

Abstract
The inexorable spread of resistance to clinically used drugs demands that we maintain a full pipeline of antibiotic candidates. As organisms have struggled to survive and compete over evolutionary history, they have developed the capacity to make a remarkably diverse array of natural products that target the cell envelope. A few have been developed for use in the clinic but most have not, and there are still an enormous number of opportunities to investigate. Substrate-binding antibiotics for Gram-positive organisms, phage-derived lysins, and outer membrane protein-targeting agents for Gram-negative organisms represent promising avenues where nature’s gifts may be repurposed for use in the clinic.
Introduction
The widespread introduction of penicillin toward the end of World War II transformed medicine, curing infections that were once death sentences and enabling invasive surgical procedures. Several new classes of antibiotics were introduced shortly thereafter, providing an arsenal of drugs that have saved countless lives over the last seventy-five years. However, resistance to all clinically-used antibiotics has emerged, and our ability to cure bacterial infections is increasingly threatened. Solving this problem is a public health priority.
Many of the world’s most successful antibiotics target the cell envelope due to its accessibility and the fact that many of its components are both essential and unique to bacteria. The cell envelope is a complex structure, consisting of the cell membrane, a peptidoglycan (PG) cell wall, a variety of associated polymers, and, in Gram-negative bacteria, a highly impermeable outer membrane (OM) [1, 2] (Figure 1A). The PG cell wall is a crosslinked polymer that encases the cytoplasmic membrane, and its assembly, reviewed in this issue by Grangeasse et al., is targeted by some of the world’s most important antibiotics. For example, penicillin and other beta-lactam antibiotics inhibit enzymes that crosslink PG chains, while vancomycin binds to Lipid II, the precursor substrate from which PG is assembled, preventing it from being used to build PG (Figure 1B). One way the drug industry has kept ahead of antibiotic resistance is by developing analogs of successful antibiotics and by using antibiotics in combination with other compounds that target important resistance mechanisms [3]. Although these approaches have been successful, high rates of antibiotic resistance reveal a pressing need for continued discovery of antibiotics with novel mechanisms of action.
Figure 1: The cell envelope, including the peptidoglycan cell wall, is a complex structure that protects bacteria from the surrounding environment.

A) Gram-positive bacteria have a single membrane surrounded by a thick layer of peptidoglycan (PG). Wall teichoic acids (WTAs) are attached covalently to PG, while lipoteichoic acids (LTAs) are anchored in the membrane. Gram-negative organisms have two membranes that sandwich a thin layer of peptidoglycan in the periplasm. The outer membrane (OM) contains outer membrane proteins (OMPs), as well as lipopolysaccharide on its outer leaflet. B) PG is synthesized by glycosyltransferases (GTs) that polymerize Lipid II and transpeptidases (TPs) that enact crosslinking. Hydrolases, including lysins, cleave PG at diverse positions.
Here, we highlight some recent discoveries of antibacterial agents that target the cell envelope. We focus on peptide-based natural products, recognizing that these antibiotics have been tailored and selected by nature for broad-spectrum activity over a long evolutionary history.
PG substrate binders
Vancomycin, which disrupts PG synthesis by binding to a PG substrate rather than an enzyme in the PG biosynthesis pathway, is the drug of choice for many drug-resistant infections caused by Gram-positive bacteria. First approved by the FDA in 1958, vancomycin binds D-Ala-D-Ala in the stem peptide of Lipid II [4] (Figure 2A), sterically blocking both the polymerization and crosslinking steps of peptidoglycan synthesis. Vancomycin has been a useful drug for so long because its mechanism makes it hard for bacteria to develop spontaneous resistance. Whereas bacteria often develop high level resistance to enzyme inhibitors through single amino acid changes in the enzyme target, altering a substrate in a biosynthetic pathway requires changing the selectivity of multiple enzymes. The probability that this would occur spontaneously through random mutation is extremely low. Vancomycin was used for more than thirty years before resistance began to spread widely due to transfer of resistance cassettes from glycopeptide-producing bacteria [5].
Figure 2: Substrate-binding antibiotics recognize various features of Lipid II, inhibiting different stages of peptidoglycan synthesis and lipid recycling.

