria, attenuated glomerulosclerosis and tubulointerstitial fibrosis, reduced infiltration of macrophages and preservation of podocyte health140. These favourable effects seem to be largely independent of any blood pressure reduction and hence may be of therapeutic value in patients with CKD who are treated with RAAS blockers. In vitro mechanistic studies have indicated that many of the antifibrotic effects of sGC stimulators and activators are coupled with cGMP-mediated suppression of transforming growth factor-β (TGFβ)–phospho-SMAD3 signalling141. Clinical trials are needed to extend these exciting preclinical findings to patients.
A randomized, double-blind, placebo-controlled, phase II study (NCT03217591) evaluated the safety and efficacy of the sGC stimulator praliciguat (IW-1973) in 140 patients with T2DM and albuminuria treated with RAAS inhibitors142. Treatment with praliciguat was not significantly associated with a reduction in albuminuria from baseline to week 8 and week 12 (the primary efficacy outcome) compared with placebo (P = 0.17). However, differences in some of the exploratory end points, including reduction in blood pressure and metabolic variables, such as haemoglobin A1c and cholesterol, favoured praliciguat treatment and support further investigation of this agent in DKD.
ACE2 activators, AT2 agonists and Mas receptor agonists
The identification and characterization of new components and pathways has led to extensive revision of the classical view of the RAAS during the past 15 years143,144. In addition to inhibiting angiotensin-converting enzyme (ACE), Ang II type 1 receptor (AT1) or the mineralocorticoid receptor, potential therapeutic approaches include stimulating ACE2 or activating AT2 or the Mas receptor. In contrast to ACE, which converts Ang I to Ang II, ACE2 convert Ang I and Ang II to Ang(1–7), which activates the Mas receptor. Ang II-mediated signalling via AT1 is associated with vasoconstriction and elevation of blood pressure, whereas activation of AT2 is associated with vasodilation and reduction of blood pressure. Accumulating evidence suggests that ACE2 activators, AT2 agonists and Mas receptor agonists have protective effects in models of cardiovascular, kidney and metabolic diseases via mechanisms that involve lowering oxidative stress, dampening inflammation and increasing NO bioactivity143,144. These preclinical findings suggest novel therapeutic strategies (Fig. 5).
Notably, cardiovascular145, kidney146 and metabolic147 diseases have been associated with increased risk of severe COVID-19 and adverse outcomes following infection with SARS-CoV-2. The virus uses ACE2 to enter host cells, which negatively impacts ACE2 function and might increase oxidative stress, reduce NO formation and signalling, alter immune cell function, impair endothelial function and induce coagulopathy148,149. Therapeutic strategies that restore ACE2−Ang(1–7)–Mas receptor signalling pathways and inhalation of NO have therefore been suggested as potential treatments for COVID-19 (refs148,150).
Inorganic nitrate and nitrite supplementation
Stimulation of the nitrate–nitrite-NO pathway via supplementation with inorganic nitrate or nitrite, either via the diet or in pill form, might enhance NO bioavailability. This approach has potential therapeutic applications in cardiovascular, metabolic and kidney disorders. Moreover, inhaled inorganic nitrite therapy has been shown to be safe and potentially efficacious in adult patients with pulmonary hypertension, heart failure with preserved ejection fraction151.
Cardiovascular effects
Considerable research efforts have focused on the cardiovascular effects of inorganic nitrate supplementation, including the effects on blood pressure, endothelial function and arterial stiffness. In 2006, the first study to report a blood pressure-lowering effect of nitrate supplementation in healthy adults showed that 0.1 mmol/kg per day of sodium nitrate reduced diastolic blood pressure by a mean of almost 4 mmHg (ref.152). A subsequent study that used an approximately threefold higher dose of dietary nitrate, in the form of beetroot juice, demonstrated a more pronounced blood pressure-lowering effect (i.e. mean reductions in systolic blood pressure of 10.4 mmHg and diastolic blood pressure of 8 mmHg), together with vasoprotective and antiplatelet properties of nitrate153. Several research groups have since confirmed the blood pressure-lowering effect of nitrate in healthy individuals. In two meta-analyses of these studies, systolic blood pressure was reduced by means of 4.1–4.8 mmHg and diastolic blood pressure was reduced by means of 1.7–2.0 mmHg (refs154,155). One of these meta-analyses also analysed the effect of nitrate intake on other cardiovascular risk factors and reported improved endothelial function, reduced arterial stiffness and reduced platelet aggregation with this intervention155.
Numerous experimental studies using various cardiovascular disease models, which are often associated with kidney and metabolic dysfunction, have also demonstrated favourable cardiovascular effects following treatment with inorganic nitrate, including antihypertensive effects and improved endothelial function8,156,157. The underlying mechanisms that contribute to such effects involve various organ systems and modulation of the RAAS, inhibition of arginase, restoration of eNOS function, dampening of sympathetic hyperactivity and anti-inflammatory and anti-oxidative effects156.
A study in 15 patients with hypertension demonstrated that acute dietary nitrate intake improved endothelial function and significantly lowered systolic and diastolic blood pressure158. The maximum blood pressure-lowering effect was observed approximately 3−4 h after nitrate ingestion, when plasma nitrite levels peaked, and the effect lasted for as long as 24 h. The first two studies investigating the cardiovascular effects of chronic nitrate supplementation in patients with hypertension were conducted independently of each other in 2015 and generated conflicting results159,160. One study showed no significant reduction in blood pressure following 1 week of nitrate supplementation, whereas the other showed a sustained blood pressure reduction following daily dietary intake of nitrate compared with placebo during a 4-week period, without any signs of tachyphylaxis160. A 2020 study showed that daily intake of nitrate for 5 weeks, in the form of either leafy green vegetables or a nitrate pill, did not significantly lower blood pressure in adults with pre-hypertension or stage 1 hypertension compared with intake of a low-nitrate control diet161. These differing findings are unlikely to be due to differences in the daily dose of nitrate, which was similar in all three studies (approximately 0.1 mmol/kg/day), and cannot be explained by patient age, body mass index or gender. However, differences in the number of simultaneous antihypertensive drugs, patient demographics, nitrate intake in the placebo group and blood pressure at the time of initiation of nitrate supplementation might be contributing factors. Another clinical trial showed that once-daily nitrate supplementation (approximately 0.1 mmol/kg/day) for 6 weeks improved vascular function in patients with hypercholesterolaemia, which was associated with a mild reduction in blood pressure162. Potential effects of nitrate supplementation on metabolic and/or kidney functions were not reported in these clinical studies.
Arterial stiffness is associated with aging and is a major risk factor for cardiovascular events such as myocardial infarction and stroke. A systematic review and meta-analysis of randomized controlled trials was conducted to estimate the effects of repeated nitrate administration (at least 3 days) on peripheral and central blood pressure and arterial stiffness in healthy individuals and in patients at increased risk of cardiovascular disease owing to obesity, hypertension, peripheral artery disease, hypercholesterolaemia and/or heart failure163. Pooled data from 45 studies using approximately 500 mg nitrate per day showed significant reductions in systolic (mean −2.91 mmHg) and diastolic blood pressure (mean −1.45 mmHg). Analysis of data from three trials that measured central (aortic) blood pressure also showed significant reductions with nitrate supplementation (mean systolic −1.6 mmHg, mean diastolic −2.0 mmHg). However, the meta-analysis found no significant differences in the effects of nitrate supplementation on blood pressure between subgroups of patients with differing health status. Notably, the reductions in blood pressure with nitrate supplementation in this meta-analysis, and in meta-analyses of data from healthy individuals154,155, are comparable with those observed in trials of reduced sodium intake and of a Dietary Approaches to Stop Hypertension (DASH) diet164,165. Analysis of data from seven trials that measured augmentation index and pulse wave velocity showed no significant effects of nitrate administration on arterial stiffness163. However, the researchers note that the number of available trials in individuals with additional cardiovascular disease risk factors (i.e. hypertension, diabetes or hyperlipidaemia) was relatively small and their analysis was likely underpowered to detect significant differences. More studies are required to draw reliable conclusions in these patient groups.
