

Abstract
Analysis of circulating tumor cells (CTCs) collected from patient's blood offers a broad range of opportunities in the field of precision oncology. With new advances in profiling technology, it is now possible to demonstrate an association between the molecular profiles of CTCs and tumor response to therapy. In this Review, we discuss mechanisms of tumor resistance to therapy and their link to phenotypic and genotypic properties of CTCs. We summarize key technologies used to isolate and analyze CTCs and discuss recent clinical studies that examined CTCs for genomic and proteomic predictors of responsiveness to therapy. We also point out current limitations that still hamper the implementation of CTCs into clinical practice. We finally reflect on how these shortcomings can be addressed with the likely contribution of multiparametric approaches and advanced data analytics.
Keywords: CTC enrichment, drug selection, microfluidics, molecular profiling, precision oncology, tumor resistance
This Review highlights the emerging role of circulating tumor cell (CTC) profiling in precision oncology. We summarize key technologies currently employed for CTC isolation and analysis and discuss recent clinical studies that examined CTCs for genomic and proteomic predictors of therapeutic response. We point out challenges associated with implementation of CTC profiling into clinical practice and reflect on potential solutions.
Abbreviations
- ADT
androgen deprivation therapy
- cfDNA
circulating cell‐free DNA
- cfRNA
circulating cell‐free RNA
- CGH
comparative genomic hybridization
- CNG, copy number gain ; CNVs
copy number variations
- CTC
circulating tumor cell
- CTSC
circulating tumor stem cell
- DEF
dielectrophoresis
- DFF
dean flow fractionation
- EMT
epithelial‐to‐mesenchymal transition
- EVs
extracellular vesicles
- FACS
fluorescence‐activated cell sorting
- FISH
fluorescence in situ hybridization
- GEDI
geometrically enhanced differential immunocapture
- ITH
intra‐tumoral heterogeneity
- LDT
laboratory‐developed test
- MACS
magnetic‐activated cell sorting
- MagRC
magnetic ranking cytometry
- mCRPC
metastatic castration‐resistant prostate cancer
- MET
mesenchymal‐to‐epithelial transition
- MICs
metastasis‐initiating cells
- MRD
minimal residual disease
- NSCLC
non‐small‐cell lung cancer
- PDAC
pancreatic ductal adenocarcinoma
- PET‐CT
positron emission tomography‐computed tomography
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- SARMS
scorpion amplification refractory mutation system
- SCBC
single‐cell barcode chip
- SCLC
small cell lung cancer
- scWestern
single‐cell Western blotting
- SERD
selective estrogen receptor downregulator
- TCGA
The Cancer Genome Atlas
- TEPs
tumor‐educated platelets
- TKI
tyrosine kinase inhibitor
1. Introduction
One of the key objectives of precision oncology is to improve diagnosis and treatment of cancer [1]. The Cancer Genome Atlas (TCGA) research network has provided comprehensive molecular profiles for hundreds of tumors, which allowed the recognition of different molecular aberrations and their functional roles in different tumors as well as the identification of potential druggable targets, which is vital for the realization of precision oncology [2]. As a result, we have witnessed an explosion of cancer therapeutics that target specific molecular aberrations driving tumors, interfere with signaling pathways, or block immune checkpoints [3].
Precision oncology has been slowly but surely changing the traditional concept of ‘one treatment fits all’ to ‘one patient, one treatment’, which heightened the need for robust molecular profiling methods for individual patients' tumors, often obtainable only through image‐guided tissues biopsies [4, 5]. However, the complex nature of tumor biology usually brings about a substantial degree of heterogeneity, which requires comprehensive analysis of the primary tumor as well as repeated sampling of its metastatic lesions. This is not feasible in clinical practice because tissue biopsies are often restricted to few sampling points and accessible sites due to the associated cost, risks and logistical hurdles [6]. Also, tumor tissue sampling might seed the tumor cells around the sampling area, leading to local dissemination [7]. The risks and caveats of solid biopsies become more pronounced when dealing with multiple metastases that require sampling from different metastatic lesions and comparing their molecular profiles to obtain a holistic view of the disease. Unfortunately, a growing body of evidence suggests that the molecular profiles of primary tumors and their metastatic lesions are not always concordant, owing to intrinsic tumor heterogeneity [8]. These pitfalls have gradually turned the tide of precision oncology toward liquid biopsies because the blood contacts most tumor sites, and thus, it became logical to speculate that liquid biopsies would better mirror heterogeneous tumors than tissues biopsies [9].
In contrast to tissue sampling, liquid biopsies are not limited by sampling frequency, tumor accessibility or the presence of clinically overt disease. The term liquid biopsy refers to sampling and detecting analytes in different biological fluids, such as blood, urine, saliva, cerebrospinal fluid, ascites, and pleural effusions [10, 11]. Analytes in the peripheral blood of cancer patients typically contain circulating tumor cells (CTCs), circulating cell‐free DNA (cfDNA), circulating cell‐free RNA (cfRNA), extracellular vesicles (EVs), tumor‐educated platelets (TEPs), proteins, and other metabolites [12]. Among these analytes, CTCs represent the most prominent liquid biopsy marker that have been broadly utilized to provide information about features of primary tumors or metastases, track evolutionary dynamics and heterogeneity of tumors, and detect the emergence of treatment resistance, residual disease, and recurrence [13]. Profiling CTCs can provide a ‘moving picture’ of longitudinal tumor progression, whereas only a ‘snapshot’ of the cancer can be gained by a conventional tissue biopsy [14].
Response to cancer therapy is conventionally determined based on diagnostic imaging and blood‐based tumor biomarkers. Diagnostic imaging techniques such as positron emission tomography‐computed tomography (PET‐CT) suffer from low sensitivity and specificity [15]. Analysis of tumor biomarkers is also hampered by low sensitivity and specificity. In addition, sampling patients with low tumor burden might lead to false negative results, whereas false‐positive findings might arise from the release of biomarkers by noncancerous tissues and comorbid diseases [16]. One of the key advantages of using CTCs as a molecular proxy to monitor cancer resistance is that they can provide quantitative information on treatment response and allow qualitative elucidation of resistance mechanisms. Unfortunately, capturing CTCs from blood samples has been technically challenging due to their low abundance (a few to hundreds per milliliter) in a high background (billions per milliliter) of blood cells [17]. With the recent advances in single‐cell technologies made during the last 5 years, profiling CTCs at the single‐cell level in peripheral blood has offered a unique minimally invasive approach to track therapeutic resistance [18].
In this Review, we survey recent studies that have leveraged CTC‐level information to predict tumor response to therapy. We discuss current limitations that still hamper the implementation of CTCs into clinical practice. We conclude by reflecting on how these shortcomings might be addressed, and in particular, on the likely contributions of multiparameter assays and advanced data analytics.
2. Tumor resistance as a challenge in precision oncology
In precision medicine, the decision to treat with targeted therapy is dictated by the presence of a specific biomarker (e.g., gene mutation or amplification) in the tumor. Unfortunately, the response of many solid tumors to targeted therapy is < 50%, owing to tumor resistance, tumor heterogeneity, or inaccuracies produced by the choice of profiling method [19]. Resistance to targeted therapy can be classified as primary or secondary resistance. Primary or intrinsic resistance describes cancer patients who do not respond to treatment at all, whereas secondary or acquired resistance involves patients who initially respond to treatment then later fail to respond as the drug‐sensitive tumor cells die and the resistant subclones continue to grow. A case in point is BRAFV600E melanoma patients who initially respond to treatment with tyrosine kinase inhibitors (e.g., vemurafenib); then, most of these patients relapse with a deadly disease due to secondary resistance [20].
Intratumor heterogeneity plays a major role in resistance to targeted therapy because a highly heterogeneous tumor would more likely harbor drug‐resistant subclones [21]. Although most targeted therapies are directed toward specific genetic aberrations, there are other factors that can contribute to resistance including epigenetic alterations that arise rapidly due to microenvironmental factors [22]. The surrounding tumor microenvironment can also contribute to tumor resistance by protecting the tumor cells from infiltrating drugs either by providing a physical barrier or through releasing paracrine signaling factors that can alter tumor cell survival [23]. This can lead to secondary resistance as the surviving tumor cells continue to grow under altered selective pressure [19]. Even exposure to therapy can impose selective pressure on the tumor, leading to the expansion of pre‐existing drug‐tolerant subclones that have survived initial therapy due to adoptive activation of alternative molecular pathways [24, 25]. Tumor resistance to therapy, whether it is intrinsic or acquired, is best studied using liquid biopsy markers to monitor tumor evolution under the selective pressure imposed by cancer therapy.
3. CTC biology and clinical implications
During metastasis, epithelial tumor cells undergo epithelial‐to‐mesenchymal transition (EMT), lose their polarity and secrete specific enzymes (e.g., cathepsin D) to digest the extracellular matrix and acquire migratory properties. Invasive tumor cells then reach the bloodstream and become CTCs. Subsequent to extravasation, CTCs can invade a new organ and undergo mesenchymal‐to‐epithelial transition (MET) [26]. There is substantial evidence that metastases can be driven by a population of CTCs with stem cell‐like features, such as self‐renewal and multipotentiality, hence referred to as circulating tumor stem cells (CTSCs) [27].
3.1. CTCs and resistance to therapy
The presence of CTCs with an EMT phenotype has been correlated with tumor relapse and resistance to therapy. For example, overexpression of CSV and plastin 3 in EMT‐positive CTCs was found to cause drug resistance in patients with metastatic colorectal cancer [28]. The acquisition of stem cell features during EMT was found to enhance the survival of the CTCs, leading to higher migratory and invasive properties as well as resistance to cancer therapy [29]. Also, the presence of CTCs with stem cell features can lead to tumor relapse following targeted therapy, such as in colorectal cancer [30]. This is because drug‐resistant CSCs might act as metastasis‐initiating cells (MICs), leading to more aggressive tumors due to the selection pressure imposed by the anticancer drug. Phenotypic switching of CTCs to a less differentiated phenotype has also been reported to correlate with tumor relapse after targeted therapy in melanoma patients [31], and the role of phenotypic switching in tumor resistance to therapy has also been reported in other types of cancer, such as breast, prostate, pancreatic, and non‐small‐cell lung cancer (NSCLC) [32]. The major mechanisms of therapeutic resistance in primary tumor cells have also been observed in CTCs, including target inactivation or mutation, improving genomic DNA repair, accelerated drug efflux, upregulation of quiescence markers, downregulation of proliferative markers, and suppression of oxygen radical formation [33]. The hypothesis that CTCs with EMT and/or stem cell phenotype represent the only route for initiating metastasis has been challenged by the finding that tumor cells can detach from the primary tumor and circulate in the blood as clusters of small cells (i.e., groups of > 2 CTCs and up to large micro‐emboli) [34]. The presence of CTCs clusters might also contribute to the metastatic potential and resistance to therapy. The ability of CTC clusters to evade cytotoxic drugs has been ascribed to lack of proliferation as indicated by Ki67 staining [35]. For instance, the overall survival after systemic therapy was significantly reduced in pancreatic ductal adenocarcinoma (PDAC) patients harboring large number of CTCs clusters (more than 30 clusters per 2 mL of blood) [36].
Expansion of CTCs from patients as cell lines or cell line‐derived xenografts might provide further clues about their therapeutic resistance and offer an opportunity to test drugs for personalized medicine. Unfortunately, CTC‐derived cell lines or xenografts require high CTC counts and might take several months, which make them unsuited for clinical practice [37]. In addition, long‐term passage of CTCs on a plate is likely to cause irreversible adaptation and clonal outgrowth [9]. Alternatively, isolation of CTCs and their downstream analysis, particularly at the single‐cell level, for molecular predictors of responsiveness to therapy can address these limitations and guide personalized oncology (Fig. 1). In the following section, we discuss CTC enrichment and analysis technologies, focusing on the approaches that have been successfully used for monitoring tumor resistance and informing drug selection.
Fig. 1.
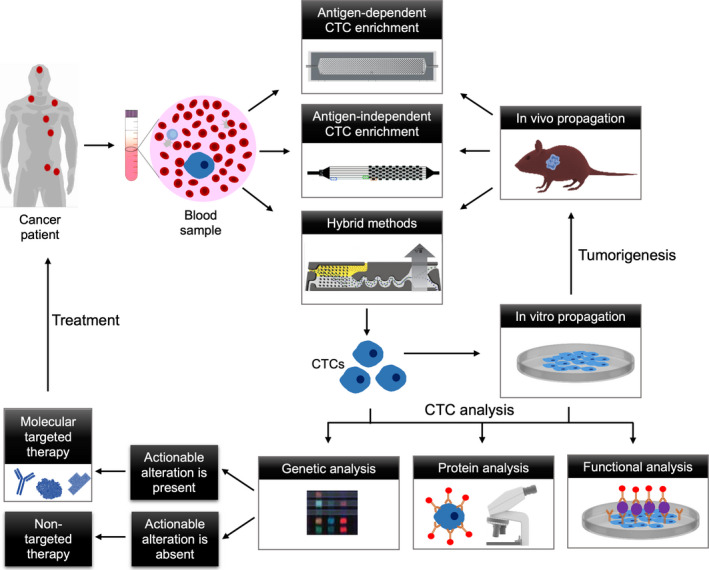
CTC analysis guiding precision oncology. To achieve this goal, CTCs are isolated from the blood of a cancer patient using antigen‐dependent or antigen‐independent CTC enrichment approaches or hybrid methods. The isolated CTCs can either be analyzed directly with single‐cell approaches or cultured in vitro before analysis. The CTCs propagated in vitro can also be xenografted to propagate in vivo then isolated using the previous approaches. The CTCs are analyzed for predictors of responsiveness to therapy using genetic, protein, and functional assays. Upon detection of actionable alterations, the patient is treated with molecular targeted therapy. In absence of molecular alterations, an appropriate therapeutic intervention is decided by the physician.
