

Abstract
Introduction:
Triggering receptors expressed on myeloid cells (TREMs) are inflammatory amplifiers with defined pathophysiological role in various infectious diseases, acute and chronic aseptic inflammation, and a variety of cancers, depicting TREMs as prominent therapeutic targets.
Areas covered:
Herein, updates from 2015–2020 are discussed to divulge the TREM ligands, as well as their peptide blockers, claimed to modulate their expression. The article also presents different strategies employed during the last five years to block interactions between TREMs and their ligands to treat various disease conditions by modulating their expression and activity.
Expert opinion:
There has been significant progress in the discovery of novel ligands and modulators of TREMs in the last five years that mainly revolved around the function of TREM molecules. A few peptides showed encouraging results to modulate the expression and activity of TREMs in pre-clinical studies, and these peptides are currently in clinical investigation. Based on the findings so far in several careful studies, we expect novel therapeutics in the near future which could have the ability to treat various disease conditions associated with TREM expression.
Keywords: ADAMs, Arthritis, Atherosclerosis, DAP12, eCIRP, Extracellular actin, HMGB-1, HSP70, Infectious diseases, PGLYRP1, Sepsis, TREM-1, TREM-2, TREM-like transcripts, TREM modulators
1. INTRODUCTION
A family of cell surface receptors, ‘TREM’ or ‘triggering receptor expressed on myeloid cells’ are group of activating receptors that are particularly expressed on myeloid cells, such as monocytes, macrophages, neutrophils, mast cells and dendritic cells (DCs). TREMs are primarily transmembrane glycoproteins having an immunoglobulin (Ig)-type fold in their extracellular domain, thus belong to Ig-superfamily. These receptors have been found as important pathological hallmarks in various inflammatory disorders of infectious as well as non-infectious disease conditions such as myocardial infarction, atherosclerosis, sepsis, gout, renal fibrosis, pancreatitis, cancer etc. [1]. Structurally, these receptors contain three units: (i) extracellular immunoglobulin domain, (ii) a transmembrane region, and (iii) short cytoplasmic domain [2] (Figure 1). In their cytoplasmic domain, there are no signaling motifs, therefore, transmembrane domains associate with the immunoreceptor tyrosine-based activation motif (ITAM)-containing molecule DAP12 (DNAX activation protein of molecular weight 12 kDa) to express their functions [3].