Overall, the current evidence for a long-term favourable cardiovascular effect of nitrate supplementation in patients with cardiovascular disease, including hypertension, is inconclusive. Hence, additional large clinical trials with different doses of nitrate would be desirable.
Metabolic effects
Impaired metabolic control with obesity and hyperglycaemia is closely coupled with increased risk of DKD, which involves complex glomerular and tubular mechanisms166,167. In addition to the well-documented therapeutic benefits of ACE inhibitors and Ang II receptor blockers in patients with kidney disease168,169, large clinical trials have shown that treatment with sodium/glucose co-transporter-2 (SGLT2) inhibitors can reduce albuminuria, risk of CKD progression and cardiovascular events in patients with T2DM and kidney disease170. The favourable effects of SGLT2 inhibition are unlikely to be solely mediated by improved glycaemic control. Experimental evidence suggests that they are likely the result of various glomerulotubular mechanisms167,171 such as modulation of the myogenic response and TGF as well as tubular reabsorption and potential modulation of renal sympathetic nerve activity172. These mechanisms could potentially also indirectly affect NO bioactivity.
Mice that lack eNOS develop hypertension173 and features that resemble metabolic syndrome (i.e. hypertension, dyslipidaemia, insulin resistance and obesity)174. In addition, eNOS deficiency in rodents is associated with kidney injury175–177 and accelerated progression of CKD178,179. Almost a decade ago, supplementation with dietary doses of nitrate was demonstrated to reverse features of metabolic syndrome in mice that lacked eNOS180. Numerous experimental studies have since confirmed that nitrate supplementation has favourable metabolic effects, which involve modulation of mitochondrial function and oxidative stress, activation of AMP-activated protein kinase (AMPK) signalling and modulation of downstream targets including sterol regulatory element-binding protein 1, acetyl-CoA carboxylase, medium-chain specific acyl-CoA dehydrogenase, mitochondrial and peroxisome proliferator-activated receptor-γ coactivator 1α7,181–184. A link between nitrate and/or nitrite supplementation and AMPK activation has also been demonstrated in experimental models of heart failure with preserved ejection fraction182 and IRI of the heart185 as well as in studies of the potential beneficial effects of this supplementation on longevity186. Knowledge of the specific mechanism(s) of AMPK activation in different cell types (for example, hepatocytes, adipocytes, skeletal muscle cells and cardiomyocytes) is limited, but studies have suggested involvement of nitrate and/or nitrite-mediated modulation of energy sensing pathways, including inhibition of target of rapamycin186, activation of sirtuin 3 (ref.182) and PKA, and modulation of mitochondria-derived ROS7,185.
Experimental evidence suggests that supplementation with nitrate might be a novel, safe and inexpensive therapeutic approach for patients with T2DM at an increased risk of developing DKD8,181,187. To date, few clinical trials have tested the potential beneficial effects of nitrate in patients with T2DM. A small trial in 27 patients with T2DM showed no significant effect of 2 weeks of nitrate supplementation (250 ml beetroot juice daily) on cardiometabolic functions (that is, blood pressure, endothelial function and insulin sensitivity)188. Another study that investigated the long-term metabolic effects of low-dose nitrate supplementation (250 mg per day for 24 weeks) in patients with T2DM found no significant difference in glycaemic control between the nitrate (n = 35) and placebo groups (n = 29)189. The reason for this lack of effect in these two studies, which contrasts with substantial experimental evidence, might be the fact that almost all the participants were receiving metformin treatment, which is known to activate AMPK190. In a mouse model of cardiometabolic disease, no additional beneficial effects on cardiovascular and metabolic parameters were observed when dietary nitrate supplementation was given in combination with metformin191, suggesting similar mechanisms of action. A phase II study that investigated the cardiometabolic effects of nitrite therapy (40 mg, three times daily) for 12 weeks in adults with stage 1–2 hypertension, metabolic syndrome and normal kidney function who were not receiving any medications that affect glucose metabolism showed that nitrite gradually lowered blood pressure during the first 8 weeks of treatment (by approximately −10 mmHg), but blood pressure levels started to return to baseline after 10–12 weeks192. Hyperinsulinaemic–euglycaemic clamp studies suggested that nitrite supplementation resulted in a trend towards decreased endogenous glucose production and improved insulin sensitivity. Strikingly, a significant improvement in carotid intima media thickness and brachial artery endothelial function was observed after 12 weeks of nitrite therapy.
Kidney effects
Patients with CKD and those with kidney failure have compromised NOS function, reduced NO bioactivity38,193 and increased cardiovascular morbidity and mortality. Moreover, a positive association between renal nitrate clearance and kidney function has been observed in patients with CKD102. Studies in adult and paediatric patients with kidney failure have shown that peritoneal dialysis and haemodialysis sessions are associated with disturbed NO homeostasis, measured as a reduction in the circulating levels of nitrate, nitrite and cGMP (a marker of NO signalling)194–197. Clinical studies are needed to investigate the therapeutic value of restoring NO homeostasis, using nitrate and/or nitrite supplementation, in these vulnerable high-risk patients.
In numerous experimental studies, chronic treatment with inorganic nitrate and nitrite has been associated with therapeutic effects such as attenuation of kidney injury and preservation of kidney blood flow and GFR in models of kidney disease with or without coexistent hypertension and metabolic disease8,181, including models with chronic pharmacological inhibition of NOS177, unilateral nephrectomy combined with a high-salt diet198, two-kidney one clip, deoxycorticosterone acetate salt, Ang II infusion199,200, ageing201 and kidney IRI202,203. Based on these studies, several mechanisms have been proposed to contribute to the favourable effects of nitrate and nitrite supplementation. These include dampening of oxidative stress via a reduction in NADPH oxidase activity, increased antioxidant capacity of superoxide dismutase, increased NO bioactivity, a reduction in Ang II sensitivity and type I angiotensin II receptor expression in the renovascular system, dampening of renal sympathetic nerve activity and modulation of immune cell phenotypes and mitochondrial function8.
In healthy adults, acute intravenous infusion of nitrite dose-dependently (0.58–5.21 μmol/kg/h) reduced blood pressure compared with placebo, but had no significant effect on GFR measured using 51Cr-EDTA clearance204,205. This blood pressure response was augmented in hypertensive compared with normotensive individuals (mean decreases in systolic blood pressure of 17 mmHg vs 10 mmHg). The researchers showed that the reduction in blood pressure was associated with a decrease in urinary levels of ENaCγ and aquaporin 2, but the effects of nitrite infusion on fractional sodium excretion were inconsistent (that is, unchanged, decreased or increased)204–206. In healthy individuals, the nitrite-mediated effects were not associated with changes in plasma or urine cGMP levels and were not significantly affected by simultaneous inhibition of XOR, ACE or carbonic anhydrase206. Moreover, dietary nitrate supplementation (approximately 0.1 mmol/kg/day) for 1 week did not significantly change eGFR (measured using creatinine clearance) compared with placebo in healthy young men207.
A systematic review and meta-analysis that investigated various modifiable lifestyle factors showed that higher vegetable intake significantly reduced the risk of CKD208. To what extent this effect might be linked to increased intake of nitrate is unknown. To date, no placebo-controlled clinical trial has investigated the effects of chronic nitrate supplementation in patients with kidney disease. However, a crossover study in patients with CKD (stages 2–4) due to hypertensive or diabetic nephropathies showed a significant reduction of blood pressure and renal resistive index 4 h after a single dose of nitrate (300 mg)209. Moreover, a prospective study with a follow-up period of almost 6 years concluded that a habitually high intake of nitrate and/or nitrite from dietary sources was independently associated with a significantly reduced risk of hypertension and CKD210.
Taken together, clinical studies have demonstrated that nitrate supplementation is associated with the lowering of blood pressure, which seems to be more pronounced in patients with hypertension. In healthy individuals this effect is not associated with significant changes in kidney function, whereas favourable effects on renal haemodynamics were observed in patients with CKD. Future longer-term, placebo-controlled, randomized trials are needed to determine if supplementation with inorganic nitrate and/or nitrite to restore NO bioactivity could be a beneficial additive treatment to slow the progression of kidney disease and associated cardiovascular and metabolic disorders. Such effects have consistently been reported in experimental studies.
