

Abstract
Over the past decade, substantial developments have been made in the detection of circulating tumor DNA (ctDNA)—cell‐free DNA (cfDNA) fragments released into the circulation from tumor cells and displaying the genetic alterations of those cells. As such, ctDNA detected in liquid biopsies serves as a powerful tool for cancer patient stratification, therapy guidance, detection of resistance, and relapse monitoring. In this Review, we describe lung cancer diagnosis and monitoring strategies using ctDNA detection technologies and compile recent evidence regarding lung cancer‐related mutation detection in liquid biopsy. We focus not only on epidermal growth factor receptor (EGFR) alterations, but also on significant co‐mutations that shed more light on novel ctDNA‐based liquid biopsy applications. Finally, we discuss future perspectives of early‐cancer detection and clonal hematopoiesis filtering strategies, with possible inclusion of microbiome‐driven liquid biopsy.
Keywords: cancer detection, cfDNA, ctDNA, liquid biopsy, LUAD, NSCLC, TMB
Liquid biopsy can greatly contribute to better detection and management of lung cancer. ctDNA detection in liquid biopsies is a powerful tool for early detection, cancer patient stratification, therapy guidance, detection of resistance, and relapse monitoring. We describe lung cancer diagnosis and monitoring strategies using ctDNA detection technologies and compile recent evidence on the identification of lung cancer‐related mutations through liquid biopsy.

Abbreviations
- AGR2
anterior gradient 2
- AKT1
AKT serine/threonine kinase 1
- ALK
ALK receptor tyrosine kinase
- ASXL1
ASXL transcriptional regulator 1
- BRAF
B‐Raf proto‐oncogene
- CAPP‐Seq
cancer profiling by deep sequencing
- CDK4
cyclin‐dependent kinase 4
- cfDNA
cell‐free DNA
- cffDNA
cell‐free fetal DNA
- CH
clonal hematopoiesis
- CNV
copy number variation
- CTC
circulating tumor cell
- ctDNA
circulating tumor DNA
- cTMB
corrected tumor mutation burden
- CTNNB1
catenin beta 1
- DNMT3A
DNA methyltransferase 3 alpha
- ds
double‐stranded
- EGFR
epidermal growth factor receptor
- ERBB2
erb‐b2 receptor tyrosine kinase 2
- FGF
fibroblastic growth factor
- FOXA1
forkhead box A1
- JAK
Janus kinase
- KEAP1
kelch‐like ECH‐associated protein 1
- KRAS
KRAS proto‐oncogene GTPase
- LCMC
Lung Cancer Mutation Consortium
- LSCC
lung squamous cell carcinoma
- LUAD
lung adenocarcinoma
- mbDNA
microbial DNA
- MDM2
MDM2 proto‐oncogene
- MEK1
serine/threonine protein kinase MEK1
- MET
MET proto‐oncogene
- mtDNA
mitochondrial DNA
- NGS
next‐generation sequencing
- NKX2‐1
NK2 homeobox 1
- NRAS
NRAS proto‐oncogene GTPase
- NSCLC
non‐small‐cell lung cancer
- OS
overall survival
- PFS
progression‐free survival
- PIK3CA
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha
- PTEN
phosphatase and tensin homolog
- RB1
RB transcriptional corepressor 1
- RET
ret proto‐oncogene
- ROS1
ROS proto‐oncogene 1
- RTK
receptor tyrosine kinase
- SEER
Surveillance, Epidemiology and End Results
- SMARCA4
SWI/SNF‐related, matrix‐associated, actin‐dependent regulator of chromatin, subfamily a, member 4
- ss
single‐stranded
- ST7
suppression of tumorigenicity 7
- STK11
serine/threonine kinase 11
- TCGA
The Cancer Genome Atlas
- TET2
tet methylcytosine dioxygenase 2
- TGFA
transforming growth factor alpha
- TKI
tyrosine kinase inhibitor
- TMB
tumor mutation burden
- TP53
tumor protein p53
- TTF‐1
transcription termination factor 1
1. Introduction
Personalized medicine—specifically precision oncology—nowadays provides molecular characterization of a patient’s tumor via tissue biopsy and can help guide treatment decisions. However, to fully implement personalization in the field of oncology, it is necessary to have an easily accessible and less invasive way to determine and follow the molecular makeup of a tumor from the moment of its detection and over the treatment of the disease. One such approach is through a liquid biopsy, where the genetic characterization of the tumor can be assessed through a biofluid sample. The term ‘liquid biopsy’ refers to tumor‐derived analytes sampled from various biological fluids, usually blood, but also other clinical specimens, such as urine, saliva, ascites, and cerebrospinal fluid [1].
The constant development of technologies to detect cell‐free DNA (cfDNA) with high sensitivity has facilitated the employment of liquid biopsies in diverse clinical applications, including in oncology. Analysis of circulating tumor DNA (ctDNA) obtained from plasma at multiple time points throughout the course of the disease allows for patient stratification for treatment (known as ‘companion diagnostics’), screening, monitoring response to the selected treatment, and detection of minimal residual disease after surgery.
In this article, we define the terms of cfDNA and ctDNA, describe their properties, and outline the historical breakthroughs in ctDNA detection. Most importantly, we then aim at summarizing the most recent state‐of‐the‐art developments in ctDNA utilization in diagnosis, including early‐stage detection, treatment selection, and follow‐up, in non‐small‐cell lung cancer (NSCLC) patients.
2. cfDNA and ctDNA: history, definitions, and properties
cfDNA refers to degraded DNA fragments of usually 167 bp length that are released into the blood [2, 3]. The specific length may result from the action of a caspase‐dependent endonuclease that cleaves DNA after a core histone and its linker. Recent studies have shown a distinct nuclear fragmentation pattern, with variable fragment lengths of cfDNA, from different tissue of origin [4, 5, 6, 7]. Several different types of cfDNA have been described in the circulation, including both double‐stranded (ds) and single‐stranded (ss) DNA particles. In humans, cfDNA originates from all cells, yet the vast majority is known to be of hematopoietic provenance [8, 9, 10, 11]. Within the ‘classical’ cfDNA, we can distinguish more specific subclasses based on its site of origin or mechanism of release, for example, mitochondrial DNA (mtDNA) [12], cell‐free fetal DNA (cffDNA) [13], extrachromosomal circular DNA [14], as well as microbial DNA (mbDNA) [15, 16, 17].
The first record of cfDNA detection in blood serum and plasma was in 1948, by Mandel and colleagues [18]. Later, in 1977, higher levels of cfDNA were detected in patients with pancreatic cancer compared with healthy controls [19], which led to the hypothesis that tumors release DNA fragments to the circulation. In 1983, Shapiro and colleagues confirmed correlations between benign vs. malignant tumors and cfDNA concentration [20]. Later, Stroun et al. [21] demonstrated that some of these DNA fragments were of tumor origin, due to their genomic instability. In the early 1990s, two independent studies noted the presence of specific KRAS proto‐oncogene GTPase (KRAS) and NRAS proto‐oncogene GTPase (NRAS) mutations in cfDNA from pancreatic adenocarcinoma [22] and patients with acute myelogenous leukemia [22, 23]. This fraction of cfDNA was later termed as ‘circulating tumor DNA’ (ctDNA).
ctDNA refers to cfDNA fragments that are released into the bloodstream from primary tumor or metastatic cells and display tumor‐specific point mutations, chromosomal rearrangements, copy‐number variation (CNV), and DNA methylation [24, 25, 26]. Importantly, ctDNA is more fragmented than cfDNA, resulting in a much higher <100 bp fraction in the plasma [27]. The 10‐bp periodicity observed for fragments smaller than 167 bp [3, 28] corresponds to a turn of the DNA helix wrapped around the histone. This might protect one part of the DNA from the nucleases present in the blood. This specific fragmentation pattern suggests that apoptosis may be a major source of cfDNA and that histones may be the key protein complex associated with DNA in the blood. The release of longer ctDNA fragments from tumor cells has been associated with necrotic cell death and occurs via active processes in living cells [11, 29, 30]. ctDNA fragments that are released into the circulation mirror the tumor status, its evolution, and the genomic alterations present in primary and/or metastatic tumors [11].
In a pan‐cancer study involving 640 patients, Bettegowda et al. [31] demonstrated that ctDNA analysis might allow monitoring of the therapeutic response, tracking resistance, and, in some cases, early detection of localized malignancies. They have also shown a correlation between tumor burden and stage of the disease. Significant differences in ctDNA levels were seen between cancer types, and the median cfDNA concentration was shown to be 100‐fold higher in patients with stage IV versus stage I disease [31].
ctDNA has a notably short half‐life in the bloodstream [32], and this characteristic is an important feature in analyzing dynamics of the mutations and tumor burden after surgery or systemic treatment throughout the disease. Thus, real‐time tumor dynamics might be monitored through ctDNA analysis for early prediction and assessment of drug response, as well as early intervention independent of detection by imaging examinations or clinical symptoms [26]. A recent study developed a mathematical model to predict the shedding rate of early‐stage NSCLC [33]. From this study, it has been estimated that there would be an average of only 1.7 genome copies of ctDNA in 15 mL of blood for lung tumors with a volume of 1 cm3.