A) The chemical structure of Lipid II. R1, R2, and R3 vary by species. In S. aureus, for example, R1 = NH2, R2 = H, and R3 = Gly5. B) When a Lipid II monomer is added to the existing PG matrix, undecaprenyl pyrophosphate is released. It is metabolized to undecaprenyl phosphate, which then gets flipped across the membrane for recycling. This lipid carrier is used in both the PG monomer, Lipid II, and in WTA precursors. Teixobactin, ramoplanin, and lysobactin bind the hydrophilic head group of Lipid II, vancomycin binds the stem peptide, and corbomycin and complestatin are proposed to bind formed PG. Bacitracin inhibits the metabolism of undecaprenyl pyrophosphate to undecaprenyl phosphate, preventing recycling of this lipid carrier, while amphomycin and friulimicin bind undecaprenyl phosphate.
Vancomycin’s substrate-binding mechanism of action was for decades thought to be very unusual, but it is now known that nature makes a remarkably diverse array of PG substrate binders [6]. These natural products, many of which are made by Gram-positive Actinobacteria [7] or Gram-negative Proteobacteria found in soil, generally contain a mix of L- and D-amino acids and have one or more rigidifying rings or crosslinks (Figure 3). Although vancomycin binds the stem peptide of Lipid II, most PG substrate binders recognize a different part of Lipid II. For example, the depsipeptides ramoplanin [8], lysobactin [9], and the recently discovered teixobactin [10] all recognize the reducing end of Lipid II, which includes the diphospho-sugar linkage (Figure 2B). Like Lipid II, other cell envelope glycopolymers such as wall teichoic acids (WTAs) are also assembled on undecaprenyl pyrophosphate (Und-PP), and many PG substrate binders can also bind to these precursors. In cells, however, many PG substrate binders lead to Lipid II accumulation [9, 11]. Because carrier lipid flux directly connects PG synthesis to assembly of other Und-P-linked substrates, Lipid II accumulation titrates away the carrier lipid used to make these substrates. In this manner, Lipid II binders may simultaneously block PG synthesis and synthesis of other pathways, but their binding to PG substrates is likely the direct cause of cell death. Some Lipid II binders, including nisin and the lysocins, have features that promote membrane perturbation, which may also contribute to their lethality [12, 13].
Figure 3: Structures of natural products that bind to Lipid II or peptidoglycan.
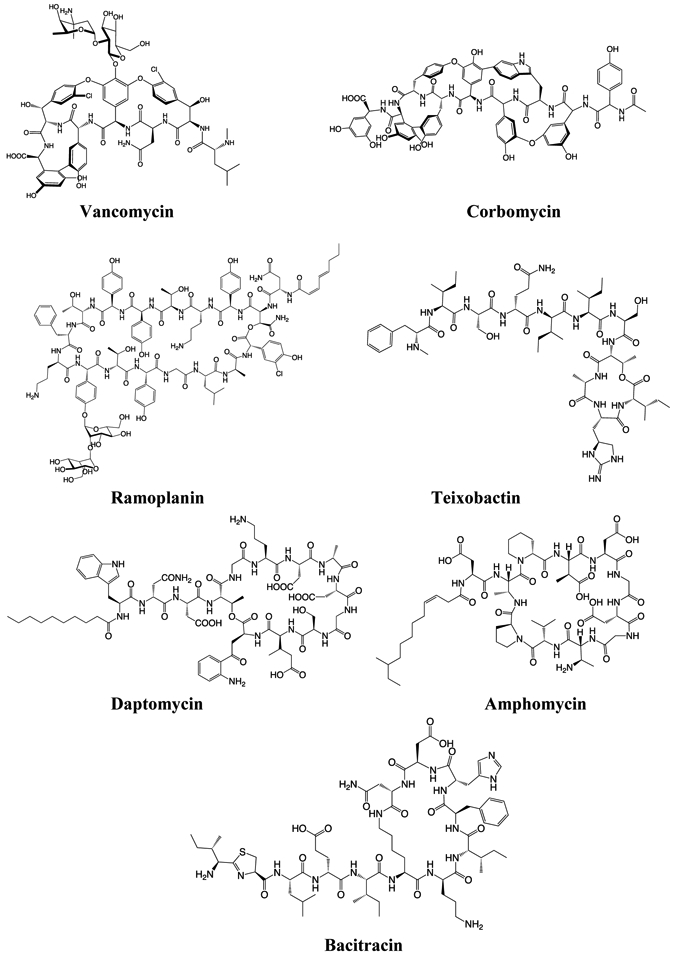
Vancomycin, corbomycin, ramoplanin, daptomycin, and amphomycin are produced by Gram-positive Actinobacteria, which include Streptomyces. Teixobactin is produced by Gram-negative Proteobacteria. Bacitracin is produced by certain strains of the Firmicute Bacillus.