Conclusions and future perspectives
Several decades have passed since the discovery of NO as the elusive endothelium-derived relaxing factor, but some controversies still exist regarding its formation and the true identity of the signalling molecule, as well as its downstream signalling and effector sites in health and disease. NO and other bioactive nitrogen oxide species have pivotal roles in multiple physiological functions, including modulation of the kidney, cardiovascular and metabolic systems. NO is classically derived from l-arginine-dependent NOS isoforms, but can also be formed endogenously via serial reduction steps of inorganic nitrate and nitrite. This nitrate–nitrite–NO pathway, which can be boosted via the diet, is of particular importance in conditions where the activity of the NOS system is reduced or non-functional (that is, hypoxia, ischaemia and low pH). Downstream signalling and functional effects are linked with both cGMP-dependent and -independent mechanisms. Reduced NO bioactivity due to compromised NO generation or increased metabolism has been associated with aging and kidney, cardiovascular and metabolic disorders, which are often coupled with increased generation of ROS leading to oxidative stress. In the kidney, NO is crucially involved in autoregulation and modulation of tubular transport, which may be of importance in the development and progression of hypertension, CKD, ischaemia–reperfusion injury and DKD. Although several experimental studies have demonstrated favourable effects of nitrate and nitrite supplementation on kidney disease and associated complications, these results await further clinical translation. Existing and future novel strategies that increase NO bioactivity and reduce oxidative stress, via both pharmacological and nutritional approaches, may have therapeutic potential to prevent and treat kidney disease and associated cardiometabolic complications. One of the challenges with such novel drug candidates is to optimize their spatial and temporal delivery to achieve the desired effects without any unwanted disturbances in normal physiological redox signalling.
Acknowledgements
M.C. sincerely thanks all co‑authors of the original articles from their groups, which are highlighted in this Review. A special thanks to E. Weitzberg and J. O. Lundberg (Karolinska Institutet) for valuable comments on the manuscript, and to S. McCann Haworth (Karolinska Institutet) for language editing before submission. M.C. receives research support from the Swedish Research Council (2016-01381, 2020-01645), the Swedish Heart and Lung Foundation (20170124, 20180568), NovoNordisk (2019#0055026) and by EFSD/Lilly European Diabetes Research Programme (2018#97012), Stockholm County Council Research Funding (ALF, 20190314), as well as Research Funds (2-560/2015) and KID funding (2-3707/2013 & 2-1930/2016) from the Karolinska Institutet, Stockholm, Sweden.
Glossary
- Nitrosylation
The formation of a nitrosyl species (X-NO, where X represents a metal centre or radical species) via a direct reaction with NO.
- Nitration
The addition of a nitronium ion (NO2+) to a nucleophilic group, leading to the generation of an X-NO2 species (formation of a nitro group).
- Transnitrosation
The transfer of NO+ from one nucleophilic centre to another.
- Transnitrosylation
The transfer of NO from one molecule to another.
- Myogenic response
The intrinsic capacity of small resistance arteries and arterioles to react (contract or dilate) in response to variations in blood pressure to keep the blood flow constant.
- Tubuloglomerular feedback
(TGF). A unique feedback system in which macula densa cells sense tubular NaCl load and communicate via purinergic signalling with the afferent arteriole, which adjusts its tone to regulate the glomerular filtration rate.
- Vascular conductance
The ease with which blood flows through a circulation (or vascular bed) at a given pressure difference (the reciprocal of resistance).
- Vascular admittance
A relative autoregulatory index that is similar to steady-state conductance (the reciprocal of resistance).
- Nitrosation
The addition of a nitrosonium ion (NO+) to a nucleophilic centre (e.g. a thiol or amine) either directly or by transfer from an NO+ donor (e.g. N2O3 or FeIINO+).
- Dietary Approaches to Stop Hypertension (DASH) diet
A diet rich in fruits, vegetables and low-fat dairy products, that was developed by the nutrition committee of the American Heart Association and has been shown to reduce blood pressure.
Author contributions
M.C. researched the data for the article, wrote the manuscript and reviewed and edited it before submission.
Competing interest
The author declares no competing interests
Footnotes
Peer review information
Nature Reviews Nephrology thanks Johannes Stegbauer, Andrew Webb and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Human Protein Atlas: http://www.proteinatlas.org/
References
- 1.GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395:709–733. doi: 10.1016/S0140-6736(20)30045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2:634–647. doi: 10.1016/S2213-8587(14)70102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whaley-Connell A, Sowers JR. Basic science: pathophysiology: the cardiorenal metabolic syndrome. J. Am. Soc. Hypertens. 2014;8:604–606. doi: 10.1016/j.jash.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangaswami J, et al. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation. 2019;139:e840–e878. doi: 10.1161/CIR.0000000000000664. [DOI] [PubMed] [Google Scholar]
- 5.Aron-Wisnewsky J, Clement K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat. Rev. Nephrol. 2016;12:169–181. doi: 10.1038/nrneph.2015.191. [DOI] [PubMed] [Google Scholar]
- 6.Yang T, Richards EM, Pepine CJ, Raizada MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018;14:442–456. doi: 10.1038/s41581-018-0018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schiffer TA, Lundberg JO, Weitzberg E, Carlstrom M. Modulation of mitochondria and NADPH oxidase function by the nitrate-nitrite-NO pathway in metabolic disease with focus on type 2 diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165811. doi: 10.1016/j.bbadis.2020.165811. [DOI] [PubMed] [Google Scholar]
- 8.Carlstrom M, Montenegro MF. Therapeutic value of stimulating the nitrate-nitrite-nitric oxide pathway to attenuate oxidative stress and restore nitric oxide bioavailability in cardiorenal disease. J. Intern. Med. 2019;285:2–18. doi: 10.1111/joim.12818. [DOI] [PubMed] [Google Scholar]
- 9.Lundberg JO, Gladwin MT, Weitzberg E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov. 2015;14:623–641. doi: 10.1038/nrd4623. [DOI] [PubMed] [Google Scholar]
- 10.Tejero J, Shiva S, Gladwin MT. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol. Rev. 2019;99:311–379. doi: 10.1152/physrev.00036.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundberg JO, Weitzberg E, Lundberg JM, Alving K. Intragastric nitric oxide production in humans: measurements in expelled air. Gut. 1994;35:1543–1546. doi: 10.1136/gut.35.11.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjamin N, et al. Stomach NO synthesis. Nature. 1994;368:502. doi: 10.1038/368502a0. [DOI] [PubMed] [Google Scholar]
- 13.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat. Med. 1995;1:804–809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 14.Bredt DS, et al. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 15.Marsden PA, et al. Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 1992;307:287–293. doi: 10.1016/0014-5793(92)80697-F. [DOI] [PubMed] [Google Scholar]