3.2. CTC enrichment strategies
Methods used to enrich CTCs are divided into antigen‐dependent, antigen‐independent approaches or a combination of both. These methods have been comprehensively reviewed elsewhere [38, 39] but a brief summary of the methods that relate to the focus of this article is provided below.
3.2.1. Antigen‐dependent CTC enrichment
This approach is usually performed using immunoaffinity techniques that utilizes a capture agent for positive selection of CTCs by targeting cell‐surface markers [40, 41], or negative selection via depletion of white blood cells (WBCs) [42].
Positive enrichment
Based on positive enrichment, the CellSearch platform is the only one to date that is approved by the Food and Drug Administration (FDA) for CTC enumeration [43]. In this method, the patient's blood is collected in a CellSave tube, which contains an anticoagulant and a proprietary fixative/stabilizer to preserve the blood for up to 96 h. The RBCs are removed by Ficoll centrifugation, and the buffy coat is then incubated with an EpCAM antibody conjugated with magnetic nanoparticles. Magnetically labeled CTCs are then isolated by applying a magnetic field and immunostained using a three‐color protocol for cytokeratin (CK) proteins, leukocyte antigen (CD45), and the nuclear stain (DAPI). The cells suspended in the magnetic chamber (MAGNEST) are then imaged with a fluorescence microscope. CTCs are identified by a distinct staining pattern (CK+/CD45−/DAPI+) and enumerated [44]. Magnetic‐activated cell sorter (MACS) is another commercially available platform for CTC isolation that relies on positive selection of CTCs [45]. However, both approaches suffer from low CTC recovery.
Several affinity‐based microfluidic devices have been developed for positive enrichment of CTCs. Microfluidics‐enabled immunoseparation involves capturing CTCs within antibody‐coated microfluidic devices. For example, the CTC‐Chip contains 78 000 microposts that are chemically conjugated with EpCAM antibody to isolate CTCs from blood (Fig. 2A) [40, 46]. The NanoVelcro chip contains a silicon nanowire substrate (SiNS) conjugated with EpCAM antibody via biotin‐streptavidin coupling (Fig. 2B) [47]. The nanostructured SiNS enhances the cell capture efficiency. The herringbone device, HBCTC‐Chip, comprises herringbone microstructures to enhance the interaction between CTCs and the antibody‐coated surface via convective mixing (Fig. 2C) [48].
Fig. 2.
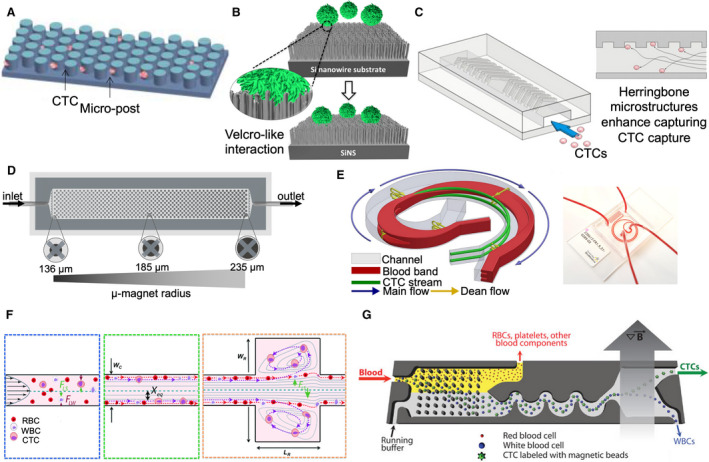
CTC enrichment approaches used for monitoring tumor resistance. (A–D) Antigen‐dependent CTC enrichment approaches. (A) Illustrative cartoon showing CTCs captured against the microposts within the CTC‐Chip. (B) The working mechanism of the nanoVelcro chip, which contains silicon nanowire substrate (SiNS) coated with EpCAM antibodies to enhance the capture efficiency of CTCs. (C) Illustration of the herringbone device (HBCTC‐Chip), featuring a microfluidic array of channels with a single inlet and outlet. (D) The microfluidic device used for magnetic ranking cytometry (MagRC) consists of 100 distinct capture zones. An array of X‐shaped microstructures creates regions of low flow velocity and circular nickel micromagnets patterned within the channel enhance the externally applied magnetic field. (E, F) Antigen‐independent CTC enrichment methods used for resistance monitoring. (E) Schematic illustration of Dean flow fractionation (DFF) principle of the antigen‐independent ClearCell Fx system (left). The target cells travel to the inner together with the blood band following the Dean flow stream in the first half curvature of the channel. Once the inner side of the channel is reached, the target cells remain focused while the blood band travels to the outer side in the second half curvature of the channel. Both target cells and nontarget cells are collected from different outlets at the end of the spiral channel. The microfluidic device utilized in the ClearCell Fx system (right) has sample and sheath inlets at one end and recovery and waste outlets at the other end. (F) Schematic of the Vortex chip featuring 8 channels. (a) At the channel inlet, the cells are randomly distributed and experience to opposing lift forces, the wall effect (F LW) and the shear‐gradient lift force (F LS). (b) As a result, the cells migrate to dynamic lateral equilibrium position (X eq), based to the channel cross section. (c) Upon entering the reservoir, the wall effect is reduced so large cells which still experience large F LW are pushed to the vortices where they become captured, whereas small cells, which do not experience sufficient F LW remain in the main flow. (G) Schematic of a hybrid CTC enrichment system, the CTC‐iChip. The blood is mixed with immunomagnetic beads and buffer before introduced into the device. CTCs are captured subsequent to inertial focusing and magnetophoresis. Figure A is reproduced with permission from Ref.[46]; Figure B is reproduced with permission from Ref. [47]; Figure C is reproduced with permission from Ref. [48]; Figure D is reproduced with permission from Ref. [41]. Figures E is reproduced with permission from Ref. [71]; Figure F is reproduced with permission from Ref. [72]; Figure G is reproduced with permission from Ref. [77]. Figure reproduced from Refs [41, 46, 47, 48, 71, 72, 77].
The captured CTCs can be released from the device by trypsinization, which might cause loss of cell‐surface receptors and subsequently confound further downstream analysis [49]. This downside has been addressed by designing smaller microfluidic devices with magnetic pores in which the CTCs are released by vertical flow centrifugation [50]. Newer iterations of the nanoVelcro device exploit competitive binding or a thermo‐responsive polymer substrate to facilitate the release of the captured CTCs. Also, nucleic acid aptamers have been used in some microfluidic sorting techniques instead of antibodies, due to their low cost, ease of modification, and simple release mechanism [51, 52].
Immunomagnetic separation involves prelabeling the cells with magnetic beads‐conjugated antibodies, followed by capturing CTCs within a microfluidic device exposed to a magnetic field [41]. This workflow has been adopted in the magnetic ranking cytometry (MagRC) technology, which can rank the cells according to the expression of cell‐surface proteins [53], intracellular proteins [54], or mRNAs [55]. The MagRC devices contain capture zones to rank the cells according to their magnetic loading (Fig. 2D). Cells with high magnetic loading are captured in earlier zones, whereas cells with low magnetic loading are captured in later zones. The cells can be simply retrieved by removing the magnetic field. Several microfluidic platforms have also exploited magnetically labeled CTCs to improve their detection speed and sensitivity, such as MagSweeper [56], IsoFlux [57], VerIFAST [58], and magnetic sifters [59].
The efficiency of CTC affinity selection is influenced by the selectivity of the antibody used for enrichment. Some antibodies are restricted to specific cancer types (e.g., EGFR‐, HER2‐expressing tumors) or could suffer from low selectivity because their target cell‐surface markers are also expressed by other cells (e.g., VCAM‐1, ICAM‐1) [60]. This limitation has been addressed by using antibody cocktails specific to several cell‐surface markers to capture all CTC subsets [48]. For example, the Adnatest is based on a combination of magnetic‐labeled antibodies specific to EpCAM, EGFR, and MUC1 [61]. The cytapheresis technology (e.g., Cell‐Collector) was introduced to overcome the problems of low CTC count in blood samples and of CTC stability during storage [62]. Cytapheresis is an invasive approach in which the blood is passed through a machine that retains EpCAM‐positive CTCs and returns the CTC‐depleted blood to the patient's bloodstream [63]. However, this method is time‐consuming and cannot be used repeatedly due to the possible contamination of blood with excess iron leaked from the magnetic nanoparticles/wires used for sorting [64].
Negative enrichment
Negative CTC selection has been sought to overcome the limitations of positive enrichment, such as heterogeneous expression of cell‐surface markers due to EMT and expression of the hallmark marker, EpCAM, by noncancerous cells in some diseases (e.g., benign colon disease). In this approach, magnetic beads conjugated with antibodies specific to CD45 and CD61 are used to deplete leukocytes and platelets, respectively [65]. However, negative selection is often followed by the isolation of circulating CD45− endothelial cells to improve the recovery of CTCs.
Overall, antigen‐dependent CTC enrichment has several advantages but might not be the method of choice if the binding between the antibody and cell‐surface receptor leads to activation of specific signaling pathways, which might complicate further analysis of intracellular proteins or mRNAs [66]. In such case, a label‐free approach that relies on the physical properties of CTCs would be a better choice.
3.2.2. Antigen‐independent CTC enrichment
Antigen‐independent, also called ‘label‐free’, CTC isolation approaches are used to isolate CTCs from the blood, depending on their size and deformability, density, and electric charge. Size‐based CTC isolation relies on microfabricated filters, such as 2D filter membranes (e.g., ISET, CellSieve, and ScreenCell), and 3D microfilters (FaCTChecker and Parsortix), to isolate single CTCs or CTC clusters based on their size and/or deformability [67]. Although CTCs (12–25 µm) are typically larger than WBCs (8–14 µm), false‐positive results might be obtained due to variations in CTC size as a result of apoptosis, clustering, or cell cycle stage [67]. Integration with flow cytometry was sought in some trials to improve the size‐based sorting of CTCs [68].
CTCs can also be isolated using hydrodynamic microfluidic devices that depend on inertial focusing to separate CTCs from other blood cells, exploiting centrifugal, Coriolis, and Euler forces to transport and manipulate liquids through their interaction with microstructures [69]. The term, inertial focusing, refers to migration of cells across streamlines into equilibrium positions within the flow cross section as they migrate downstream in a microchannel. However, the purity of CTCs is usually low due to the size overlap between CTCs and WBCs. Also, cell–cell interactions might affect the cell‐focusing behavior and reduce the separation efficiency. Recently, an inertial focusing based labyrinth device was demonstrated to be capable of isolating single CTCs and CTC clusters from the blood of NSCLC patients with 100% and 96% cell recovery, respectively [70]. Other inertial focusing platforms that can isolate viable CTCs have also been introduced. The ClearCell Fx platform is an automated hydrodynamic centrifugation system that employs Dean flow fractionation (DFF) to separate between viable CTCs and other blood cells (Fig. 2E) [71]. The Vortex method hydrodynamically traps CTCs in side channels at high flow rates (Fig. 2F) [72].
Density‐based CTC isolation technologies (e.g., Ficoll gradient centrifugation), employ gradient centrifugation to isolate CTCs from other hematopoietic components according to their sedimentation rate. CTCs and mononuclear cells have lower buoyant densities (< 1.077 g·mL−1) unlike RBCs and neutrophils (> 1.077 g·mL−1) and can thus be extracted from the top layer. The advantage of this method is that it offers a simple way to isolate CTCs. However, the sensitivity of this system is poor due to the leakage of CTCs to the plasma layer or the formation of large CTC aggregates settling to the bottom of the gradient. This problem was solved using the OncoQuick gradient system (Greiner Bio‐One, Frickenhausen, Germany), which includes a porous barrier above the density gradient to prevent mixing with the whole blood [65]. This system has been demonstrated to achieve higher CTC enrichment compared to the standard Ficoll gradient method.
Electric charge‐based approaches exploit dielectrophoresis (DEP) to isolate CTCs based on their distinct dielectric properties, leading different dielectrophoretic migration compared to other cell types [73]. DEP separation techniques can achieve a single‐cell level purification. However, the process is slow because the separation requires a low fluid velocity, resulting in low sample throughput. A commercially available DEPArray technology combining the precision of microfluidics and power of microelectronics, has been utilized to isolate CTCs from breast and colon cancer patients [74, 75].
3.2.3. Combined approaches
The choice of a particular CTC enrichment technology is driven primarily by the analyte to be measured. For example, molecular profiling of CTCs would require technologies capable of isolating pure CTCs to improve the quality of molecular information gleaned from the assay. Eliminating the need for sample preprocessing prior to CTC separation is also crucial to avoid cross‐contamination and reduce sample loss. Antigen‐independent CTC enrichment approaches entail less complicated workflows, but the CTC purity is typically low. Antigen‐driven approaches permit the isolation of defined CTC phenotypes, making them compatible with molecular assays. However, these approaches require highly specific antibodies or even antibody cocktails as well as a robust release strategy.