Daptomycin, an important drug used to treat resistant Gram-positive infections, has now been shown to form a complex with Lipid II. Daptomycin has an unusual history: clinical trials were initially abandoned due to muscle toxicity, but a perspicacious pharmacologist later recognized that dosing adjustments could solve the toxicity problem and brought it back to the clinic [14]. Daptomycin gained FDA approval in 2003 even though its mechanism of action was unknown. Initial studies suggested that daptomycin inhibits PG synthesis [15], and its structure resembles those of other calcium-dependent cyclic lipopeptides like amphomycin [16] and friulimicin [17], which bind Und-P (Figure 2B). All these molecules also share some structural features with bacitracin, a cyclic peptide that inhibits dephosphorylation of Und-PP, preventing its recycling [18]. However, in vitro studies failed to demonstrate a specific target for daptomycin in the PG synthesis or recycling pathways. Focus turned to daptomycin’s membrane-disrupting properties as a likely mechanism of cell death [19], but a unique target to explain its specificity for bacteria was not identified. Reconciling many of the prior results, Grein et al. have now shown that daptomycin-Ca2+ binds Lipid II, but that complexation requires the membrane lipid phosphatidylglycerol [20]. This requirement may explain why it took so long to show an interaction with Lipid II. Recent bioinformatic analyses have also uncovered other novel calcium-dependent antibiotics, the malacidins [21] and cadasides [22], further demonstrating the wealth of such molecules that exist in nature.
Lipid II binders represent promising avenues for clinical development because, as with vancomycin, resistance cannot easily develop through spontaneous mutations to alter the target. Other mechanisms of resistance are possible, of course, including strategies to modify the antibiotics themselves. There are widespread D-stereospecific peptidases that can inactivate D-amino acid-containing peptide antibiotics [23]. Moreover, low levels of resistance to substrate binders can arise through cell envelope changes that limit access of these antibiotics to their targets. Even modest reductions in pathogen susceptibility to substrate binders can have serious consequences in the clinic [24, 25]. Nevertheless, substrate binders remain exceptionally attractive candidates for new antibiotics because the probability of spontaneous high-level resistance is extremely low.
A recent discovery suggests that some natural products may target fully formed PG. In 2020, the antibiotic complestatin and a novel compound, corbomycin, were reported to bind PG and proposed to inhibit cell growth by sterically blocking access of multiple cell wall hydrolases to their substrates [26] (Figure 2B). Cell wall hydrolases are important in PG maturation, remodeling, and daughter cell separation [27]. They are typically not individually essential; however, deleting a number of cell wall hydrolases can result in growth defects. The proposed mechanism of action for complestatin and corbomycin is novel and merits further investigation, although previous data demonstrating broad effects of complestatin, including inhibition of complement, may preclude its clinical use [28]. It is not known if corbomycin will have the same undesired effects. One potential limitation of corbomycin is that it would not be useful in combination with beta-lactams or other cell wall synthesis inhibitors. By blocking hydrolases, corbomycin prevents the cell wall lytic activity that these drugs rely on for killing. This limitation would likely apply to other pan-selective cell wall hydrolase inhibitors. However, because removing certain cell wall hydrolases can increase susceptibility to beta-lactam antibiotics, agents that selectively target these hydrolases might be useful adjuncts to beta-lactam therapy [29, 30]. The bulgecin natural products, which inhibit lytic transglycosylases important in Gram-negative bacteria, are attracting attention for this use [31, 32].
Lysins as drugs
Some cell wall hydrolases may also be used as therapeutic agents themselves. Bacteria and bacteriophages have weaponized hydrolases to lyse competitors or their hosts [33], raising the possibility that we, too, could deploy them as antibiotics. Bacteriophages are a particularly rich source of bacteriolytic proteins and peptides; most encode lysins, PG hydrolases that degrade the cell wall, although some produce lytic agents with other molecular targets in the PG synthesis pathway [34, 35] (Figure 1B). As potential treatments for bacterial infections, lysins are appealing because they are specific for PG and rapidly cause lysis [36]. In degrading PG rather than inhibiting an enzyme target, lysins, like substrate-binding antibiotics, may be slow to engender resistance.