Figure 1:
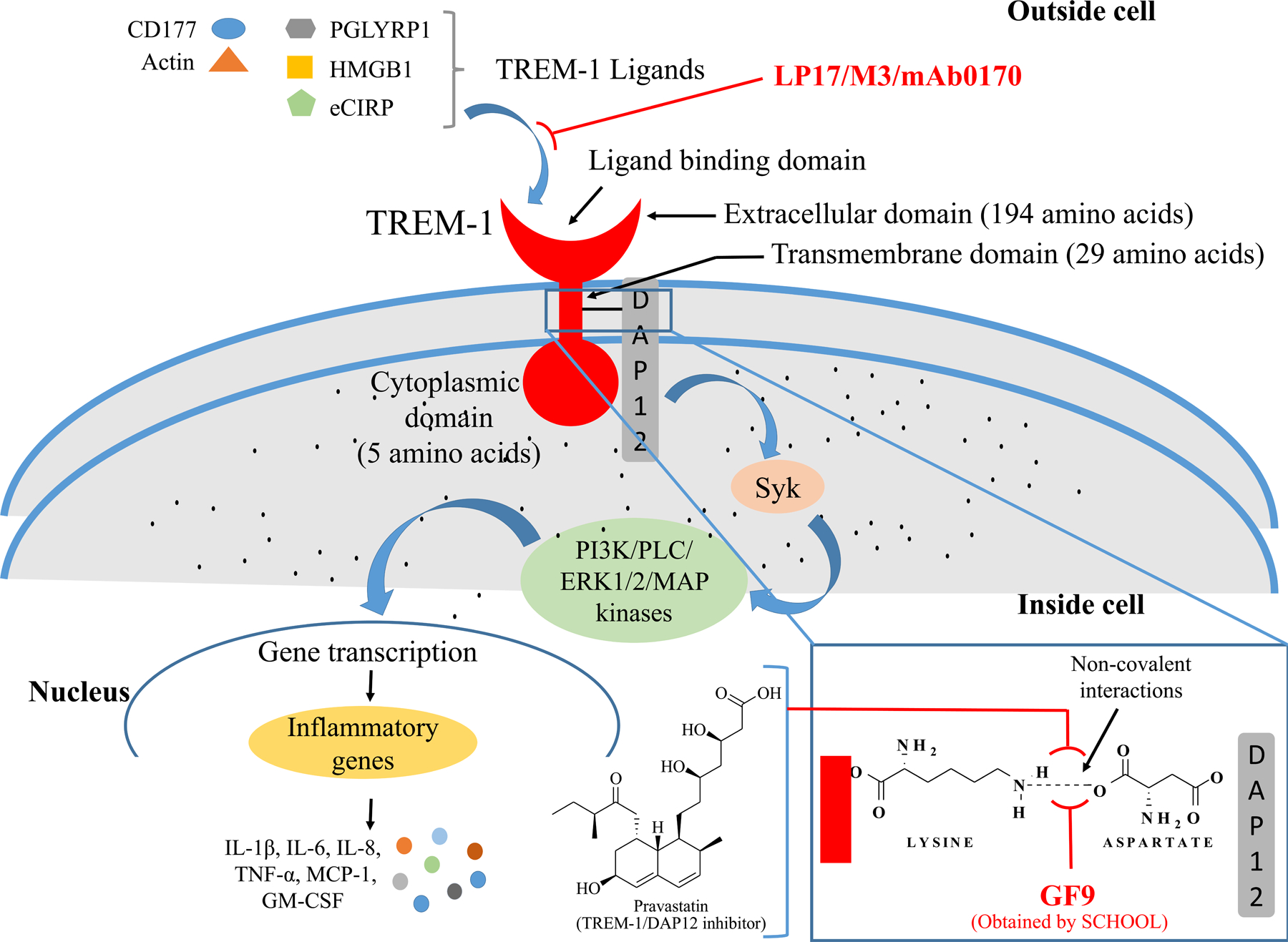
Schematic illustration of TREM-1 pathway: TREM-1 is an immunoglobulin superfamily receptor located on the surface of plasma membrane. Its extracellular domain consisting of 194 amino acids is mainly responsible for ligand binding. Transmembrane domain consisting of 29 amino acids generally associates with DAP12, phosphorylation of which results in the activation of Syk that triggers the activation of PI3K, PLC, ERK, MAPK, and NF-κB, to regulate inflammatory gene transcription. This activation further increases the production of cytokines and chemokines (IL-1β, IL-6, IL-8, TNF-α, MCP-1, GM-CSF). Inhibition of ligand binding to TREM-1 by various peptides, small molecules and antibodies or blocking the TREM-1/DAP12 interaction is beneficial in the treatment of inflammatory diseases.
So far, five TREM isoforms, TREM-1 to TREM-5, have been discovered, and human TREM isoforms share low sequence homology with each other and are located on chromosome 6P21.1 [2,4]. Various related receptors, TREM-like transcripts (TLT-1 to TLT-5) also map to this region of the genome and share V-domain of the Ig-superfamily. TLTs also consist of ITAM in their cytoplasmic tail to express their functions [5]. Among all five isoforms of TREM, TREM-1 and TREM-2 are the best-characterized receptors that play divergent roles in several infectious and inflammatory diseases. In this critical review article, we focused on these two receptors in significant details to highlight their expression, signaling and their modulatory significance in various diseases.
2. TREM-1 expression and signaling
TREM-1 isoform is the most characterized member of TREM family that is expressed on mature monocytes, macrophages and neutrophils and function as a crucial amplifier of inflammatory responses [6–8]. However, in addition to myeloid cells, TREM-1 is also present on the surface of non-myeloid cell types including epithelial and endothelial cells [9–11]. Several recent studies revealing the presence of TREM-1 in different cell types and species are presented in Table 1 [12–19]. Human immune cells expressing a high expression of TREM-1 are monocytes, neutrophils, DCs, granulocytes and natural killer (NK) cells. Immune cells expressing low levels of TREM-1 include T cells and all isoforms of B cells [5,20,21].