- 16.Xie QW, et al. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 1992;256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 17.Chartrain NA, et al. Molecular cloning, structure, and chromosomal localization of the human inducible nitric oxide synthase gene. J. Biol. Chem. 1994;269:6765–6772. doi: 10.1016/S0021-9258(17)37441-0. [DOI] [PubMed] [Google Scholar]
- 18.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012;33:829–837. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 20.Papapetropoulos A, Hobbs AJ, Topouzis S. Extending the translational potential of targeting NO/cGMP-regulated pathways in the CVS. Br. J. Pharmacol. 2015;172:1397–1414. doi: 10.1111/bph.12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ. Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ischiropoulos H. Protein tyrosine nitration — an update. Arch. Biochem. Biophys. 2009;484:117–121. doi: 10.1016/j.abb.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 23.Villacorta L, Gao Z, Schopfer FJ, Freeman BA, Chen YE. Nitro-fatty acids in cardiovascular regulation and diseases: characteristics and molecular mechanisms. Front. Biosci. 2016;21:873–889. doi: 10.2741/4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleschyov AL. The NO-heme signaling hypothesis. Free Radic. Biol. Med. 2017;112:544–552. doi: 10.1016/j.freeradbiomed.2017.08.025. [DOI] [PubMed] [Google Scholar]
- 25.Vedernikov YP, Mordvintcev PI, Malenkova IV, Vanin AF. Similarity between the vasorelaxing activity of dinitrosyl iron cysteine complexes and endothelium-derived relaxing factor. Eur. J. Pharmacol. 1992;211:313–317. doi: 10.1016/0014-2999(92)90386-I. [DOI] [PubMed] [Google Scholar]
- 26.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 27.Moller MN, et al. Detection and quantification of nitric oxide-derived oxidants in biological systems. J. Biol. Chem. 2019;294:14776–14802. doi: 10.1074/jbc.REV119.006136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolden C, King SB, Kim-Shapiro DB. Reactions between nitrosopersulfide and heme proteins. Free Radic. Biol. Med. 2016;99:418–425. doi: 10.1016/j.freeradbiomed.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy ME, Sies H. Reversible conversion of nitroxyl anion to nitric oxide by superoxide dismutase. Proc. Natl Acad. Sci. USA. 1991;88:10860–10864. doi: 10.1073/pnas.88.23.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl Acad. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heiss EH, Dirsch VM. Regulation of eNOS enzyme activity by posttranslational modification. Curr. Pharm. Des. 2014;20:3503–3513. doi: 10.2174/13816128113196660745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma NM, Patel KP. Post-translational regulation of neuronal nitric oxide synthase: implications for sympathoexcitatory states. Expert Opin. Ther. Targets. 2017;21:11–22. doi: 10.1080/14728222.2017.1265505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 2012;287:4411–4418. doi: 10.1074/jbc.R111.285742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J. Mol. Cell. Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 35.Mount PF, Power DA. Nitric oxide in the kidney: functions and regulation of synthesis. Acta Physiol. 2006;187:433–446. doi: 10.1111/j.1748-1716.2006.01582.x. [DOI] [PubMed] [Google Scholar]
- 36.Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol. Med. 2000;6:347–373. doi: 10.1007/BF03401781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baylis C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2008;294:F1–F9. doi: 10.1152/ajprenal.00424.2007. [DOI] [PubMed] [Google Scholar]
- 38.Baylis C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006;2:209–220. doi: 10.1038/ncpneph0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farah C, Michel LYM, Balligand JL. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018;15:292–316. doi: 10.1038/nrcardio.2017.224. [DOI] [PubMed] [Google Scholar]
- 40.Eissa NT, et al. Alternative splicing of human inducible nitric-oxide synthase mRNA. tissue-specific regulation and induction by cytokines. J. Biol. Chem. 1996;271:27184–27187. doi: 10.1074/jbc.271.43.27184. [DOI] [PubMed] [Google Scholar]
- 41.Saur D, Paehge H, Schusdziarra V, Allescher HD. Distinct expression of splice variants of neuronal nitric oxide synthase in the human gastrointestinal tract. Gastroenterology. 2000;118:849–858. doi: 10.1016/S0016-5085(00)70171-5. [DOI] [PubMed] [Google Scholar]
- 42.Lu D, et al. Salt sensitive splice variant of nNOS expressed in the macula densa cells. Am. J. Physiol. Renal Physiol. 2010;298:F1465–F1471. doi: 10.1152/ajprenal.00650.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jarry A, et al. Expression of NOS1 and soluble guanylyl cyclase by human kidney epithelial cells: morphological evidence for an autocrine/paracrine action of nitric oxide. Kidney Int. 2003;64:170–180. doi: 10.1046/j.1523-1755.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 44.Goligorsky MS, Brodsky SV, Noiri E. Nitric oxide in acute renal failure: NOS versus NOS. Kidney Int. 2002;61:855–861. doi: 10.1046/j.1523-1755.2002.00233.x. [DOI] [PubMed] [Google Scholar]
- 45.Yoo KH, Thornhill BA, Forbes MS, Chevalier RL. Inducible nitric oxide synthase modulates hydronephrosis following partial or complete unilateral ureteral obstruction in the neonatal mouse. Am. J. Physiol. Renal Physiol. 2010;298:F62–F71. doi: 10.1152/ajprenal.00234.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heemskerk S, et al. Upregulation of renal inducible nitric oxide synthase during human endotoxemia and sepsis is associated with proximal tubule injury. Clin. J. Am. Soc. Nephrol. 2006;1:853–862. doi: 10.2215/CJN.00490206. [DOI] [PubMed] [Google Scholar]
- 47.Udi S, et al. Dual inhibition of cannabinoid CB1 receptor and inducible NOS attenuates obesity-induced chronic kidney disease. Br. J. Pharmacol. 2020;177:110–127. doi: 10.1111/bph.14849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bautista-Garcia P, et al. Chronic inhibition of NOS-2 ameliorates renal injury, as well as COX-2 and TGF-beta 1 overexpression in 5/6 nephrectomized rats. Nephrol. Dial. Transpl. 2006;21:3074–3081. doi: 10.1093/ndt/gfl444. [DOI] [PubMed] [Google Scholar]
- 49.Kelm M. Nitric oxide metabolism and breakdown. Biochim. Biophys. Acta. 1999;1411:273–289. doi: 10.1016/S0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 50.Dejam A, et al. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- 51.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 52.Rhodes P, et al. The L-arginine:nitric oxide pathway is the major source of plasma nitrite in fasted humans. Biochem. Biophys. Res. Commun. 1995;209:590–596. doi: 10.1006/bbrc.1995.1541. [DOI] [PubMed] [Google Scholar]
- 53.Lundberg JO, Govoni M. Inorganic nitrate is a possible source for systemic generation of nitric oxide. Free Radic. Biol. Med. 2004;37:395–400. doi: 10.1016/j.freeradbiomed.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 54.Qin L, et al. Sialin (SLC17A5) functions as a nitrate transporter in the plasma membrane. Proc. Natl Acad. Sci. USA. 2012;109:13434–13439. doi: 10.1073/pnas.1116633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edwards DA, Fletcher K, Rowlands EN. Antagonism between perchlorate, iodide, thiocyanate, and nitrate for secretion in human saliva; analogy with the iodide trap of the thyroid. Lancet. 1954;266:498–499. doi: 10.1016/S0140-6736(54)91196-4. [DOI] [PubMed] [Google Scholar]
- 56.Duncan C, et al. Chemical generation of nitric oxide in the mouth from the enterosalivary circulation of dietary nitrate. Nat. Med. 1995;1:546–551. doi: 10.1038/nm0695-546. [DOI] [PubMed] [Google Scholar]