In general, detection of ctDNA requires the presence of typical mutations that can be readily detected by simple sequencing techniques, proving the presence of tumor. With the advent of genomic information from the most recent cancer genome sequencing studies, it has become clear that practically all cancers of every type harbor somatic alterations. Cancer somatic mutations occur at minor frequencies in normal cell populations and therefore provide impeccably specific biomarkers from a biological perspective [34]. Some historic studies provide temporal analyses of the total tumor burden, as well as identifying specific mutations that appear during therapy [29, 35, 36, 37, 38, 39, 40].
Identification of somatic mutations within white blood cells might be a recurring source of discordance between tumor and total cfDNA genotyping. This phenomenon is called clonal hematopoiesis (CH) and is an aging‐related phenomenon whereby nonmalignant hematopoietic stem and progenitor cells acquire somatic alterations that can confer a selective advantage [41]. CH mutations are similar to the mutations detected in plasma and may involve both canonical CH genes, such as DNA methyltransferase 3 alpha (DNMT3A); tet methylcytosine dioxygenase 2 (TET2); ASXL transcriptional regulator 1 (ASXL1); and Janus kinase (JAK), and driver mutations related to tumors, such as KRAS, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha (PIK3CA), and epidermal growth factor receptor (EGFR) mutations [42, 43, 44, 45]. Because hematopoietic cells are the primary source of cfDNA [46] and contribute somatic variants to the cfDNA pool [43, 44], several approaches to distinguish mutations derived from CH from their tumor‐derived counterparts have been proposed [42, 44, 47]. Studies that indicated that CH and tumor cfDNA have fragments of a different size distribution might also help to distinguish between the two [48, 49, 50]. Chan et al. [51] elaborate elegantly on further clinical implications of the CH phenomenon.
In addition to considering CH before implementing ctDNA analysis in the clinic, other strict guidelines and standard operating procedures need to be formulated. Factors like preanalytical standardization need to be well optimized. For instance, the CANCER‐ID consortium was funded between public and private sector units with the aim of establishing standard protocols for and clinical validation of ctDNA‐ and circulating tumor cell (CTC)‐based biomarkers. Lampignano et al. [52] compare the preanalytical and analytical workflows of cfDNA‐based techniques, and Grölz et al. [53] explain the importance of preserving whole‐blood specimens after blood drawn for use as liquid biopsies, and summarize preservation solutions that are currently available. Through entities like CANCER‐ID or the International Liquid Biopsy Standardization Alliance (ILSA) [54], the importance of working toward the global use of liquid biopsy in oncology practice is being well recognized.
3. A glimpse at lung cancer
Lung cancer remains the most common cause of cancer death worldwide, with an estimated 1.8 million deaths each year [55]. About 85% of patients are histologically grouped as NSCLC, of which lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LSCC) are the most common subtypes [56, 57]. Until 2004, all NSCLC subtypes—LUAD, LSCC, and large cell carcinoma—were treated in the same manner with chemotherapy (cisplatin or carboplatin in combination with either docetaxel, paclitaxel, gemcitabine, or vinorelbine). The FDA approval of gefitinib—the first EGFR inhibitor—was a game changer in NSCLC treatment, leading to stratification of patients with activating EGFR mutations to targeted therapy [58].
Lung cancer treatment is stage‐specific. Early‐stage disease can be cured by surgical resection, while locally advanced disease demands multimodal treatment, including chemotherapy, radiotherapy, and surgery for chosen cases. Intrinsically, due to the usually late prognosis, NSCLC is a metastatic disease (henceforth, stage IV or metastatic is applied to NSCLC or LUAD) with gloomy prognosis. Its response rate is about 30%, progression‐free survival (PFS) 4–6 months, and median overall survival (OS) about 12 months; however, toxicity usually applies to all treatment strategies. In the past, some physicians withheld chemotherapy on the basis of patients’ age (with the cutoff arbitrarily set at 70 years) [57].
3.1. Early detection
Although the understanding of lung cancer pathobiology has significantly improved over the last few decades, poor disease prognosis is partially attributed to late stages at diagnosis, given that there are very few early symptoms [59]. When diagnosed at an early stage, patients with NSCLC have a 5‐year survival rate of about 71%. For patients diagnosed with stage IV disease, it is less than 2% [60]. Early diagnosis may thus improve patient outcome [61], especially if ctDNA‐guided adjuvant therapy administration reaches the bedside, as proposed in the TRACERx consortium study [62, 63]. Therefore, there is still a considerable need to develop noninvasive integrative biomarkers for early detection of lung cancer.
Despite the fact that several alterations have been noted in histologically normal bronchial epithelium specimens of smokers [64], early diagnosis in NSCLC remains low. Molecular changes are detected in hyperplasia and dysplasia, stages that precede carcinoma in situ and invasive carcinoma [57]. Recent studies show that aneuploidy and driver mutations also precede cancer diagnosis by several years [65]. Driver fusion oncogenes in LUAD could arise in the early decades of life, with a long latency period before diagnosis [66].
In 2018, Cohen et al. [67] developed CancerSEEK—a multianalyte test that can detect eight human cancer types through determination of the levels of circulating proteins and mutations in ctDNA. For lung cancer, the probability of detection reached 75%. It seems that ctDNA combined with protein biomarkers might serve as an opportunity to detect cancers before they metastasize, when it is not yet evident radiologically. At this stage, patients can be cured in up to 50% of cases with systemic therapies. Nonetheless, further studies should be performed for the possible implementation of CancerSEEK in the clinical practice.
As the majority of cfDNA originates from hematopoietic cells, these CH mutations are detected in plasma and, without appropriate controls, can be incorrectly attributed to tumor. High‐sensitivity cfDNA analyses have identified CH mutations in 60–90% of individuals without cancer and shown them to be age‐related [43, 44]. To avoid calling these false‐positive CH mutations, white blood cell controls, fragment length discrimination, CH‐associated variant filtering, and deep‐error controlled sequencing are required (Fig. 1).
Fig. 1.
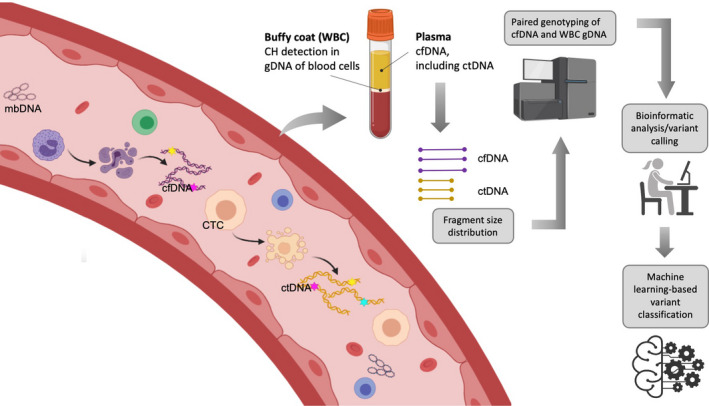
Strategies to filter clonal hematopoiesis (CH) from tumor‐derived mutated circulating free DNA (cfDNA). Plasma‐derived cfDNA, including circulating tumor DNA (ctDNA), is subjected to fragment size analysis to better discriminate between tumor‐ and non‐tumor‐derived cfDNA. Then, along with white blood cell (WBC)‐derived genomic DNA (gDNA), cfDNA is sequenced and analyzed via a rigorous bioinformatic pipeline. CH may be excluded by filtering nonsynonymous mutations except for the positive selection analysis, mutational signature analysis, and genes canonically associated with CH [48].
To address this issue, in their latest discovery, Chabon et al. [48] implemented certain improvements to cancer profiling by deep sequencing (CAPP‐Seq) [68] of ctDNA analysis at lung cancer screening. Although very low levels of ctDNA were detected in early‐stage lung cancer patients, its presence was confirmed as strongly prognostic. The mutational profile of total cfDNA from lung cancer patients and risk‐matched controls revealed nonrecurrent CH, and the mutations were detected on longer cfDNA fragments. A machine‐learning method called ‘lung cancer likelihood in plasma’ (Lung‐CLiP) allowed for robust discrimination of early‐stage lung cancer patients from risk‐matched controls [48]. These findings give hope for employment of ctDNA‐based screening methods into the clinical practice, hence reducing cancer‐related mortality by increasing the early‐detection rate.
3.2. Personalized treatment for NSCLC
Targetable oncogenic drivers account for almost 25% of LUADs, of which EGFR mutations are the most frequent [69]. The use of targeted therapies has reduced lung cancer mortality [70]. Based on Surveillance, Epidemiology and End Results (SEER) cancer registries, among men, incidence‐based mortality from NSCLC decreased by 6.3% annually from 2013 through 2016. Similar patterns were found among women with NSCLC [70].
The Lung Cancer Mutation Consortium (LCMC) performed the first large‐scale study in LUADs for detection of oncogenic driver mutations in the United States. Multiplex genotyping for mutation detection was performed in several sites, using any of three methods: matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry (Sequenom, Arizona Research Laboratories), multiplex single‐nucleotide extension sequencing (SNaPshot, Applied Biosystems), or Sanger sequencing with peptide nucleic acid probes [71]. The LCMC prioritized genotyping EGFR, KRAS, erb‐b2 receptor tyrosine kinase 2 (ERBB2), AKT serine/threonine kinase 1 (AKT1), B‐Raf proto‐oncogene (BRAF), serine/threonine protein kinase MEK1 (MEK1), NRAS, PIK3CA, ALK receptor tyrosine kinase (ALK), and MET proto‐oncogene (MET). KRAS mutations were the most frequent, found in 182 of 733 specimens (25%), followed by sensitizing EGFR mutations (exon 19 deletions, L858R, L861Q, and G719X) in 122 of 733 specimens (17%). This kind of EGFR mutation results in sensitivity to tyrosine kinase inhibitors (TKIs). ALK rearrangements occurred in 57 of 733 specimens (8%). The median survival of patients with each of the five most common oncogenic drivers ranged from 2.0 years (mutations in two genes) to 4.3 years (ALK‐rearranged tumors). The 260 patients with an oncogenic driver and treatment with a targeted drug had a median survival of 3.5 years; the 318 patients with a driver and no targeted therapy had 2.4 years median survival; and the 360 patients with no driver identified had 2.1 years (P < 0.001) [71].