Both antigen‐dependent and antigen‐independent CTC enrichment approaches have their own advantages and limitations and hence combining both methods has been pursued in an attempt to improve overall CTC purity and recovery. For instance, using a geometrically enhanced differential immunocapture (GEDI) chip, which sorts CTCs based on their size and immunoaffinity properties, can improve cell recovery, yielding CTC counts much higher than the CellSearch system [76]. Also, the CTC‐iChip can isolate CTCs from large volumes of blood by micropillar arrays, hydrodynamic size‐based sorting, and magnetophoresis, permitting the recovery of pure cells which are suitable for cell culture and downstream profiling (Fig. 2G) [77].
It is worth noting that for CTC analysis at the single‐cell level, CTC enrichment might be combined with another isolation step using fluorescence‐activated cell sorting (FACS) [78], laser microdissection [79], or manual [80] and automated micromanipulation [81] to isolate single CTCs.
3.3. Molecular profiling of CTCs
In addition to CTC enrichment, comprehensive molecular and functional profiling of isolated CTCs is highly desirable to characterize their metastatic potential and resistance to therapy [82].
3.3.1. Genetic analysis of CTCs
Quantitative real‐time polymerase chain reaction (qRT‐PCR) utilizes primers designed to target specific genes of interest and has been used to investigate the transcriptional landscape of CTCs from different types of cancer [48]. However, the method is susceptible to amplification bias and is unable to distinguish between viable and apoptotic cells [82]. There is also a significant loss of mRNA molecules during purification and processing steps.
Fluorescence in situ hybridization (FISH) is a cytogenetic technique that uses fluorescent probes to specifically detect the presence of specific DNA or RNA sequences in individual cells [83]. FISH might require labeling with multiple fluorescent probes to attain a sufficient signal, which might preclude accurate quantification. However, a quantifiable, dual‐colorimetric RNA‐in situ hybridization (ISH) technique was introduced to characterize EMT in CTCs collected from breast cancer patients [26].
Comparative genomic hybridization (CGH) is a cytogenetic method for analyzing copy number variations (CNVs) relative to ploidy level in the DNA of a test sample compared to a reference sample. Each DNA sample is labeled with different fluorescent molecules and the difference in fluorescence ratio is used to determine the DNA copy number along the chromosome [84]. CGH is used to detect unbalanced chromosomal changes and structural rearrangements inside the cell. A more specific form of CGH based on the use of DNA array, thus referred to as (ACGH), has been used to reveal copy number aberrations among single CTCs collected from patients with metastatic colorectal cancer [85].
Next‐generation sequencing is used for in‐depth genomic analysis of CTCs. Single‐cell RNA sequencing includes reverse transcription, cDNA amplification, and preparation of a sequencing library. After transcription, minute amounts of cDNA might be amplified, leading to amplification biases [86]. Third‐generation sequencing methods alleviated this constraint, allowing for sequencing of nonamplified cDNAs, which is tremendously valuable for CTC analysis at the single‐cell level [87]. This powerful approach has been used to evaluate the inter‐ and intra‐tumoral heterogeneity (ITH) in CTCs, showing that ITH is a result of clonal evolution and presence of cancer stem cells [88]. Also, it was demonstrated that CTCs retrieved from prostate cancer patients shared 90% and 70% of the mutations present in the primary tumor and metastatic sites [89]. This study suggested that CTCs might originate from both the primary tumor and its metastatic lesions.
3.3.2. Protein‐level analysis for CTCs
Immunostaining is the most popular approach for analyzing proteins in CTCs. For instance, epithelial CTCs are commonly identified based on the presence of specific proteins, such as EpCAM, CK proteins, and absence of CD45 [44]. In addition, immunostaining for more biomarkers has been used for identification of CTCs in specific types of cancer, such as EGFR in prostate cancer and HER2 in breast cancer. The method is somewhat limited by the availability of specific antibodies. Several single‐cell approaches have been developed to analyze proteins in CTCs, such as MagRC [90], single‐cell barcode chip (SCBC) [91], and single‐cell western blotting (scWestern) [92].
3.3.3. Functional analysis of CTCs
The invasion assay is used to evaluate the invasiveness of CTCs based on their ability to digest a fluorescently labeled adhesion matrix. For example, this assay was used in conjunction with the MagRC platform to investigate the invasiveness of CTCs with low EpCAM expression. This study revealed the dynamic heterogeneity of CTCs and their phenotypic changes by mimicking the multistep metastatic cascade [93].
The EPISPOT assay is used to analyze tumor‐specific proteins released by CTCs [94]. In this assay, the cells are seeded and cultured in EPISPOT plate, which contains a nitrocellulose membrane coated with an antibody specific to the secreted protein of interest. During an incubation period (24–48 h), the secreted protein is captured by the immobilized antibody. The cells are then washed off and the secreted protein is detected using a fluorescently labeled secondary antibody. The fluorescent immunospots are then counted and used to estimate the number of viable CTCs that actively secrete proteins. The EPISPOT assay was used to demonstrate that viable epithelial tumor cells secrete full‐length CK19 and that CK19‐secreting cells might constitute a highly metastatic subset of breast cancer cells [95].
3.4. Monitoring drug resistance and informing drug selection
Currently, selection of targeted therapy in patients with metastatic cancer is solely based on the analysis of a primary tumor. A metastatic relapse might be encountered many years after tumor resection, while the data available from the resected primary tumor could be outdated. On the contrary, CTC analysis offers a means for interrogating the tumor repeatedly over the course of treatment. Evidence that CTCs can be used as a surrogate for the tumor tissue to monitor drug resistance came from two main observations. First, implantation of CTCs collected from patients into immunodeficient mice led to the formation of CTC‐derived explant models that recapitulate the donor's response to chemotherapy [37]. Second, molecular analysis of individual CTCs showed distinct somatic gene copy‐number profiles in patients with chemosensitive and chemorefractory SCLC [96]. In the following sections, we focus on how CTC analysis can contribute to elucidation of therapeutic resistance and effective personalized cancer therapy.
3.4.1. CTC count and therapeutic response
The CellSearch system has been used in several studies to investigate whether CTC enumeration carried out at baseline and during chemotherapy might be predictive of patient's response to treatment. In 2008, a landmark article demonstrated the prognostic value of CTC count in patients with metastatic castration‐resistant prostate cancer (mCRPC) when conducted before initiating a new treatment [97]. Although the baseline CTC count did not appear to be predictive of treatment response, the change in CTC count during treatment has offered a reliable marker of response to treatment in different clinical settings. In the IMMC38 study, mCRPC patients with high baseline CTC counts that changed to low counts after chemotherapy had better clinical outcomes than those patients whose baseline CTC counts remained high even after treatment [97]. These findings were later confirmed by a randomized phase III study that led to the approval of the prostate cancer drug, abiraterone [98]. Similarly, in metastatic breast cancer patients, a drop in CTC count after 4 weeks of treatment with first‐line therapy correlated with improved survival [99]. The prognostic value of CTC count has also been established in other types of cancer, such as lung and colorectal cancers. A clinical study revealed that metastatic colorectal cancer patients with low CTC counts after therapy had extended progression‐free and overall survival [100]. Also, the reduction of CTC count in SCLC patients after several cycles of chemotherapy was found to be a strong predictor of overall survival [101].
Variations in CTC count after drug exposure can also be used to study the pharmacodynamics of new drugs during early‐phase clinical trials. For example, a phase I trial of the PARP inhibitor, niraparib, has exploited changes in CTC count and nuclear γH2AX expression as endpoints of antitumor activity on a cohort of mCRPC patients [102]. A phase I study of ARQ197, a selective inhibitor of the hepatocyte growth factor receptor c‐MET, evaluated the changes in CTC count in patients with different tumors [103]. A phase I trial of EZN‐4176, a second‐generation antisense oligonucleotide to exon 4 of the androgen receptor, has utilized the variations in CTC count to evaluate the drug efficacy in prostate cancer patients [104].
The relationship between the number of CTC clusters and drug resistance was also investigated [105]. CTCs were collected from patients with early‐stage, locally advanced, or refractory metastatic breast cancer. Patient‐derived CTCs were cultured in an integrated microfluidic system incorporating tapered microfabricated wells for drug testing in situ (Fig. 3A,B). It was found that cluster formation correlated inversely with increased drug concentration (Fig. 3C). The method permitted determination of the median inhibitory concentrations after two weeks of treatment, allowing for rapid intervention upon detection of drug resistance or tolerance.
Fig. 3.
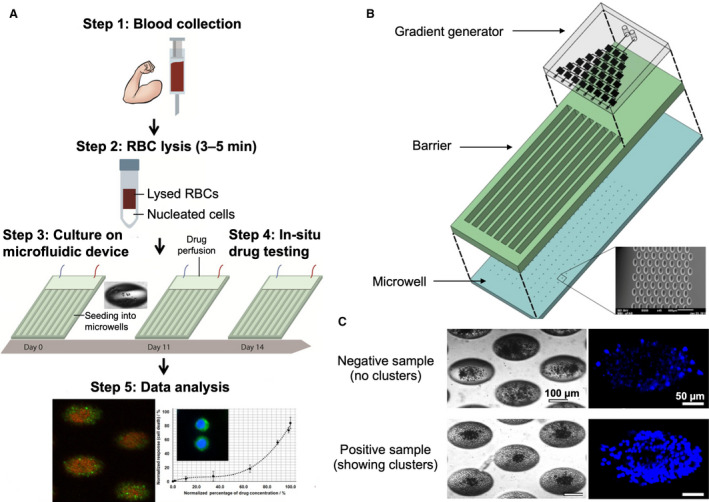
CTC cluster assay for drug screening. (A) Schematic depicting the cluster assay for anticancer drug screening. Blood samples are first lysed to remove red blood cells (RBCs), and the nucleated cell fraction is then seeded into an integrated microwell‐based microfluidic device. Drugs are introduced directly in situ, and the CTC clusters are generated within 2 weeks. (B) Three‐dimensional layout of the drug assay device showing the gradient generator, barrier, and microwells. (C) Representative bright‐field and fluorescent images of negative and positive samples. Nuclei were stained using Hoechst dye. Figures adapted with permission from Ref. [105].
Although several researchers have independently confirmed the prognostic value of CTC count alterations, no consensus has been reached on the optimal cutoff threshold for clinical prognostication [106]. Several factors are likely to contribute to this problem. First, several CTC detection technologies with different cutoffs for CTC enumeration have been used in different clinical studies, which made it difficult to compare the results and draw firm conclusions. Second, CTC isolation methods that target epithelial markers, such as EpCAM, might underestimate the CTC count in metastatic diseases [107]. Third, the lack of studies showing that switching of treatment in patients with persistent CTCs might have an effect on either progression‐free or overall survival. For example, a phase III clinical trial by the Southwest Oncology Group (SWOG S0500 study) found that changing therapy according to CellSearch results did not affect either progression‐free or overall survival of cancer patients [108]. For these reasons, studies that investigated the prognostic value of CTC count are relatively old and no new studies have been pursued. However, molecular characterization of CTCs at the genotypic and phenotypic levels has the potential to identify predictive biomarkers of response to personalized therapies.
3.4.2. Genomic aberrations and resistance
CTCs provide a valuable source for studying the genomic aberrations in cancer patients. Genomic‐level assays can be used to analyze CTCs for the presence or absence of key signaling oncogenic mutations. However, the driver mutation landscape usually changes as a result of the outgrowth of drug‐resistant subclones after exposure to systemic therapy. It is thus necessary to assess the molecular indicators of resistance in CTCs at the time of starting new line of targeted therapy and repeatedly over the course of treatment to guide clinicians on when to stop or change the treatment plan. As a case in point, a study on 254 breast cancer patients revealed the presence of HER2 overexpression in almost one‐third of the patients who had no HER2 overexpression in the primary tumor [109]. This finding had a remarkable clinical impact as overexpression of HER2 is a biomarker of response to HER2‐targeting drugs in breast cancer [110]. Another study revealed the presence of activating mutations in the EGFR‐encoding gene in CTCs collected from lung cancer patients receiving EGFR‐targeting tyrosine kinase inhibitors (TKIs). Interestingly, some of these mutations were absent in the primary tumor, indicating the de novo emergence of these mutations [111]. Both examples highlighted the superior predictive value of CTC assays compared to conventional tissue biopsies.
EGFR mutations can affect the outcome of EGFR TKIs in lung cancer patients. In one study, CTCs were isolated from NSCLC patients using the CTC‐chip and EGFR mutations were analyzed using the Scorpion Amplification Refractory Mutation System (SARMS) [111]. Analysis of CTCs isolated from EGFR‐mutant patients demonstrated a decrease in CTC count and improved radiological response within a week after starting a course of treatment with the EGFR TKI, gefitinib. In addition, analysis of CTCs obtained from patients with increased CTC count after treatment revealed the presence of EGFR T790M mutation, which was found responsible of resistance to EGFR TKIs. In a subsequent study, The CTC‐Chip was used to isolate the CTCs from NSCLC patients recurring after treatment with first‐generation EGFR TKIs [112]. The CTCs isolated from 76% of the patients were genotyped and the EGFR T790M mutation was identified in 50% of the cases. Interestingly, the mutational status of CTCs and primary tumors was concordant in only 57% of the patients, providing further evidence that molecular profiling of CTCs would better reflect the heterogeneous nature of the tumor. The emergence of EGFR T790M mutation in CTCs isolated from NSCLC patients treated with EGFR TKIs was also demonstrated using a different approach for CTC enrichment based on gradient centrifugation and negative enrichment prior to molecular characterization with droplet digital PCR [113]. Another study assessed the predictive value of CTCs harboring EGFR DelEx19 mutation in NSCLC patients [114]. The CTCs were isolated using gradient centrifugation and molecularly analyzed with qRT‐PCR. It was noted that 50% of the patients receiving EGFR TKIs were responsive to treatment and turned negative for EGFR‐mutant CTCs.