Table 1:
Expression of TREM-1 in various disease conditions
| Disease | Expression | References | |
|---|---|---|---|
| Human | Mouse | ||
| Atherosclerosis |
Soluble form: ND Circulating: Enhanced, on monocytes with oxLDL Tissue: Enhanced mRNA and protein in severe condition |
Soluble form: Increased during high fat diet Circulating: Enhanced, myeloid cells after high cholesterol/high fat diet Tissue: Enhanced mRNA in aortic arch after high fat diet |
[12–14] |
| Myocardial Infarction |
Soluble form: Enhanced and can cause mortality and recurrence of cardiovascular events Circulating: ND Tissue: Enhanced number of TREM-1 expressing granulocytes |
Soluble form: Increased Circulating: Increased Tissue: Enhanced mRNA/protein by infiltrating granulocytes |
[15] |
| Mesenteric Ischemia-Reperfusion Injury |
Soluble form: ND Circulating: ND Tissue: ND |
Soluble form: Increased in plasma Circulating: ND Tissue: ND |
[16] |
| Gout |
Soluble form: Enhanced in synovial fluid Circulating: Enhanced, on monocytes in synovial fluid Tissue: Presence of TREM-1-expressing macrophages in tophaceous tissue |
Soluble form: ND Circulating: ND Tissue: ND |
[17] |
| Renal Ischemia-Reperfusion Injury |
Soluble form: ND Circulating: ND Tissue: mRNA |
Soluble form: Increased in plasma Circulating: Increased on monocytes Tissue: Increased mRNA/protein |
[18] |
| Psoriasis |
Soluble form: Increased in Plasma Circulating: ND Tissue: Increased number of TREM-1 expressing myeloid cells |
Soluble form: ND Circulating: Enhanced in splenic cells Tissue: Enhanced expression of mRNA |
[19] |
ND, not detected; oxLDL, oxidized low density lipoproteins
Multiple membrane-bound receptors exist in a soluble form in clinical samples of patients with inflammatory disease and soluble form of receptors of TREM family have also been identified [22]. A general belief is that the production of sTREM-1 (17.5 kDa) may occur through two alternate pathways, such as alternate splicing and proteolytic cleavage of membrane bound receptors by metalloproteinases (MMPs) [23,24]. sTREM-1 was first observed in the plasma of patients with sepsis and in the bronchial lavage of pneumonia patients [25,26]. So far, its elevated levels have been found in various inflammatory diseases including inflammatory bowel disease, inflammatory rheumatoid arthritis (RA), intra-abdominal infections, pneumonia, pleural effusion, and lung cancer [27] (Table 1). Recent studies also indicate that TREM expression on monocytes in peripheral blood could help to predict the occurrence of disease in glioma patients [28]. In addition to the serum, an increased level of sTREM-1 was also found in the cerebral spinal fluid of patients with bacterial meningitis suggesting the role of TREM-1 signaling in the underlying pathogenesis of central nervous system (CNS) infections [29,30].
Number of studies revealed its role in various disease conditions. The significant relation between TREM-1 and vitamin D receptors has also been observed by Nguyen et al. [31]. Elevated level of TREM-1 in keratinocytes in swine was observed when treated with vitamin D sufficient diet, which suggest a significant role of vitamin D in signaling of TREM-1 pathway. Similarly, the relationship between elevated TREM-1 and plaque stability in symptomatic and asymptomatic patients with carotid stenosis was determined [32]. TREM-1 expression was found significantly increased in atherosclerotic plaques from carotid arteries of symptomatic patients as compared to asymptomatic patients. Further, TREM-1 decoy receptor and TREM-1 antibodies reduced tumor necrosis factor alpha (TNF-α)-induced expression of TREM-1, suggesting its potential role in the destabilization of atherosclerotic plaque. Pathophysiological role of TREM-1 in obesity-induced insulin resistance has also been reported [34]. TREM-1 has also been found in the pathophysiology of various cardiovascular diseases [13,15,35]. The role of TREM-1 as a potential therapeutic target in various diseases is summarized in Table 1.