Other nationwide studies were performed following the LCMC study [71], including a genomic screening network (LC‐SCRUM‐Japan) and the French nationwide IFCT‐InCa project, as well as a project in Cologne, Germany (reviewed in ref. [69]). All these initiatives paved the way for the current genomic classification of LUAD and set the basis for the genomic classification of LSCC [69, 72].
Recently, the TARGET study used a ctDNA assay involving a panel of 641 cancer‐associated genes. For the first 100 patients, ctDNA data showed good concordance with tissue (78% concordance) and identified potentially actionable mutations in 41% of patients. Of these patients, 11 of 41 (27%) went on to a matched therapy [73].
3.2.1. EGFR‐mutant LUAD genomic assessment
Targeted next‐generation sequencing in tumor tissue from metastatic NSCLC is gradually becoming more used for the identification of targetable driver mutations and gene fusions. However, the first question that comes up when a driver actionable mutation is detected is: What is the meaning of the other, often co‐occurring, mutations and gene alterations reported?
EGFR‐mutant NSCLC was analyzed using the MSK‐IMPACT assay, a clinical test that detects mutations, copy‐number alterations, and select fusions involving 341 (version 1), 410 (version 2), or 468 (version 3) cancer‐associated genes [74]. The median number of co‐mutations was 5 (range 0–19) in samples prior to EGFR TKI therapy. The most frequent co‐occurring mutations were tumor protein p53 (TP53) (60% n = 119), PIK3CA (12%, n = 23), catenin beta 1 (CTNNB1) (9%, n = 18), and RB transcriptional corepressor 1 (RB1) (10%, n = 19). The most frequent concurrent amplifications were EGFR (22%, n = 45), NK2 homeobox 1/transcription termination factor 1 (NKX2‐1/TTF‐1) (15%, n = 29), MDM2 proto‐oncogene (MDM2) (12%, n = 23), cyclin‐dependent kinase 4 (CDK4) (10%, n = 21), and forkhead box A1 (FOXA1) (10%, n = 20) [74]. FOXA1 is a transcription factor that is frequently mutated in prostate, breast, bladder, and salivary gland tumors [75]. The median PFS was 11 months; however, if the TP53 mutation was also present, then the median PFS was reduced (6 months) [74]. PFS was 5 months when pretreatment MET amplification (2%, n = 4) was noted. The presence of TP53 alterations was also associated with shorter survival [74]. The TP53 concurrent mutation also predicted shorter PFS to EGFR TKI in EGFR‐mutated NSCLC [76]. More recent studies [77] reconfirm the Helena Yu co‐mutation plot of genomic alterations [74] and help to gain insights on co‐acquired alterations through the evolution of TKI therapy in NSCLC patients [77]. PIK3CA mutations showed a domain‐dependent effect on PFS. Mutations in the kinase domain (Y1021H and H1047R), helical domain (E542K), and C2 domain (N345K) were associated with poorer PFS, while mutations in the p85‐binding domain (R88Q, R108H, and K111E) were associated with an improved survival [77]. Intriguingly, multiple driver mutations occur in the same gene, especially in PIK3CA (10% of samples) and EGFR (10%) [78]. In fact, The Cancer Genome Atlas (TCGA) research network showed that the principal driver alterations in NSCLC (i.e., LUAD)—either EGFR mutations or KRAS mutations—include the co‐occurrence of several others, commonly with TP53 mutations, as well as serine/threonine kinase 11 (STK11) and kelch‐like ECH‐associated protein 1 (KEAP1) mutations and alterations [79].
3.2.2. Detection of EGFR mutations in ctDNA
In our group [80], EGFR mutations in ctDNA were assessed in a large cohort of 1026 NSCLC patients, and sensitizing EGFR mutations were found in 113 patients (11%). More than 50% of samples had <10 pg mutated genomes per µL, with allelic fractions below 0.25%. Patients treated first line with TKI had an objective response rate of 72% and a median PFS of 11 months. Of the 105 patients screened after progression to EGFR TKIs, sensitizing mutations were found in 56%, and the acquired T790M mutation was found in 35%. Detection of EGFR mutations in plasma ctDNA was used as a selection criterion for first‐line gefitinib in patients with LUAD (BENEFIT study) [81]. The objective response rate among 188 patients was 72%, while median PFS was 9.5 months. Of 167 patients with available blood samples, 147 (88%) had clearance of EGFR mutations in ctDNA at week 8, and median PFS was longer for these patients than for the 20 patients whose EGFR mutations persisted at week 8 (11 months vs 2.1 months, P < 0.0001). From baseline next‐generation sequencing (NGS) data in 179 patients [using an ultra‐deep (20 000×) 168‐gene panel named LungPlasma (Burning Rock Biotech, Guangzhou, China)], three subgroups were seen: those with EGFR mutations alone (n = 58), those with mutations in EGFR and tumor‐suppressor genes [TP53 (65%), RB1 (8%) or phosphatase and tensin homolog (PTEN), (3%) (n = 97)], and those with mutations in EGFR and oncogenes [MET, ERBB2, KRAS, BRAF, ret proto‐oncogene (RET) or ROS proto‐oncogene 1 (ROS1) (n = 24)]. Corresponding median PFS in these subgroups was 13.2 months, 9.3 months, and 4.7 months, respectively [81].
An earlier seminal study deciphered the evolution and clinical impact of co‐occurring genetic alterations in 1122 EGFR‐mutated NSCLC patients by means of the Guardant 360 ctDNA assay, which covered single‐nucleotide variants, small insertions/deletions (indels), gene rearrangements/fusions, and copy‐number gains across 68 clinically relevant cancer genes [82]. PIK3CA and CTNNB1 mutations were noted in tumor tissue, as described above [74]. Interestingly, concurrent genomic alterations detectable in EGFR‐mutant NSCLC patients encoding the EGFR C797S mutation were also found [82]. Using new versions of the Guardant360 ctDNA assay, covering 73 cancer‐related genes, two studies have reported great concordance between the ctDNA assay and tissue‐based clinical genotyping. In the United States, 282 patients were evaluated in the NILE study (Non‐Invasive versus Invasive Lung Evaluation) [83]. In tissue‐positive patients, the biomarker was identified alone (12/60) or concordant with ctDNA (48/60), with an 80% ctDNA sensitivity. Importantly, the ctDNA median turnaround time was faster than tissue (9 vs 15 days; P < 0.0001) [83]. We conducted a similar prospective study in Spain [84] in 186 NSCLC patients, confirming that the Guardant360 NGS ctDNA assay was not inferior to standard‐of‐care tissue testing in detecting recommended biomarkers, further confirming that ctDNA‐based first‐line therapy produces outcomes similar to tissue‐based testing [84].
As confirmed in multiple studies, detection of EGFR mutations in plasma serves as a satisfactory surrogate for tissue biopsy. Significant weight should be put on the co‐occurring genetic alterations, since their detection serves as an important prognostic marker.
3.2.3. Significance of EGFR L858R mutation in ctDNA
We carried out a large‐scale ctDNA screening of patients with NSCLC for EGFR mutations in Spain. EGFR mutations were found in 350 of 2015 patients (16.6%). For 217 patients who received erlotinib, the adjusted hazard ratios for the duration of PFS were 2.94 for men (P < 0.001); 1.92 for the presence of the L858R mutation, as compared with a deletion in exon 19 (P = 0.02); and 1.68 for the presence of the L858R mutation in paired serum DNA, as compared with the absence of the mutation (P = 0.02) [85]. After examining the ctDNA of 97 patients included in the EURTAC phase 3 trial [86] using a peptide nucleic acid‐mediated 5’ nuclease real‐time polymerase chain reaction (TaqMan) assay, it was determined that median OS was shorter in patients with the L858R mutation in ctDNA than in those with the exon 19 deletion (13.7 vs 30 months, P < 0.01). Moreover, univariate analysis of patients with EGFR mutations in ctDNA identified the L858R mutation in tumor tissue or in ctDNA as a marker of shorter OS (hazard ratio, 2.70, P < 0.001) and PFS (hazard ratio, 2.04, P = 0.008). For patients with the L858R mutation detected primarily in tissue, median OS was 13.7 months for patients with the L858R mutation in ctDNA and 27.7 months for those with no mutation detected (P = 0.03). The conclusion was that the L858R mutation in ctDNA might be a novel surrogate prognostic marker [87].