Mutations in KRAS—a protein downstream of EGFR—block the efficacy of EGFR‐targeting therapy. Meta‐analysis of the mutational status of KRAS in CTCs and tumor tissues revealed discrepancies in colorectal cancer patients [115]. Isolation of CTCs from colorectal cancer patients using the CellSearch system followed by their comprehensive genetic analysis with ACGH revealed mutations in known driver genes, such as KRAS, APC, and PIK3CA [85]. These mutations were also observed in the primary and metastatic tumors of the same patients. Remarkably, high level of amplification of CDK8, a potential target for CDK inhibitors, was observed in some of the isolated CTCs.
BRAF mutations have been utilized as predictors of response to targeted therapies in melanoma and colorectal cancer. For example, CTCs were detected in smaller numbers in patients with metastatic BRAFv600E melanoma and declined after BRAF‐targeted therapy [116]. In this study, CTCs were isolated using the HBCTC‐Chip; however, the low purity of CTCs precluded further molecular analysis. In a following study, the molecular signature of melanoma‐derived CTCs was revealed [117]. CTCs were isolated from metastatic BRAFv600E melanoma patients using the CTC‐iChip. A panel of 19 specific RNA transcripts was quantitatively analyzed with digital PCR, providing a CTC score. A decrease in CTC score within 7 weeks of treatment with immune checkpoint inhibitors was found to correlate with improvement in progression‐free and overall survival.
In breast cancer, resistance to HER2‐targeting therapy can occur as a result of PIK3CA mutations, which has been reported in 15.9% of breast cancer patients [118]. However, PIK3CA mutations were detected more frequently in metastatic breast cancer patients with HER2‐negative tumors and HER2‐positive CTCs [119]. Also, ESR1 mutations have emerged as acquired endocrine resistance mechanism in estrogen receptor (ER)‐positive metastatic breast cancer patients [120]. In a proof‐of‐concept study, CTCs were isolated from ER‐positive breast cancer patients using the CTC‐iChip. These CTCs were used to generate CTC lines, which were screened for mutations in a panel of 1000 annotated cancer genes using next‐generation sequencing [121]. Sequencing results revealed mutations in PIK3CA, ESR1, and FGFR2 genes, among others. Drug sensitivity testing of one CTC line harboring activating mutations in PIK3CA and FGFR2 revealed responsiveness to PIK3CA and FGFR2 inhibitors, whereas combined inhibition exhibited a cooperative effect.
Mutations in the androgen receptor (AR) gene were identified in CTCs from CRPC patients. Amplification of AR leads to overexpression of AR protein and hypersensitization of prostate cancer cells to residual androgens in patients receiving drug‐induced castration therapy. Mutations in the AR gene were associated with resistance to hormone therapy and blockade therapy was found to improve the survival rate of CRPC patients [122]. A protocol has been developed to combine CTC enumeration with the CellSearch system and characterization of AR amplification by FISH in an integrated CellSearch cartridge [123]. Using this protocol, AR amplifications were readily observed in CRPC patients. Interestingly, marked heterogeneity in AR gene copy numbers was observed in CTCs collected from the same patient. A similar observation was reported when ACGH analysis was carried out on CTCs isolated by FACS and immunomagnetic enrichment [124].
The most significant predictive value of CTCs has been highlighted by several reports demonstrating the correlation between the existence of AR splice variant 7 (AR‐V7)—coding for a truncated and constitutively active AR—and resistance to anti‐AR therapy [125, 126]. A case in point is a clinical study on two groups of CRPC patients receiving either abiraterone or enzalutamide [126]. Using the AdnaTest platform, the CTCs were isolated and AR‐V7 was detected with PCR. None of the patients harboring AR‐V7pos CTCs had a 50% PSA response rate compared to 68% and 53% of the patients with AR‐V7neg CTCs and progressing on abiraterone or enzalutamide, respectively. Using the same methodology, the presence of AR‐V7pos CTCs was observed in 46% of CRPC patients receiving taxanes [125]. However, the PSA response rate did not vary significantly between the AR‐V7pos and AR‐V7neg patients. Also, the progression‐free survival of AR‐V7pos patients treated with taxanes was longer than the patients treated with abiraterone or enzalutamide from the previous study. Admittedly, this indirect comparison between patients from different cohorts has to be interpreted more carefully, particularly because patients treated with taxanes had more advanced disease.
The AR splice variants 1, 3, 4, 7, and 12 were also analyzed by RNA sequencing of single CTCs isolated using the CTC‐iChip followed by cell picking by a micromanipulator [127]. This study revealed heterogeneous expression levels of the different splice variants between and within prostate cancer patients progressing on an AR inhibitor. These splice variants were not detected in the primary tumors, indicating that alternative splicing occurs during disease progression. AR‐V7 was the most frequently formed variant in 36% of the CTCs and 73% of the patients. Retrospective analysis of CTCs indicated the activation of noncanonical Wnt signaling and that heterogeneity in signaling pathways among single CTCs might have contributed to treatment failure. Analysis of AR‐V7 in CTCs isolated from prostate cancer patients has also been demonstrated using the MagRC platform [55]. Compared to other microfluidic platforms, the MagRC platform enabled the isolation of CTCs and analysis of specific mRNAs in one step and did not require any genotyping modalities. Instead, mRNA expression is determined by enumerating magnetically ranked CTCs and their distribution in the MagRC device, without interference from nonspecifically captured hematopoietic cells.
In addition to AR, TMPRSS2‐ERG rearrangements, resulting from fusion of an ERG oncogene and AR‐driven TMPRSS2 promoter, have been detected in more than 50% of prostate cancer patients [128]. The finding that patients with specific ERG rearrangements were more sensitive to anti‐AR therapy has shed light into the potential predictive value of these rearrangements and called for further trials [129]. In this study, CTCs were isolated from prostate cancer patients using the CellSearch system and characterized by FISH. Patients harboring TMPRSS2:ERG rearrangements responded better to abiraterone in terms of PSA response than patients in whom rearrangements were absent. However, the results shown by another study could not confirm such correlation [130]. The CellSearch system was also used for CTC enrichment from mCRPC patients, but the expression of TMRSS2:ERG was determined with qRT‐PCR. Few years later, the potential role of TMPRSS2:ERG as predictive marker to taxane therapy was confirmed [131]. However, further studies are still needed to clarify the discordance between the results and allow for prospective evaluation of the predictive value of TMPRSS2:ERG rearrangements.
Some studies assessed the possibility of using CTCs as a surrogate to detect other genomic rearrangements, such as ALK and ROS1, in lung cancer patients. In one study, CTCs were isolated from NSCLC patients using the ClearCell Fx system and ALK rearrangement patterns were detected with FISH [132]. It was noted that ALKpos CTC counts were higher in ALKpos patients compared to ALKneg patients and healthy donors. In addition, the study revealed an association between the count of ALKpos CTCs and response to the ALK inhibitor, crizotinib. Furthermore, a copy number gain (CNG) of oncogenic ALK in CTCs was found to contribute to disease progression despite treatment with crizotinib. In this respect, ALK CNG has also been hypothesized as a possible mechanism of resistance to ALK inhibitors in other studies [133, 134]. Using a different enrichment approach, the NanoVelcro chip was used to isolate CTCs from NSCLC patients and ALK rearrangements were detected with FISH. A correlation between the number of ALKpos CTCs and disease progression was demonstrated in patients treated with crizotinib [135].
The ISET filtration system and the filter adapted (FA)‐FISH method were used to detect ROS1 rearrangements in CTCs obtained from four NSCLC patients treated with crizotinib [136]. In this study, ROS1pos CTCs were detected in all patients with ROS1 rearrangement previously detected by a tumor biopsy. The number of ROS1pos CTCs was reduced in two patients who responded to crizotinib treatment, whereas no change was observed in one nonresponsive patient to crizotinib. In addition, the ROS1 CNG was assessed and it was found that number of ROS1 copies in ROS1pos CTCs had increased in two patients experiencing disease progression despite treatment.
3.4.3. Altered proteins and resistance
Surface proteins of CTCs have been established as suitable biomarkers for monitoring tumor resistance while targeted therapies directed toward these proteins have been approved in several types of cancer, such as breast, prostate, and lung cancers. Also, several single‐cell assays have been developed to analyze proteins in CTCs, despite their low yield in most cancer patients [41, 137]. In breast cancer, some of the most successful therapeutic agents are directed toward ER and HER2. Breast cancer patients with ER‐positive primary tumors can harbor ER‐negative CTCs [33]. Also, HER2‐positive CTCs were detected in patients with HER2‐negative primary tumors [109]. The discordance between ER or HER2 expression in CTCs and primary tumors was suggested as a potential cause of breast cancer resistance to endocrine or targeted therapy, respectively.
In one study, CTCs were isolated from metastatic breast cancer patients using the CellSearch system and two markers of endocrine sensitivity (ER and BCL2) were analyzed by immunostaining [138]. The study demonstrated a decrease in the number of ERpos CTCs during treatment with the selective ER downregulator (SERD), fulvestrant. The data also suggested a correlation between ER and BCL2 expression in CTCs, which supported the notion that ERneg CTCs might lack ER signaling.
Two multicenter studies have evaluated the predictive value of HER2pos CTCs in patients with HER2neg breast cancer and revealed higher survival under HER2‐targeted therapy with trastuzumab [139, 140]. A third study showed that the presence of HER2pos CTCs was not associated with tumor size, hormone receptor status, axillary lymph node involvement or histopathological grading [141]. In these studies, the CTCs were isolated using the CellSearch System and HER2 was detected with immunostaining. Besides other findings [142, 143], these reports suggested the need for determining the HER2 status in CTCs from higher number of patients in clinical trials to assess the efficacy of HER2‐targeted therapy.
PD‐1/PD‐L1 expression has been recognized as a suitable predictive biomarker for monitoring tumor response to checkpoint blockade immunotherapy, which has been approved in several types of cancer, such as NSCLC, breast, gastrointestinal, and head and neck cancer [144]. A correlation between PD‐L1 expression in CTCs and primary tumors was reported [145]. The expression of PD‐L1 in CTCs has been utilized as a biomarker for response to PD‐1 checkpoint inhibitors, such as nivolumab and pembrolizumab. In one study, the CellSearch system was used to isolate the CTCs from NSCLC patients treated with nivolumab and PD‐L1 positivity was detected with immunostaining [146]. After 6 months of treatment, patients with PD‐L1neg CTCs were found responsive to therapy, whereas the persistence of PD‐L1pos CTCs was associated with lack of response. Another study evaluated the expression of PD‐1/PD‐L1 in ISET filtration‐enriched CTCs isolated from NSCLC patients before and after chemotherapy [147]. The results showed a decrease in the number of PD‐1pos CTCs associated with an increase in the number of PD‐L1pos CTCs after three cycles of chemotherapy, suggesting that chemotherapy might have eliminated PD‐1pos CTCs. Several reports have confirmed the presence of PD‐L1pos CTCs in NSCLC patients using different enrichment technologies, such as the Vortex method and ClearCell FX system [148, 149]. In addition to qualitative PD‐L1 detection, one study revealed a correlation between the abundance of PD‐L1 in CTCs and the response to PD‐1/PD‐L1 blockade therapy in patients with advanced gastrointestinal tumors [150]. Immunofluorescence was used to quantify PD‐L1 expression in magnetically enriched CTCs. Prior to treatment, the abundance of PD‐L1 in CTCs was found to serve as a predictor of the therapeutic response.
Although the majority of protein assays have been applied to cell‐surface proteins, analysis of intracellular proteins is particularly beneficial for assessing the tumor response to therapy because intracellular oncoproteins are responsible of the initiation and progression of tumors [151]. For example, the subcellular localization of AR has been used as biomarker of response to abiraterone, enzalutamide, and taxanes. After activation by androgens, AR is translocated from the cytoplasm to the nucleus to act as a transcription factor for target genes. Thus, the nuclear localization of AR in tumor cells indicates an active AR signaling status [152]. In one study, it was demonstrated that the absence of nuclear AR in mCRPC patients treated with docetaxel correlated with clinical response, as assessed by immunostaining of AR in CTCs enriched using the CellSearch system [153]. This was ascribed to the ability of taxanes to inhibit ligand‐induced AR nuclear translocation and downstream transcriptional activation of AR target genes, such as PSA. Overall, 71% of the patients who responded to taxanes had cytoplasmic AR, whereas 72% of the CTCs sampled from patients with disease progression have exhibited nuclear AR. In another report, the AR abundance in CTCs obtained from CRPC patients progressing on abiraterone was determined using the ImageStreamX system, which is based on microscopy and flow cytometry [154]. The median AR staining intensity was found to be three times higher in patients progressing on abiraterone compared to patients who were abiraterone‐naïve, whereas no difference in AR subcellular localization was observed. AR signaling was studied by others to investigate the response of mCRPC patients to ADT [155]. In one study, CTCs were captured using the CTC‐Chip and analyzed with immunostaining for PSA and its membrane‐bound form, PSMA. Accordingly, the captured CTCs were assigned into three different categories with respect to AR signaling, including AR‐off (PSAneg/PSMApos), AR‐mixed (PSApos/PSMApos), and AR‐on (PSApos/PSMAneg). Initiation of first‐line ADT induced a marked phenotypic switch from AR‐on to AR‐off CTCs, followed by the disappearance of CTCs after three months. By contrast, secondary hormonal therapy resulted in a variable response. In addition, the presence of AR‐on and AR‐mixed CTCs in patients treated with abiraterone was associated with adverse clinical outcome. Interestingly, a correlation between AR activation and upregulation of PSA and PSMA was observed, which made it possible to predict the outcome of AR‐based therapy based on PSA and PSMA levels.