A bacteriophage lysin, PlySs2 (also called CF-301 or exebecase), is currently undergoing Phase 3 clinical trials as an addition to the standard of care for treating S. aureus bacteremia, including endocarditis (https://clinicaltrials.gov/ct2/show/NCT04160468). PlySs2 was originally identified in 2013 from a Streptococcus suis phage [37]. It has an N-terminal CHAP (cysteine-histidine-dependent amidohydrolases/peptidases) domain and a C-terminal SH3b cell wall-binding domain, an architecture characteristic of some Gram-positive cell wall hydrolases. Unlike many other identified lysins, PlySs2 has relatively broad activity, lysing a range of Gram-positive organisms including S. aureus and several Streptococcus species, some of the most common causes of endocarditis. PlySs2 is particularly exciting because it disrupts biofilms, which pose a formidable barrier to antibiotics in many clinical infections [38]. Notably, the precise enzymatic activity of PlySs2 has not been determined. Preliminary studies showed that it acts as a D-Ala-Gly endopeptidase to cleave stem peptide crossbridges, but this activity cannot alone explain its broad cellular activity, including that against organisms lacking this linkage in their stem peptides [39]. As has been demonstrated for other phage lysins, the CHAP domain may possess both endopeptidase and amidase activity. Recent advances that make it possible to obtain defined PG substrates from a wide range of bacteria should enable studies to precisely determine this hydrolase’s substrate preferences [27].
Although bacteriophage lysins have shown promise for treating Gram-positive infections, the OM barrier prevents most of them from being effective against Gram-negative organisms. Efforts to improve the delivery of lysins to the periplasm have included adding hydrophobic tails or linking the enzymes to bacteriocins that facilitate transport across the OM [40]. Some lysins, however, naturally possess amphipathic tails that allow them access to the periplasm [41]. Many of these hydrophobic Gram-negative lysins bind serum components, which may limit their use to superficial or lung infections. Yet promise remains, and new Gram-negative-targeting lysins are under development [36, 42].
Outer membrane-targeting antibiotics
The OM not only blocks access of lysins to their targets in the periplasm, but restricts access of many antibiotics that work well against Gram-positive organisms. Strategies to compromise the OM barrier are therefore highly sought, and attention is now being focused on the machinery that assembles the OM. One essential machine in the OM is Bam, for β-Barrel assembly machine, which is described in more detail by Kahne et al. in this same issue. The central component of Bam is itself a β-barrel protein called BamA, which is required to assemble the OM translocon (LptDE) that puts lipopolysaccharide in the outer leaflet of the membrane to create a highly impermeable barrier [43, 44, 45, 46, 47]. BamA consists of five periplasmic, N-terminal polypeptide transport-associated (POTRA) domains and a 16-stranded ß-barrel OM protein with eight loops exposed to the cell surface. The N-terminus of the ß-barrel holds the C-terminus of ß-barrel substrates as folding proceeds [48, 49, 50]. The ability of BamA to open at the seam between its N- and C-terminal β-strands is crucial to allow binding of substrates [51] (Figure 4A). Strategies to disrupt BamA gating or to prevent substrate binding or release from BamA could lead to new treatments for Gram-negative infections.
Figure 4: BamA catalyzes folding and insertion of beta-barrel proteins into the OM and is a highly promising antibiotic target.

A) As part of the Bam complex, BamA folds ß-barrels for insertion into the outer membrane. A gate within BamA opens to engage the substrate, with the N-terminal side of the BamA ß-barrel forming hydrogen bonds with the C-terminus of the substrate. Darobactin is proposed to stabilize the closed conformation of BamA such that the substrate cannot be inserted into this gate. B) The structure of darobactin.
The discovery of a BamA-binding antibody with activity against some E. coli strains provided initial validation that it is possible to target BamA [52]. Other evidence that BamA can be targeted is supplied by the example of lectin-like bacteriocins like LlpA, which are secreted by Proteobacteria [53]. These proteins contain two lectin domains, one of which binds rhamnose in lipopolysaccharide, while the second seems to have a different function important for strain selectivity. Though these toxins’ mechanism of action is not yet fully clear, killing is dependent on particular polymorphisms within BamA, and they are suggested to interact directly with Bam. Similar to the lectin-like bacteriocins, a series of new chimeric peptidomimetic antibiotics are proposed to target both BamA and lipopolysaccharide for binding [54]. Recent work has also identified a peptide antibiotic that apparently inhibits BamA. Darobactin, a ribosomally-encoded peptide isolated from the nematode symbiont Photorhabdus khaini, has activity against a broad range of Gram-negative organisms (Figure 4B). Darobactin may stabilize BamA in a closed-gate conformation, preventing substrate engagement for folding [55]. BamA and the OM translocon are both attractive targets because they are essential and accessible, and agents that target them from outside would not be subject to efflux pumps [56]. Moreover, by compromising the OM barrier, OM-targeting drugs are expected to synergize well with other antibiotics.