The FLAURA phase 3 trial of osimertinib vs gefitinib in first line also showed a shorter PFS and OS in EGFR‐mutant NSCLC harboring the L858R mutation in tumor tissue [88, 89]. Anterior gradient 2 (AGR2), a disulfide isomerase that promotes lung tumorigenesis, is significantly expressed (using TCGA datasets) in EGFR L858R airway tumors induced by transforming growth factor alpha (TGFA; an EGFR ligand), but not in normal lung. Experimental data points out that AGR2 may contribute to the growth of EGFR L858R airway lung tumors induced by TGFA. In addition, TGFA induces the expression of AGR2 in human EGFR‐mutant LUAD. As explained below, TGFA‐mediated fibrosis associated with EGFR‐mutant lung tumors in vivo may induce growth factors (e.g., FGF) that confer resistance to EGFR TKIs, gefitinib, erlotinib, afatinib, and osimertinib. It has been posited that TGFA is a therapeutic target for recurrent EGFR‐mutant lung cancer (e.g., osimertinib‐resistant LUAD) [90]. Although the biological background of L858R mutations has not been elucidated, it is remarkable that multiple driver mutations in the same EGFR gene can occur for L858R, in contrast with the EGFR exon 19 deletion [91]. A number of rare (minor) mutations are found in extracellular and transmembrane domains, together with the L858R major driver mutation. These findings lead us to speculate that multiple mutations in the same oncogene cooperate to potentiate its tumor‐promoting activity [90].
3.2.4. Landscape of acquired EGFR mutations, other mutations, and gene fusions in EGFR‐mutant NSCLC
EGFR‐mutant NSCLC patients treated with TKI often develop acquired EGFR mutations that, until now, were only examined in re‐biopsies. As an example, a patient that harbored EGFR exon 19 deletion was treated with afatinib [92]. After progression, the second biopsy still revealed the presence of EGFR exon 19 deletion and the acquired EGFR exon 20 T790M. Treatment was then switched to osimertinib (a mutant‐specific third‐generation EGFR TKI). When the third biopsy was performed, EGFR exon 19 deletion was pervasively identified, with disappearance of EGFR exon 20 T790M and emergent EGFR exon 20 C797S (acquired with osimertinib). The patient was treated with gefitinib (EGFR inhibitor, first generation) which induced tumor response once more [92].
Previously, it was suggested that combining first‐ and third‐generation TKIs in first‐line therapy could be crucial, because neither a T790M, nor a C797S mutation, alone, would be able to drive resistance to the combination [93]. For T790M‐positive, erlotinib‐resistant NSCLCs that develop a C797S mutation following therapy with a third‐generation TKI, the configuration of the T790M and C797S mutations influences how the cells can respond to therapy. If the two mutations are in trans (on separate alleles), then the combination of first‐ and third‐generation TKIs can restore EGFR inhibition. Conversely, if the two mutations are in cis (on the same allele), the cells are refractory to any of the EGFR TKIs, as well as the combination of first‐ and third‐generation inhibitors [93]. It was foreseen that clinical assessment of the cis versus trans configuration can be examined by NGS, since T790M and C797S mutations are in close enough proximity to coexist on a significant number of individual sequencing reads [93, 94]. Interestingly, it was shown that, at a concentration of 1 µmol·L−1, afatinib can inhibit mutant EGFR with C797S in the absence of T790M [93].
Osimertinib is the preferred first‐line therapy for EGFR‐mutant NSCLC [88, 89]; however, resistance unavoidably develops in patients. Resistance is mediated by acquired secondary mutations in EGFR. In addition to C797S, others also occur, such as L718Q [95]. Analysis of ctDNA data from patients disclosed that L718Q mutations usually appear in the context of an L858R driver mutation [96]. This adds evidence to the observation that additional mutations occur in the presence of EGFR L858R, rather than in EGFR exon 19 deletion [78].
On the same lines as Niederst’s observations [93], treatment in mice revealed that both erlotinib and afatinib caused regression of osimertinib‐resistant C797S‐containing tumors, whereas only afatinib was effective in L718Q mutant tumors. Combination of first‐line osimertinib plus erlotinib could prevent the emergence of secondary mutations in EGFR [96].
A novel EGFR G724S mutation, causing resistance to osimertinib, occurs with exon 19 deletion, but not L858R. In addition, the exon 19 deletion/G724S retains sensitivity to afatinib, but not to erlotinib [97]. EGFR C724S was identified in 4% of postosimertinib patients treated with first‐line osimertinib, whereas C797X was identified in 29% of postosimertinib patients treated with later‐line osimertinib [98].
Targeted NGS for 416 cancer‐related genes was carried out in 93 osimertinib‐resistant NSCLC patient samples, mainly in ctDNA, and matched pretreatment samples of 12 patients. A co‐mutation plot of postosimertinib‐treated patients revealed two subgroups of patients: those with major EGFR tertiary mutations at the positions of L718/G719, L792, and G796/C797 (identified with a frequency of 9.7%, 10.8%, and 24.7%, respectively). In most cases, mutations were also noted in EGFR T790M and TP53 [99]. Almost all patients without EGFR resistance mutations showed TP53, MET, KRAS, or PIK3CA mutations [99].
Median OS after osimertinib progression (osimertinib given as second‐line in EGFR T790M mutant NSCLC patents) was 5.4 months in 40 patients from the AURA study, whose plasma was available after disease progression [100]. Twelve (30%) of these had the T790M mutation (four of whom also had C797S). Patients without detectable EGFR‐activating mutations in plasma before treatment had the best overall and postprogression survival (22.4 months and 10.8 months, respectively). Loss of T790M but presence of EGFR‐activating mutations in plasma was associated with the shortest PFS (median 2.6 months). Importantly, in 22 postprogression tumor samples, one squamous cell and two small‐cell transformations were seen. In addition, the number of patients was small and T790M was found in 50% of samples, C797S in 17%, MET amplification in 50%, BRAF mutations in 8%, and KRAS mutations in 8%.
Once we have the data from a ctDNA analysis, what should we do with it? What do we do when facing the multiplicity of genomic aberrations described? Is it a basic rule in medicine to bear in mind their frequency? Secondly, are the acquired driver co‐alterations druggable? A recent study has shed light on these issues [98]. MSK‐Fusion Solid, a custom RNAseq panel, was used to detect fusions in cases where no resistance mechanism was identified by NGS and sufficient tissue was available. Among 62 patients, histological squamous transformation was identified in 15% of first‐line osimertinib cases and 14% of later‐line cases. Nineteen percent of patients treated with first‐line osimertinib had off‐target genetic resistance (2 MET amplifications, 1 KRAS mutation, 1 RET fusion, and 1 BRAF fusion), whereas 4% had an acquired EGFR mutation (EGFR G724S). Patients with squamous transformation acquired PIK3CA mutation, chromosome 3q amplification, and fibroblastic growth factor (FGF) amplification. The compound mutation EGFR S768 + V769L and the mutation MET H1094Y were also identified. Longitudinal analysis of two patients, who received later‐line osimertinib with emergence of ALK fusion, revealed that one patient acquired an EGFR C797S mutation and lost the ALK fusion after treatment with osimertinib and alectinib. The other patient acquired the ALK G1202R mutation after treatment with osimertinib and alectinib [98]. Recent studies confirm the validity of ctDNA analysis for response assessment to osimertinib [101] and others regarding the prognostic impact of TP53 mutations, suggesting that EGFR‐mutant and TP53 wild‐type patients may benefit from the combination of EGFR TKI with bevacizumab [102].
Great progress has also been made in the use of ALK inhibitors for the treatment of patients with ALK‐positive NSCLC, from crizotinib to second‐generation ALK inhibitors, including alectinib, brigatinib and ensartinib, and lorlatinib, a third‐generation ALK inhibitor [103]. The combination of ALK and MET inhibitors is emerging as a plausible efficient combination, since MET amplification has been detected in 15% of tumor biopsies from patients relapsing on next‐generation ALK inhibitors, including 12% and 22% of biopsies from patients progressing on second‐generation inhibitors or lorlatinib, respectively [104]. Also, two tumor specimens harbored suppression of tumorigenicity 7 (ST7)‐MET rearrangement [105]. A recent phase 2 study, VISION, evaluates tepotinib (a MET inhibitor) for the treatment of NSCLC with MET exon 14 skipping mutations. PFS with tepotinib was 11 months (tissue biopsy, n = 60), 8.5 months (liquid biopsy, n = 66) and 8.5 months (combined biopsy, n = 60) [106]. In addition, for MET exon 14 skipping mutations, patients treated with crizotinib (MET inhibitor; PROFILE 1001), PFS was significantly shorter in patients with MET mutations detected in ctDNA versus nondetected in ctDNA, with a median of 3.6 months vs 7.8 months (hazard ratio = 2.27, P = 0.06) [107].
It is clear that targeted NGS in tissue and plasma provides complementary theranostic information in the current era of single targeted therapy. However, the genomic evaluation could become complex with the cornucopia of genomic alterations that are being developed, and tracking plasma is the most suitable advance that the practitioner has, although it poses multiple hurdles on how to treat the patient. One appealing case described an EGFR L858R patient that also displayed EGFR G719S in four different regions of the resected lung tumor, in different allelic proportions, as well as SWI/SNF‐related, matrix‐associated, actin‐dependent regulator of chromatin, subfamily a, member 4 (SMARCA4) [108]. At the time of relapse, she received afatinib and a liver metastasis biopsy was performed where L858R was found in an allelic fraction of 14%, together with other gene mutations. After 2 years of afatinib, a resection of an ovarian metastasis revealed mutation of the EGFR gene in six different regions: L858R, G719S (allelic fraction range, 40–50%), and EGFR C797S (allelic fraction range 9–14%) in five of the six areas of the ovary. SMARCA4 mutation was found in all six ovarian areas analyzed, with an allelic fraction of more than 40%. After the ovary metastasis resection, the patient continued afatinib for more than 2 years without recurrence [108]. Almonertinib is a third‐generation EGFR TKI with high selectivity for EGFR‐sensitizing and T790M resistance mutations that show great inhibitory activity against T790M, T790M/L858R, and T790M/Del19 [109]. The secondary objective of a recently opened multicenter, open‐phase II clinical study (NCT04841811), in patients with unresectable stage III NSCLC is to assess the safety of different treatment decisions guided by ctDNA monitoring [110].