Unlike intracellular proteins that are expressed by tumor cells as well as normal cells, genetically altered oncoproteins are produced only by tumor cells and thus proteins harboring tumor‐specific fusions or mutations have become targets to molecular therapeutics. For example, the mutated BRCA2 protein has been utilized as a predictive biomarker in patients with pancreatic, breast, or ovarian cancer [156]. BRAC2 mutations result in impaired homology‐directed DNA repair, making these tumors sensitive to DNA repair‐targeting drugs, such as PARP inhibitors [157]. In 2018, the FDA approved the PARP inhibitor, olaparib, for treatment of patients with advanced breast and ovarian cancers harboring BRCA mutations [158]. More recently, the MagRC platform was featured as a technology capable of analyzing intracellular proteins in the nucleus, mitochondria and cytoplasm of rare tumor cells, without interference from hematopoietic cells [54]. The approach relied on the use of magnetic amplification reagents to facilitate sensitive detection of low‐abundance proteins (Fig. 4A,B). After labeling the target protein with the magnetic reagents, the protein‐harboring cells were magnetically ranked using a MagRC device featuring eight capture zones. The number of captured cells as well as their distribution among the capture zones were used to determine the protein level (Fig. 4C). This approach was proven capable of tracking the expression of a variety of therapeutic proteins in CTCs isolated from orthotopic prostate cancer xenografts as well as from mCRPC patients. Remarkably, the method was used to detect mutated BRCA2 protein in CTCs isolated from pancreatic tumor xenografts (Fig. 4D) and predict how mice with drug‐sensitive and drug‐resistant tumors would respond to the PARP inhibitor, olaparib (Fig. 4E).
Fig. 4.
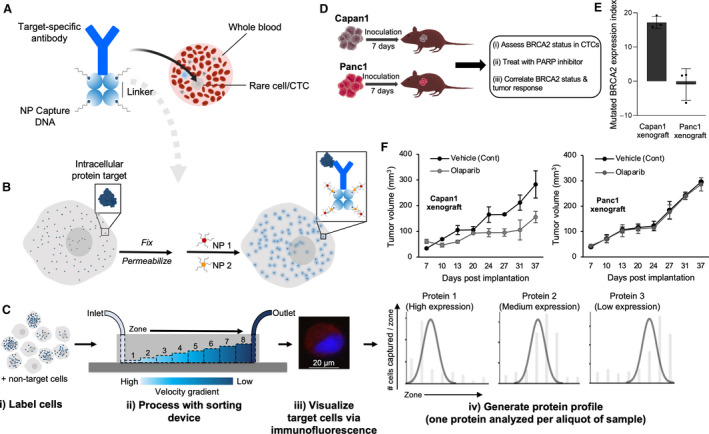
Analysis of intracellular proteins using the MagRC approach. (A) An antibody specific for the target intracellular protein is tagged with streptavidin then modified with biotin‐labeled ssDNAs using biotin–streptavidin coupling. (B) The cells expressing the target intracellular protein are fixed, permeabilized, and incubated with the intracellular protein‐specific antibody modified with ssDNAs. The ssDNAs are then hybridized with two capture probes (CP1 and CP2), which are composed of complementary DNA sequences modified at one end with MNPs. Aggregates of MNPs are thus formed and trapped within the cells that express the intracellular protein. (C) The cells are sorted using a microfluidic device featuring eight capture zones, immunostained, and counted to generate a profile characteristic for the target protein. (D) Schematic illustration for therapeutic protein analysis in xenografted mice. (E) Analysis of mutated BRCA2 protein in CTCs captured from the blood of mice bearing either Capan1 (with mutated BRCA2) or Panc1 (with wild‐type BRCA2) xenograft. The analysis was carried out at day 7 after tumor formation. After tumor formation, the mice were randomly divided into control and treated groups. Mice in the treated group received 50 mg·kg−1 olaparib, whereas mice in the control group received only the vehicle. (F) Tumor volume is plotted against duration of treatment for Capan1 and Panc1 xenografts. Figure reprinted with permission from Ref. [54].
4. CTC analysis in clinical care
Regulation of tumor biomarker tests is a complex process and even tests approved by the FDA may not have established clinical utility [159]. Nevertheless, there are many commercially available tests that have not been submitted to the FDA for approval but are instead marketed as laboratory‐developed tests (LDTs). Although LDTs must be certified, these tests do not follow the same guidelines applied by the FDA to other diagnostics and their analytical and/or clinical validity might not be thoroughly reviewed [159]. The semi‐automated CellSearch platform (Menarini Silicon Biosystems Inc., Huntington Valley, PA, USA), while not a microfluidic technology, is the only FDA‐cleared method for CTC detection in patients with metastatic breast, prostate, or colorectal cancer [43]. The analytical and clinical validity of CTC enumeration have been verified for prognostication of patients with metastatic breast, prostate, or colorectal cancer, as well as patients with nonmetastatic breast cancer treated with chemotherapy [160]. Clinical studies have demonstrated reduced progression‐free and overall survival for patients with metastatic breast, prostate, and colorectal cancer harboring ≥ 5 CTCs, ≥ 5 CTCs, or ≥ 3 CTCs per 7.5 mL blood, respectively [97, 99, 100]. In other cancers, such as pancreatic [161] and ovarian [162], the CTC counts were lower, and the test has not been approved by FDA.
Although more than a decade has passed since its FDA approval, the CellSearch platform has not been widely adopted in recent tumor resistance studies due to the reports showing that patients with high CTC counts might not benefit from switching treatment plans [163]. In addition, the recovery of CTCs by the CellSearch system is relatively low compared to other technologies that can recover orders of magnitude higher CTC counts from smaller sample volumes [90]. Furthermore, the CellSearch platform cannot be utilized to monitor mesenchymal CTCs with downregulated EpCAM and/or CK expression, which have been implicated in therapeutic resistance [26]. Notably, the CellSearch platform could only detect CTCs in the blood of only 32% of 101 metastatic NSCLC patients [164].
In addition, there is a FDA‐approved test for the constitutively active AR‐V7 associated with prostate cancer resistance to ADT using a commercially available platform from Epic Sciences [165]. There are also several ongoing clinical trials in which therapeutic decisions are made according to the molecular or phenotypic characteristics of CTCs obtained from different types of cancer, such as DETECT‐IV in breast cancer and CABA‐V7 in prostate cancer [6]. Also, several platforms for antigen‐independent CTC enrichment are currently under evaluation but are not yet approved for clinical practice [9]. The ‘no cell left behind’ platform from Epic Sciences can capture all nucleated cells from a clinical specimen on slides. The captured cells are then analyzed with multiplexed immunofluorescence assays using proprietary algorithms to identify CTCs, followed by physical picking of single cells for further downstream analysis [166].
5. Conclusions
CTC‐based liquid biopsy is currently entering a third wave of clinical development, following the establishment of laboratory platforms and promising results from evaluative studies. CTCs have been shown to preserve the molecular identity of primary tumors while yielding a renewable source of informative material. Profiling CTCs may provide early surrogate endpoints for clinical outcomes, thereby reducing the long follow‐up periods and substantial costs associated with clinical trials on neoadjuvant and/or adjuvant therapies. CTCs are also amenable to rapid phenotypic and functional readouts, that when coupled with iterative computational approaches, will contribute to a systematic understanding of the mechanisms of tumor progression and therapeutic resistance.
One of the key advantages of using CTCs for liquid biopsy is that this approach offers an opportunity to perform a range of analyses at the DNA, RNA, and protein level, as well as permitting functional studies. Extending the range of information extractable from CTCs by interrogating multiple analytes would likely improve their diagnostic power in terms of both specificity and sensitivity. In addition, multidimensional assays of several liquid biopsy markers would provide a more effective means to predicting tumor response to therapy. For instance, ctDNA‐based tests are more sensitive than CTC assays in identifying gene mutations in cancer patients [167] and can be used to detect minimal residual disease (MRD) or clinical relapse in an early stage [168]. As complementary tools, interrogation of CTCs and ctDNA allowed for robust monitoring tumor response to targeted therapy in NSCLC patients [169]. Integrative analysis of other liquid biopsy markers such cfRNAs and EVs is foreseen to advance the field of precision oncology in the near future, whereas other candidates such as TEPs and tumor metabolites are likely to contribute in the longer term.
Developments in assay technology must be accompanied by implementing new statistical tools that utilize high‐dimensional machine learning approaches to process the large amount of data obtainable from multiparametric assays. Machine learning approaches have already been applied to liquid biopsy biomarkers to identify ovarian cancer patients using neural networks [170]. Nevertheless, the most advanced methods applied to CTC assays were mainly dedicated to the identification and enumeration of cells [171]. Machine learning approaches offer an opportunity to discover cancer‐specific signatures in CTCs, exploiting publicly available data sets from research networks (e.g., TCGA) for simulation experiments. In addition, adopting machine learning strategies can reduce the cost of CTC assays by reducing the depth of analysis required (e.g., depth of sequencing), as well as the quantity of reagents needed. Admittedly, the recent popularity of machine learning programmes has raised ethical and legal concerns. Addressing these issues necessitates either the use of privacy‐oriented algorithms that do not disclose private clinical data or the future availability of machine learning programmes in research institutes to avoid sharing the data with third parties.
The next frontier for CTC‐based tests for resistance monitoring is to address the need for predictive biomarkers for immunotherapy. Immune checkpoint inhibitors have offered the most durable response in NSCLC and melanoma, although it is still difficult to predict the tumor response to most of these inhibitors. This outlook is actually based on a recent report which demonstrated that expansion of T‐cell clones within the tumor is paralleled by their expansion in peripheral blood, which can assist in predicting tumor infiltration and therapeutic response [172]. Consistent with this finding, two recent reports have demonstrated the presence of large numbers of T‐cell clones in the peripheral blood of melanoma patients responding to immune checkpoint blockade [173, 174].
Whatever the clinical question that needs to be addressed or the CTC profiling technology, CTC‐based assays would need to go through thousands of tests for validation to ensure that the assay can respond to the clinical question with sufficient statistical significance. If the CTC assay is based on a microfluidic device, then these devices must be amenable to large scale production at reasonable cost to generate assays that are highly reproducible. A sample‐to‐answer microfluidic system would be highly amenable to clinical practice since it can improve the processing throughput and eliminate the need for manual handling of samples. Several companies are currently developing integrated platforms that combine sorting and downstream analysis.
Another major challenge that still faces the approval of liquid biopsy tests is that tissue biopsies are still adopted by the regulatory agencies as the gold standard. However, the value of tissue biopsies as a reference is questionable because CTCs can be derived from metastatic lesions that were not biopsied. In addition, occult metastases, which are not detectable by current imaging techniques, might contribute to the pool of CTCs in patients. Thus, CTCs might reveal different molecular landscapes than the reference primary tumor, leading to test failure.
To conclude, future CTC assay developments will require orchestrated efforts from preclinical and clinical researchers, spanning a range of disciplines such as basic biology, molecular biology, assay technology, and data analytics. Although the CTC tools we have in hand should keep us busy for the near future, it is up to the next generation of CTC assays to devise solutions that can revolutionize the practice of precision oncology and clinical management of cancer patients.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank the Canadian Institutes of Health Research (Grant #FDN‐148415), the Natural Sciences and Engineering Research Council of Canada (Grant #2016‐06090), the Province of Ontario though the Ministry of Research, Innovation and Science (Grant #RE05‐009), and the National Cancer Institute of the National Institutes of Health (Grant #1R33CA204574). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the other funding agencies.