Conclusion and future directions
The cell envelope, including the PG cell wall synthesis machinery, still holds a wealth of untapped targets. For example, members of the SEDS (for shape, elongation, division, and sporulation) family of proteins, which includes the Rod complex protein RodA and the divisome complex protein FtsW, were recently shown to have PG polymerase activity [57, 58]. FtsW is conserved and essential in all bacteria, and RodA is required in rod-shaped organisms, which include many problematic Gram-negative pathogens. These proteins represent important new targets for investigation, and recently obtained structural information may facilitate development of inhibitors [59]. It is conceivable that molecules that inhibit these targets are found in nature [60]. Lipid II flippases have also emerged as promising targets for which natural product inhibitors are known [35, 61, 62]. In addition to PG, other polymers like teichoic acids in Gram-positive organisms [63, 64] and lipopolysaccharide in Gram-negative organisms [65, 66] are intrinsic resistance factors. Targeting them in combination with other drugs may be a valuable strategy [3]. To combat antibiotic resistance, it is crucial that we continue to uncover new targets, pursue drugs with novel mechanisms of action, and explore compound combinations that can overcome, or mitigate the spread of, antibiotic resistance.
Highlights.
Nature produces abundant cell envelope-targeting antibiotics.
Substrate-binding antibiotics are diverse, plentiful, and slow to cause resistance.
Peptidoglycan-degrading lysins are an alternative to small molecule antibiotics.
Outer membrane proteins can be directly targeted to kill Gram-negative bacteria.
Acknowledgements
We acknowledge the National Institutes of Health Grants R01 AI148752 (to S. W.) and T32GM007753 (to J. E. P.) for support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank David Tomasek for help in designing Figure 4.
Footnotes
Competing interests
The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Silhavy TJ, Kahne D, Walker S: The Bacterial Cell Envelope. Cold Spring Harb Perspect Biol 2010, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajagopal M, Walker S: Envelope Structures of Gram-Positive Bacteria. Curr Top Microbiol Immunol 2017, 404:1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Theuretzbacher U, Outterson K, Engel A, Karlén A: The global preclinical antibacterial pipeline. Nature Reviews Microbiology 2020, 18:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine DP: Vancomycin: A History. Clin Infect Dis 2006, 42:S5–S12. [DOI] [PubMed] [Google Scholar]
- 5.Bugg TDH, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT: Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30:10408–10415. [DOI] [PubMed] [Google Scholar]
- 6.Grein F, Schneider T, Sahl H-G: Docking on Lipid II—A Widespread Mechanism for Potent Bactericidal Activities of Antibiotic Peptides. Journal of Molecular Biology 2019, 431:3520–3530. [DOI] [PubMed] [Google Scholar]
- 7.van Bergeijk DA, Terlouw BR, Medema MH, van Wezel GP: Ecology and genomics of Actinobacteria: new concepts for natural product discovery. Nature Reviews Microbiology 2020, 18:546–558. [DOI] [PubMed] [Google Scholar]
- 8.Hu Y, Helm JS, Chen L, Ye X-Y, Walker S: Ramoplanin Inhibits Bacterial Transglycosylases by Binding as a Dimer to Lipid II. J Am Chem Soc 2003, 125:8736–8737. [DOI] [PubMed] [Google Scholar]
- 9.Lee W, Schaefer K, Qiao Y, Srisuknimit V, Steinmetz H, Müller R, Kahne D, Walker S: The Mechanism of Action of Lysobactin. J Am Chem Soc 2016, 138:100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S, et al. : A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiao Y, Lebar MD, Schirner K, Schaefer K, Tsukamoto H, Kahne D, Walker S: Detection of Lipid-Linked Peptidoglycan Precursors by Exploiting an Unexpected Transpeptidase Reaction. J Am Chem Soc 2014, 136:14678–14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiedemann I, Breukink E, van Kraaij C, Kuipers OP, Bierbaum G, de Kruijff B, Sahl HG: Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. The Journal of Biological Chemistry 2001, 276:1772–1779. [DOI] [PubMed] [Google Scholar]