Targeted NGS ctDNA assays are warranted to manage the complexity of genomic alterations; however, intrinsic and acquired resistance also involves the identification of pathways that influence sensitivity, such as in KRAS G12C‐mutant NSCLC, where the combination of the KRAS G12C inhibitor, PI3K inhibitor, and SHP2 inhibitor caused tumor regression in mouse models with acquired resistance to AMG510 [111]. Co‐treatment in EGFR‐mutant NSCLC is highly recommended to improve PFS and OS. We and other groups have shown that targeting STAT3, YAP1, and SHP2 could have great synergism in vitro and in vivo in EGFR‐mutant cell line models [112, 113, 114] and that, by preventing NF‐κB signaling activation, the formation of acquired EGFR mutations could be prohibited [115].
3.2.5. ctDNA as a biomarker for immunotherapy
Most recently, the introduction of immunotherapy with promising responses in subgroups of cancer patients preceded the search for biomarkers to better stratify patients. This raised the question of whether studying a tumor microenvironment from the tissue biopsy was crucial for predictive biomarker discovery or whether a biomarker could be reasonably sought in a peripheral blood sample. It has been demonstrated that the latter may be achieved via analysis of tumor mutation burden (TMB) in ctDNA, adding another liquid biopsy biomarker to a constantly expanding list of blood tests for the management of cancer. An interesting approach to combine two liquid biopsy biosources would be to complement TMB detected on ctDNA with PD‐L1 assessment on CTCs [116]. The recently opened BESPOKE clinical trial (NCT04761783) is to examine the impact of SIGNATERA™ (a personalized and tumor‐informed 16‐plex NGS assay to detect ctDNA) on clinical decision‐making regarding immunotherapy for treatment of solid tumors [117].
Interestingly, fusion‐driven LUADs often have SETD2 mutations, which are not seen in LUADs with EGFR, KRAS, BRAF, or MET mutations [66]. This finding is of interest, since ALK or ROS1 fusion‐driven LUADs show poor benefit with immune checkpoint inhibitors (anti‐PD‐1 or anti‐PD‐L1 monoclonal antibodies) [118, 119]. SETD2 methyltransferase mediates STAT1 methylation on lysine 525, being an essential signaling event for interferon‐alpha‐dependent antiviral immunity [120]. The current management of metastatic NSCLC has evolved from chemotherapy to chemotherapy plus immune checkpoint inhibitors in a large number of NSCLC patients with no identifiable driver oncogene mutations or fusions [56, 121].
Despite the progress in immunotherapy, not all NSCLC patients respond. Whole‐exome sequencing of 104 patients treated with immune checkpoint inhibitors identified that corrected TMB (cTMB) adjusted for tumor purity predicted the benefit of immunotherapy, as well as smoke‐related mutational signature and human leukocyte antigen status. However, mutations in receptor tyrosine kinase (RTK) genes were indicative of no response [122].
4. Conclusions and perspectives
In the hope of overcoming several obstacles and challenges resulting from classical tissue biopsy‐based diagnosis, liquid biopsy emerged as a robust tool for ctDNA monitoring and disease detection and monitoring. Importantly, liquid biopsies trump tissue biopsies when there is insufficient material for testing or its quality is unsatisfactory. In addition, a patient’s poor performance status and tumor accessibility are often a substantial concern.
Tumors are highly heterogeneous, and sampling in its entirety is challenging: How well does a small tissue biopsy sample represent the whole tumor? In patients with multiple metastases, to gain a holistic view of the disease biopsy, samples should be collected from all of the (known) metastatic sites (Fig. 2). Since the blood reaches most tumor sites in patients with advanced cancers, it is perhaps reasonable to speculate that blood‐based liquid biopsies might better reflect tumor heterogeneity. Moreover, tumors evolve over time and can modify their molecular fingerprint, making clinical decisions based on historical biopsy data insignificant. The limitation of acquiring tissue biopsy samples longitudinally to determine disease response or monitor relapse is also reduced by the liquid biopsy approach, since multiple samples can be collected noninvasively over time (Fig. 2).
Fig. 2.
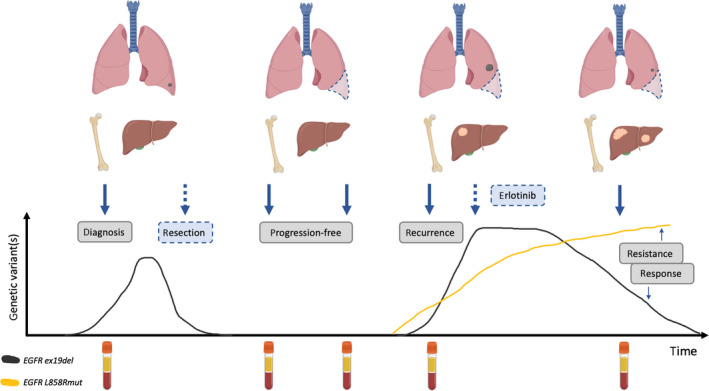
ctDNA‐based liquid biopsy in clinical use. Lung cancer was diagnosed at early stage, EGFR exon 19 deletion (ex19del) was detected, and the tumor was subjected to resection. Subsequently, two liquid biopsies detected no mutated circulating tumor DNA (ctDNA), indicating no progression of the disease. At the recurrence, EGFR ex19del and L858R mutation (L858Rmut) was detected in ctDNA and erlotinib was administered to the patient. L858R mutant cells metastasized to the liver and developed resistance to erlotinib (yellow line). Tumor derived from the EGFR ex19del clone responded well to the therapy (gray line). Solid arrows indicate liquid biopsy sample collection time points; dashed arrows indicate treatment procedures.
Although early‐detection strategies based on ctDNA are promising, numerous hindrances must be addressed before they can be robustly applied in the clinic. False‐positive results can be precarious for any screening assay. Experience thus far suggests that benign tumors and non‐neoplastic conditions do not generally give rise to ctDNA [29], so the ‘overdiagnosis’ of benign tumors is not likely to pose a major problem. Moreover, strategies to filter out CH signal are being developed (Fig. 1) [34, 35, 36].
ctDNA can be robustly detected in plasma when a significant number of copies of mutant ctDNA are shed into blood. However, when the amounts of ctDNA are too low, analysis of individual mutant loci might result in a negative result because of sampling background noise even when using an assay with perfect analytical sensitivity [123]. It is much easier to detect a single mutation in the follow‐up of an advanced disease and more demanding in the early stage where higher depth for detecting a broad panel of mutations requires more ctDNA input. Where the amount ctDNA isolated is not sufficient for an NGS analysis, alternative platforms, such as NanoString nCounter, can be of service [124].
A revolutionary approach for cancer management was proposed by Wan and colleagues [125], in which tumor‐guided personalized ctDNA screening is performed in longitudinally collected plasma samples. Such patient‐specific mutation lists provide an opportunity for highly sensitive monitoring from a range of sequencing data types using methods for signal aggregation, weighting, and error suppression [125]. All this makes ctDNA still the most useful technique for companion diagnostics approaches where the mutation is druggable.
Despite its potential, ctDNA analysis is not suitable to diagnose all cancers, since some tumor types (e.g., gliomas and sarcomas) are poor ctDNA shedders. Moreover, ctDNA‐based assays are applicable in tumors with higher TMB, whereas, for example, glioblastoma multiforme or pancreatic adenocarcinoma are barely detectable in blood, especially at earlier stages, due to their low shedding and TMB. Thus, Poore et al. [17] have approached this limitation: They addressed the possibility of using mbDNA to discriminate between cancer and healthy patients. In short, their findings suggest that mbDNA may serve as a biomarker for cancer diagnosis and detection even for low‐TMB cancers. This new path in liquid biopsy cancer diagnostics has been opened by highlighting another clinical application of microbiome‐based assays [17, 126].
In conclusion, ctDNA‐based liquid biopsies can be a powerful tool for cancer diagnosis, monitoring, prognosis, and individualized treatment and can completely change the current paradigms of cancer management, especially in a multianalyte setting. However, considerable research and development are still needed to improve the isolation, enrichment, and downstream analysis of all circulating biomarkers. In our view, in current clinical practice, ctDNA measurements need to be combined with standard‐of‐care approaches and ideally combined with other blood‐based biosources.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank Stephanie Davies for language editing. This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie grant agreement no 765492.
