

Abstract
Liquid biopsy, a minimally invasive approach, is a highly powerful clinical tool for the real‐time follow‐up of cancer and overcomes many limitations of tissue biopsies. Epigenetic alterations have a high potential to provide a valuable source of innovative biomarkers for cancer, owing to their stability, frequency, and noninvasive accessibility in bodily fluids. Numerous DNA methylation markers are now tested in circulating tumor DNA (ctDNA) as potential biomarkers, in various types of cancer. DNA methylation in combination with liquid biopsy is very powerful in identifying circulating epigenetic biomarkers of clinical importance. Blood‐based epigenetic biomarkers have a high potential for early detection of cancer since DNA methylation in plasma can be detected early during cancer pathogenesis. In this review, we summarize the latest findings on DNA methylation markers in ctDNA for early detection, prognosis, minimal residual disease, risk of relapse, treatment selection, and resistance, for breast, prostate, lung, and colorectal cancer.
Keywords: cell‐free DNA, circulating tumor cells, circulating tumor DNA, DNA methylation, methylation‐specific PCR, minimal residual disease
Blood‐based epigenetic biomarkers present in circulating tumor DNA (ctDNA) have a high potential for early detection of cancer, since DNA methylation in plasma can be detected early during cancer pathogenesis. In this review, we summarize the latest findings on DNA methylation markers in ctDNA for early detection, prognosis, minimal residual disease, risk of relapse, treatment selection, and resistance in patients with breast, prostate, lung, and colorectal cancer.
Abbreviations
- AA
abiraterone acetate
- BPH
benign prostatic hyperplasia
- cfDNA
cell‐free DNA
- CRC
colorectal cancer
- CTCs
circulating tumor cells
- ctDNA
circulating tumor DNA
- ddMSP
droplet digital methylation‐specific PCR
- ddPCR
droplet digital PCR
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
- mCRPC
metastatic castration‐resistant prostate cancer
- MRD
minimal residual disease
- MS‐ddPCR
methylation‐specific droplet digital PCR
- MSP
methylation‐specific PCR
- NSCLC
non‐small‐cell lung cancer
- OS
overall survival
- qMSP
quantitative methylation‐specific PCR
- SB
sodium bisulfite
- TCGA
Cancer Genome Atlas
1. Introduction
Liquid biopsy is a highly powerful clinical tool for the real‐time follow‐up of cancer patients that overcomes many limitations of tissue biopsies [1, 2]. Liquid biopsy approaches include the enumeration and molecular characterization of circulating tumor cells (CTCs), and the analysis of circulating tumor DNA (ctDNA), circulating miRNAs, and tumor‐derived extracellular vesicles that are shed from primary tumors and metastatic sites into peripheral blood. The major advantage of liquid biopsy analysis is that it is minimally invasive and can provide real‐time information on tumor characteristics in regular time intervals. ctDNA can be detected in the biological fluids of cancer patients as a small fraction of cell‐free DNA (cfDNA) that is usually detected at very low concentrations. ctDNA analysis is mainly focused on the detection of cancer‐specific mutations that are highly important for therapeutic treatment of and disease monitoring in cancer patients [3, 4, 5]. ctDNA analysis was recently shown to be highly promising for early cancer detection: CancerSEEK is a newly developed blood test that can detect eight common cancer types through the quantitation of circulating protein levels and mutations in cfDNA [6]. It is, however, important to point out that, although ctDNA analysis in most cases is targeted as it requires prior knowledge of the mutations to be analyzed, ctDNA in plasma is not only a subfraction of cfDNA but also a mixture of fragmented alleles derived from different cancer deposits.
Epigenetic alterations have a high potential to provide a valuable source of innovative biomarkers for cancer, since they are stable, highly frequent for specific genes, and can also be detected in biological fluids in a minimally invasive way. A variety of studies on DNA methylation markers focus on the evaluation of their clinical significance as novel epigenetic biomarkers [7]. Among epigenetic alterations, DNA methylation is very important in cancer since it affects gene expression in a similar way to how mutations affect gene functions [8]. The fact that DNA methylation is playing a crucial role in all types of cancer makes it ideal as a source of a variety of tumor biomarkers [9, 10].
Analysis of DNA methylation markers in primary tumors has already shown impressive results. The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov) is an important source of information for DNA methylation biomarkers in many types of cancer. In a recent study, homogenous groups of secondary breast cancers to matched cohorts of primary breast cancers that were included in the TCGA were compared in order to identify specific gene expression and DNA methylation signatures [11]. Significant differences were identified in gene expression and DNA methylation signatures in invasive ductal carcinomas and in invasive lobular carcinomas; these differences were found to be important for tumor growth and proliferation.
The characterization of molecular alterations specific for different disease subtypes is highly important [11]. A recent study focused on an integrative molecular analysis in all available TCGA tumor specimens in 33 different types of cancer; this study has shown that samples were clustered according to histology, type of tissue, and anatomic origin [12]. This study has clearly shown that clustering based on DNA methylation data was highly influenced by the cell type, emphasizing the important role of the cell of origin [12]. Based on this, similarities at the molecular level among different cancer types that are related in terms of histology or anatomy could be used to develop a universal approach for the analysis of cancer; such an analysis is highly important in developing strategies for future therapeutic development [13].
DNA methylation in combination with liquid biopsy is very powerful in identifying circulating epigenetic biomarkers of clinical importance. A recent study has shown that DNA methylation analysis in ctDNA could provide information on the tissue of origin, thus contributing to the improvement of survival of cancer patients for a variety of tumors [14]. Compared to other classes of molecular biomarkers, such as mRNA and proteins, DNA methylation has many advantages since it is chemically stable and can be detected in clinical samples that are kept for a long time even under nonideal storage conditions [15]. Moreover, the incidence of aberrant methylation of specific CpG islands is high in tumor samples and can thus be easily detected by using genome‐wide screening technologies, in contrast to cancer‐specific mutations, which are not only rare but are also spread at many different positions in tumor DNA [15].
Despite all these advantages, up to now clinical tests are commercially available only for a very low number of DNA methylation‐based biomarkers (only 14 at present) [16, 17], and even a lower number of specific DNA methylation‐based tests are approved by the Food and Drug Administration (FDA) or European Medicines Agency as in vitro diagnostic (IVD) tests [7].
Changes in DNA methylation patterns in plasma are known to arise early during cancer pathogenesis [18]. Blood‐based DNA methylation tests are thus now being explored to develop tests for early cancer diagnosis [19]. ctDNA methylation analysis was used to stratify patients with primary central nervous system tumors [20] and successfully profile melanoma progression to brain metastasis [21]. According to a recent study, DNA methylation profiling could be used for a histomolecular stratification of patients with brain metastases [22].
Large‐scale targeted methylation sequencing of plasma cfDNA has a high potential for early cancer diagnosis. Recently, a novel assay targeting 9223 CpG sites that are consistently hypermethylated according to TCGA was developed and validated in plasma cfDNA from patients with advanced colorectal cancer (CRC), non‐small‐cell lung carcinoma (NSCLC), breast cancer, and melanoma [23]. According to the results presented, this method could not only detect these types of cancer with high accuracy, but, moreover, methylation scores in plasma cfDNA were correlated with treatment outcomes [23].
Another recent study has shown that cfDNA methylation analysis in plasma could provide some patterns that can detect and discriminate common primary intracranial tumors [24]. In renal cell carcinoma, a novel assay based on a combination of methylated cfDNA immunoprecipitation and high‐throughput sequencing was shown to detect early‐stage tumors and classify patients across all stages of the disease [25]. Based on a sensitive, immunoprecipitation‐based methodology for DNA methylation analysis of cfDNA, tumor‐specific changes were revealed and used further for early cancer detection and classification [26]. It was also shown that methylation profiling has the potential to track evolutionary changes in ctDNA [27].
In this review, we summarize the latest findings on DNA methylation markers in ctDNA for early detection, prognosis, minimal residual disease (MRD), risk of relapse, treatment selection, and resistance, for breast, prostate, lung, and CRC.
2. DNA methylation markers in ctDNA
The main analytical methodologies used for the analysis of ctDNA methylation markers are based either (a) on PCR following sodium bisulfite (SB) treatment that converts all nonmethylated cytocines to uracils and subsequently through PCR to thymines, which include methylation‐specific PCR (MSP), real‐time MSP, and droplet digital MSP (ddMSP) or (b) on a large‐scale omic approaches that include genome‐wide DNA methylation analysis, pyrosequencing, methyl‐BEAMing, array‐based genome‐wide DNA methylation analysis, highly multiplexed targeted next‐generation sequencing, and bisulfite sequencing. ctDNA methylation markers can provide information on early detection, prognosis, MRD, and therapy response in various types of cancer (Fig. 1).
Fig. 1.