- 57.Lundberg JO, Weitzberg E. Biology of nitrogen oxides in the gastrointestinal tract. Gut. 2013;62:616–629. doi: 10.1136/gutjnl-2011-301649. [DOI] [PubMed] [Google Scholar]
- 58.Helms CC, Liu X, Kim-Shapiro DB. Recent insights into nitrite signaling processes in blood. Biol. Chem. 2016;398:319–329. doi: 10.1515/hsz-2016-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blood AB. The medicinal chemistry of nitrite as a source of nitric oxide signaling. Curr. Top. Med. Chem. 2017;17:1758–1768. doi: 10.2174/1568026617666161116145046. [DOI] [PubMed] [Google Scholar]
- 60.Webb A, et al. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc. Natl Acad. Sci. USA. 2004;101:13683–13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gautier C, van Faassen E, Mikula I, Martasek P, Slama-Schwok A. Endothelial nitric oxide synthase reduces nitrite anions to NO under anoxia. Biochem. Biophys. Res. Commun. 2006;341:816–821. doi: 10.1016/j.bbrc.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 62.Castello PR, David PS, McClure T, Crook Z, Poyton RO. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006;3:277–287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 63.van Faassen EE, et al. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med. Res. Rev. 2009;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crawford JH, et al. Hypoxia, red blood cells, and nitrite regulate NO−dependent hypoxic vasodilation. Blood. 2006;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlstrom M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol. Rev. 2015;95:405–511. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Persson AE, Lai EY, Gao X, Carlstrom M, Patzak A. Interactions between adenosine, angiotensin II and nitric oxide on the afferent arteriole influence sensitivity of the tubuloglomerular feedback. Front. Physiol. 2013;4:187. doi: 10.3389/fphys.2013.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johns EJ, Kopp UC, DiBona GF. Neural control of renal function. Compr. Physiol. 2011;1:731–767. doi: 10.1002/cphy.c100043. [DOI] [PubMed] [Google Scholar]
- 68.Just A, Arendshorst WJ. Nitric oxide blunts myogenic autoregulation in rat renal but not skeletal muscle circulation via tubuloglomerular feedback. J. Physiol. 2005;569:959–974. doi: 10.1113/jphysiol.2005.094888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi Y, Wang X, Chon KH, Cupples WA. Tubuloglomerular feedback-dependent modulation of renal myogenic autoregulation by nitric oxide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006;290:R982–R991. doi: 10.1152/ajpregu.00346.2005. [DOI] [PubMed] [Google Scholar]
- 70.Lai EY, Wellstein A, Welch WJ, Wilcox CS. Superoxide modulates myogenic contractions of mouse afferent arterioles. Hypertension. 2011;58:650–656. doi: 10.1161/HYPERTENSIONAHA.111.170472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ichihara A, Navar LG. Neuronal NOS contributes to biphasic autoregulatory response during enhanced TGF activity. Am. J. Physiol. 1999;277:F113–F120. doi: 10.1152/ajprenal.1999.277.1.F113. [DOI] [PubMed] [Google Scholar]
- 72.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown R, et al. Abolished tubuloglomerular feedback and increased plasma renin in adenosine A1 receptor-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;281:R1362–1367. doi: 10.1152/ajpregu.2001.281.5.R1362. [DOI] [PubMed] [Google Scholar]
- 74.Sun D, et al. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc. Natl Acad. Sci. USA. 2001;98:9983–9988. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Inscho EW, Cook AK, Imig JD, Vial C, Evans RJ. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J. Clin. Invest. 2003;112:1895–1905. doi: 10.1172/JCI18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu R, et al. Purinergic receptor signaling at the basolateral membrane of macula densa cells. J. Am. Soc. Nephrol. 2002;13:1145–1151. doi: 10.1097/01.ASN.0000014827.71910.39. [DOI] [PubMed] [Google Scholar]
- 77.Ollerstam A, Persson AE. Macula densa neuronal nitric oxide synthase. Cardiovasc. Res. 2002;56:189–196. doi: 10.1016/S0008-6363(02)00536-9. [DOI] [PubMed] [Google Scholar]
- 78.Wilcox CS, et al. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc. Natl Acad. Sci. USA. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu R, Carretero OA, Ren Y, Garvin JL. Increased intracellular pH at the macula densa activates nNOS during tubuloglomerular feedback. Kidney Int. 2005;67:1837–1843. doi: 10.1111/j.1523-1755.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- 80.Vallon V, et al. Feedback control of glomerular vascular tone in neuronal nitric oxide synthase knockout mice. J. Am. Soc. Nephrol. 2001;12:1599–1606. doi: 10.1681/ASN.V1281599. [DOI] [PubMed] [Google Scholar]
- 81.Tojo A, Onozato ML, Fujita T. Role of macula densa neuronal nitric oxide synthase in renal diseases. Med. Mol. Morphol. 2006;39:2–7. doi: 10.1007/s00795-006-0310-2. [DOI] [PubMed] [Google Scholar]
- 82.Thorup C, Persson AE. Impaired effect of nitric oxide synthesis inhibition on tubuloglomerular feedback in hypertensive rats. Am. J. Physiol. 1996;271:F246–F252. doi: 10.1152/ajprenal.1996.271.2.F246. [DOI] [PubMed] [Google Scholar]
- 83.Welch WJ, Tojo A, Wilcox CS. Roles of NO and oxygen radicals in tubuloglomerular feedback in SHR. Am. J. Physiol. Renal Physiol. 2000;278:F769–776. doi: 10.1152/ajprenal.2000.278.5.F769. [DOI] [PubMed] [Google Scholar]
- 84.Ollerstam A, Pittner J, Persson AE, Thorup C. Increased blood pressure in rats after long-term inhibition of the neuronal isoform of nitric oxide synthase. J. Clin. Invest. 1997;99:2212–2218. doi: 10.1172/JCI119394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pallone TL, Silldorff EP. Pericyte regulation of renal medullary blood flow. Exp. Nephrol. 2001;9:165–170. doi: 10.1159/000052608. [DOI] [PubMed] [Google Scholar]
- 86.Sendeski MM, et al. Functional characterization of isolated, perfused outermedullary descending human vasa recta. Acta Physiol. 2013;208:50–56. doi: 10.1111/apha.12084. [DOI] [PubMed] [Google Scholar]
- 87.Cao C, et al. Intrinsic nitric oxide and superoxide production regulates descending vasa recta contraction. Am. J. Physiol. Renal Physiol. 2010;299:F1056–F1064. doi: 10.1152/ajprenal.00070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pittner J, Wolgast M, Casellas D, Persson AE. Increased shear stress-released NO and decreased endothelial calcium in rat isolated perfused juxtamedullary nephrons. Kidney Int. 2005;67:227–236. doi: 10.1111/j.1523-1755.2005.00073.x. [DOI] [PubMed] [Google Scholar]
- 89.Imig JD, Roman RJ. Nitric oxide modulates vascular tone in preglomerular arterioles. Hypertension. 1992;19:770–774. doi: 10.1161/01.HYP.19.6.770. [DOI] [PubMed] [Google Scholar]
- 90.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu. Rev. Physiol. 2010;73:359–376. doi: 10.1146/annurev-physiol-012110-142247. [DOI] [PubMed] [Google Scholar]
- 91.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am. J. Physiol. Renal Physiol. 2002;282:F777–F784. doi: 10.1152/ajprenal.00334.2001. [DOI] [PubMed] [Google Scholar]
- 92.Satoh N, et al. Effects of nitric oxide on renal proximal tubular Na+ transport. BioMed. Res. Int. 2017;2017:6871081. doi: 10.1155/2017/6871081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bahadoran Z, Carlstrom M, Mirmiran P, Ghasemi A. Nitric oxide: to be or not to be an endocrine hormone? Acta Physiol. 2020;229:e13443. doi: 10.1111/apha.13443. [DOI] [PubMed] [Google Scholar]
- 94.Boron, W. F. & Boulpaep, E. L. Medical Physiology, a Cellular and Molecular Approach 3rd edn (Elsevier, 2016).