References
- 1. Kumar‐Sinha C & Chinnaiyan AM (2018) Precision oncology in the age of integrative genomics. Nat Biotechnol 36, 46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C & Stuart JM (2013) The cancer genome atlas Pan‐cancer analysis project. Nat Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scott AM, Wolchok JD & Old LJ (2012) Antibody therapy of cancer. Nat Rev Cancer 12, 278–287. [DOI] [PubMed] [Google Scholar]
- 4. La Thangue NB & Kerr DJ (2011) Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat Rev Clin Oncol 8, 587–596. [DOI] [PubMed] [Google Scholar]
- 5. Basik M, Aguilar‐Mahecha A, Rousseau C, Diaz Z, Tejpar S, Spatz A, Greenwood CM & Batist G (2013) Biopsies: next‐generation biospecimens for tailoring therapy. Nat Rev Clin Oncol 10, 437–450. [DOI] [PubMed] [Google Scholar]
- 6. Keller L & Pantel K (2019) Unravelling tumour heterogeneity by single‐cell profiling of circulating tumour cells. Nat Rev Cancer 19, 553–567. [DOI] [PubMed] [Google Scholar]
- 7. Joosse SA, Beyer B, Gasch C, Nastaly P, Kuske A, Isbarn H, Horst LJ, Hille C, Gorges TM, Cayrefourcq L et al. (2020) Tumor‐associated release of prostatic cells into the blood after transrectal ultrasound‐guided biopsy in patients with histologically confirmed prostate cancer. Clin Chem 66, 161–168. [DOI] [PubMed] [Google Scholar]
- 8. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr & Kinzler KW (2013) Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kilgour E, Rothwell DG, Brady G & Dive C (2020) Liquid biopsy‐based biomarkers of treatment response and resistance. Cancer Cell 37, 485–495. [DOI] [PubMed] [Google Scholar]
- 10. Wan JCM, Massie C, Garcia‐Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R & Rosenfeld N (2017) Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17, 223–238. [DOI] [PubMed] [Google Scholar]
- 11. Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, Ptak J, Brem H, Chaichana K, Gallia GL et al. (2015) Detection of tumor‐derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA 112, 9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heitzer E, Haque IS, Roberts CES & Speicher MR (2019) Current and future perspectives of liquid biopsies in genomics‐driven oncology. Nat Rev Genet 20, 71–88. [DOI] [PubMed] [Google Scholar]
- 13. Siravegna G, Marsoni S, Siena S & Bardelli A (2017) Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14, 531–548. [DOI] [PubMed] [Google Scholar]
- 14. Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DWY, Marass F, Gale D et al. (2015) Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun 6, 8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Challapalli A & Aboagye EO (2016) Positron emission tomography imaging of tumor cell metabolism and application to therapy response monitoring. Front Oncol 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhavsar T, McCue P & Birbe R (2013) Molecular diagnosis of prostate cancer: are we up to age? Semin Oncol 40, 259–275. [DOI] [PubMed] [Google Scholar]
- 17. Krivacic RT, Ladanyi A, Curry DN, Hsieh HB, Kuhn P, Bergsrud DE, Kepros JF, Barbera T, Ho MY, Chen LB et al. (2004) A rare‐cell detector for cancer. Proc Natl Acad Sci USA 101, 10501–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Labib M & Kelley SO (2020) Single‐cell analysis targeting the proteome. Nat Rev Chem 4, 143–158. [DOI] [PubMed] [Google Scholar]
- 19. Holohan C, Van Schaeybroeck S, Longley DB & Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 20. Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M & Stuart DD (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marusyk A, Almendro V & Polyak K (2012) Intra‐tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer 12, 323–334. [DOI] [PubMed] [Google Scholar]
- 22. Brown R, Curry E, Magnani L, Wilhelm‐Benartzi CS & Borley J (2014) Poised epigenetic states and acquired drug resistance in cancer. Nat Rev Cancer 14, 747–753. [DOI] [PubMed] [Google Scholar]
- 23. Gilbert LA & Hemann MT (2010) DNA damage‐mediated induction of a chemoresistant niche. Cell 143, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen DX, Bos PD & Massague J (2009) Metastasis: from dissemination to organ‐specific colonization. Nat Rev Cancer 9, 274–284. [DOI] [PubMed] [Google Scholar]
- 25. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G et al. (2012) Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 486, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM et al. (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pattabiraman DR & Weinberg RA (2014) Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov 13, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yokobori T, Iinuma H, Shimamura T, Imoto S, Sugimachi K, Ishii H, Iwatsuki M, Ota D, Ohkuma M, Iwaya T et al. (2013) Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial‐mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res 73, 2059–2069. [DOI] [PubMed] [Google Scholar]
- 29. Adorno‐Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D, Hubert C, Venere M, Mulkearns‐Hubert E, Sinyuk M et al. (2015) Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res 75, 924–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Sousa MF, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, Anderson JE, Storm EE, Modrusan Z, Koeppen H et al. (2017) A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 543, 676–680. [DOI] [PubMed] [Google Scholar]
- 31. Tsao SC, Wang J, Wang Y, Behren A, Cebon J & Trau M (2018) Characterising the phenotypic evolution of circulating tumour cells during treatment. Nat Commun 9, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hata AN, Niederst MJ, Archibald HL, Gomez‐Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V et al. (2016) Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paoletti C, Muniz MC, Thomas DG, Griffith KA, Kidwell KM, Tokudome N, Brown ME, Aung K, Miller MC, Blossom DL et al. (2015) Development of circulating tumor cell‐endocrine therapy index in patients with hormone receptor‐positive breast cancer. Clin Cancer Res 21, 2487–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H et al. (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, Priest LJ, Greystoke A, Zhou C, Morris K et al. (2012) Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small‐cell lung cancer. J Clin Oncol 30, 525–532. [DOI] [PubMed] [Google Scholar]
- 36. Chang MC, Chang YT, Chen JY, Jeng YM, Yang CY, Tien YW, Yang SH, Chen HL, Liang TY, Wang CF et al. (2016) Clinical significance of circulating tumor microemboli as a prognostic marker in patients with pancreatic ductal adenocarcinoma. Clin Chem 62, 505–513. [DOI] [PubMed] [Google Scholar]
- 37. Hodgkinson CL, Morrow CJ, Li Y, Metcalf RL, Rothwell DG, Trapani F, Polanski R, Burt DJ, Simpson KL, Morris K et al. (2014) Tumorigenicity and genetic profiling of circulating tumor cells in small‐cell lung cancer. Nat Med 20, 897–903. [DOI] [PubMed] [Google Scholar]
- 38. Green BJ, Saberi Safaei T, Mepham A, Labib M, Mohamadi RM & Kelley SO (2016) Beyond the capture of circulating tumor cells: next‐generation devices and materials. Angew Chem Int Ed Engl 55, 1252–1265. [DOI] [PubMed] [Google Scholar]
- 39. Jackson JM, Witek MA, Kamande JW & Soper SA (2017) Materials and microfluidics: enabling the efficient isolation and analysis of circulating tumour cells. Chem Soc Rev 46, 4245–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A et al. (2007) Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Poudineh M, Aldridge PM, Ahmed S, Green BJ, Kermanshah L, Nguyen V, Tu C, Mohamadi RM, Nam RK, Hansen A et al. (2017) Tracking the dynamics of circulating tumour cell phenotypes using nanoparticle‐mediated magnetic ranking. Nat Nanotechnol 12, 274–281. [DOI] [PubMed] [Google Scholar]
- 42. Karabacak NM, Spuhler PS, Fachin F, Lim EJ, Pai V, Ozkumur E, Martel JM, Kojic N, Smith K, Chen PI et al. (2014) Microfluidic, marker‐free isolation of circulating tumor cells from blood samples. Nat Protoc 9, 694–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW et al. (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351, 781–791. [DOI] [PubMed] [Google Scholar]
- 44. Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW & Terstappen LW (2004) Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 10, 6897–6904. [DOI] [PubMed] [Google Scholar]
- 45. Blumke K, Bilkenroth U, Schmidt U, Melchior A, Fussel S, Bartel F, Heynemann H, Fornara P, Taubert H, Wirth MP et al. (2005) Detection of circulating tumor cells from renal carcinoma patients: experiences of a two‐center study. Oncol Rep 14, 895–899. [PubMed] [Google Scholar]
- 46. Sequist LV, Nagrath S, Toner M, Haber DA & Lynch TJ (2009) The CTC‐chip: an exciting new tool to detect circulating tumor cells in lung cancer patients. J Thorac Oncol 4, 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin M, Chen JF, Lu YT, Zhang Y, Song J, Hou S, Ke Z & Tseng HR (2014) Nanostructure embedded microchips for detection, isolation, and characterization of circulating tumor cells. Acc Chem Res 47, 2941–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK et al. (2010) Isolation of circulating tumor cells using a microvortex‐generating herringbone‐chip. Proc Natl Acad Sci USA 107, 18392–18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Praharaj PP, Bhutia SK, Nagrath S, Bitting RL & Deep G (2018) Circulating tumor cell‐derived organoids: current challenges and promises in medical research and precision medicine. Biochim Biophys Acta Rev Cancer 1869, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fischer JC, Niederacher D, Topp SA, Honisch E, Schumacher S, Schmitz N, Zacarias Fohrding L, Vay C, Hoffmann I, Kasprowicz NS et al. (2013) Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc Natl Acad Sci USA 110, 16580–16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao W, Cui CH, Bose S, Guo D, Shen C, Wong WP, Halvorsen K, Farokhzad OC, Teo GS, Phillips JA et al. (2012) Bioinspired multivalent DNA network for capture and release of cells. Proc Natl Acad Sci USA 109, 19626–19631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Labib M, Zamay AS, Muharemagic D, Chechik AV, Bell JC & Berezovski MV (2012) Electrochemical differentiation of epitope‐specific aptamers. Anal Chem 84, 2548–2556. [DOI] [PubMed] [Google Scholar]
- 53. Labib M, Green B, Mohamadi RM, Mepham A, Ahmed SU, Mahmoudian L, Chang IH, Sargent EH & Kelley SO (2016) Aptamer and antisense‐mediated two‐dimensional isolation of specific cancer cell subpopulations. J Am Chem Soc 138, 2476–2479. [DOI] [PubMed] [Google Scholar]
- 54. Labib M, Wang Z, Ahmed SU, Mohamadi RM, Duong B, Green B, Sargent EH & Kelley SO (2020) Tracking the expression of therapeutic protein targets in rare cells by antibody‐mediated nanoparticle labelling and magnetic sorting. Nat Biomed Eng, in press 5, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Labib M, Mohamadi RM, Poudineh M, Ahmed SU, Ivanov I, Huang CL, Moosavi M, Sargent EH & Kelley SO (2018) Single‐cell mRNA cytometry via sequence‐specific nanoparticle clustering and trapping. Nat Chem 10, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh KH, Yu W, Xiao W, Davis MM, Pease RF, Mindrinos MN et al. (2009) Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci USA 106, 3970–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Harb W, Fan A, Tran T, Danila DC, Keys D, Schwartz M & Ionescu‐Zanetti C (2013) Mutational analysis of circulating tumor cells using a novel microfluidic collection device and qPCR assay. Transl Oncol 6, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Casavant BP, Guckenberger DJ, Berry SM, Tokar JT, Lang JM & Beebe DJ (2013) The VerIFAST: an integrated method for cell isolation and extracellular/intracellular staining. Lab Chip 13, 391–396. [DOI] [PubMed] [Google Scholar]
- 59. Earhart CM, Hughes CE, Gaster RS, Ooi CC, Wilson RJ, Zhou LY, Humke EW, Xu L, Wong DJ, Willingham SB et al. (2014) Isolation and mutational analysis of circulating tumor cells from lung cancer patients with magnetic sifters and biochips. Lab Chip 14, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Witek MA, Aufforth RD, Wang H, Kamande JW, Jackson JM, Pullagurla SR, Hupert ML, Usary J, Wysham WZ, Hilliard D et al. (2017) Discrete microfluidics for the isolation of circulating tumor cell subpopulations targeting fibroblast activation protein alpha and epithelial cell adhesion molecule. NPJ Precis Oncol 1, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Danila DC, Samoila A, Patel C, Schreiber N, Herkal A, Anand A, Bastos D, Heller G, Fleisher M & Scher HI (2016) Clinical validity of detecting circulating tumor cells by AdnaTest assay compared with direct detection of tumor mRNA in stabilized whole blood, as a biomarker predicting overall survival for metastatic castration‐resistant prostate cancer patients. Cancer J 22, 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gorges TM, Penkalla N, Schalk T, Joosse SA, Riethdorf S, Tucholski J, Lucke K, Wikman H, Jackson S, Brychta N et al. (2016) Enumeration and molecular characterization of tumor cells in lung cancer patients using a novel in vivo device for capturing circulating tumor cells. Clin Cancer Res 22, 2197–2206. [DOI] [PubMed] [Google Scholar]
- 63. Vermesh O, Aalipour A, Ge TJ, Saenz Y, Guo Y, Alam IS, Park SM, Adelson CN, Mitsutake Y, Vilches‐Moure J et al. (2018) An intravascular magnetic wire for the high‐throughput retrieval of circulating tumour cells in vivo. Nat Biomed Eng 2, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim TH, Wang Y, Oliver CR, Thamm DH, Cooling L, Paoletti C, Smith KJ, Nagrath S & Hayes DF (2019) A temporary indwelling intravascular aphaeretic system for in vivo enrichment of circulating tumor cells. Nat Commun 10, 1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rosenberg R, Gertler R, Friederichs J, Fuehrer K, Dahm M, Phelps R, Thorban S, Nekarda H & Siewert JR (2002) Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry 49, 150–158. [DOI] [PubMed] [Google Scholar]