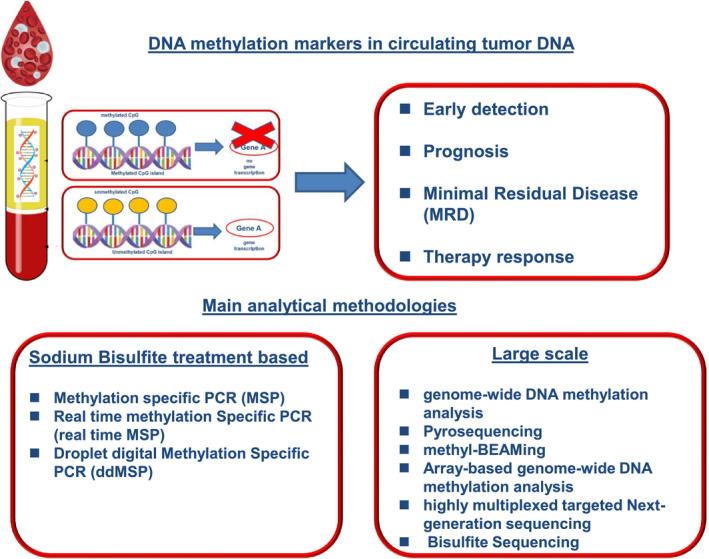
ctDNA methylation markers can provide information on early detection, prognosis, MRD, and therapy response. The main analytical methodologies are based either (a) on PCR following SB conversion or (b) on a large‐scale omic approach.
2.1. Breast cancer
2.1.1. Early detection
Detection of methylated ctDNA in the peripheral blood of cancer patients is highly promising for the discovery of highly sensitive and specific molecular biomarkers for screening and early detection. Recently, by using genome‐wide DNA methylation analysis, novel cfDNA biomarkers that could differentiate breast cancer patients from healthy individuals were identified for sporadic breast cancer [28]. According to a recent review based on 14 studies, cfDNA methylation analysis is highly promising for both early detection and disease relapse [29]. Methylation of SPAG6, NKX2‐6, ITIH5, and PER1 genes was detected early in serum of breast cancer patients [30].
Toward the same direction, another study has evaluated the prognostic significance of promoter methylation of seven genes (APC, BRCA1, CCND2, FOXA1, PSAT1, RASSF1A, and SCGB3A1) in primary tissues and cfDNA of breast cancer patients [31]. According to the results reported, methylation of APC, FOXA1, RASSF1A, or SCGB3A1 could discriminate noncancerous from cancerous tissues with an accuracy of 95%. The same study has shown that high PSAT1 methylation levels were associated with longer disease‐free survival (DFS), and that high FOXA1 methylation levels were associated with shorter DFS, while a combination of APC, FOXA1, and RASSF1A methylation provided a sensitivity, specificity, and accuracy of over 70% [31].
According to a very recent epigenome‐wide study, the DNA methylation profile in blood starts to change when breast cancer becomes invasive, and these changes can be detected years before the tumor is clinically detected, since differences in DNA methylation patterns are a consequence and not the cause of breast cancer [32]. Early detection of primary breast cancer through the analysis of epigenetic biomarkers in cfDNA by droplet digital PCR (ddPCR) was shown to give accurate results, comparable to mammography screening [33].
2.1.2. Prognosis
SOX17 promoter methylation was detected in plasma ctDNA in patients with operable breast cancer, after surgical removal of the primary tumor [34]. DNA methylation of five cancer‐related genes (KLK10, SOX17, WNT5A, MSH2, and GATA3) was evaluated as a prognostic marker in a study that included 150 and 16 breast cancer patients under adjuvant and neoadjuvant therapy, respectively, 34 patients with metastatic disease and 35 healthy volunteers by quantitative MSP (QMSP) [35]. According to the results presented in this study, GATA3 methylation was detected in all patient groups but not in the healthy control population. In the metastatic setting, WNT5A methylation was correlated with poor prognosis and a shorter overall survival (OS), SOX17 methylation to shorter PFS and OS, and KLK10 methylation to relapse. When at least 3 or 4 of these genes were methylated, a shorter OS and no response to therapy were documented in this group [35]. Genome‐wide methylation and QMSP have shown that CCND2 promoter hypermethylation was detected in 40.9% of breast tumors and 44.4% of plasma circulating cfDNA of patients, and could serve as a potential diagnostic and prognostic marker in breast cancer [36].
2.1.3. Minimal residual disease
In operable breast cancer, there is a high risk of recurrence and disease progression after surgery and therapeutic treatment. Early detection of MRD during a disease‐free follow‐up period would greatly improve patient outcomes. However, MRD monitoring in breast cancer through DNA methylation markers in ctDNA analysis is still not well established [37]. In breast cancer, ctDNA assays for mutation detection are not as yet ideal for MRD detection since different mutations can be present in the same gene in individual tumors, while DNA methylation markers of specific genes are universally present in cells of a common type and easier to be identified. In a very recent study, the detection and quantification of breast‐specific DNA methylation patterns in cfDNA were shown to have a sensitivity of 80% and a specificity of 97% for the detection of localized disease; an increase in cfDNA levels was associated with aggressive disease and a decrease was detected after neoadjuvant chemotherapy, while the detection of cfDNA after the completion of chemotherapy was an indication of MRD [38].
2.1.4. Therapy response and resistance
In 25–30% of hormone‐positive metastatic breast cancer patients treated with endocrine therapy, adaptive mechanisms emerge through alterations in the estrogen receptor ligand‐binding domain [39, 40]. Recently, ESR1 methylation in CTCs and corresponding plasma ctDNA was shown to be a potential biomarker for response to endocrine therapy; moreover, a high concordance of ESR1 methylation in CTCs and paired plasma ctDNA was shown in the metastatic setting [39]. Another similar study verified these results, by showing that ESR1 epigenetic status, as evaluated by methylation‐specific ddPCR (MS‐ddPCR), is indicative of resistance to endocrine therapy [40]. TBCRC 005, a multisite prospective study, revealed the prognostic significance of a panel of 10 cfDNA methylation markers for survival outcomes in metastatic breast cancer. However, the clinical utility of this assay for risk stratification and disease monitoring should be further confirmed [41]. In triple‐negative breast cancer, results of the GeparSixto trial have shown that MGMT promoter methylation was not related to different chemotherapy response rates [42].
In Table 1, we summarize the main studies on ctDNA methylation markers in breast cancer.
Table 1.
DNA methylation markers in breast cancer.