- 95.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu. Rev. Physiol. 2011;73:359–376. doi: 10.1146/annurev-physiol-012110-142247. [DOI] [PubMed] [Google Scholar]
- 96.Broere A, et al. Human renal and systemic hemodynamic, natriuretic, and neurohumoral responses to different doses of L-NAME. Am. J. Physiol. 1998;275:F870–F877. doi: 10.1152/ajprenal.1998.275.6.F870. [DOI] [PubMed] [Google Scholar]
- 97.Bech JN, Nielsen CB, Pedersen EB. Effects of systemic NO synthesis inhibition on RPF, GFR, UNa, and vasoactive hormones in healthy humans. Am. J. Physiol. 1996;270:F845–F851. doi: 10.1152/ajprenal.1996.270.5.F845. [DOI] [PubMed] [Google Scholar]
- 98.Shirai A, et al. Angiotensin II dose-dependently stimulates human renal proximal tubule transport by the nitric oxide/guanosine 3′,5′-cyclic monophosphate pathway. J. Am. Soc. Nephrol. 2014;25:1523–1532. doi: 10.1681/ASN.2013060596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hyndman KA, et al. Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension. 2013;62:91–98. doi: 10.1161/HYPERTENSIONAHA.113.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wagner DA, Schultz DS, Deen WM, Young VR, Tannenbaum SR. Metabolic fate of an oral dose of 15N-labeled nitrate in humans: effect of diet supplementation with ascorbic acid. Cancer Res. 1983;43:1921–1925. [PubMed] [Google Scholar]
- 101.Green LC, et al. Nitrate biosynthesis in man. Proc. Natl Acad. Sci. USA. 1981;78:7764–7768. doi: 10.1073/pnas.78.12.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Williams JK, et al. Renal nitrate clearance in chronic kidney disease. Nitric Oxide. 2020;97:16–19. doi: 10.1016/j.niox.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 103.Sundqvist ML, Lundberg JO, Weitzberg E, Carlstrom M. Renal handling of nitrate in women and men with elevated blood pressure. Acta Physiol. 2021 doi: 10.1111/apha.13637. [DOI] [PubMed] [Google Scholar]
- 104.Godfrey M, Majid DS. Renal handling of circulating nitrates in anesthetized dogs. Am. J. Physiol. 1998;275:F68–F73. doi: 10.1152/ajprenal.1998.275.1.F68. [DOI] [PubMed] [Google Scholar]
- 105.Suto T, et al. Acute changes in urinary excretion of nitrite + nitrate do not necessarily predict renal vascular NO production. Kidney Int. 1995;48:1272–1277. doi: 10.1038/ki.1995.411. [DOI] [PubMed] [Google Scholar]
- 106.Chobanyan-Jurgens K, et al. Renal carbonic anhydrases are involved in the reabsorption of endogenous nitrite. Nitric Oxide. 2012;26:126–131. doi: 10.1016/j.niox.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 107.Rahma M, et al. Effects of furosemide on the tubular reabsorption of nitrates in anesthetized dogs. Eur. J. Pharmacol. 2001;428:113–119. doi: 10.1016/S0014-2999(01)01357-7. [DOI] [PubMed] [Google Scholar]
- 108.Wall SM, Lazo-Fernandez Y. The role of pendrin in renal physiology. Annu. Rev. Physiol. 2015;77:363–378. doi: 10.1146/annurev-physiol-021014-071854. [DOI] [PubMed] [Google Scholar]
- 109.Thumova M, et al. Pendrin protein abundance in the kidney is regulated by nitric oxide and cAMP. Am. J. Physiol. Renal Physiol. 2012;303:F812–F820. doi: 10.1152/ajprenal.00577.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yarovaya N, et al. Sialin, an anion transporter defective in sialic acid storage diseases, shows highly variable expression in adult mouse brain, and is developmentally regulated. Neurobiol. Dis. 2005;19:351–365. doi: 10.1016/j.nbd.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 111.Jiang F, et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat. Rev. Cardiol. 2014;11:413–426. doi: 10.1038/nrcardio.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barnes M, Brisbois EJ. Clinical use of inhaled nitric oxide: local and systemic applications. Free Radic. Biol. Med. 2020;152:422–431. doi: 10.1016/j.freeradbiomed.2019.11.029. [DOI] [PubMed] [Google Scholar]
- 113.Ruan SY, et al. Inhaled nitric oxide therapy and risk of renal dysfunction: a systematic review and meta-analysis of randomized trials. Crit. Care. 2015;19:137. doi: 10.1186/s13054-015-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Daiber A, Munzel T. Organic nitrate therapy, nitrate tolerance, and nitrate-induced endothelial dysfunction: emphasis on redox biology and oxidative stress. Antioxid. Redox Signal. 2015;23:899–942. doi: 10.1089/ars.2015.6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Antoniades C, et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009;30:1142–1150. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- 116.Yang J, Gonon AT, Sjoquist PO, Lundberg JO, Pernow J. Arginase regulates red blood cell nitric oxide synthase and export of cardioprotective nitric oxide bioactivity. Proc. Natl Acad. Sci. USA. 2013;110:15049–15054. doi: 10.1073/pnas.1307058110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu X, Xu X, Shang R, Chen Y. Asymmetric dimethylarginine (ADMA) as an important risk factor for the increased cardiovascular diseases and heart failure in chronic kidney disease. Nitric Oxide. 2018;78:113–120. doi: 10.1016/j.niox.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Caldwell RW, Rodriguez PC, Toque HA, Narayanan SP, Caldwell RB. Arginase: a multifaceted enzyme important in health and disease. Physiol. Rev. 2018;98:641–665. doi: 10.1152/physrev.00037.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kovamees O, et al. Arginase inhibition improves microvascular endothelial function in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2016;101:3952–3958. doi: 10.1210/jc.2016-2007. [DOI] [PubMed] [Google Scholar]
- 120.Shemyakin A, et al. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation. 2012;126:2943–2950. doi: 10.1161/CIRCULATIONAHA.112.140335. [DOI] [PubMed] [Google Scholar]
- 121.Ashmore T, et al. Dietary nitrate increases arginine availability and protects mitochondrial complex I and energetics in the hypoxic rat heart. J. Physiol. 2014;592:4715–4731. doi: 10.1113/jphysiol.2014.275263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pernow J, Jung C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc. Res. 2013;98:334–343. doi: 10.1093/cvr/cvt036. [DOI] [PubMed] [Google Scholar]
- 123.Szabo C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2012;17:68–80. doi: 10.1089/ars.2011.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kasinath BS, Feliers D, Lee HJ. Hydrogen sulfide as a regulatory factor in kidney health and disease. Biochem. Pharmacol. 2018;149:29–41. doi: 10.1016/j.bcp.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 125.Li Z, Polhemus DJ, Lefer DJ. Evolution of hydrogen sulfide therapeutics to treat cardiovascular disease. Circ. Res. 2018;123:590–600. doi: 10.1161/CIRCRESAHA.118.311134. [DOI] [PubMed] [Google Scholar]
- 126.Altaany Z, Yang G, Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell Mol. Med. 2013;17:879–888. doi: 10.1111/jcmm.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coletta C, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl Acad. Sci. USA. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pardue S, et al. Hydrogen sulfide stimulates xanthine oxidoreductase conversion to nitrite reductase and formation of NO. Redox Biol. 2020;34:101447. doi: 10.1016/j.redox.2020.101447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bucci M, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler. Thromb. Vasc. Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- 130.Cortese-Krott MM, et al. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl Acad. Sci. USA. 2015;112:E4651–E4660. doi: 10.1073/pnas.1509277112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kukreja RC, Salloum FN, Das A. Cyclic guanosine monophosphate signaling and phosphodiesterase-5 inhibitors in cardioprotection. J. Am. Coll. Cardiol. 2012;59:1921–1927. doi: 10.1016/j.jacc.2011.09.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brown KE, Dhaun N, Goddard J, Webb DJ. Potential therapeutic role of phosphodiesterase type 5 inhibition in hypertension and chronic kidney disease. Hypertension. 2014;63:5–11. doi: 10.1161/HYPERTENSIONAHA.113.01774. [DOI] [PubMed] [Google Scholar]
- 133.Rodriguez-Iturbe B, et al. Early treatment with cGMP phosphodiesterase inhibitor ameliorates progression of renal damage. Kidney Int. 2005;68:2131–2142. doi: 10.1111/j.1523-1755.2005.00669.x. [DOI] [PubMed] [Google Scholar]