- 13.Santiago M, Lee W, Fayad AA, Coe KA, Rajagopal M, Do T, Hennessen F, Srisuknimit V, Müller R, Meredith TC, et al. : Genome-wide mutant profiling predicts the mechanism of a Lipid II binding antibiotic. Nat Chem Biol 2018, 14:601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenstein BI, Oleson FB, Baltz RH: Daptomycin: From the Mountain to the Clinic, with Essential Help from Francis Tally, MD. Clin Infect Dis 2010, 50:S10–S15. [DOI] [PubMed] [Google Scholar]
- 15.Allen NE, Hobbs JN, Alborn WE: Inhibition of peptidoglycan biosynthesis in gram-positive bacteria by LY146032. Antimicrob Agents Chemother 1987, 31:1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka H, Iwai Y, Ōiwa R, Shinohara S, Shimizu S, Oka T, Ōmura S: Studies on bacterial cell wall inhibitors: II. Inhibition of peptidoglycan synthesis in vivo and in vitro by amphomycin. Biochimica et Biophysica Acta (BBA) - General Subjects 1977, 497:633–640. [DOI] [PubMed] [Google Scholar]
- 17.Schneider T, Gries K, Josten M, Wiedemann I, Pelzer S, Labischinski H, Sahl H-G: The lipopeptide antibiotic Friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob Agents Chemother 2009, 53:1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siewert G, Strominger JL: Bacitracin: An inhibitor of the dephosphorylation of lipid pyrophosphate, an intermediate in the biosynthesis of the peptidoglycan of bacterial cell walls. Proc Natl Acad Sci USA 1967, 57:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silverman JA, Perlmutter NG, Shapiro HM: Correlation of Daptomycin Bactericidal Activity and Membrane Depolarization in Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2003, 47:2538–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *20.Grein F, Müller A, Scherer KM, Liu X, Ludwig KC, Klöckner A, Strach M, Sahl H-G, Kubitscheck U, Schneider T: Ca 2+ -Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids. Nature Communications 2020, 11:1455.* Using a combination of microscopy and biochemistry, the authors demonstrate that daptomycin-Ca2+ binds to Lipid II, but only in the presence of phosphatidylglycerol. This finding clarifies previous results that suggested that daptomycin inhibits peptidoglycan synthesis but failed to identify a specific target.
- *21.Hover BM, Kim S-H, Katz M, Charlop-Powers Z, Owen JG, Ternei MA, Maniko J, Estrela AB, Molina H, Park S, et al. : Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nature Microbiology 2018, 3:415–422.* A new class of calcium-dependent antibiotics was identified using bioinformatics to find calcium-binding motifs in the DNA of soil bacteria. This tool can be applied to the identification of further such molecules.
- 22.Wu C, Shang Z, Lemetre C, Ternei MA, Brady SF: Cadasides, Calcium-Dependent Acidic Lipopeptides from the Soil Metagenome That Are Active against Multidrug-Resistant Bacteria. J Am Chem Soc 2019, 141:3910–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.Li Y-X, Zhong Z, Hou P, Zhang W-P, Qian P-Y: Resistance to nonribosomal peptide antibiotics mediated by d -stereospecific peptidases. Nature Chemical Biology 2018, 14:381–387.* The authors use a genome-mining approach to identify widespread D-stereospecific peptidases that can inactivate antibiotics frequently lauded for not generating resistance. If transferred to pathogens, these peptidases could confer clinical resistance.
- 24.Gardete S, Tomasz A: Mechanisms of vancomycin resistance in Staphylococcus aureus. J Clin Invest 2014, 124:2836–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller WR, Bayer AS, Arias CA: Mechanism of Action and Resistance to Daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harb Perspect Med 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *26.Culp EJ, Waglechner N, Wang W, Fiebig-Comyn AA, Hsu Y-P, Koteva K, Sychantha D, Coombes BK, Van Nieuwenhze MS, Brun YV, et al. : Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 2020, 578:582–587.* By looking for genes lacking known resistance factors, the authors identified two natural product antibiotics that seem to bind to peptidoglycan, broadly preventing the activity of cell wall hydrolases. This interesting mechanism of action merits further study.