| DNA methylation markers evaluated | Type of sample/number of patients/controls | DNA methylation markers—of clinical significance | Methodology | Ref |
|---|---|---|---|---|
| Early detection | ||||
| 38 differentially methylated CpG positionsSelected marker: CYFIP1 | Leukocytes/sporadic breast cancer: 22 + 80/healthy women: 10 + 80 | Methylation at CYFIP1 was identified as a novel epigenetic biomarker candidate for sporadic breast cancer |
Genome‐wide DNA methylation analysis Illumina methylation arrays |
[28] |
| SPAG6, NKX2‐6, ITIH5, PER1 |
cfDNA serum test cohort (n = 103), serum validation cohort (n = 368), plasma cohort (n = 125) |
Novel biomarker candidates: SPAG6, NKX2‐6, and PER1 DCIS: SPAG6 and ITIH5 showed 63% sensitivity for early invasive tumor (pT1, pN0): SPAG6 and ITIH5 showed 51% sensitivity and 80% specificity DCIS detection—serum validation cohort: NKX2‐6 and ITIH5, 50% sensitivity DCIS detection—plasma cohort: SPAG6, PER1, and ITIH5 64% sensitivity |
TCGA/human methylation 450 BeadChip data/pyrosequencing | [30] |
| APC, BRCA1, CCND2, FOXA1, PSAT1, RASSF1A, and SCGB3A1 |
cfDNA cohort #1, n = 137 cfDNA cohort #2, n = 44 |
APC, FOXA1, RASSF1A, and SCGB3A1 discriminated normal from cancerous tissue with high accuracy (95.55%) High PSAT1 methylation levels associated with longer DFS Higher FOXA1 methylation levels associated with shorter DFS APC, FOXA1, and RASSF1A in cfDNA disclosed a sensitivity, specificity, and accuracy over 70%. |
Multiplex QMSP | [31] |
| 9601 CpG markers were identified associated with invasive breast cancer | Prospectively collected blood DNA samples from the Sister Study 1552 cases, 224 subcohort | DNA methylation profile in blood starts to change when breast cancer gets invasive | Epigenome‐wide study using Infinium HumanMethylation 450 Bead Chips | [32] |
| JAK3, RASGRF1, CPXM1, SHF, DNM3, CAV2, HOXA10, B3GNT5, ST3GAL6, DACH1, P2RX3, and chr8:23572595 |
Breast tissues: 56 microdissected cfDNA: 34 cell lines, and 29 blood samples from healthy volunteers (HVs, control group) cfDNA samples from 80 HVs and 87 cancer patients |
The best detection model adopted four methylation markers (RASGRF1, CPXM1, HOXA10, and DACH1) and two parameters (cfDNA concentration and the mean of 12 methylation markers) The area under the receiver operating characteristic curve for cancer normal discrimination was 0.916 and 0.876 in the training and validation dataset, respectively |
Array‐based genome‐wide DNA methylation analysis | [33] |
| An independent dataset of 53 HVs and 58 BC patients. |
The sensitivity and the specificity of the model were 0.862 (stages 0‐I 0.846, IIA 0.862, IIB‐III 0.818, metastatic BC 0.935) and 0.827, respectively Early detection of primary breast cancer through the analysis of epigenetic biomarkers was shown to give accurate results, comparable to mammography screening |
cfDNA: ddMSP | ||
| Prognosis | ||||
| SOX17 |
79 primary breast tumors, 114 paired samples of DNA isolated from CTCs 114 samples of cfDNA, 60 healthy individuals |
There was a significant correlation between SOX17 methylation in cfDNA and CTCs in patients with early breast cancer (P = 0.008), but not in patients with verified metastasis (P = 0.283) SOX17 promoter is highly methylated in primary breast tumors, in CTCs isolated from patients with breast cancer, and in corresponding cfDNA samples |
MSP | [34] |
| KLK10, SOX17, WNT5A, MSH2, GATA3 | Plasma cfDNA: 150 breast cancer patients under adjuvant therapy | The methylation of WNT5A was statistically significantly correlated with greater tumor size and poor prognosis characteristics and in advanced‐stage disease with shorter OS | Quantitative MSP | [35] |
| 16 breast cancer patients under neoadjuvant therapy | In the metastatic group, also SOX17 methylation was significantly correlated with the incidence of death, shorter PFS, and OS | |||
| 34 patients with metastatic disease | KLK10 methylation was significantly correlated with unfavorable clinicopathological characteristics and relapse, whereas in the adjuvant group to shorter DFI | |||
| 35 healthy volunteers | Methylation of at least 3 or 4 genes was significantly correlated with shorter OS and no pharmacotherapy response, respectively | |||
| CCND2 | 93 tumors and paired adjacent normal tissues of breast cancer patients circulating cfDNA : 18 breast cancer patients |
40.9% of breast tumors 44.4% of plasma circulating cfDNA CCND2 promoter hypermethylation is an independent poor prognostic factor |
Genome‐wide methylation and QMSP | [36] |
| Ten cfDNA methylation markers | Serum samples from 141 women at baseline, at week 4, and at first restaging | Prognostic significance for survival outcomes in metastatic breast cancer | Quantitative multiplex assay (cMethDNA) | [41] |
| Therapy response | ||||
| ESR1 |
CTCs, ctDNA: 65 primary breast tumors FFPE, EpCAM+ CTC fractions (122 patients and 30 healthy donors; HD), plasma ctDNA (108 patients and 30HD) CTCs, CellSearch, and paired plasma ctDNA for 58 patients with breast cancer |
ESR1 methylation was detected in:
ESR1 methylation was highly concordant in 58 paired DNA samples, isolated from CTCs (CellSearch) and corresponding plasma ESR1 methylation was observed in 10/36 (27.8%) CTC‐positive samples and was associated with lack of response to treatment (P = 0.023, Fisher's exact test) |
Real‐time MSP | [39] |
| ESR1 | ctDNA: 49 women with hormone receptor‐positive HER2‐negative MBC were prospectively enrolled before treatment start and after 3 months | An epigenetic characterization strategy based on ctDNA is capable of being integrated in the current clinical workflow to give useful insights on treatment sensitivity | MS‐ddPCR | [40] |
2.2. Prostate cancer
2.2.1. Early detection
Numerous diagnostic tests based on prostate‐specific antigen (PSA) have been developed so far to improve early detection of prostate cancer; however, minimally invasive tests with better specificity and sensitivity are still needed to further improve diagnosis and risk stratification. Tests based on DNA methylation markers in cfDNA are highly promising for early detection since they are minimally invasive, highly specific, and can be detected at very early stages of the disease. These epigenetic alterations can be detected in both plasma and urine of prostate cancer patients [43]. In localized and de novo metastatic prostate cancer, three genes, DOCK2, HAPLN3, and FBXO30, were found to be specifically hypermethylated in prostate cancer tissues using MS‐ddPCR [44]. In plasma cfDNA, methylation of these three genes was detected in 61.5% of metastatic prostate cancer patients and was associated with significantly shorter time to progression to metastatic castration‐resistant prostate cancer (mCRPC), indicating a potential usefulness for the identification of hormone‐naïve metastatic prostate cancer patients who could benefit from intensified treatment [44]. It was also recently reported that DNA methylation in regions of chromosome 8q24 may be associated with the risk of developing prostate cancer [45]. Another study has shown that the detection of promoter methylation of MCAM, ERα, and ERβ genes in serum ctDNA could be utilized as a combined biomarker for the early detection of prostate cancer, with a sensitivity and specificity almost equal to and better than serum PSA, respectively [46].
The quantification of DNA methylation levels of another set of eight genes (APC, FOXA1, GSTP1, HOXD3, RARβ2, RASSF1A, SEPT9, and SOX17) in plasma, based on a multiplex QMSP, was evaluated for the early detection of lung, prostate, and CRC [47]. Out of these eight genes, only two, SEPT9 and SOX17, were methylated in all three cancers; FOXA1, RARβ2, and RASSF1A methylation was detected in lung and prostate cancer with 64% sensitivity and 70% specificity; and methylation of GSTP1 and SOX17 could discriminate lung cancer from prostate cancer with 93% specificity [47]. A robust validation of these studies in large prospective cohorts would be of high importance since positive findings could reduce the numbers of unnecessary prostate biopsies.
2.2.2. Prognosis
In localized intermediate‐grade prostate cancer, current clinically established prognostic markers, such as PSA, lack sensitivity and specificity in distinguishing aggressive from indolent disease; toward this direction, the identification of novel prognostic methylation biomarkers for prostate cancer is highly important [48]. In a recent prospective study, the prognostic significance of promoter methylation of GSTP1 and APC in ctDNA was evaluated in castration‐resistant prostate cancer. According to the results reported, pretreatment detection of DNA methylation of these markers is prognostic for worse OS [49]. In another study, DNA methylation of eight genes was evaluated by pyrosequencing in prostate cancer and results were used in relation to the Gleason score for patient stratification. According to the results presented, DNA methylation based on five genes in relation to the Gleason score could predict metastatic lethal progression and is highly promising for risk stratification of patients in the advanced stage [50].
2.2.3. Minimal residual disease
Promoter methylation of two genes, namely ST6GALNAC3 and ZNF660, after being evaluated in 705 prostate cancer tissues and 110 nonmalignant tissue samples, was reported to be cancer‐specific with an area under curve in ROC curves of 0.917–0.995 and 0.846–0.903 [51]. In the same study, hypermethylation of ZNF660 was significantly associated with biochemical recurrence, indicating a potential utility for the stratification of low/intermediate‐grade cases into indolent or more aggressive subtypes [51]. In the same study, using ddPCR, promoter methylation of these two genes (ST6GALNAC3 and ZNF660) and additionally CCDC181 and HAPLN3 was evaluated in ctDNA of 27 patients with prostate cancer and 10 patients with benign prostate hyperplasia using MS‐ddPCR [51]. ctDNA analysis based on methylation of three of these genes (ST6GALNAC3, CCDC181, and HAPLN3) could detect prostate cancer with 100% diagnostic specificity and 67% diagnostic sensitivity [51].
2.2.4. Therapy resistance and response
DNA methylation is highly important in prostate cancer initiation and progression. Epigenetic alterations that regulate prostate cancer progression could provide a source of biomarkers indicating resistance or response to specific therapies [52]. Abiraterone acetate (AA) is administered to patients with mCRPC, and the identification of predictive biomarkers to this drug is highly important. A recent study that was performed in plasma cfDNA in a group of 108 samples from 33 prostate cancer patients treated with AA provided a list of DNA methylation‐based predictive biomarkers for response to AA treatment [53]. When serially collected cfDNA samples from mCRPC patients under androgen deprivation therapy were analyzed by genome‐wide methylation analysis, maintenance of changes in DNA methylation profiles indicated a longer time to clinical progression, while the detection of DNA methylation markers for neuroendocrine CRPC indicated a faster time to clinical progression [54]. A recently developed cfDNA methylation assay specific for prostate cancer, called mDETECT, was evaluated in a relatively low number of plasma samples sequentially collected from prostate cancer patients who were beginning androgen deprivation therapy; this test was based on a highly multiplexed targeted next‐generation sequencing of PCR products, comprising 46 PCR probes to 40 regions [53]. Results of this test were comparable to PSA values at different time points [55].