- 66. Munz M, Baeuerle PA & Gires O (2009) The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 69, 5627–5629. [DOI] [PubMed] [Google Scholar]
- 67. Ferreira MM, Ramani VC & Jeffrey SS (2016) Circulating tumor cell technologies. Mol Oncol 10, 374–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Au SH, Edd J, Stoddard AE, Wong KHK, Fachin F, Maheswaran S, Haber DA, Stott SL, Kapur R & Toner M (2017) Microfluidic isolation of circulating tumor cell clusters by size and asymmetry. Sci Rep 7, 2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou J, Kulasinghe A, Bogseth A, O'Byrne K, Punyadeera C & Papautsky I (2019) Isolation of circulating tumor cells in non‐small‐cell‐lung‐cancer patients using a multi‐flow microfluidic channel. Microsyst Nanoeng 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zeinali M, Lee M, Nadhan A, Mathur A, Hedman C, Lin E, Harouaka R, Wicha MS, Zhao L, Palanisamy N et al. (2020) High‐throughput label‐free isolation of heterogeneous circulating tumor cells and CTC clusters from non‐small‐cell lung cancer patients. Cancers (Basel) 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee Y, Guan G & Bhagat AA (2018) ClearCell(R) FX, a label‐free microfluidics technology for enrichment of viable circulating tumor cells. Cytometry A 93, 1251–1254. [DOI] [PubMed] [Google Scholar]
- 72. Sollier E, Go DE, Che J, Gossett DR, O'Byrne S, Weaver WM, Kummer N, Rettig M, Goldman J, Nickols N et al. (2014) Size‐selective collection of circulating tumor cells using Vortex technology. Lab Chip 14, 63–77. [DOI] [PubMed] [Google Scholar]
- 73. Adams AA, Okagbare PI, Feng J, Hupert ML, Patterson D, Gottert J, McCarley RL, Nikitopoulos D, Murphy MC & Soper SA (2008) Highly efficient circulating tumor cell isolation from whole blood and label‐free enumeration using polymer‐based microfluidics with an integrated conductivity sensor. J Am Chem Soc 130, 8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Peeters DJ, De Laere B, Van den Eynden GG, Van Laere SJ, Rothe F, Ignatiadis M, Sieuwerts AM, Lambrechts D, Rutten A, van Dam PA et al. (2013) Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from Cell Search enriched blood samples using dielectrophoretic cell sorting. Br J Cancer 108, 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fabbri F, Carloni S, Zoli W, Ulivi P, Gallerani G, Fici P, Chiadini E, Passardi A, Frassineti GL, Ragazzini A et al. (2013) Detection and recovery of circulating colon cancer cells using a dielectrophoresis‐based device: KRAS mutation status in pure CTCs. Cancer Lett 335, 225–231. [DOI] [PubMed] [Google Scholar]
- 76. Kirby BJ, Jodari M, Loftus MS, Gakhar G, Pratt ED, Chanel‐Vos C, Gleghorn JP, Santana SM, Liu H, Smith JP et al. (2012) Functional characterization of circulating tumor cells with a prostate‐cancer‐specific microfluidic device. PLoS One 7, e35976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen PI, Morgan B, Trautwein J et al. (2013) Inertial focusing for tumor antigen‐dependent and ‐independent sorting of rare circulating tumor cells. Sci Transl Med 5, 179ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bhagwat N, Dulmage K, Pletcher CH Jr, Wang L, DeMuth W, Sen M, Balli D, Yee SS, Sa S, Tong F et al. (2018) An integrated flow cytometry‐based platform for isolation and molecular characterization of circulating tumor single cells and clusters. Sci Rep 8, 5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Park ES, Yan JP, Ang RA, Lee JH, Deng X, Duffy SP, Beja K, Annala M, Black PC, Chi KN et al. (2018) Isolation and genome sequencing of individual circulating tumor cells using hydrogel encapsulation and laser capture microdissection. Lab Chip 18, 1736–1749. [DOI] [PubMed] [Google Scholar]
- 80. Muller C, Holtschmidt J, Auer M, Heitzer E, Lamszus K, Schulte A, Matschke J, Langer‐Freitag S, Gasch C, Stoupiec M et al. (2014) Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med 6, 247ra101. [DOI] [PubMed] [Google Scholar]
- 81. Rihawi K, Alfieri R, Fiorentino M, Fontana F, Capizzi E, Cavazzoni A, Terracciano M, La Monica S, Ferrarini A, Buson G et al. (2019) MYC amplification as a potential mechanism of primary resistance to crizotinib in ALK‐rearranged non‐small cell lung cancer: a brief report. Transl Oncol 12, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kalinich M, Bhan I, Kwan TT, Miyamoto DT, Javaid S, LiCausi JA, Milner JD, Hong X, Goyal L, Sil S et al. (2017) An RNA‐based signature enables high specificity detection of circulating tumor cells in hepatocellular carcinoma. Proc Natl Acad Sci USA 114, 1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Briley WE, Bondy MH, Randeria PS, Dupper TJ & Mirkin CA (2015) Quantification and real‐time tracking of RNA in live cells using sticky‐flares. Proc Natl Acad Sci USA 112, 9591–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Barrett MT, Scheffer A, Ben‐Dor A, Sampas N, Lipson D, Kincaid R, Tsang P, Curry B, Baird K, Meltzer PS et al. (2004) Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci USA 101, 17765–17770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl‐Geigl J, Mauermann O, Lackner C et al. (2013) Complex tumor genomes inferred from single circulating tumor cells by array‐CGH and next‐generation sequencing. Cancer Res 73, 2965–2975. [DOI] [PubMed] [Google Scholar]
- 86. Deng Q, Ramskold D, Reinius B & Sandberg R (2014) Single‐cell RNA‐seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. [DOI] [PubMed] [Google Scholar]
- 87. Heather JM & Chain B (2016) The sequence of sequencers: the history of sequencing DNA. Genomics 107, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hu Z, Ding J, Ma Z, Sun R, Seoane JA, Scott Shaffer J, Suarez CJ, Berghoff AS, Cremolini C, Falcone A et al. (2019) Quantitative evidence for early metastatic seeding in colorectal cancer. Nat Genet 51, 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz‐Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R et al. (2014) Whole‐exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol 32, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Labib M, Philpott DN, Wang Z, Nemr C, Chen JB, Sargent EH & Kelley SO (2020) Magnetic ranking cytometry: profiling rare cells at the single‐cell level. Acc Chem Res 53, 1445–1457. [DOI] [PubMed] [Google Scholar]
- 91. Shi Q, Qin L, Wei W, Geng F, Fan R, Shin YS, Guo D, Hood L, Mischel PS & Heath JR (2012) Single‐cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc Natl Acad Sci USA 109, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hughes AJ, Spelke DP, Xu Z, Kang CC, Schaffer DV & Herr AE (2014) Single‐cell western blotting. Nat Methods 11, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Green BJ, Kermanshah L, Labib M, Ahmed SU, Silva PN, Mahmoudian L, Chang IH, Mohamadi RM, Rocheleau JV & Kelley SO (2017) Isolation of phenotypically distinct cancer cells using nanoparticle‐mediated sorting. ACS Appl Mater Interfaces 9, 20435–20443. [DOI] [PubMed] [Google Scholar]
- 94. Jin A, Ozawa T, Tajiri K, Obata T, Kondo S, Kinoshita K, Kadowaki S, Takahashi K, Sugiyama T, Kishi H et al. (2009) Rapid and efficient single‐cell manipulation method for screening antigen‐specific antibody‐secreting cells from human peripheral blood. Nat Med 15, 1088–1092. [DOI] [PubMed] [Google Scholar]
- 95. Alix‐Panabieres C, Vendrell JP, Slijper M, Pelle O, Barbotte E, Mercier G, Jacot W, Fabbro M & Pantel K (2009) Full‐length cytokeratin‐19 is released by human tumor cells: a potential role in metastatic progression of breast cancer. Breast Cancer Res 11, R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez‐Gutierrez F, Li Y, Burt DJ, Antonello J, Morrow C et al. (2017) Molecular analysis of circulating tumor cells identifies distinct copy‐number profiles in patients with chemosensitive and chemorefractory small‐cell lung cancer. Nat Med 23, 114–119. [DOI] [PubMed] [Google Scholar]
- 97. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, Doyle GV, Terstappen LW, Pienta KJ & Raghavan D (2008) Circulating tumor cells predict survival benefit from treatment in metastatic castration‐resistant prostate cancer. Clin Cancer Res 14, 6302–6309. [DOI] [PubMed] [Google Scholar]
- 98. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F et al. (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364, 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, Doyle GV, Matera J, Allard WJ, Miller MC et al. (2005) Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol 23, 1420–1430. [DOI] [PubMed] [Google Scholar]
- 100. Cohen SJ, Punt CJ, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, Picus J, Morse M, Mitchell E, Miller MC et al. (2008) Relationship of circulating tumor cells to tumor response, progression‐free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol 26, 3213–3221. [DOI] [PubMed] [Google Scholar]
- 101. Hiltermann TJN, Pore MM, van den Berg A, Timens W, Boezen HM, Liesker JJW, Schouwink JH, Wijnands WJA, Kerner G, Kruyt FAE et al. (2012) Circulating tumor cells in small‐cell lung cancer: a predictive and prognostic factor. Ann Oncol 23, 2937–2942. [DOI] [PubMed] [Google Scholar]
- 102. Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A et al. (2013) The poly(ADP‐ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose‐escalation trial. Lancet Oncol 14, 882–892. [DOI] [PubMed] [Google Scholar]
- 103. Yap TA, Olmos D, Brunetto AT, Tunariu N, Barriuso J, Riisnaes R, Pope L, Clark J, Futreal A, Germuska M et al. (2011) Phase I trial of a selective c‐MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol 29, 1271–1279. [DOI] [PubMed] [Google Scholar]
- 104. Bianchini D, Omlin A, Pezaro C, Lorente D, Ferraldeschi R, Mukherji D, Crespo M, Figueiredo I, Miranda S, Riisnaes R et al. (2013) First‐in‐human Phase I study of EZN‐4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration‐resistant prostate cancer. Br J Cancer 109, 2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Khoo BL, Grenci G, Jing T, Lim YB, Lee SC, Thiery JP, Han J & Lim CT (2016) Liquid biopsy and therapeutic response: circulating tumor cell cultures for evaluation of anticancer treatment. Sci Adv 2, e1600274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Foy V, Fernandez‐Gutierrez F, Faivre‐Finn C, Dive C & Blackhall F (2017) The clinical utility of circulating tumour cells in patients with small cell lung cancer. Transl Lung Cancer Res 6, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lindsay CR, Faugeroux V, Michiels S, Pailler E, Facchinetti F, Ou D, Bluthgen MV, Pannet C, Ngo‐Camus M, Bescher G et al. (2017) A prospective examination of circulating tumor cell profiles in non‐small‐cell lung cancer molecular subgroups. Ann Oncol 28, 1523–1531. [DOI] [PubMed] [Google Scholar]
- 108. Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland‐Jones B, Srkalovic G, Tejwani S, Schott AF, O'Rourke MA, Lew DL et al. (2014) Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol 32, 3483–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fehm T, Muller V, Aktas B, Janni W, Schneeweiss A, Stickeler E, Lattrich C, Lohberg CR, Solomayer E, Rack B et al. (2010) HER2 status of circulating tumor cells in patients with metastatic breast cancer: a prospective, multicenter trial. Breast Cancer Res Treat 124, 403–412. [DOI] [PubMed] [Google Scholar]
- 110. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello‐Gruszfeld A, Crown J, Chan A, Kaufman B et al. (2006) Lapatinib plus capecitabine for HER2‐positive advanced breast cancer. N Engl J Med 355, 2733–2743. [DOI] [PubMed] [Google Scholar]
- 111. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW et al. (2008) Detection of mutations in EGFR in circulating lung‐cancer cells. N Engl J Med 359, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sundaresan TK, Sequist LV, Heymach JV, Riely GJ, Janne PA, Koch WH, Sullivan JP, Fox DB, Maher R, Muzikansky A et al. (2016) Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood‐based analyses. Clin Cancer Res 22, 1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. He J, Tan W & Ma J (2017) Circulating tumor cells and DNA for real‐time EGFR detection and monitoring of non‐small‐cell lung cancer. Future Oncol 13, 787–797. [DOI] [PubMed] [Google Scholar]
- 114. Breitenbuecher F, Hoffarth S, Worm K, Cortes‐Incio D, Gauler TC, Kohler J, Herold T, Schmid KW, Freitag L, Kasper S et al. (2014) Development of a highly sensitive and specific method for detection of circulating tumor cells harboring somatic mutations in non‐small‐cell lung cancer patients. PLoS One 9, e85350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu Y, Meucci S, Sheng L & Keilholz U (2017) Meta‐analysis of the mutational status of circulation tumor cells and paired primary tumor tissues from colorectal cancer patients. Oncotarget 8, 77928–77941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Luo X, Mitra D, Sullivan RJ, Wittner BS, Kimura AM, Pan S, Hoang MP, Brannigan BW, Lawrence DP, Flaherty KT et al. (2014) Isolation and molecular characterization of circulating melanoma cells. Cell Rep 7, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hong X, Sullivan RJ, Kalinich M, Kwan TT, Giobbie‐Hurder A, Pan S, LiCausi JA, Milner JD, Nieman LT, Wittner BS et al. (2018) Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc Natl Acad Sci USA 115, 2467–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Schneck H, Blassl C, Meier‐Stiegen F, Neves RP, Janni W, Fehm T & Neubauer H (2013) Analysing the mutational status of PIK3CA in circulating tumor cells from metastatic breast cancer patients. Mol Oncol 7, 976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gasch C, Oldopp T, Mauermann O, Gorges TM, Andreas A, Coith C, Muller V, Fehm T, Janni W, Pantel K et al. (2016) Frequent detection of PIK3CA mutations in single circulating tumor cells of patients suffering from HER2‐negative metastatic breast cancer. Mol Oncol 10, 1330–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Angus L, Beije N, Jager A, Martens JW & Sleijfer S (2017) ESR1 mutations: moving towards guiding treatment decision‐making in metastatic breast cancer patients. Cancer Treat Rev 52, 33–40. [DOI] [PubMed] [Google Scholar]