- 27.Do T, Page JE, Walker S: Uncovering the activities, biological roles, and regulation of bacterial cell wall hydrolases and tailoring enzymes. J Biol Chem 2020, 295:3347–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneko I, Kamoshida K, Takahashi S: Complestatin, a potent anti-complement substance produced by Streptomyces lavendulae. J Antibiot 1989, 42:236–241. [DOI] [PubMed] [Google Scholar]
- 29.Korsak D, Liebscher S, Vollmer W: Susceptibility to Antibiotics and β-Lactamase Induction in Murein Hydrolase Mutants of Escherichia coli. Antimicrob Agents Chemother 2005, 49:1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Do T, Schaefer K, Santiago AG, Coe KA, Fernandes PB, Kahne D, Pinho MG, Walker S: Staphylococcus aureus cell growth and division are regulated by an amidase that trims peptides from uncrosslinked peptidoglycan. Nat Microbiol 2020, doi: 10.1038/s41564-019-0632-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams AH, Wheeler R, Thiriau C, Haouz A, Taha M-K, Boneca IG: Bulgecin A: The Key to a Broad-Spectrum Inhibitor That Targets Lytic Transglycosylases. Antibiotics (Basel) 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dik DA, Madukoma CS, Tomoshige S, Kim C, Lastochkin E, Boggess WC, Fisher JF, Shrout JD, Mobashery S: Slt, MltD, and MltG of Pseudomonas aeruginosa as Targets of Bulgecin A in Potentiation of β-Lactam Antibiotics. ACS Chem Biol 2019, 14:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harding CJ, Huwiler SG, Somers H, Lambert C, Ray LJ, Till R, Taylor G, Moynihan PJ, Sockett RE, Lovering AL: A lysozyme with altered substrate specificity facilitates prey cell exit by the periplasmic predator Bdellovibrio bacteriovorus. Nat Commun 2020, 11:4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernhardt TG, Wang I-N, Struck DK, Young R: A Protein Antibiotic in the Phage Qβ Virion: Diversity in Lysis Targets. Science 2001, 292:2326–2329. [DOI] [PubMed] [Google Scholar]
- 35.Chamakura KR, Sham L-T, Davis RM, Min L, Cho H, Ruiz N, Bernhardt TG, Young R: A viral protein antibiotic inhibits lipid II flippase activity. Nat Microbiol 2017, 2:1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vázquez R, García E, García P: Phage Lysins for Fighting Bacterial Respiratory Infections: A New Generation of Antimicrobials. Front Immunol 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gilmer DB, Schmitz JE, Euler CW, Fischetti VA: Novel Bacteriophage Lysin with Broad Lytic Activity Protects against Mixed Infection by Streptococcus pyogenes and Methicillin-Resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2013, 57:2743–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuch R, Khan BK, Raz A, Rotolo JA, Wittekind M: Bacteriophage Lysin CF-301, a Potent Antistaphylococcal Biofilm Agent. Antimicrobial Agents and Chemotherapy 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lood R, Molina H, Fischetti VA: Determining bacteriophage endopeptidase activity using either fluorophore-quencher labeled peptides combined with liquid chromatography-mass spectrometry (LC-MS) or Förster resonance energy transfer (FRET) assays. PLOS ONE 2017, 12:e0173919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heselpoth RD, Euler CW, Schuch R, Fischetti VA: Lysocins: Bioengineered Antimicrobials That Deliver Lysins across the Outer Membrane of Gram-Negative Bacteria. Antimicrob Agents Chemother 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raz A, Serrano A, Hernandez A, Euler CW, Fischetti VA: Isolation of Phage Lysins That Effectively Kill Pseudomonas aeruginosa in Mouse Models of Lung and Skin Infection. Antimicrob Agents Chemother 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghose C, Euler CW: Gram-Negative Bacterial Lysins. Antibiotics (Basel) 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D: Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 2005, 121:235–245. [DOI] [PubMed] [Google Scholar]
- 44.Wu T, McCandlish AC, Gronenberg LS, Chng S-S, Silhavy TJ, Kahne D: Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. PNAS 2006, 103:11754–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bos MP, Tefsen B, Geurtsen J, Tommassen J: Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. PNAS 2004, 101:9417–9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakelar J, Buchanan SK, Noinaj N: The structure of the β-barrel assembly machinery complex. Science 2016, 351:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, et al. : Structural basis of outer membrane protein insertion by the BAM complex. Nature 2016, 531:64–69. [DOI] [PubMed] [Google Scholar]
- 48.Lee J, Tomasek D, Santos TM, May MD, Meuskens I, Kahne D: Formation of a β-barrel membrane protein is catalyzed by the interior surface of the assembly machine protein BamA. eLife 2019, 8:e49787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doyle MT, Bernstein HD: Bacterial outer membrane proteins assemble via asymmetric interactions with the BamA β-barrel. Nature Communications 2019, 10:3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *50.Tomasek D, Rawson S, Lee J, Wzorek JS, Harrison SC, Li Z, Kahne D: Structure of a nascent membrane protein as it folds on the BAM complex. Nature 2020, 583:473–478.* A cryo-EM structure of BamA folding a substrate suggests a mechanism for how substrates are folded and released into the membrane in the absence of an exogenous energy source.