The transmembrane domain of human TREM-1 contains a lysine amino acid residue 217 that interacts with amino acid aspartate in DAP12 and transduces downstream signaling [7,8]. TREM-1 signaling is activated by binding of various ligands that initiates the association and phosphorylation of ITAM of DAP12. Phosphorylation of DAP12 by Src family kinases results in the activation of non-receptor tyrosine kinase Syk that in turn, triggers the activation of signaling molecules phosphatidylinositol 3-kinases (PI3K), phospholipase C (PLC), extracellular signal-regulated kinases (ERK1/2) and mitogen-activated protein (MAP) kinases regulating nuclear factor (NF-κB) activation and functioning of inflammatory genes [1,7,34]. The activation of inflammatory genes further increases the production of pro-inflammatory cytokines including interleukin (IL)-1β, IL-6, IL-8, TNF-α, monocyte chemoattractant protein (MCP)-1, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [36] (Figure 1). TREM-1 signaling also regulates calcium influx via phosphorylation of Syk which further triggers MAPK/ERK pathway [37]. In addition to cytokines and chemokines, TREM-1 also controls the degranulation and production of reactive oxygen species (ROS). Specifically, TREM-1 accelerates TLT-2 ligation that can increase the production of ROS in polymorphonuclear neutrophils [38]. A recent study indicated that TREM-1 is generally activated by multimerization which required a two-step process comprising of upregulation followed by aggregation of TREM-1 at the cell surface. The study also confirmed that the ectodomain of TREM-1 can aggregate in a concentration-dependent manner and adapter protein DAP12 stabilizes TREM-1 multimerization, which is also mediated via its various endogenous ligands [39].
3. TREM-1 ligands and peptide blockers
3.1. TREM-1 decoy receptors
The compounds or molecules that can bind to the surface of TREM-1 are still a matter of concern. Until 2006, no endogenous ligand of TREM-1 had been identified [40]. There was a general belief in biologists that the neutralization of TREM-1 with a TREM-1-Fc fusion protein impaired the ability of serum samples of septic patients to activate monocytes in vitro and this activation can be blocked by using TREM-1 antibody [40,41]. TREM-1 can specifically bind to platelets from healthy patients and neutrophils from patients with sepsis, which suggest the presence of unknown TREM-1 ligands [38,40]. In 2006, Mariani et al. developed methods to detect various TREM-1 ligands of unknown identity [40].
To date, multiple putative ligands have been discovered that trigger TREM-1 signaling on the surface of platelets. Haselmayer et al. [38] identified membrane-bound ligand which can interact with TREM-1 and can amplify lipopolysaccharide (LPS) -induced neutrophil activation.
The hyperactivation of inflammatory responses can be mitigated by blocking TREM-1 signaling using small molecules and peptide mimetics. Particularly, sTREM-1 can bind to TREM-1 ligands and further block their recruitment to membrane-bound TREM-1 [42,43]. However, due to the high degree of degradation of sTREM-1 their use as therapeutic agents is very limited. Intriguingly, sTREM-1 has been so far used to design small peptides to inhibit the interactions between the ligands and membrane-bound TREM-1. Although the concept of TREM/sTREM is known and developed within last two decades, its application in clinical practice is yet to be discovered. There is still a hope to develop novel therapeutics by targeting TREM-1 in chronic inflammatory diseases since the infiltration of macrophages with upregulated TREM-1 in the pathogenesis of chronic inflammatory diseases is a major hallmark of the disease process. These findings further support TREM-1 as a potential therapeutic target [28,44,45]. In view of no definitive therapy with high TREM-1 expression, it is imperative to design novel small molecules which effectively and efficiently target TREM-1 to suppress inflammation.
The classic examples of designed peptides are LR12 and LP17 that were derived from human or mouse TREM-1 complementarity determining regions (CDR) 2 and 3, respectively. CDR-3 corresponds to ‘F’ β strand of extracellular domain of TREM-1, tyrosine residue of which may be responsible to mediate TREM-1 homodimerization and thus disruption in the homeostasis of the physiological processes [4,43]. Peptides LR12 and LP17 block the binding of sTREM-1 to a ligand from mice peritoneal exudate cells [42–46]. Various clinical trials [47,48] have been completed in the recent past to access the safety, tolerability, and pharmacokinetics of LR12 (also known as Nangibotide) in healthy male subjects after its pre-clinical success [49–52]. Clinical studies revealed that LR12 is safe even at the highest dose tested (10 mg/kg) with mild adverse effects which were considered unrelated to the treatment. Pharmacokinetic studies indicated that its blood concentrations increased in a dose-dependent manner, with the clearance of 6.6 L/kg in healthy subjects of weight 70 kg and thus displayed short effective half-life of about 3 minutes. Even after peptide administration for 28 days, no anti-circulating anti-drug antibodies were found suggesting its safety [47].