In Table 2, we summarize the main studies on ctDNA methylation markers in prostate cancer.
Table 2.
DNA methylation markers in prostate cancer.
| DNA methylation markers tested | Type of sample/number of patients/controls | Selected DNA methylation markers of clinical significance | Methodology | Ref |
|---|---|---|---|---|
| Early detection | ||||
| DNA methylome | Four metastatic treatment‐naïve prostate cancer (PCa) patients urine and plasma | Urine and plasma are viable surrogates for tumor tissue biopsies, capturing up to 39.40% and 64.14% of tumor‐specific methylation alterations, respectively | Infinium® Methylation EPIC BeadChip (Illumina) | [43] |
|
63 CpG sites located nearby the cancer susceptibility SNPs at 8q24 or in promoter, exon 2, exon 3, or 30 regions for MYC |
694 prostate cancer cases including 172 aggressive cases (stage III/IV or Gleason score >8) 516 nonaggressive cases (stage I/II and Gleason score >8) 703 controls |
8q24 DNA methylation levels may be associated with prostate cancer risk 8 CpG sites whose DNA methylation levels were associated with the risk of overall prostate cancer The most significant CpG site overall was located at Chr8:128428897 in POU5F1B When the cases were stratified by disease aggressiveness, two moderately correlated CpG sites in MYC (Chr8:128753187 and Chr8:128753154) were identified that were specifically associated with the risk of aggressive but not nonaggressive prostate cancer |
Targeted pyrosequencing assays | [45] |
| SSBP2, MCAM, ERα, ERβ, APC, CCND2, MGMT, GSTP1, p16, and RARβ2 | 84 serum samples from PC, 30 controls 7 cases diagnosed as high‐grade prostatic intraepithelial neoplasia | MCAM, ERα, and Erβ combined biomarker for the early detection of prostate cancer, a combination marker panel of MCAM, ERα, and ERβ increased the sensitivity to 75% and the specificity became 70% for the minimally invasive early detection test of PC with a sensitivity and specificity almost equal and better than serum PSA | QMSP | [46] |
| Prognosis | ||||
| DOCK2, HAPLN3, and FBXO30 |
cfDNA plasma samples 36 healthy controls 61 benign prostatic hyperplasia (BPH) 102 localized PCa 65 de novo mPCa patients |
ctDNA methylation of DOCK2, HAPLN3, and/or FBXO30 was detected in 61.5% (40/65) of de novo mPCa patients ctDNA methylation of DOCK2, HAPLN3, and/or FBXO30 was markedly increased in high‐volume compared to low‐volume mPCa (89.3% (25/28) vs 32.1% (10/31), P < 0.001) Detection of methylated ctDNA was associated with significantly shorter time to progression to metastatic castration‐resistant PCa, independent of tumor volume Methylated ctDNA (DOCK2/HAPLN3/FBXO30) may be potentially useful for identification of hormone‐naïve mPCa patients who could benefit from intensified treatment. |
MS‐ddPCR/cfDNA | [44] |
| APC, FOXA1, GSTP1, HOXD3, RARβ2, RASSF1A, SEPT9, and SOX17 |
Circulating cfDNA 121 PCa 136 asymptomatic donors' plasma samples |
FOXA1, RARβ2, and RASSF1A detected PCa with 64% sensitivity and 70% specificity | Multiplex QMSP/cfDNA plasma | [47] |
| GSTP1 and APC | Plasma cfDNA prospective study, 50 CRPC patients Control group 10 healthy age‐matched men 10 men aged under 35 10 healthy women | Prognostic significance, OS | MSP | [49] |
| CpG methylation of eight biomarkers previously identified using the HumanMethylation 450 array |
Training dataset: 366 men with no evidence of recurrence and 58 who developed metastasis or died of PCa Testing dataset: 29 cases with metastatic lethal PCa Comparison group: 29 cases who remained recurrence‐free for at least 5 years postsurgery |
Five CpGs in relation to the Gleason score could predict metastatic lethal progression and is highly promising for risk stratification of patients in the advanced stage | Pyrosequencing | [50] |
| MRD | ||||
| ST6GALNAC3 and ZNF660 in primary tissues ST6GALNAC3, ZNF660, CCDC181, and HAPLN3 promoter methylation in liquid biopsies |
705 prostate cancer tissues, 110 nonmalignant tissue samples Liquid biopsies (serum): 27 patients with prostate cancer 10 patients with BPH (control) |
In tissues, hypermethylation of ST6GALNAC3 and ZNF660 was highly cancer‐specific with AUC of ROC curve analysis of 0.917–0.995 and 0.846–0.903, respectively |
Primary tissues: MS qPCR or methylation array Liquid Biopsies: ddMSP analysis |
[51] |
| Therapy response | ||||
| 485 577 cytosines interrogated by the microarray in each sample | Plasma cfDNA in a group of 108 samples from 33 prostate cancer patients treated with AA | AA differentially modified positions: 26 874 cytosines were differentially modified when comparing AA‐sensitive with the AA‐resistant patients DNA methylation‐based predictive biomarkers for response to AA treatment | Infinium HumanMethylation 450K BeadChip | [53] |
| cfDNA methylome analysis | 45 plasma cfDNA serially collected cfDNA samples from 16 mCRPC patients under androgen deprivation therapy (12 enzalutamide‐treated 4 abiraterone‐treated) |
RUNX3, RGS12, and FBP1 GSTP1, TBX15, AOX1 NPBWR1, ZSCAN12, PCDHGA11, PHOX2A, TBX10, TEX28, TKTL1, and TSPAN32 B3GNTL1, FAM19A5, INPP5A, MAD1L1, MCF2L, MYT1L, and PRDM16 Monitoring the cfDNA methylome during therapy in mCRPC may serve as predictive marker of response to androgen targeting agents |
Genome‐wide methylation analysis (cfMeDIP‐seq) | [54] |
| 40 regions were identified that were each methylated between 47% and 94% of TCGA patient samples | Seven patients with biochemical recurrence that were initiating androgen deprivation therapy (ADT) |
Overall 86% of patients were positive for 20 or more of these regions, and only 6.6% of patients had 5 or less probes positive mDETECT levels seemed to anticipate rising PSA levels, suggesting it may be able to provide an earlier indication of tumor progression, as well as tracking tumor burden in a PSA‐negative tumor |
mDETECT: highly multiplexed targeted next‐generation sequencing of targeted PCR products comprising of 46 PCR probes to 40 regions |
[55] |
2.3. Lung cancer
During the last 20 years, numerous studies have shown the importance of epigenetic alterations in lung cancer. DNA methylation of specific genes has been shown to play an important role in lung cancer pathogenesis, and can provide novel biomarkers for early detection, prognosis, and prediction of response to specific treatments [56]. In parallel, liquid biopsy analysis has a significant contribution to the management of lung cancer patients [57] and the detection of EGFR mutations in plasma cfDNA is now used on a routine basis for the stratification of NSCLC patients [58, 59]. Beyond gene mutations, alterations in DNA methylation of specific genes can provide novel epigenetic biomarkers in NSCLC that could be used for early detection, prognosis, and prediction of response to specific therapies [9, 60, 61]. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from NSCLC patients was first reported in 1999 by Esteller et al. [62]. Since then, DNA methylation was shown in many studies to be an ideal source of candidate biomarkers since it is stable, easy to detect, and can be detected at high percentages in tumor samples [9, 63]. In lung cancer, a variety of genes were found to be methylated in various types of samples, including tissues, plasma, sputum, and even bronchoscopic washings/brushings [9]. Liquid biopsy analysis of DNA methylation markers is highly promising for diagnosis, prognosis, risk assessment, and disease monitoring [64].
2.3.1. Early detection
Plasma‐based detection of cancer‐specific DNA methylation markers may provide a simple cost‐effective method for the early detection of lung cancer and can be used for noninvasive diagnostics and monitoring. A very recent review on the main approaches to develop biomarkers for the early detection of lung cancer, with considerations of detection of rare tumor events, focused on DNA methylation‐based detection in plasma and sputum [65]. In a recent study, analysis of DNA methylation markers in plasma of NSCLC patients using DNA methylation‐specific qPCR could distinguish lung cancer patients from healthy controls with high sensitivity and specificity [66].