- 134.Andersson KE. PDE5 inhibitors - pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018;175:2554–2565. doi: 10.1111/bph.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scheele W, et al. Phosphodiesterase type 5 inhibition reduces albuminuria in subjects with overt diabetic nephropathy. J. Am. Soc. Nephrol. 2016;27:3459–3468. doi: 10.1681/ASN.2015050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Buys ES, et al. Discovery and development of next generation sGC stimulators with diverse multidimensional pharmacology and broad therapeutic potential. Nitric Oxide. 2018;78:72–80. doi: 10.1016/j.niox.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 137.Sandner P, et al. Soluble guanylate cyclase stimulators and activators. Handb. Exp. Pharmacol. 2019;264:355–394. doi: 10.1007/164_2018_197. [DOI] [PubMed] [Google Scholar]
- 138.Pieske B, et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the soluble guanylate cyclase stimulator in heart failure patients with preserved EF (SOCRATES-PRESERVED) study. Eur. Heart J. 2017;38:1119–1127. doi: 10.1093/eurheartj/ehw593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Armstrong PW, et al. Vericiguat in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2020;382:1883–1893. doi: 10.1056/NEJMoa1915928. [DOI] [PubMed] [Google Scholar]
- 140.Stasch JP, Schlossmann J, Hocher B. Renal effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Curr. Opin. Pharmacol. 2015;21:95–104. doi: 10.1016/j.coph.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 141.Schinner E, et al. Inhibition of the TGFbeta signalling pathway by cGMP and cGMP-dependent kinase I in renal fibrosis. FEBS Open Bio. 2017;7:550–561. doi: 10.1002/2211-5463.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hanrahan JP, et al. Effects of the soluble guanylate cyclase stimulator praliciguat in diabetic kidney disease: a randomized placebo-controlled clinical trial. Clin. J. Am. Soc. Nephrol. 2020;16:59–69. doi: 10.2215/CJN.08410520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Horiuchi M, Iwanami J, Mogi M. Regulation of angiotensin II receptors beyond the classical pathway. Clin. Sci. 2012;123:193–203. doi: 10.1042/CS20110677. [DOI] [PubMed] [Google Scholar]
- 144.Seva Pessoa B, et al. Key developments in renin-angiotensin-aldosterone system inhibition. Nat. Rev. Nephrol. 2013;9:26–36. doi: 10.1038/nrneph.2012.249. [DOI] [PubMed] [Google Scholar]
- 145.Bae S, Kim SR, Kim MN, Shim WJ, Park SM. Impact of cardiovascular disease and risk factors on fatal outcomes in patients with COVID-19 according to age: a systematic review and meta-analysis. Heart. 2021;107:373–380. doi: 10.1136/heartjnl-2020-317901. [DOI] [PubMed] [Google Scholar]
- 146.Council E-E, Group EW. Chronic kidney disease is a key risk factor for severe COVID-19: a call to action by the ERA-EDTA. Nephrol. Dial. Transpl. 2021;36:87–94. doi: 10.1093/ndt/gfaa314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lim S, Bae JH, Kwon HS, Nauck MA. COVID-19 and diabetes mellitus: from pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021;17:11–30. doi: 10.1038/s41574-020-00435-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ni W, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care. 2020;24:422. doi: 10.1186/s13054-020-03120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Perico L, et al. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021;17:46–64. doi: 10.1038/s41581-020-00357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fang W, et al. The role of NO in COVID-19 and potential therapeutic strategies. Free Radic. Biol. Med. 2020;163:153–162. doi: 10.1016/j.freeradbiomed.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Simon MA, et al. Acute hemodynamic effects of inhaled sodium nitrite in pulmonary hypertension associated with heart failure with preserved ejection fraction. JCI Insight. 2016;1:e89620. doi: 10.1172/jci.insight.89620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Larsen FJ, Ekblom B, Sahlin K, Lundberg JO, Weitzberg E. Effects of dietary nitrate on blood pressure in healthy volunteers. N. Engl. J. Med. 2006;355:2792–2793. doi: 10.1056/NEJMc062800. [DOI] [PubMed] [Google Scholar]
- 153.Webb AJ, et al. Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension. 2008;51:784–790. doi: 10.1161/HYPERTENSIONAHA.107.103523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ashor AW, Lara J, Siervo M. Medium-term effects of dietary nitrate supplementation on systolic and diastolic blood pressure in adults: a systematic review and meta-analysis. J. Hypertens. 2017;35:1353–1359. doi: 10.1097/HJH.0000000000001305. [DOI] [PubMed] [Google Scholar]
- 155.Jackson JK, Patterson AJ, MacDonald-Wicks LK, Oldmeadow C, McEvoy MA. The role of inorganic nitrate and nitrite in cardiovascular disease risk factors: a systematic review and meta-analysis of human evidence. Nutr. Rev. 2018;264:355–394. doi: 10.1093/nutrit/nuy005. [DOI] [PubMed] [Google Scholar]
- 156.Carlstrom M, Lundberg JO, Weitzberg E. Mechanisms underlying blood pressure reduction by dietary inorganic nitrate. Acta Physiol. 2018;224:e13080. doi: 10.1111/apha.13080. [DOI] [PubMed] [Google Scholar]
- 157.Omar SA, Webb AJ, Lundberg JO, Weitzberg E. Therapeutic effects of inorganic nitrate and nitrite in cardiovascular and metabolic diseases. J. Intern. Med. 2016;279:315–336. doi: 10.1111/joim.12441. [DOI] [PubMed] [Google Scholar]
- 158.Ghosh SM, et al. Enhanced vasodilator activity of nitrite in hypertension: critical role for erythrocytic xanthine oxidoreductase and translational potential. Hypertension. 2013;61:1091–1102. doi: 10.1161/HYPERTENSIONAHA.111.00933. [DOI] [PubMed] [Google Scholar]
- 159.Bondonno CP, et al. Absence of an effect of high nitrate intake from beetroot juice on blood pressure in treated hypertensive individuals: a randomized controlled trial. Am. J. Clin. Nutr. 2015;102:368–375. doi: 10.3945/ajcn.114.101188. [DOI] [PubMed] [Google Scholar]
- 160.Kapil V, Khambata RS, Robertson A, Caulfield MJ, Ahluwalia A. Dietary nitrate provides sustained blood pressure lowering in hypertensive patients: a randomized, phase 2, double-blind, placebo-controlled study. Hypertension. 2015;65:320–327. doi: 10.1161/HYPERTENSIONAHA.114.04675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sundqvist ML, et al. A randomized clinical trial of the effects of leafy green vegetables and inorganic nitrate on blood pressure. Am. J. Clin. Nutr. 2020;111:749–756. doi: 10.1093/ajcn/nqaa024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Velmurugan S, et al. Dietary nitrate improves vascular function in patients with hypercholesterolemia: a randomized, double-blind, placebo-controlled study. Am. J. Clin. Nutr. 2016;103:25–38. doi: 10.3945/ajcn.115.116244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li D, et al. Repeated administration of inorganic nitrate on blood pressure and arterial stiffness: a systematic review and meta-analysis of randomized controlled trials. J. Hypertens. 2020;38:2122–2140. doi: 10.1097/HJH.0000000000002524. [DOI] [PubMed] [Google Scholar]
- 164.Sacks FM, et al. Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001;344:3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 165.Appel LJ, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N. Engl. J. Med. 1997;336:1117–1124. doi: 10.1056/NEJM199704173361601. [DOI] [PubMed] [Google Scholar]
- 166.Camara NO, Iseki K, Kramer H, Liu ZH, Sharma K. Kidney disease and obesity: epidemiology, mechanisms and treatment. Nat. Rev. Nephrol. 2017;13:181–190. doi: 10.1038/nrneph.2016.191. [DOI] [PubMed] [Google Scholar]
- 167.Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020;16:317–336. doi: 10.1038/s41581-020-0256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 1993;329:1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 169.Maschio G, et al. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N. Engl. J. Med. 1996;334:939–945. doi: 10.1056/NEJM199604113341502. [DOI] [PubMed] [Google Scholar]
- 170.Perkovic V, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019;380:2295–2306. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 171.Wilcox CS. Antihypertensive and renal mechanisms of SGLT2 (sodium-glucose linked transporter 2) inhibitors. Hypertension. 2020;75:894–901. doi: 10.1161/HYPERTENSIONAHA.119.11684. [DOI] [PubMed] [Google Scholar]
- 172.Herat LY, et al. SGLT2 inhibitor-induced sympathoinhibition: a novel mechanism for cardiorenal protection. JACC Basic. Transl. Sci. 2020;5:169–179. doi: 10.1016/j.jacbts.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Huang PL, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 174.Huang PL. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Forbes MS, Thornhill BA, Park MH, Chevalier RL. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am. J. Pathol. 2007;170:87–99. doi: 10.2353/ajpath.2007.060610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Heeringa P, et al. Lack of endothelial nitric oxide synthase aggravates murine accelerated anti-glomerular basement membrane glomerulonephritis. Am. J. Pathol. 2000;156:879–888. doi: 10.1016/S0002-9440(10)64957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kanematsu Y, et al. Dietary doses of nitrite restore circulating nitric oxide level and improve renal injury in L-NAME-induced hypertensive rats. Am. J. Physiol. Renal Physiol. 2008;295:F1457–F1462. doi: 10.1152/ajprenal.00621.2007. [DOI] [PubMed] [Google Scholar]