References
- 1. Pantel K & Alix‐Panabières C (2010) Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med 16, 398–406. [DOI] [PubMed] [Google Scholar]
- 2. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl H et al. (2005) Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA 102, 16368–16373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, Zheng YW, Leung TY, Lau TK, Cantor CR et al. (2010) Maternal plasma DNA sequencing reveals the genome‐wide genetic and mutational profile of the fetus. Sci Transl Med 2, 61ra91. [DOI] [PubMed] [Google Scholar]
- 4. Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, Jensen S, Medina JE, Hruban C, White JR et al. (2019) Genome‐wide cell‐free DNA fragmentation in patients with cancer. Nature 570, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Snyder MW, Kircher M, Hill AJ, Daza RM & Shendure J (2016) Cell‐free DNA comprises an in vivo nucleosome footprint that informs its tissues‐of‐origin. Cell 164, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun K, Jiang P, Cheng SH, Cheng THT, Wong J, Wong VWS, Ng SSM, Ma BBY, Leung TY, Chan SL et al. (2019) Orientation‐aware plasma cell‐free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res 29, 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, Abete L, Pristauz G, Petru E, Geigl JB et al. (2016) Inferring expressed genes by whole‐genome sequencing of plasma DNA. Nat Genet 48, 1273–1278. [DOI] [PubMed] [Google Scholar]
- 8. Elzanowska J, Semira C & Costa‐Silva B (2020) DNA in extracellular vesicles: biological and clinical aspects. Mol Oncol 15, 1701–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lam WKJ, Gai W, Sun K, Wong RSM, Chan RWY, Jiang P, Chan NPH, Hui WWI, Chan AWH, Szeto CC et al. (2017) DNA of erythroid origin is present in human plasma and informs the types of anemia. Clin Chem 63, 1614–1623. 10.1373/clinchem.2017.272401. [DOI] [PubMed] [Google Scholar]
- 10. Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, Samet Y, Maoz M, Druid H, Arner P et al. (2018) Comprehensive human cell‐type methylation atlas reveals origins of circulating cell‐free DNA in health and disease. Nat Commun 9, 5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thierry AR, El Messaoudi S, Gahan PB, Anker P & Stroun M (2016) Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 35, 347–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernandes J, Michel V, Camorlinga‐Ponce M, Gomez A, Maldonado C, De Reuse H, Torres J & Touati E (2014) Circulating mitochondrial DNA level, a noninvasive biomarker for the early detection of gastric cancer. Cancer Epidemiol Biomarkers Prev 23, 2430–2438. [DOI] [PubMed] [Google Scholar]
- 13. Alberry M, Maddocks D, Jones M, Abdel Hadi M, Abdel‐Fattah S, Avent N & Soothill PW (2007) Free fetal DNA in maternal plasma in anembryonic pregnancies: confirmation that the origin is the trophoblast. Prenat Diagn 27, 415–418. [DOI] [PubMed] [Google Scholar]
- 14. Kumar P, Dillon LW, Shibata Y, Jazaeri AA, Jones DR & Dutta A (2017) Normal and cancerous tissues release extrachromosomal circular DNA (eccDNA) into the circulation. Mol Cancer Res 15, 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li SK, Leung RK, Guo HX, Wei JF, Wang JH, Kwong KT, Lee SS, Zhang C & Tsui SK (2012) Detection and identification of plasma bacterial and viral elements in HIV/AIDS patients in comparison to healthy adults. Clin Microbiol Infect 18, 1126–1133. [DOI] [PubMed] [Google Scholar]
- 16. Moustafa A, Xie C, Kirkness E, Biggs W, Wong E, Turpaz Y, Bloom K, Delwart E, Nelson KE, Venter JC et al. (2017) The blood DNA virome in 8,000 humans. PLoS Pathog 13, e1006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ et al. (2020) Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Mandel P & Metais P (1948) Nuclear acids in human blood plasma. C R Seances Soc Biol Fil 142, 241–243. [PubMed] [Google Scholar]
- 19. Leon SA, Shapiro B, Sklaroff DM & Yaros MJ (1977) Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37, 646–650. [PubMed] [Google Scholar]
- 20. Shapiro B, Chakrabarty M, Cohn EM & Leon SA (1983) Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 51, 2116–2120. [DOI] [PubMed] [Google Scholar]
- 21. Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C & Beljanski M (1989) Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 46, 318–322. [DOI] [PubMed] [Google Scholar]
- 22. Sorenson GD, Pribish DM, Valone FH, Memoli VA, Bzik DJ & Yao SL (1994) Soluble normal and mutated DNA sequences from single‐copy genes in human blood. Cancer Epidemiol Biomarkers Prev 3, 67–71. [PubMed] [Google Scholar]
- 23. Vasioukhin V, Anker P, Maurice P, Lyautey J, Lederrey C & Stroun M (1994) Point mutations of the N‐ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol 86, 774–779. [DOI] [PubMed] [Google Scholar]
- 24. Corcoran RB & Chabner BA (2018) Application of cell‐free DNA analysis to cancer treatment. N Engl J Med 379, 1754–1765. [DOI] [PubMed] [Google Scholar]
- 25. Heitzer E, Ulz P & Geigl JB (2015) Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem 61, 112–123. [DOI] [PubMed] [Google Scholar]
- 26. Oliveira KCS, Ramos IB, Silva JMC, Barra WF, Riggins GJ, Palande V, Pinho CT, Frenkel‐Morgenstern M, Santos SEB, Assumpcao PP et al. (2020) Current perspectives on circulating tumor DNA, precision medicine, and personalized clinical management of cancer. Mol Cancer Res 18, 517–528. [DOI] [PubMed] [Google Scholar]
- 27. Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, Gongora C & Thierry AR (2011) High fragmentation characterizes tumour‐derived circulating DNA. PLoS One 6, e23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, Wong GL, Chan SL, Mok TS, Chan HL et al. (2015) Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA 112, E1317–E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA et al. (2008) Circulating mutant DNA to assess tumor dynamics. Nat Med 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stroun M, Lyautey J, Lederrey C, Olson‐Sand A & Anker P (2001) About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 313, 139–142. [DOI] [PubMed] [Google Scholar]
- 31. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM et al. (2014) Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 6, 224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kustanovich A, Schwartz R, Peretz T & Grinshpun A (2019) Life and death of circulating cell‐free DNA. Cancer Biol Ther 20, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Avanzini S, Kurtz DM, Chabon JJ, Moding EJ, Hori SS, Gambhir SS, Alizadeh AA, Diehn M & Reiter JG (2020) A mathematical model of ctDNA shedding predicts tumor detection size. Sci Adv 6, 10.1126/sciadv.abc4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA & Kinzler KW (2013) Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler‐Araujo B et al. (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368, 1199–1209. [DOI] [PubMed] [Google Scholar]
- 36. Diaz LA Jr, Sausen M, Fisher GA & Velculescu VE (2013) Insights into therapeutic resistance from whole‐genome analyses of circulating tumor DNA. Oncotarget 4, 1856–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA et al. (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holdhoff M, Schmidt K, Donehower R & Diaz LA Jr (2009) Analysis of circulating tumor DNA to confirm somatic KRAS mutations. In J Natl Cancer Inst 101, 1284–1285. [DOI] [PubMed] [Google Scholar]
- 39. Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O'Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B et al. (2012) Detection of chromosomal alterations in the circulation of cancer patients with whole‐genome sequencing. Sci Transl Med 4, 162ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS et al. (2013) Non‐invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112. [DOI] [PubMed] [Google Scholar]
- 41. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP & Ebert BL (2015) Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu Y, Ulrich BC, Supplee J, Kuang Y, Lizotte PH, Feeney NB, Guibert NM, Awad MM, Wong KK, Jänne PA et al. (2018) False‐positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res 24, 4437–4443. [DOI] [PubMed] [Google Scholar]
- 43. Liu J, Chen X, Wang J, Zhou S, Wang CL, Ye MZ, Wang XY, Song Y, Wang YQ, Zhang LT et al. (2019) Biological background of the genomic variations of cf‐DNA in healthy individuals. Ann Oncol 30, 464–470. [DOI] [PubMed] [Google Scholar]
- 44. Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, Abida W, Juluru K, De Bruijn I, Hou C et al. (2019) High‐intensity sequencing reveals the sources of plasma circulating cell‐free DNA variants. Nat Med 25, 1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xia L, Li Z, Zhou B, Tian G, Zeng L, Dai H, Li X, Liu C, Lu S, Xu F et al. (2017) Statistical analysis of mutant allele frequency level of circulating cell‐free DNA and blood cells in healthy individuals. Sci Rep 7, 7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lui YY, Chik KW, Chiu RW, Ho CY, Lam CW & Lo YM (2002) Predominant hematopoietic origin of cell‐free DNA in plasma and serum after sex‐mismatched bone marrow transplantation. Clin Chem 48, 421–427. [PubMed] [Google Scholar]