2.3.2. Prognosis
When DNA methylation of APC, HOXA9, RARβ2, and RASSF1A was evaluated by QMSP in lung cancer, it was found that HOXA9 and RASSF1A had higher methylation levels in small‐cell lung cancer than in NSCLC, with HOXA9 methylation levels displaying a sensitivity of 63.8%, and RASSF1A displaying a specificity of 96.2% for small‐cell lung cancer detection in ctDNA [67]. Additionally, HOXA9 methylation levels were higher in squamous cell carcinoma than in adenocarcinoma [67]. Very recently, the detection of KMT2C (MLL3) promoter methylation was detected in plasma cfDNA of NSCLC patients at both early and advanced stages, but not in plasma of healthy individuals [68]. Promoter methylation of this gene in plasma cfDNA needs to be further evaluated in a large and well‐defined patient cohort [68]. Promoter methylation of two other genes, namely BRMS1 and SOX17, in plasma ctDNA from NSCLC patients has also been shown to be of prognostic significance [69, 70].
2.3.3. Therapy response and resistance
Up to now, only a few studies have evaluated the potential of DNA methylation markers in ctDNA of lung cancer patients for therapy resistance. Recently, a combined detection of somatic mutations and DNA methylation markers in plasma cfDNA was used to evaluate response to osimertinib in NSCLC patients positive for the T790M EGFR mutation [71]. According to the results presented, DNA methylation levels were significantly higher in the plasma samples of patients with somatic mutations than in patients without mutations and healthy controls; a decrease in DNA methylation levels was associated with better treatment efficacy, while an increase indicated disease progression [71].
In Table 3, we summarize the main studies on ctDNA methylation markers in lung cancer.
Table 3.
DNA methylation markers in lung cancer.
| DNA methylation markers tested | Type of sample/number of patients/controls |
Selected DNA methylation markers of clinical significance |
Methodology | Ref |
|---|---|---|---|---|
| Early detection | ||||
| Set of 10 marker loci |
Liquid biopsy test plasma cfDNA NSCLC patients: 18 healthy: 47 |
Distinguish lung cancer patients from healthy controls with high sensitivity and specificity | Real‐time MSP | [66] |
| APC, HOXA9, RARβ2, RASSF1A |
152 tissue samples 129 plasma samples; 28 benign lesions of lung |
HOXA9 and RASSF1A displayed higher methylation levels in SCLC than in NSCLC HOXA9 methylation levels sensitivity: 63.8% for SCLC detection in ccfDNA RASSF1A methylation levels specificity: 96.2% for SCLC detection in ccfDNA |
Quantitative MSP | [67] |
| KMT2C (MLL3) |
Operable NSCLC: 48 fresh frozen NSCLC tissues, 48 adjacent non‐neoplastic tissues, 48 matched plasma samples Metastatic NSCLC: 91 NSCLC plasma samples; 60 plasma samples from HD |
In metastatic NSCLC, KMT2C promoter methylation in plasma cfDNA was related to worse PFS worse OS | Real‐time MSP | [68] |
| Prognosis | ||||
| HOXA9, KRTAP8‐1, CCND1, TULP2 |
TCGA: 338 tissue samples from lung adenocarcinoma patients including 149 nonmalignant ones Tumor samples and matched adjacent lung samples from 25 patients |
Methylation of HOXA9, KRTAP8‐1, CCND1, and TULP2 has great potential for the early recognition of lung adenocarcinoma | Pyrosequencing | [60] |
| MGMT, p16, DAP kinase, GSTP1 | Normal lung, primary NSCLC, and corresponding serum were obtained from each of the 22 patients |
First report on the detection of aberrant promoter hypermethylation of tumor suppressor genes Abnormal promoter hypermethylation of tumor suppressor genes is readily detectable in the serum DNA of cancer patients using MSP analysis |
MSP | [62] |
| SOX17 |
Operable NSCLC: 57 primary tumors and paired adjacent noncancerous tissues and in ctDNA isolated from 48 corresponding plasma samples Advanced NSCLC: Plasma from 74 patients with and 49 healthy individuals |
Detection of SOX17 promoter methylation in plasma provides prognostic information | Real‐time MSP | [69] |
| Breast cancer metastasis suppressor 1 (BRMS1) | 57 NSCLC tumors and adjacent noncancerous tissues, cfDNA, 48 corresponding plasma samples, cfDNA isolated from plasma of 74 patients with advanced NSCLC and 24 healthy individuals. | Methylation of BRMS1 promoter in cfDNA isolated from plasma of NSCLC patients provides important prognostic information | Real‐time MSP | [70] |
| Therapy response | ||||
| Combined detection of somatic mutations and DNA methylation markers | 85 longitudinal plasma samples obtained from 8 stage IV osimertinib‐treated EGFR T790 M‐positive lung adenocarcinoma patients |
The methylation levels were significantly higher in the plasma samples of patients with detectable somatic mutations than patients without somatic mutations and healthy controls. A decrease in DNA methylation levels was associated with the efficacy of treatment, while an increase was indicating disease progression |
Bisulfite sequencing | [71] |
2.4. Colorectal cancer
2.4.1. Early detection
In CRC, a blood‐based test based on real‐time MSP detection of methylated Septin9 in DNA obtained from peripheral blood samples has been FDA‐approved for early detection; however, a positive result should still be verified by colonoscopy or sigmoidoscopy [17]. Methylated Septin9 also has a high potential to be used as a routine biomarker for CRC recurrence monitoring, especially in combination with contrast‐enhanced computed tomography [72]. Another approach to identify potential serum methylation biomarkers for the detection of advanced CRC is to use pooled samples; unsupervised clustering has shown that cfDNA methylation patterns can distinguish advanced neoplasia from healthy controls [73]. Methylated ctDNA markers are highly promising for the development of a blood‐based CRC screening liquid biopsy test [74, 75, 76].
2.4.2. Prognosis
RASSF1A promoter methylation was reported as a prognostic biomarker in patients with stage II and III CRC receiving oxaliplatin‐based chemotherapy, when investigated by MSP in 108 CRC patients before and after chemotherapy and 78 healthy controls [77]. DNA methylation of BCAT1 and IKZF1 was recently reported in ctDNA of CRC patients, and it was shown to be related to CRC stage; after surgery, these DNA methylation markers were not detected, indicating a possible role of these markers on the adequacy of surgical resection [78].
2.4.3. Therapy response and resistance
In a prospective study, a combined detection of NPY methylation along with tumor‐specific mutations in ctDNA could give similar results to radiographic evaluation, showing that this combined liquid biopsy approach can be used for the follow‐up of mCRC patients during treatment [79]. According to a recent study analysis, a five‐gene methylation panel (EYA4, GRIA4, ITGA4, MAP3K14 ‐AS1, and MSC) in cfDNA using ddPCR can be used in cases where patient‐specific mutations cannot be detected for monitoring tumor burden dynamics in liquid biopsy under different therapeutic regimens [80]. The detection of MGMT methylation in plasma ctDNA could be used as a predictive biomarker of response to alkylating agents [81].
In Table 4, we summarize the main studies on ctDNA methylation markers in CRC.
Table 4.
DNA methylation markers in CRC.