- 121. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z et al. (2014) Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mostaghel EA, Plymate SR & Montgomery B (2014) Molecular pathways: targeting resistance in the androgen receptor for therapeutic benefit. Clin Cancer Res 20, 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Swennenhuis JF, Tibbe AG, Levink R, Sipkema RC & Terstappen LW (2009) Characterization of circulating tumor cells by fluorescence in situ hybridization. Cytometry A 75, 520–527. [DOI] [PubMed] [Google Scholar]
- 124. Magbanua MJ, Sosa EV, Scott JH, Simko J, Collins C, Pinkel D, Ryan CJ & Park JW (2012) Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA et al. (2015) Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration‐resistant prostate cancer. JAMA Oncol 1, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Antonarakis ES, Nakazawa M & Luo J (2014) Resistance to androgen‐pathway drugs in prostate cancer. N Engl J Med 371, 2234. [DOI] [PubMed] [Google Scholar]
- 127. Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J et al. (2015) RNA‐Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 349, 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G et al. (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Attard G, de Bono JS, Logothetis CJ, Fizazi K, Mukherjee SD, Joshua AM, Schrijvers D, van den Eertwegh AJ, Li W, Molina A et al. (2015) Improvements in radiographic progression‐free survival stratified by ERG gene status in metastatic castration‐resistant prostate cancer patients treated with abiraterone acetate. Clin Cancer Res 21, 1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Danila DC, Anand A, Sung CC, Heller G, Leversha MA, Cao L, Lilja H, Molina A, Sawyers CL, Fleisher M & Scher HI (2011) TMPRSS2‐ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration‐resistant prostate cancer patients treated with abiraterone acetate. Eur Urol 60, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Reig O, Marin‐Aguilera M, Carrera G, Jimenez N, Pare L, Garcia‐Recio S, Gaba L, Pereira MV, Fernandez P, Prat A et al. (2016) TMPRSS2‐ERG in blood and docetaxel resistance in metastatic castration‐resistant prostate cancer. Eur Urol 70, 709–713. [DOI] [PubMed] [Google Scholar]
- 132. Tan CL, Lim TH, Lim T, Tan DS, Chua YW, Ang MK, Pang B, Lim CT, Takano A, Lim AS et al. (2016) Concordance of anaplastic lymphoma kinase (ALK) gene rearrangements between circulating tumor cells and tumor in non‐small cell lung cancer. Oncotarget 7, 23251–23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pailler E, Adam J, Barthelemy A, Oulhen M, Auger N, Valent A, Borget I, Planchard D, Taylor M, Andre F et al. (2013) Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK‐positive non‐small‐cell lung cancer. J Clin Oncol 31, 2273–2281. [DOI] [PubMed] [Google Scholar]
- 134. Pailler E, Oulhen M, Borget I, Remon J, Ross K, Auger N, Billiot F, Ngo Camus M, Commo F, Lindsay CR et al. (2017) Circulating tumor cells with aberrant ALK copy number predict progression‐free survival during crizotinib treatment in ALK‐rearranged non‐small cell lung cancer patients. Cancer Res 77, 2222–2230. [DOI] [PubMed] [Google Scholar]
- 135. He W, Xu D, Wang Z, Wu H, Xiang X, Tang B, Jiang W, Cui Y, Wang H, Jiang N et al. (2019) Three‐dimensional nanostructured substrates enable dynamic detection of ALK‐rearrangement in circulating tumor cells from treatment‐naive patients with stage III/IV lung adenocarcinoma. J Transl Med 17, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Pailler E, Auger N, Lindsay CR, Vielh P, Islas‐Morris‐Hernandez A, Borget I, Ngo‐Camus M, Planchard D, Soria JC, Besse B et al. (2015) High level of chromosomal instability in circulating tumor cells of ROS1‐rearranged non‐small‐cell lung cancer. Ann Oncol 26, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sinkala E, Sollier‐Christen E, Renier C, Rosas‐Canyelles E, Che J, Heirich K, Duncombe TA, Vlassakis J, Yamauchi KA, Huang H et al. (2017) Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat Commun 8, 14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Paoletti C, Larios JM, Muniz MC, Aung K, Cannell EM, Darga EP, Kidwell KM, Thomas DG, Tokudome N, Brown ME et al. (2016) Heterogeneous estrogen receptor expression in circulating tumor cells suggests diverse mechanisms of fulvestrant resistance. Mol Oncol 10, 1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Riethdorf S, Muller V, Zhang L, Rau T, Loibl S, Komor M, Roller M, Huober J, Fehm T, Schrader I et al. (2010) Detection and HER2 expression of circulating tumor cells: prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin Cancer Res 16, 2634–2645. [DOI] [PubMed] [Google Scholar]
- 140. Ignatiadis M, Rothé F, Chaboteaux C, Durbecq V, Rouas G, Criscitiello C, Metallo J, Kheddoumi N, Singhal SK, Michiels S et al. (2011) HER2‐positive circulating tumor cells in breast cancer. PLoS One 6, e15624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Jaeger BAS, Neugebauer J, Andergassen U, Melcher C, Schochter F, Mouarrawy D, Ziemendorff G, Clemens M, Abel EV, Heinrich G et al. (2017) The HER2 phenotype of circulating tumor cells in HER2‐positive early breast cancer: a translational research project of a prospective randomized phase III trial. PLoS One 12, e0173593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Beije N, Onstenk W, Kraan J, Sieuwerts AM, Hamberg P, Dirix Y, Brouwer A, de Jongh FE, Jager A, Seynaeve CM et al. (2016) Prognostic impact of HER2 and ER status of circulating tumor cells in metastatic breast cancer patients with a HER2‐negative primary tumor. Neoplasia 18, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wallwiener M, Hartkopf AD, Riethdorf S, Nees J, Sprick MR, Schonfisc B, Taran FA, Heil J, Sohn C, Pantel K et al. (2015) The impact of HER2 phenotype of circulating tumor cells in metastatic breast cancer: a retrospective study in 107 patients. BMC Cancer 15, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Xu‐Monette ZY, Zhang M, Li J & Young KH (2017) PD‐1/PD‐L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol 8, 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Guibert N, Delaunay M, Lusque A, Boubekeur N, Rouquette I, Clermont E, Mourlanette J, Gouin S, Dormoy I, Favre G et al. (2018) PD‐L1 expression in circulating tumor cells of advanced non‐small cell lung cancer patients treated with nivolumab. Lung Cancer 120, 108–112. [DOI] [PubMed] [Google Scholar]
- 146. Nicolazzo C, Raimondi C, Mancini M, Caponnetto S, Gradilone A, Gandini O, Mastromartino M, Del Bene G, Prete A, Longo F et al. (2016) Monitoring PD‐L1 positive circulating tumor cells in non‐small cell lung cancer patients treated with the PD‐1 inhibitor Nivolumab. Sci Rep 6, 31726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kallergi G, Vetsika EK, Aggouraki D, Lagoudaki E, Koutsopoulos A, Koinis F, Katsarlinos P, Trypaki M, Messaritakis I, Stournaras C et al. (2018) Evaluation of PD‐L1/PD‐1 on circulating tumor cells in patients with advanced non‐small cell lung cancer. Ther Adv Med Oncol 10, 1758834017750121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kulasinghe A, Kapeleris J, Kimberley R, Mattarollo SR, Thompson EW, Thiery JP, Kenny L, O'Byrne K & Punyadeera C (2018) The prognostic significance of circulating tumor cells in head and neck and non‐small‐cell lung cancer. Cancer Med 7, 5910–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Dhar M, Wong J, Che J, Matsumoto M, Grogan T, Elashoff D, Garon EB, Goldman JW, Sollier Christen E, Di Carlo D et al. (2018) Evaluation of PD‐L1 expression on vortex‐isolated circulating tumor cells in metastatic lung cancer. Sci Rep 8, 2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yue C, Jiang Y, Li P, Wang Y, Xue J, Li N, Li D, Wang R, Dang Y, Hu Z et al. (2018) Dynamic change of PD‐L1 expression on circulating tumor cells in advanced solid tumor patients undergoing PD‐1 blockade therapy. Oncoimmunology 7, e1438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Stratton MR, Campbell PJ & Futreal PA (2009) The cancer genome. Nature 458, 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. van Soest RJ, van Royen ME, de Morree ES, Moll JM, Teubel W, Wiemer EA, Mathijssen RH, de Wit R & van Weerden WM (2013) Cross‐resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration‐resistant prostate cancer. Eur J Cancer 49, 3821–3830. [DOI] [PubMed] [Google Scholar]
- 153. Darshan MS, Loftus MS, Thadani‐Mulero M, Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel‐Vos C, Shen R, Tagawa ST et al. (2011) Taxane‐induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 71, 6019–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Reyes EE, VanderWeele DJ, Isikbay M, Duggan R, Campanile A, Stadler WM, Vander Griend DJ & Szmulewitz RZ (2014) Quantitative characterization of androgen receptor protein expression and cellular localization in circulating tumor cells from patients with metastatic castration‐resistant prostate cancer. J Transl Med 12, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, Ulman M, Smas ME, Lord JB, Brannigan BW, Trautwein J et al. (2012) Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov 2, 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van't Veer L, Garber JE, Evans G, Isaacs C, Daly MB, Matloff E et al. (2002) Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 346, 1616–1622. [DOI] [PubMed] [Google Scholar]
- 157. Rowe BP & Glazer PM (2010) Emergence of rationally designed therapeutic strategies for breast cancer targeting DNA repair mechanisms. Breast Cancer Res 12, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Eriksson I, Wettermark B & Bergfeldt K (2018) Real‐world use and outcomes of olaparib: a population‐based cohort study. Target Oncol 13, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hayes DF (2018) Precision medicine and testing for tumor biomarkers‐are all tests born equal? JAMA Oncol 4, 773–774. [DOI] [PubMed] [Google Scholar]
- 160. Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, Silberstein JL, Taylor MN, Maughan BL, Denmeade SR et al. (2017) Clinical significance of androgen receptor splice variant‐7 mRNA detection in circulating tumor cells of men with metastatic castration‐resistant prostate cancer treated with first‐ and second‐line abiraterone and enzalutamide. J Clin Oncol 35, 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Khoja L, Backen A, Sloane R, Menasce L, Ryder D, Krebs M, Board R, Clack G, Hughes A, Blackhall F et al. (2012) A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br J Cancer 106, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ghazani AA, Castro CM, Gorbatov R, Lee H & Weissleder R (2012) Sensitive and direct detection of circulating tumor cells by multimarker micro‐nuclear magnetic resonance. Neoplasia 14, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Riethdorf S, O'Flaherty L, Hille C & Pantel K (2018) Clinical applications of the cell search platform in cancer patients. Adv Drug Deliv Rev 125, 102–121. [DOI] [PubMed] [Google Scholar]
- 164. Krebs MG, Sloane R, Priest L, Lancashire L, Hou JM, Greystoke A, Ward TH, Ferraldeschi R, Hughes A, Clack G et al. (2011) Evaluation and prognostic significance of circulating tumor cells in patients with non‐small‐cell lung cancer. J Clin Oncol 29, 1556–1563. [DOI] [PubMed] [Google Scholar]
- 165. Scher HI, Graf RP, Schreiber NA, Jayaram A, Winquist E, McLaughlin B, Lu D, Fleisher M, Orr S, Lowes L et al. (2018) Assessment of the validity of nuclear‐localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration‐resistant prostate cancer. JAMA Oncol 4, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Keomanee‐Dizon K, Shishido SN & Kuhn P (2020) Circulating tumor cells: high‐throughput imaging of CTCs and bioinformatic analysis. Recent Results Cancer Res 215, 89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Guibert N, Pradines A, Farella M, Casanova A, Gouin S, Keller L, Favre G & Mazieres J (2016) Monitoring KRAS mutations in circulating DNA and tumor cells using digital droplet PCR during treatment of KRAS‐mutated lung adenocarcinoma. Lung Cancer 100, 1–4. [DOI] [PubMed] [Google Scholar]
- 168. Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, Knudsen M, Nordentoft I, Wu HT, Tin AS et al. (2019) Analysis of plasma cell‐free DNA by ultradeep sequencing in patients with stages I to III colorectal cancer. JAMA Oncol 5, 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhang Y, Zheng H, Zhan Y, Long M, Liu S, Lu J, Zang H & Fan S (2018) Detection and application of circulating tumor cell and circulating tumor DNA in the non‐small cell lung cancer. Am J Cancer Res 8, 2377–2386. [PMC free article] [PubMed] [Google Scholar]
- 170. Elias KM, Fendler W, Stawiski K, Fiascone SJ, Vitonis AF, Berkowitz RS, Frendl G, Konstantinopoulos P, Crum CP, Kedzierska M et al. (2017) Diagnostic potential for a serum miRNA neural network for detection of ovarian cancer. Elife 6, e28932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Nitta N, Sugimura T, Isozaki A, Mikami H, Hiraki K, Sakuma S, Iino T, Arai F, Endo T, Fujiwaki Y et al. (2018) Intelligent image‐activated cell sorting. Cell 175, 266–276, e213. [DOI] [PubMed] [Google Scholar]
- 172. Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O'Gorman WE, Au‐Yeung A et al. (2020) Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. [DOI] [PubMed] [Google Scholar]
- 173. Fairfax BP, Taylor CA, Watson RA, Nassiri I, Danielli S, Fang H, Mahe EA, Cooper R, Woodcock V, Traill Z et al. (2020) Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat Med 26, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Valpione S, Galvani E, Tweedy J, Mundra PA, Banyard A, Middlehurst P, Barry J, Mills S, Salih Z, Weightman J et al. (2020) Immune‐awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer 1, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]