- 47. Li BT, Janku F, Jung B, Hou C, Madwani K, Alden R, Razavi P, Reis‐Filho JS, Shen R, Isbell JM et al. (2019) Ultra‐deep next‐generation sequencing of plasma cell‐free DNA in patients with advanced lung cancers: results from the Actionable Genome Consortium. Ann Oncol 30, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, Schroers‐Martin J, Nabet BY, Chen B, Chaudhuri AA et al. (2020) Integrating genomic features for non‐invasive early lung cancer detection. Nature 580, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leal A, van Grieken NCT, Palsgrove DN, Phallen J, Medina JE, Hruban C, Broeckaert MAM, Anagnostou V, Adleff V, Bruhm DC et al. (2020) White blood cell and cell‐free DNA analyses for detection of residual disease in gastric cancer. Nat Commun 11, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marass F, Stephens D, Ptashkin R, Zehir A, Berger MF, Solit DB, Diaz LA & Tsui DWY (2020) Fragment size analysis may distinguish clonal hematopoiesis from tumor‐derived mutations in cell‐free DNA. Clin Chem 66, 616–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chan HT, Chin YM, Nakamura Y & Low SK (2020) Clonal hematopoiesis in liquid biopsy: from biological noise to valuable clinical implications. Cancers (Basel) 12, 2277. 10.3390/cancers12082277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lampignano R, Neumann MHD, Weber S, Kloten V, Herdean A, Voss T, Groelz D, Babayan A, Tibbesma M, Schlumpberger M et al. (2020) Multicenter evaluation of circulating cell‐free DNA extraction and downstream analyses for the development of standardized (Pre)analytical work flows. Clin Chem 66, 149–160. [DOI] [PubMed] [Google Scholar]
- 53. Grölz D, Hauch S, Schlumpberger M, Guenther K, Voss T, Sprenger‐Haussels M & Oelmüller U (2018) Liquid biopsy preservation solutions for standardized pre‐analytical workflows‐venous whole blood and plasma. Curr Pathobiol Rep 6, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Connors D, Allen J, Alvarez JD, Boyle J, Cristofanilli M, Hiller C, Keating S, Kelloff G, Leiman L, McCormack R et al. (2020) International liquid biopsy standardization alliance white paper. Crit Rev Oncol Hematol 156, 103112. [DOI] [PubMed] [Google Scholar]
- 55. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A & Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71, 209–249. [DOI] [PubMed] [Google Scholar]
- 56. Herbst RS, Morgensztern D & Boshoff C (2018) The biology and management of non‐small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- 57. Spira A & Ettinger DS (2004) Multidisciplinary management of lung cancer. N Engl J Med 350, 379–392. [DOI] [PubMed] [Google Scholar]
- 58. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- 59. Gridelli C, Rossi A, Carbone DP, Guarize J, Karachaliou N, Mok T, Petrella F, Spaggiari L & Rosell R (2015) Non‐small‐cell lung cancer. Nat Rev Dis Primers 1, 15009. [DOI] [PubMed] [Google Scholar]
- 60. Goldstraw P, Chansky K, Crowley J, Rami‐Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P, Mitchell A & Bolejack V (2016) The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol 11, 39–51. [DOI] [PubMed] [Google Scholar]
- 61. Cassim S, Chepulis L, Keenan R, Kidd J, Firth M & Lawrenson R (2019) Patient and carer perceived barriers to early presentation and diagnosis of lung cancer: a systematic review. BMC Cancer 19, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abbosh C, Birkbak NJ, Wilson GA, Jamal‐Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal R et al. (2017) Phylogenetic ctDNA analysis depicts early‐stage lung cancer evolution. Nature 545, 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li BT, Stephens D, Chaft JE, Rudin CM, Jones DR, Rusch VW, Rimner A & Isbell JM (2017) Liquid biopsy for ctDNA to revolutionize the care of patients with early stage lung cancers. Ann Transl Med 5, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wistuba II, Mao L & Gazdar AF (2002) Smoking molecular damage in bronchial epithelium. Oncogene 21, 7298–7306. [DOI] [PubMed] [Google Scholar]
- 65. Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu K et al. (2020) The evolutionary history of 2,658 cancers. Nature 578, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee JJ, Park S, Park H, Kim S, Lee J, Youk J, Yi K, An Y, Park IK, Kang CH et al. (2019) Tracing oncogene rearrangements in the mutational history of lung adenocarcinoma. Cell 177, 1842–1857.e1821. [DOI] [PubMed] [Google Scholar]
- 67. Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A et al. (2018) Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science 359, 926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C et al. (2016) Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 34, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rosell R & Karachaliou N (2016) Large‐scale screening for somatic mutations in lung cancer. Lancet 387, 1354–1356. [DOI] [PubMed] [Google Scholar]
- 70. Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, Mariotto AB, Lowy DR & Feuer EJ (2020) The effect of advances in lung‐cancer treatment on population mortality. N Engl J Med 383, 640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Varella‐Garcia M, Franklin WA, Aronson SL, Su PF et al. (2014) Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311, 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rosell R, Karachaliou N & Arrieta O (2020) Novel molecular targets for the treatment of lung cancer. Curr Opin Oncol 32, 37–43. [DOI] [PubMed] [Google Scholar]
- 73. Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, Smith N, Villa S, Dransfield J, Clipson A et al. (2019) Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med 25, 738–743. [DOI] [PubMed] [Google Scholar]
- 74. Yu HA, Suzawa K, Jordan E, Zehir A, Ni A, Kim R, Kris MG, Hellmann MD, Li BT, Somwar R et al. (2018) Concurrent alterations in EGFR‐mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res 24, 3108–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Parolia A, Cieslik M, Chu SC, Xiao L, Ouchi T, Zhang Y, Wang X, Vats P, Cao X, Pitchiaya S et al. (2019) Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kim ES, Roy UB, Ersek JL, King J, Smith RA, Martin N, Martins R, Moore A, Silvestri GA & Jett J (2019) Updates regarding biomarker testing for non‐small cell lung cancer: considerations from the national lung cancer roundtable. J Thorac Oncol 14, 338–342. [DOI] [PubMed] [Google Scholar]
- 77. Jin Y, Bao H, Le X, Fan X, Tang M, Shi X, Zhao J, Yan J, Xu Y, Quek K et al. (2020) Distinct co‐acquired alterations and genomic evolution during TKI treatment in non‐small‐cell lung cancer patients with or without acquired T790M mutation. Oncogene 39, 1846–1859. [DOI] [PubMed] [Google Scholar]
- 78. Saito Y, Koya J, Araki M, Kogure Y, Shingaki S, Tabata M, McClure MB, Yoshifuji K, Matsumoto S, Isaka Y et al. (2020) Landscape and function of multiple mutations within individual oncogenes. Nature 582, 95–99. [DOI] [PubMed] [Google Scholar]
- 79. Network CGAR (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mayo‐de‐Las‐Casas C, Jordana‐Ariza N, Garzón‐Ibañez M, Balada‐Bel A, Bertrán‐Alamillo J, Viteri‐Ramírez S, Reguart N, Muñoz‐Quintana MA, Lianes‐Barragan P, Camps C et al. (2017) Large scale, prospective screening of EGFR mutations in the blood of advanced NSCLC patients to guide treatment decisions. Ann Oncol 28, 2248–2255. [DOI] [PubMed] [Google Scholar]
- 81. Wang Z, Cheng Y, An T, Gao H, Wang K, Zhou Q, Hu Y, Song Y, Ding C, Peng F et al. (2018) Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first‐line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single‐arm, multicentre clinical trial. Lancet Respir Med 6, 681–690. [DOI] [PubMed] [Google Scholar]
- 82. Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, McGranahan N, Wilson GA, Birkbak NJ, Olivas VR et al. (2017) Evolution and clinical impact of co‐occurring genetic alterations in advanced‐stage EGFR‐mutant lung cancers. Nat Genet 49, 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Leighl NB, Page RD, Raymond VM, Daniel DB, Divers SG, Reckamp KL, Villalona‐Calero MA, Dix D, Odegaard JI, Lanman RB et al. (2019) Clinical utility of comprehensive cell‐free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non‐small cell lung cancer. Clin Cancer Res 25, 4691–4700. [DOI] [PubMed] [Google Scholar]
- 84. Palmero R, Taus A, Viteri S, Majem M, Carcereny E, Garde‐Noguera J, Felip E, Nadal E, Malfettone A, Sampayo M et al. (2021) Biomarker discovery and outcomes for comprehensive cell‐free circulating tumor DNA versus standard‐of‐care tissue testing in advanced non–small‐cell lung cancer. JCO Precis Oncol 5, 93–102. 10.1200/PO.20.00241 [DOI] [PubMed] [Google Scholar]
- 85. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez‐Vivanco G, Isla D, Provencio M et al. (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361, 958–967. [DOI] [PubMed] [Google Scholar]
- 86. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia‐Gomez R, Pallares C, Sanchez JM et al. (2012) Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 13, 239–246. [DOI] [PubMed] [Google Scholar]
- 87. Karachaliou N, Mayo‐de las Casas C, Queralt C, de Aguirre I, Melloni B, Cardenal F, Garcia‐Gomez R, Massuti B, Sánchez JM, Porta R et al. (2015) Association of EGFR L858R mutation in circulating free DNA with survival in the EURTAC Trial. JAMA Oncol 1, 149–157. [DOI] [PubMed] [Google Scholar]
- 88. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, Zhou C, Reungwetwattana T, Cheng Y, Chewaskulyong B et al. (2020) Overall survival with osimertinib in untreated. N Engl J Med 382, 41–50. [DOI] [PubMed] [Google Scholar]
- 89. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T et al. (2018) Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med 378, 113–125. [DOI] [PubMed] [Google Scholar]