- 178.Nakayama T, et al. Endothelial injury due to eNOS deficiency accelerates the progression of chronic renal disease in the mouse. Am. J. Physiol. Renal Physiol. 2009;296:F317–F327. doi: 10.1152/ajprenal.90450.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kang DH, Nakagawa T, Feng L, Johnson RJ. Nitric oxide modulates vascular disease in the remnant kidney model. Am. J. Pathol. 2002;161:239–248. doi: 10.1016/S0002-9440(10)64175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Carlstrom M, et al. Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice. Proc. Natl Acad. Sci. USA. 2010;107:17716–17720. doi: 10.1073/pnas.1008872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lundberg JO, Carlstrom M, Weitzberg E. Metabolic effects of dietary nitrate in health and disease. Cell Metab. 2018;28:9–22. doi: 10.1016/j.cmet.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 182.Lai YC, et al. SIRT3-AMP-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation. 2016;133:717–731. doi: 10.1161/CIRCULATIONAHA.115.018935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McNally BD, et al. Inorganic nitrate promotes glucose uptake and oxidative catabolism in white adipose tissue through the XOR-catalyzed nitric oxide pathway. Diabetes. 2020;69:893–901. doi: 10.2337/db19-0892. [DOI] [PubMed] [Google Scholar]
- 184.Singamsetty S, et al. Inorganic nitrite improves components of the metabolic syndrome independent of weight change in a murine model of obesity and insulin resistance. J. Physiol. 2015;593:3135–3145. doi: 10.1113/JP270386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kamga Pride C, et al. Nitrite activates protein kinase A in normoxia to mediate mitochondrial fusion and tolerance to ischaemia/reperfusion. Cardiovasc. Res. 2014;101:57–68. doi: 10.1093/cvr/cvt224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Moretti CH, et al. Dietary nitrite extends lifespan and prevents age-related locomotor decline in the fruit fly. Free Radic. Biol. Med. 2020;160:860–870. doi: 10.1016/j.freeradbiomed.2020.09.018. [DOI] [PubMed] [Google Scholar]
- 187.Bahadoran Z, Ghasemi A, Mirmiran P, Azizi F, Hadaegh F. Beneficial effects of inorganic nitrate/nitrite in type 2 diabetes and its complications. Nutr. Metab. 2015;12:16. doi: 10.1186/s12986-015-0013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gilchrist M, et al. Effect of dietary nitrate on blood pressure, endothelial function, and insulin sensitivity in type 2 diabetes. Free Radic. Biol. Med. 2013;60:89–97. doi: 10.1016/j.freeradbiomed.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 189.Bahadoran Z, et al. Effect of inorganic nitrate on metabolic parameters in patients with type 2 diabetes: A 24-week randomized double-blind placebo-controlled clinical trial. Nitric Oxide. 2020;107:58–65. doi: 10.1016/j.niox.2020.12.005. [DOI] [PubMed] [Google Scholar]
- 190.Zhou G, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cordero-Herrera I, et al. Head-to-head comparison of inorganic nitrate and metformin in a mouse model of cardiometabolic disease. Nitric Oxide. 2020;97:48–56. doi: 10.1016/j.niox.2020.01.013. [DOI] [PubMed] [Google Scholar]
- 192.Hughan KS, et al. Effects of oral sodium nitrite on blood pressure, insulin sensitivity, and intima-media arterial thickening in adults with hypertension and metabolic syndrome. Hypertension. 2020;76:866–874. doi: 10.1161/HYPERTENSIONAHA.120.14930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Reddy YS, et al. Nitric oxide status in patients with chronic kidney disease. Indian J. Nephrol. 2015;25:287–291. doi: 10.4103/0971-4065.147376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bryan NS, et al. Acute effects of hemodialysis on nitrite and nitrate: potential cardiovascular implications in dialysis patients. Free Radic. Biol. Med. 2013;58:46–51. doi: 10.1016/j.freeradbiomed.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 195.Carlstrom M, et al. Peritoneal dialysis impairs nitric oxide homeostasis and may predispose infants with low systolic blood pressure to cerebral ischemia. Nitric Oxide. 2016;58:1–9. doi: 10.1016/j.niox.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 196.Carlstrom M, et al. Plasma nitrate/nitrite removal by peritoneal dialysis might predispose infants with low blood pressure to cerebral ischaemia. Clin. Kidney J. 2015;8:215–218. doi: 10.1093/ckj/sfv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Heredia Martinez A, et al. Removal of nitrate and nitrite by hemodialysis in end-stage renal disease and by sustained low-efficiency dialysis in acute kidney injury. Nitric Oxide. 2020;98:33–40. doi: 10.1016/j.niox.2020.02.004. [DOI] [PubMed] [Google Scholar]
- 198.Carlstrom M, et al. Dietary nitrate attenuates oxidative stress, prevents cardiac and renal injuries, and reduces blood pressure in salt-induced hypertension. Cardiovasc. Res. 2011;89:574–585. doi: 10.1093/cvr/cvq366. [DOI] [PubMed] [Google Scholar]
- 199.Gao X, et al. NADPH oxidase in the renal microvasculature is a primary target for blood pressure-lowering effects by inorganic nitrate and nitrite. Hypertension. 2015;65:161–170. doi: 10.1161/HYPERTENSIONAHA.114.04222. [DOI] [PubMed] [Google Scholar]
- 200.Guimaraes DD, et al. Dietary nitrate reduces blood pressure in rats with angiotensin II-induced hypertension via mechanisms that involve reduction of sympathetic hyperactivity. Hypertension. 2019;73:839–848. doi: 10.1161/HYPERTENSIONAHA.118.12425. [DOI] [PubMed] [Google Scholar]
- 201.Hezel M, et al. Dietary nitrate improves age-related hypertension and metabolic abnormalities in rats via modulation of angiotensin II receptor signaling and inhibition of superoxide generation. Free Radic. Biol. Med. 2016;99:87–98. doi: 10.1016/j.freeradbiomed.2016.07.025. [DOI] [PubMed] [Google Scholar]
- 202.Tripatara P, et al. Nitrite-derived nitric oxide protects the rat kidney against ischemia/reperfusion injury in vivo: role for xanthine oxidoreductase. J. Am. Soc. Nephrol. 2007;18:570–580. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 203.Yang T, et al. Dietary nitrate attenuates renal ischemia-reperfusion injuries by modulation of immune responses and reduction of oxidative stress. Redox Biol. 2017;13:320–330. doi: 10.1016/j.redox.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rosenbaek JB, et al. Effect of sodium nitrite on renal function and sodium and water excretion and brachial and central blood pressure in healthy subjects: a dose-response study. Am. J. Physiol. Renal Physiol. 2017;313:F378–F387. doi: 10.1152/ajprenal.00400.2016. [DOI] [PubMed] [Google Scholar]
- 205.Rosenbaek JB, et al. Effects of sodium nitrite on renal function and blood pressure in hypertensive vs. healthy study participants: a randomized, placebo-controlled, crossover study. J. Hypertens. 2018;36:666–679. doi: 10.1097/HJH.0000000000001598. [DOI] [PubMed] [Google Scholar]
- 206.Rosenbaek JB, Pedersen EB, Bech JN. The effect of sodium nitrite infusion on renal function, brachial and central blood pressure during enzyme inhibition by allopurinol, enalapril or acetazolamide in healthy subjects: a randomized, double-blinded, placebo-controlled, crossover study. BMC Nephrol. 2018;19:244. doi: 10.1186/s12882-018-1035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Carpentier A, Stragier S, Brejeon C, Poortmans JR. Nitrate supplementation, exercise, and kidney function: are there detrimental effects? Med. Sci. Sports Exerc. 2015;47:1519–1522. doi: 10.1249/MSS.0000000000000548. [DOI] [PubMed] [Google Scholar]
- 208.Kelly JT, et al. Modifiable lifestyle factors for primary prevention of CKD: a systematic review and meta-analysis. J. Am. Soc. Nephrol. 2021;32:239–253. doi: 10.1681/ASN.2020030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kemmner S, et al. Dietary nitrate load lowers blood pressure and renal resistive index in patients with chronic kidney disease: a pilot study. Nitric Oxide. 2017;64:7–15. doi: 10.1016/j.niox.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 210.Bahadoran Z, et al. Association between dietary intakes of nitrate and nitrite and the risk of hypertension and chronic kidney disease: tehran lipid and glucose study. Nutrients. 2016;8:811. doi: 10.3390/nu8120811. [DOI] [PMC free article] [PubMed] [Google Scholar]