| DNA methylation markers tested | Type of sample/number of patients/controls | Selected DNA methylation markers of clinical significance | Methodology | Ref |
|---|---|---|---|---|
| Early detection | ||||
| Septin9 |
93 patients with CRC and 94 individuals with no evidence of disease 135 patients with CRC, 91 healthy controls, 169 patients with adenomatous polyps 81 patients with hyperplastic polyps |
FDA‐approved for early detection of CRC a positive result should still be verified by colonoscopy or sigmoidoscopy discriminated between patients with CRC and healthy controls with high clinical sensitivity and specificity in pivotal case–control studies |
Real‐time MSP Epi proColon® 2.0 CE |
[17] |
| Septin9 | 650 plasma samples |
Routine biomarker for CRC recurrence monitoring, especially in combination with contrast‐enhanced computed tomography mSEPT9 analysis might be popularized as a routine biomarker for CRC screening. The combined detection of mSEPT9 and CECT can play an important role for recurrence monitoring |
Real‐time MSP Epi proColon® 2.0 CE |
[72] |
| 866 836 CpG positions across the genome | Serum samples from 20 individuals with no colorectal findings, 20 patients with advanced adenomas, 20 patients with CRC (stages I and II) |
cfDNA methylation patterns can distinguish advanced neoplasia from healthy controls The differential methylation analysis revealed 1384 CpG sites with at least 10% difference in the methylation level between no colorectal findings controls and advanced neoplasia, the majority of which were hypomethylated |
Methylation levels of 866 836 CpG positions across the genome using the MethylationEPIC array Unsupervised clustering |
[73] |
| Prognosis | ||||
| RASSF1A | 108 CRC patients before and after chemotherapy and 78 healthy controls | Promoter methylation of RASSF1A can influence sensitivity to oxaliplatin‐based chemotherapy, which can be used to predict outcomes for patients with stage II and stage III CRC | Real‐time MSP | [77] |
| MRD | ||||
| BCAT1 and IKZF1 |
91 cancer tissues 187 cfDNA samples |
Significant methylation of either BCAT1 or IKZF1 was seen in 86/91 (94.5%) cancer tissues ctDNA methylated in BCAT1 or IKZF1 was detected in 116 (62.0%) cases at diagnosis and was significantly more likely to be detected with later stage and distal tumor location BCAT1 and IKZF1 related to CRC stage while after surgery these DNA methylation markers were not detected, indicating a possible role of these markers on the adequacy of surgical resection |
Real‐time multiplex PCR assay | [78] |
| Therapy response | ||||
| NPY | 24 metastatic CRC patients | Prospective study, a combined detection of NPY methylation along with tumor‐specific mutations in ctDNA could give similar results to radiographic evaluation, showing that this combined liquid biopsy approach can be used for the follow‐up of mCRC patients during treatment | Droplet‐based digital PCR (ddPCR) | [79] |
| EYA4, GRIA4, ITGA4, MAP3K14 ‐AS1, MSC |
85 tissue DNA 182 cfDNA from mCRC patients |
87% of mCRC patients (87%) showed positivity in at least one marker EYA4: 67%, GRIA4: 71.3%, ITGA4: 69.2%, MAP3K14 ‐AS1: 69.8%, MSC: 62.1% Methylation can be used as a universal test to circumvent the absence of patient‐specific mutations for monitoring tumor burden dynamics via liquid biopsy |
Genome‐wide methylation microarrays ddPCR |
[80] |
| MGMT | 60 metastatic CRC tissue samples | Predictive biomarker of response to alkylating agents |
Methyl‐BEAMing Pyrosequencing |
(81) |
3. ctDNA methylation assays: standardization and pre‐analytical considerations
Pre‐analytical conditions could significantly affect the results of DNA methylation analyses in liquid biopsies. For this reason, standardization of pre‐analytical conditions and implementation of quality control steps is extremely important for reliable liquid biopsy analysis, and a prerequisite for routine applications in the clinic. In a recent systematic study, the stability of DNA methylation in plasma and SB‐converted DNA under different storage conditions was evaluated [63]. In the same study, the reliability of whole‐genome amplification procedures for SB‐converted DNA samples was checked by real‐time MSP for ACTB, SOX17, and BRMS1. According to this study, plasma and SB‐converted DNA samples are stable and can be used safely for MSP when kept at −80 °C [63].
Different blood collection tubes and cfDNA isolation methods can influence the cfDNA amount and the detection of promoter methylation [82, 83]. Recently, there has been an international effort to standardize liquid biopsy procedures and protocols, such as the International Liquid Biopsy Standardization Alliance (https://fnih.org/what‐we‐do/biomarkers‐consortium/programs/ilsa)—who recently provided a white paper focused on the independent liquid biopsy‐ and standardization‐based programs [84]—the European Liquid Biopsy Society (https://www.uke.de/english/departments‐institutes/institutes/tumor‐biology/european‐liquid‐biopsy‐society‐elbs/index.html) and the International Society of Liquid Biopsy (https://www.isliquidbiopsy.org/).
4. Conclusions and future perspectives
ctDNA methylation analysis has the potential to improve early cancer detection, which could lead to a substantial reduction in cancer‐related mortality. Biomarkers based on DNA methylation in ctDNA have a huge potential to be used for screening, early diagnosis, and in predicting and monitoring the response to specific therapies [85]. However, despite considerable interest in the field of discovering and developing novel biomarkers, a lot of improvements are still necessary. Toward the discovery of novel reliable biomarkers that can be measured with high specificity and sensitivity at an acceptable cost in the routine clinical setting, epigenetic alterations in liquid biopsy samples are highly promising. However, further research is required to determine which of these methylated ctDNA markers are the most accurate when applied to large cohorts of patients.
Acknowledgements
This study has been financially supported by the European Union and Greek National Funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH–CREATE–INNOVATE (Project Code: T1RCI‐02935) and the ERA‐NET on Translational Cancer Research (TRANSCAN‐2) Project PROLIPSY ‘Minimally and non‐invasive methods for early detection and/or progression of cancer’.
References
- 1. Lianidou E & Pantel K (2019) Liquid biopsies. Genes Chromosomes Cancer 58, 219–232. [DOI] [PubMed] [Google Scholar]
- 2. Lianidou E & Hoon D (2017) Circulating tumor cells and circulating tumor DNA. In The Tietz Textbook of Clinical Chemistry and Molecular Diagnostics (Nader R, Horrath AR & Wittwer C, eds), pp. 1111–1144.Elsevier Ltd, Amsterdam. [Google Scholar]
- 3. Keller L, Belloum Y, Wikman H & Pantel K (2021) Clinical relevance of blood‐based ctDNA analysis: mutation detection and beyond. Br J Cancer 124, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Corcoran RB & Chabner BA (2018) Application of cell‐free DNA analysis to cancer treatment. N Engl J Med 379, 1754–1765. [DOI] [PubMed] [Google Scholar]
- 5. Alix‐Panabières C & Pantel K (2016) Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 6, 479–491. [DOI] [PubMed] [Google Scholar]
- 6. Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A et al. (2018) Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science 359, 926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. García‐Giménez JL, Seco‐Cervera M, Tollefsbol TO, Romá‐Mateo C, Peiró‐Chova L, Lapunzina P & Pallardó FV (2017) Epigenetic biomarkers: current strategies and future challenges for their use in the clinical laboratory. Crit Rev Clin Lab Sci 54, 529–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dor Y & Cedar H (2018) Principles of DNA methylation and their implications for biology and medicine. Lancet (London, England) 392, 777–786. [DOI] [PubMed] [Google Scholar]
- 9. Balgkouranidou I, Liloglou T & Lianidou ES (2013) Lung cancer epigenetics: emerging biomarkers. Biomark Med 7, 49–58. [DOI] [PubMed] [Google Scholar]
- 10. Mastoraki S & Lianidou E (2017) DNA and histone methylation in lung cancer. In the DNA and Histone Methylation as Cancer Targets (Kaneda A & Tsukada Y, eds), pp. 403–436.Humana Press, Cham. [Google Scholar]
- 11. Graff‐Baker AN, Orozco JIJ, Marzese DM, Salomon MP, Hoon DSB & Goldfarb M (2018) Epigenomic and transcriptomic characterization of secondary breast cancers. Ann Surg Oncol 25, 3082–3087. [DOI] [PubMed] [Google Scholar]
- 12. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V et al. (2018) Cell‐of‐origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R et al. (2018) The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep 23, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moran S, Martínez‐Cardús A, Sayols S, Musulén E, Balañá C, Estival‐Gonzalez A, Moutinho C, Heyn H, Diaz‐Lagares A, de Moura MC et al. (2016) Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol 17, 1386–1395. [DOI] [PubMed] [Google Scholar]
- 15. Lianidou ES (2016) Gene expression profiling and DNA methylation analyses of CTCs. Mol Oncol 10, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L et al. (2018) Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol 15, 459–466. [DOI] [PubMed] [Google Scholar]