- 90. Tomoshige K, Guo M, Tsuchiya T, Fukazawa T, Fink‐Baldauf IM, Stuart WD, Naomoto Y, Nagayasu T & Maeda Y (2018) An EGFR ligand promotes EGFR‐mutant but not KRAS‐mutant lung cancer in vivo. Oncogene 37, 3894–3908. [DOI] [PubMed] [Google Scholar]
- 91. Kim EY, Cho EN, Park HS, Hong JY, Lim S, Youn JP, Hwang SY & Chang YS (2016) Compound EGFR mutation is frequently detected with co‐mutations of actionable genes and associated with poor clinical outcome in lung adenocarcinoma. Cancer Biol Ther 17, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chic N, Mayo‐de‐Las‐Casas C & Reguart N (2017) Successful treatment with gefitinib in advanced non‐small cell lung cancer after acquired resistance to osimertinib. J Thorac Oncol 12, e78–e80. [DOI] [PubMed] [Google Scholar]
- 93. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, Sequist LV & Engelman JA (2015) The allelic context of the C797S mutation acquired upon treatment with third‐generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res 21, 3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y et al. (2015) Acquired EGFR C797S mutation mediates resistance to AZD9291 in non‐small cell lung cancer harboring EGFR T790M. Nat Med 21, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, Lagrasta CA, Bottarelli L, Osipova G, Capelletto E et al. (2016) L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR‐mutated NSCLC. J Thorac Oncol 11, e121–e123. [DOI] [PubMed] [Google Scholar]
- 96. Starrett JH, Guernet AA, Cuomo ME, Poels KE, VanAlderwereltVan Rosenburgh IK, Nagelberg A, Farnsworth D, Price KS, Khan H et al. (2020) Drug sensitivity and allele specificity of first‐line osimertinib resistance. Cancer Res 80, 2017–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Brown BP, Zhang YK, Westover D, Yan Y, Qiao H, Huang V, Du Z, Smith JA, Ross JS, Miller VA et al. (2019) On‐target resistance to the mutant‐selective EGFR inhibitor osimertinib can develop in an allele‐specific manner dependent on the original EGFR‐activating mutation. Clin Cancer Res 25, 3341–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, Chang JC, Paik PK, Offin M, Arcila ME et al. (2020) Tumor analyses reveal squamous transformation and off‐target alterations as early resistance mechanisms to first‐line osimertinib in EGFR‐mutant lung cancer. Clin Cancer Res 26, 2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, Bao H, Tong X, Wang X, Shao YW et al. (2018) Investigating novel resistance mechanisms to third‐generation EGFR tyrosine kinase inhibitor osimertinib in non‐small cell lung cancer patients. Clin Cancer Res 24, 3097–3107. [DOI] [PubMed] [Google Scholar]
- 100. Lin CC, Shih JY, Yu CJ, Ho CC, Liao WY, Lee JH, Tsai TH, Su KY, Hsieh MS, Chang YL et al. (2018) Outcomes in patients with non‐small‐cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study. Lancet Respir Med 6, 107–116. [DOI] [PubMed] [Google Scholar]
- 101. Beagan JJ, Bach S, van Boerdonk RA, van Dijk E, Thunnissen E, van den Broek D, Weiss J, Kazemier G, Pegtel DM, Bahce I et al. (2020) Circulating tumor DNA analysis of EGFR‐mutant non‐small cell lung cancer patients receiving osimertinib following previous tyrosine kinase inhibitor treatment. Lung Cancer 145, 173–180. [DOI] [PubMed] [Google Scholar]
- 102. Cheng Y, Ma L, Liu Y, Zhu J, Xin Y, Liu X, Wang Y, Zhang T, Yang C, Wang S et al. (2020) Comprehensive characterization and clinical impact of concomitant genomic alterations in EGFR‐mutant NSCLCs treated with EGFR kinase inhibitors. Lung Cancer 145, 63–70. [DOI] [PubMed] [Google Scholar]
- 103. Shaw AT, Bauer TM, de Marinis F, Felip E, Goto Y, Liu G, Mazieres J, Kim DW, Mok T, Polli A et al. (2020) First‐line lorlatinib or crizotinib in advanced. N Engl J Med 383, 2018–2029. [DOI] [PubMed] [Google Scholar]
- 104. Dagogo‐Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, Chin E, Ackil J, Farago AF, Hata AN et al. (2019) Treatment with next‐generation ALK inhibitors fuels plasma. Clin Cancer Res 25, 6662–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, Lo YC, Li YY, Lamberti G, Nguyen T et al. (2020) Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14‐mutant NSCLC. Clin Cancer Res 26, 2615–2625. [DOI] [PubMed] [Google Scholar]
- 106. Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, Mazieres J, Viteri S, Senellart H, Van Meerbeeck J et al. (2020) Tepotinib in non‐small‐cell lung cancer with MET exon 14 skipping mutations. N Engl J Med 383, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Drilon A, Clark JW, Weiss J, Ou SI, Camidge DR, Solomon BJ, Otterson GA, Villaruz LC, Riely GJ, Heist RS et al. (2020) Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med 26, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kunimasa K, Hirotsu Y, Miyashita Y, Goto T, Amemiya K, Mochizuki H, Samamoto I, Ohki T, Oyama T, Honma K et al. (2020) Multiregional sequence revealed SMARCA4 R1192C mutant clones acquired EGFR C797S mutation in the metastatic site of an EGFR‐mutated NSCLC patient. Lung Cancer 148, 28–32. [DOI] [PubMed] [Google Scholar]
- 109. Yang JC, Camidge DR, Yang CT, Zhou J, Guo R, Chiu CH, Chang GC, Shiah HS, Chen Y, Wang CC et al. (2020) Safety, efficacy, and pharmacokinetics of almonertinib (HS‐10296) in pretreated patients With EGFR‐mutated advanced NSCLC: a multicenter, open‐label, phase 1 trial. J Thorac Oncol 15, 1907–1918. [DOI] [PubMed] [Google Scholar]
- 110. ctDNA guiding treatment after almonertinib induction therapy for EGFRm+ NSCLC in the MDT diagnostic model. https://ClinicalTrials.gov/show/NCT04841811
- 111. Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R & Ebi H (2020) Epithelial‐to‐mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C‐mutant non‐small cell lung cancer. Clin Cancer Res 26, 5962–5973. [DOI] [PubMed] [Google Scholar]
- 112. Chaib I, Karachaliou N, Pilotto S, Codony Servat J, Cai X, Li X, Drozdowskyj A, Servat CC, Yang J, Hu C et al. (2017) Co‐activation of STAT3 and YES‐associated protein 1 (YAP1) pathway in EGFR‐mutant NSCLC. J Natl Cancer Inst 109, djx014. 10.1093/jnci/djx014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ito M, Codony‐Servat C, Karachaliou N & Rosell R (2019) Targeting PKCι‐PAK1 in EGFR‐mutation positive non‐small cell lung cancer. Transl Lung Cancer Res 8, 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Karachaliou N, Chaib I, Cardona AF, Berenguer J, Bracht JWP, Yang J, Cai X, Wang Z, Hu C, Drozdowskyj A et al. (2018) Common co‐activation of AXL and CDCP1 in EGFR‐mutation‐positive non‐smallcell lung cancer associated with poor prognosis. EBioMedicine 29, 112–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. El Kadi N, Wang L, Davis A, Korkaya H, Cooke A, Vadnala V, Brown NA, Betz BL, Cascalho M, Kalemkerian GP et al. (2018) The EGFR T790M mutation is acquired through AICDA‐mediated deamination of 5‐methylcytosine following TKI treatment in lung cancer. Cancer Res 78, 6728–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Heidrich I, Ačkar L, Mossahebi Mohammadi P & Pantel K (2021) Liquid biopsies: potential and challenges. Int J Cancer 148, 528–545. [DOI] [PubMed] [Google Scholar]
- 117. BESPOKE study of ctDNA guided immunotherapy. https://ClinicalTrials.gov/show/NCT04761783
- 118. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR et al. (2016) EGFR mutations and ALK rearrangements are associated with low response rates to PD‐1 pathway blockade in non‐small cell lung cancer: a retrospective analysis. Clin Cancer Res 22, 4585–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR et al. (2015) Co‐occurring genomic alterations define major subsets of KRAS‐mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 5, 860–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chen K, Liu J, Liu S, Xia M, Zhang X, Han D, Jiang Y, Wang C & Cao X (2017) Methyltransferase SETD2‐mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 170, 492–506.e414. [DOI] [PubMed] [Google Scholar]
- 121. Hanna NH, Temin S & Masters G (2020) Therapy for stage IV non‐small‐cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update summary. JCO Oncol Pract 16, e844–e848. [DOI] [PubMed] [Google Scholar]
- 122. Anagnostou V, Niknafs N, Marrone K, Bruhm DC, White JR, Naidoo J, Hummelink K, Monkhorst K, Lalezari F, Lanis M et al. (2020) Multimodal genomic features predict outcome of immune checkpoint blockade in non‐small‐cell lung cancer. Nat Cancer 1, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wan JCM, Massie C, Garcia‐Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R & Rosenfeld N (2017) Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17, 223–238. [DOI] [PubMed] [Google Scholar]
- 124. Giménez‐Capitán A, Bracht J, García JJ, Jordana‐Ariza N, García B, Garzón M, Mayo‐de‐Las‐Casas C, Viteri‐Ramirez S, Martinez‐Bueno A, Aguilar A et al. (2021) Multiplex detection of clinically relevant mutations in liquid biopsies of cancer patients using a hybridization‐based platform. Clin Chem 67, 554–563. [DOI] [PubMed] [Google Scholar]
- 125. Wan JCM, Heider K, Gale D, Murphy S, Fisher E, Mouliere F, Ruiz‐Valdepenas A, Santonja A, Morris J, Chandrananda D et al. (2020) ctDNA monitoring using patient‐specific sequencing and integration of variant reads. Sci Transl Med 12, eaaz8084. 10.1126/scitranslmed.aaz8084 [DOI] [PubMed] [Google Scholar]
- 126. Newsome RC & Jobin C (2020) Microbiome‐derived liquid biopsy: new hope for cancer screening? Clin Chem 67, 463–465. [DOI] [PMC free article] [PubMed] [Google Scholar]