- 51.Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, Buchanan SK: Lateral Opening and Exit Pore Formation Are Required for BamA Function. Structure 2014, 22:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Storek KM, Auerbach MR, Shi H, Garcia NK, Sun D, Nickerson NN, Vij R, Lin Z, Chiang N, Schneider K, et al. : Monoclonal antibody targeting the β-barrel assembly machine of Escherichia coli is bactericidal. PNAS 2018, 115:3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghequire MGK, Swings T, Michiels J, Buchanan SK, Mot RD: Hitting with a BAM: Selective Killing by Lectin-Like Bacteriocins. mBio 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luther A, Urfer M, Zahn M, Müller M, Wang S-Y, Mondal M, Vitale A, Hartmann J-B, Sharpe T, Monte FL, et al. : Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576:452–458. [DOI] [PubMed] [Google Scholar]
- *55.Imai Y, Meyer KJ, Iinishi A, Favre-Godal Q, Green R, Manuse S, Caboni M, Mori M, Niles S, Ghiglieri M, et al. : A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576:459–464.* The authors identified darobactin, a novel peptidic antibiotic, that seems to bind to BamA, stabilizing a closed gate conformation that prevents substrate folding.
- 56.Hart EM, Mitchell AM, Konovalova A, Grabowicz M, Sheng J, Han X, Rodriguez-Rivera FP, Schwaid AG, Malinverni JC, Balibar CJ, et al. : A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. PNAS 2019, 116:21748–21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ: SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 2016, 537:634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *58.Taguchi A, Welsh MA, Marmont LS, Lee W, Sjodt M, Kruse AC, Kahne D, Bernhardt TG, Walker S: FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nature Microbiology 2019, 4:587–594.* This work shows that the universally-conserved divisome protein FtsW is a peptidoglycan polymerase and that it requires complexation with a divisome penicillin-binding protein (PBP) for polymerase activity. SEDS-PBP complexes are promising antibiotic targets.
- *59.Sjodt M, Rohs PDA, Gilman MSA, Erlandson SC, Zheng S, Green AG, Brock KP, Taguchi A, Kahne D, Walker S, et al. : Structural coordination of polymerization and crosslinking by a SEDS–bPBP peptidoglycan synthase complex. Nature Microbiology 2020, 5:813–820.* This work provides a crystal structure of RodA complexed with a penicillin binding protein, lending insight into how they work together to polymerize and crosslink peptidoglycan. This information may facilitate the design of antibiotics to target RodA or other SEDS proteins.
- 60.Emami K, Guyet A, Kawai Y, Devi J, Wu LJ, Allenby N, Daniel RA, Errington J: RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nature Microbiology 2017, 2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sham L-T, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N: MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345:220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chu J, Vila-Farres X, Inoyama D, Ternei M, Cohen LJ, Gordon EA, Reddy BVB, Charlop-Powers Z, Zebroski HA, Gallardo-Macias R, et al. : Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat Chem Biol 2016, 12:1004–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown S, Santa Maria JP, Walker S: Wall teichoic acids of gram-positive bacteria. Annual Review of Microbiology 2013, 67:313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Percy MG, Gründling A: Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu Rev Microbiol 2014, 68:81–100. [DOI] [PubMed] [Google Scholar]
- 65.Zhang G, Meredith TC, Kahne D: On the essentiality of lipopolysaccharide to Gram-negative bacteria. Current Opinion in Microbiology 2013, 16:779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bertani B, Ruiz N: Function and biogenesis of lipopolysaccharides. EcoSal Plus 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]