- 17. Lamb YN & Dhillon S (2017) Epi proColon® 2.0 CE: a blood‐based screening test for colorectal cancer. Mol Diagn Ther 21, 225–232. [DOI] [PubMed] [Google Scholar]
- 18. Leygo C, Williams M, Jin HC, Chan MWY, Chu WK, Grusch M & Cheng YY (2017) DNA methylation as a noninvasive epigenetic biomarker for the detection of cancer. Dis Markers 2017, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Widschwendter M, Jones A, Evans I, Reisel D, Dillner J, Sundström K, Steyerberg EW, Vergouwe Y, Wegwarth O, Rebitschek FG et al. (2018) Epigenome‐based cancer risk prediction: rationale, opportunities and challenges. Nat Rev Clin Oncol 15, 292–309. [DOI] [PubMed] [Google Scholar]
- 20. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE et al. (2018) DNA methylation‐based classification of central nervous system tumours. Nature 555, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, Kiyohara E, Wang J, Kawas NP, Donovan NC et al. (2014) Epigenome‐wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet 23, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Orozco JIJ, Knijnenburg TA, Manughian‐Peter AO, Salomon MP, Barkhoudarian G, Jalas JR, Wilmott JS, Hothi P, Wang X, Takasumi Y et al. (2018) Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat Commun 9, 4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, Samet Y, Maoz M, Druid H, Arner P et al. (2018) Comprehensive human cell‐type methylation atlas reveals origins of circulating cell‐free DNA in health and disease. Nat Commun 9, 5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nassiri F, Chakravarthy A, Feng S, Shen SY, Nejad R, Zuccato JA, Voisin MR, Patil V, Horbinski C, Aldape K et al. (2020) Detection and discrimination of intracranial tumors using plasma cell‐ free DNA methylomes. Nat Med 26, 1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, Chakravarthy A, Shen SY, Bakouny Z, Boccardo F et al. (2020) Detection of renal cell carcinoma using plasma and urine cell‐free DNA methylomes. Nat Med 26, 1041–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T et al. (2018) Sensitive tumour detection and classification using plasma cell‐free DNA methylomes. Nature 563, 579–583. [DOI] [PubMed] [Google Scholar]
- 27. Cresswell GD, Nichol D, Spiteri I, Tari H, Zapata L, Heide T, Maley CC, Magnani L, Schiavon G, Ashworth A et al. (2020) Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking. Nat Commun 11, 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cappetta M, Fernandez L, Brignoni L, Artagaveytia N, Bonilla C, López M, Esteller M, Bertoni B & Berdasco M (2021) Discovery of novel DNA methylation biomarkers for non‐invasive sporadic breast cancer detection in the Latino population. Mol Oncol 15, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheuk IW, Shin VY & Kwong A (2017) Detection of methylated circulating DNA as noninvasive biomarkers for breast cancer diagnosis. J Breast Cancer 20, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mijnes J, Tiedemann J, Eschenbruch J, Gasthaus J, Bringezu S, Bauerschlag D, Maass N, Arnold N, Weimer J, Anzeneder T et al. (2019) SNiPER: a novel hypermethylation biomarker panel for liquid biopsy based early breast cancer detection. Oncotarget 10, 6494–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Salta S, Nunes P, Fontes‐Sousa M, Lopes P, Freitas M, Caldas M, Antunes L, Castro F, Antunes P, Palma de Sousa S et al. (2018) A DNA methylation‐based test for breast cancer detection in circulating cell‐free DNA. J Clin Med 7, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu Z, Sandler DP & Taylor JA (2020) Blood DNA methylation and breast cancer: a prospective case‐cohort analysis in the sister study. J Natl Cancer Inst 112, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uehiro N, Sato F, Pu F, Tanaka S, Kawashima M, Kawaguchi K, Sugimoto M, Saji S & Toi M (2016) Circulating cell‐free DNA‐based epigenetic assay can detect early breast cancer. Breast Cancer Res 18, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chimonidou M, Strati A, Malamos N, Georgoulias V & Lianidou ES (2013) SOX17 promoter methylation in circulating tumor cells and matched cell‐free DNA isolated from plasma of patients with breast cancer. Clin Chem 59, 270–279. [DOI] [PubMed] [Google Scholar]
- 35. Panagopoulou M, Karaglani M, Balgkouranidou I, Biziota E, Koukaki T, Karamitrousis E, Nena E, Tsamardinos I, Kolios G, Lianidou E et al. (2019) Circulating cell‐free DNA in breast cancer: size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 38, 3387–3401. [DOI] [PubMed] [Google Scholar]
- 36. Hung CS, Wang SC, Yen YT, Lee TH, Wen WC & Lin RK (2018) Hypermethylation of CCND2 in lung and breast cancer is a potential biomarker and drug target. Int J Mol Sci 19, 3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin SY, Orozco JIJ & Hoon DSB (2018) Detection of minimal residual disease and its clinical applications in melanoma and breast cancer patients. Adv Exp Med Biol 1100, 83–95. [DOI] [PubMed] [Google Scholar]
- 38. Moss J, Zick A, Grinshpun A, Carmon E, Maoz M, Ochana BL, Abraham O, Arieli O, Germansky L, Meir K et al. (2020) Circulating breast‐derived DNA allows universal detection and monitoring of localized breast cancer. Ann Oncol 31, 395–403. [DOI] [PubMed] [Google Scholar]
- 39. Mastoraki S, Strati A, Tzanikou E, Chimonidou M, Politaki E, Voutsina A, Psyrri A, Georgoulias V & Lianidou E (2018) ESR1 methylation: a liquid biopsy‐based epigenetic assay for the follow‐up of patients with metastatic breast cancer receiving endocrine treatment. Clin Cancer Res 24, 1500–1510. [DOI] [PubMed] [Google Scholar]
- 40. Gerratana L, Basile D, Franzoni A, Allegri L, Viotto D, Corvaja C, Bortot L, Bertoli E, Buriolla S, Targato G et al. (2020) Plasma‐based longitudinal evaluation of ESR1 epigenetic status in hormone receptor‐positive HER2‐negative metastatic breast cancer. Front Oncol 10, 550185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Visvanathan K, Fackler MS, Zhang Z, Lopez‐Bujanda ZA, Jeter SC, Sokoll LJ, Garrett‐Mayer E, Cope LM, Umbricht CB, Euhus DM et al. (2017) Monitoring of serum DNA methylation as an early independent marker of response and survival in metastatic breast cancer: TBCRC 005 prospective biomarker study. J Clin Oncol 35, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jank P, Gehlhaar C, Bianca L, Caterina F, Andreas S, Karn T, Marmé F, Sinn HP, van Mackelenbergh M, Sinn B et al. (2020) MGMT promoter methylation in triple negative breast cancer of the GeparSixto trial. PLoS One 15, e0238021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Silva R, Moran B, Russell NM, Fahey C, Vlajnic T, Manecksha RP, Finn SP, Brennan DJ, Gallagher WM & Perry AS (2020) Evaluating liquid biopsies for methylomic profiling of prostate cancer. Epigenetics 15, 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bjerre MT, Nørgaard M, Larsen OH, Jensen SØ, Strand SH, Østergren P, Fode M, Fredsøe J, Ulhøi BP, Mortensen MM et al. (2020) Epigenetic analysis of circulating tumor DNA in localized and metastatic prostate cancer: evaluation of clinical biomarker potential. Cells 9, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barry KH, Moore LE, Sampson JN, Koutros S, Yan L, Meyer A, Reddy M, Oler AJ, Cook MB, Fraumeni JF Jr et al. (2017) Prospective study of DNA methylation at chromosome 8q24 in peripheral blood and prostate cancer risk. Br J Cancer 116, 1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brait M, Banerjee M, Maldonado L, Ooki A, Loyo M, Guida E, Izumchenko E, Mangold L, Humphreys E, Rosenbaum E et al. (2017) Promoter methylation of MCAM, ERα and ERβ in serum of early stage prostate cancer patients. Oncotarget 8, 15431–15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Constâncio V, Nunes SP, Moreira‐Barbosa C, Freitas R, Oliveira J, Pousa I, Oliveira J, Soares M, Dias CG, Dias T et al. (2019) Early detection of the major male cancer types in blood‐based liquid biopsies using a DNA methylation panel. Clin Epigenetics 11, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lam D, Clark S, Stirzaker C & Pidsley R (2020) Advances in prognostic methylation biomarkers for prostate cancer. Cancers (Basel) 12, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hendriks RJ, Dijkstra S, Smit FP, Vandersmissen J, Van de Voorde H, Mulders PFA, van Oort IM, Van Criekinge W & Schalken JA (2018) Epigenetic markers in circulating cell‐free DNA as prognostic markers for survival of castration‐resistant prostate cancer patients. Prostate 78, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao S, Leonardson A, Geybels MS, McDaniel AS, Yu M, Kolb S, Zong H, Carter K, Siddiqui J, Cheng A et al. (2018) A five‐CpG DNA methylation score to predict metastatic‐lethal outcomes in men treated with radical prostatectomy for localized prostate cancer. Prostate 78, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haldrup C, Pedersen AL, Øgaard N, Strand SH, Høyer S, Borre M, Ørntoft TF & Sørensen KD (2018) Biomarker potential of ST6GALNAC3 and ZNF660 promoter hypermethylation in prostate cancer tissue and liquid biopsies. Mol Oncol 12, 545–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ge R, Wang Z, Montironi R, Jiang Z, Cheng M, Santoni M, Huang K, Massari F, Lu X, Cimadamore A et al. (2020) Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann Oncol 31, 470–479. [DOI] [PubMed] [Google Scholar]
- 53. Gordevičius J, Kriščiūnas A, Groot DE, Yip SM, Susic M, Kwan A, Kustra R, Joshua AM, Chi KN, Petronis A et al. (2018) Cell‐free DNA modification dynamics in abiraterone acetate‐treated prostate cancer patients. Clin Cancer Res 24, 3317–3324. [DOI] [PubMed] [Google Scholar]
- 54. Peter MR, Bilenky M, Isserlin R, Bader GD, Shen SY, De Carvalho DD, Hansen AR, Hu P, Fleshner NE, Joshua AM et al. (2020) Dynamics of the cell‐free DNA methylome of metastatic prostate cancer during androgen‐targeting treatment. Epigenomics 12, 1317–1332. [DOI] [PubMed] [Google Scholar]
- 55. Carson JJK, Di Lena MA, Berman DM, Siemens DR & Mueller CR (2020) Development and initial clinical correlation of a DNA methylation‐based blood test for prostate cancer. Prostate 80, 1038–1042. [DOI] [PubMed] [Google Scholar]
- 56. Duruisseaux M & Esteller M (2018) Lung cancer epigenetics: from knowledge to applications. Semin Cancer Biol 51, 116–128. [DOI] [PubMed] [Google Scholar]
- 57. Abbosh C, Birkbak NJ, Wilson GA, Jamal‐Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal R et al. (2017) Phylogenetic ctDNA analysis depicts early‐stage lung cancer evolution. Nature 545, 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Sandoval‐Tan J, Ladrera G, Thongprasert S, Srimuninnimit V, Liao M et al. (2015) Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first‐line intercalated Erlotinib and chemotherapy. Clin Cancer Res 21, 3196–3203. [DOI] [PubMed] [Google Scholar]
- 59. Sequist LV, Goldman JW, Wakelee HA, Camidge DR, Yu HA, Varga A, Solomon B, Oxnard GR, Ou SHI, Papadimitrakopoulou V et al. (2015) Efficacy of rociletinib (CO‐1686) in plasma‐genotyped T790M‐positive non‐small cell lung cancer (NSCLC) patients (pts). J Clin Oncol 33, 8001–8001. [Google Scholar]
- 60. Shen N, Du J, Zhou H, Chen N, Pan Y, Hoheisel JD, Jiang Z, Xiao L, Tao Y & Mo X (2019) A diagnostic panel of DNA methylation biomarkers for lung adenocarcinoma. Front Oncol 9, 1281–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leal A, Sidransky D & Brait M (2019) Tissue and cell‐free DNA‐based epigenomic approaches for cancer detection. Clin Chem 66, 105–116. [DOI] [PubMed] [Google Scholar]
- 62. Esteller M, Sanchez‐Cespedes M, Rosell R, Sidransky D, Baylin SB & Herman JG (1999) Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non‐small cell lung cancer patients. Cancer Res 59, 67–70. [PubMed] [Google Scholar]
- 63. Zavridou M, Mastoraki S, Strati A, Tzanikou E, Chimonidou M & Lianidou E (2018) Evaluation of preanalytical conditions and implementation of quality control steps for reliable gene expression and DNA methylation analyses in liquid biopsies. Clin Chem 64, 1522–1533. [DOI] [PubMed] [Google Scholar]
- 64. Mastoraki S & Lianidou E (2017) DNA and histone methylation in lung cancer. In DNA and Histone Methylation as Cancer Targets (Kaneda A, Tsukada TY, eds), pp. 403–436.Humana Press, Springer, New York, NY. [Google Scholar]
- 65. Herman JG & Farooq M (2020) Noninvasive diagnostics for early detection of lung cancer: challenges and potential with a focus on changes in DNA methylation. Cancer Epidemiol Biomarkers Prev 29, 2416–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vrba L, Oshiro MM, Kim SS, Garland LL, Placencia C, Mahadevan D, Nelson MA & Futscher BW (2020) DNA methylation biomarkers discovered in silico detect cancer in liquid biopsies from non‐small cell lung cancer patients. Epigenetics 15, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nunes SP, Diniz F, Moreira‐Barbosa C, Constâncio V, Silva AV, Oliveira J, Soares M, Paulino S, Cunha AL, Rodrigues J et al. (2019) Subtyping lung cancer using DNA methylation in liquid biopsies. J Clin Med 8, 1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mastoraki S, Balgkouranidou I, Tsaroucha E, Klinakis A, Georgoulias V & Lianidou E (2020) KMT2C promoter methylation in plasma‐circulating tumor DNA is a prognostic biomarker in non‐small cell lung cancer. Mol Oncol. 10.1002/1878-0261.12848. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Balgkouranidou I, Chimonidou M, Milaki G, Tsaroucha E, Kakolyris S, Georgoulias V & Lianidou E (2016) SOX17 promoter methylation in plasma circulating tumor DNA of patients with non‐small cell lung cancer. Clin Chem Lab Med 54, 1385–1393. [DOI] [PubMed] [Google Scholar]
- 70. Balgkouranidou I, Chimonidou M, Milaki G, Tsarouxa EG, Kakolyris S, Welch DR, Georgoulias V & Lianidou ES (2014) Breast cancer metastasis suppressor‐1 promoter methylation in cell‐free DNA provides prognostic information in non‐small cell lung cancer. Br J Cancer 110, 2054–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xia S, Ye J, Chen Y, Lizaso A, Huang L, Shi L, Su J, Han‐Zhang H, Chuai S, Li L et al. (2019) Parallel serial assessment of somatic mutation and methylation profile from circulating tumor DNA predicts treatment response and impending disease progression in osimertinib‐treated lung adenocarcinoma patients. Transl Lung Cancer Res 8, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sun J, Fei F, Zhang M, Li Y, Zhang X & Zhu Sand Zhang S (2019) The role of mSEPT9 in screening, diagnosis, and recurrence monitoring of colorectal cancer. BMC Cancer 19, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gallardo‐Gómez M, Moran S, Páez de la Cadena M, Martínez‐Zorzano VS, Rodríguez‐Berrocal FJ, Rodríguez‐Girondo M, Esteller M, Cubiella J, Bujanda L, Castells A et al. (2018) A new approach to epigenome‐wide discovery of non‐invasive methylation biomarkers for colorectal cancer screening in circulating cell‐free DNA using pooled samples. Clin Epigenetics 10, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Petit J, Carroll G, Gould T, Pockney P, Dun M & Scott RJ (2018) Cell‐free DNA as a diagnostic blood‐based biomarker for colorectal cancer: a systematic review. J Surg Res 236, 184–197. [DOI] [PubMed] [Google Scholar]
- 75. Worm Ørntoft MB (2018) Review of blood‐based colorectal cancer screening: how far are circulating cell‐free DNA methylation markers from clinical implementation? Clin Colorectal Cancer 17, e415–e433. [DOI] [PubMed] [Google Scholar]
- 76. Danese E, Montagnana M & Lippi G (2019) Circulating molecular biomarkers for screening or early diagnosis of colorectal cancer: which is ready for prime time? Ann Transl Med 7, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sun X, Yuan W, Hao F & Zhuang W (2017) Promoter methylation of RASSF1A indicates prognosis for patients with stage II and III colorectal cancer treated with oxaliplatin‐based chemotherapy. Med Sci Monit 23, 389–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Symonds EL, Pedersen SK, Murray DH, Jedi M, Byrne SE, Rabbitt P, Baker RT, Bastin D & Young GP (2018) Circulating tumour DNA for monitoring colorectal cancer‐a prospective cohort study to assess relationship to tissue methylation, cancer characteristics and surgical resection. Clin Epigenetics 10, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Boeckx N, Op de Beeck K, Beyens M, Deschoolmeester V, Hermans C, De Clercq P, Garrigou S, Normand C, Monsaert E, Papadimitriou K et al. (2018) Mutation and methylation analysis of circulating tumor DNA can be used for follow‐up of metastatic colorectal cancer patients. Clin Colorectal Cancer 17, e369–e379. [DOI] [PubMed] [Google Scholar]
- 80. Barault L, Amatu A, Siravegna G, Ponzetti A, Moran S, Cassingena A, Mussolin B, Falcomatà C, Binder AM, Cristiano C et al. (2018) Discovery of methylated circulating DNA biomarkers for comprehensive non‐invasive monitoring of treatment response in metastatic colorectal cancer. Gut 67, 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Barault L, Amatu A, Bleeker FE, Moutinho C, Falcomatà C, Fiano V, Cassingena A, Siravegna G, Milione M, Cassoni P et al. (2015) Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann Oncol 26, 1994–1999. [DOI] [PubMed] [Google Scholar]
- 82. Barták BK, Kalmár A, Galamb O, Wichmann B, Nagy ZB, Tulassay Z, Dank M, Igaz P & Molnár B (2019) Blood collection and cell‐free DNA isolation methods influence the sensitivity of liquid biopsy analysis for colorectal cancer detection. Pathol Oncol Res 25, 915–923. [DOI] [PubMed] [Google Scholar]
- 83. Distler J, Tetzner R, Weiss G, König T, Schlegel A & Bagrowski M (2016) Evaluation of different blood collection tubes and blood storage conditions for the preservation and stability of cell‐free circulating DNA for the analysis of the methylated (m)SEPT9 colorectal cancer screening marker. Adv Exp Med Biol 924, 175–178. [DOI] [PubMed] [Google Scholar]
- 84. Connors D, Allen J, Alvarez JD, Boyle J, Cristofanilli M, Hiller C, Keating S, Kelloff G, Leiman L, McCormack R et al. (2020) International liquid biopsy standardization alliance white paper. Crit Rev Oncol Hematol 156, 103112. [DOI] [PubMed] [Google Scholar]
- 85. Berdasco M & Esteller M (2019) Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet 20, 109–127. [DOI] [PubMed] [Google Scholar]