Treatment with LP17 diminished LPS-mediated induction of pro-inflammatory mediators such as TNF-α and IL-1β suggesting that LP17 can significantly block cellular TREM-1 signaling [43]. In vivo administration of LP17 also showed significant improvement in the treatment of sepsis, rheumatoid arthritis, inflammatory bowel disease, and cancer [46,53]. The conservative addition of one or more amino acids in the amino acid sequence of LP17 can also be referred to as putative peptide blockers of TREM-1 [42]. LL-37 has also been found to be a modulator of TREM-1 expression and inhibits microbial inflammation but only tested under in vitro conditions [54].
3.2. TREM-1 ligands
After the discovery of TREM-1 in 2000, no endogenous ligand of TREM-1 or sTREM-1 had been identified for several years. However, in last decade several potential ligands of TREM-1 have been proposed, as discussed below.
3.2.1. CD177
CD177 antigen is a glycosylphosphatidylinositol-linked N-glycosylated cell surface glycoprotein that was first identified by Lalezari et al. in 1971 in a case of neonatal alloimmune neutropenia and was also found to play a critical role in neutrophil activation [55]. In 2018, Colonna et al. provided evidence of CD177 as an endogenous sTREM-1 binding ligand [56]. To confirm the binding, HEK293 cells (human embryonic kidney cells) transfected with CD177 were incubated with sTREM-1 for 45 minutes followed by immediate analysis. The results revealed the binding of sTREM-1 to HEK293 cells transfected with CD177. Anti-monoclonal antibody (R33) of CD177 blocked the binding of TREM-1 ligands to neutrophils and in vitro inhibited the transendothelial migration of human neutrophils [56].
3.2.2. Extracellular actin
Actin is a family of multi-functional globular proteins present in all eukaryotic cells which has been found to participate in various cellular processes, including muscle contraction, cell division and cytokinesis, cell motility, cell signaling etc. It was observed that actin could activate the inflammatory response by interacting through TREM-1 [57]. There was a controversy on the presence of actin on the cell surface since actin is a cellular cytoskeleton protein. Besides its presence in the cytoplasm, its distribution was also detected on the surface of platelets in the resting state [58]. Therefore, platelets provide surface actin for TREM-1 recognition to activate signaling. In 2017, Fu et al. found that recombinant actin can directly interact with the recombinant TREM-1 extracellular domain and enhance inflammatory response when injected in wild type mice but not in TREM-1−/− mice. This amplification of inflammatory response could be inhibited by peptide LP17. It was confirmed that the extracellular actin is co-localized with TREM-1 in the lung tissues of septic mice [57].
3.2.3. PGLYRP1, HMGB1 and HSP70
Peptidoglycan receptor protein 1 (PGLYRP1) is also a TREM-1 ligand identified by Read and colleagues [59]. These receptors are primarily found in bacterial wall and thus play a pivotal role in maintaining the bacterial morphology. PGLYRP1 also binds to sTREM-1. The crosslinking of PGLYRP1 by peptidoglycans is required to stimulate TREM-1 which signifies its importance as a TREM-1 ligand in various infectious diseases. Similar to TREM-1, PGLYRP1 mRNA expression is upregulated in tissues following the inflammatory diseases. Findings support the beneficial effect of its activation in various inflammatory diseases. Due to the antibacterial activity of PGLTYRP1, it maintains beneficial healthy intestinal microbiome and protecting mice from experimentally induced ulcerative colitis. PGLYRP1−/− mice were found most sensitive to ulcerative colitis and variants in its gene are associated to ulcerative colitis in human [60,61]. It is still unclear if this is happening due to the bactericidal property of PGLYRP1 or involves TREM-1 signaling.
Stennicke and colleagues [62] identified TREM-1 antibodies that can specifically block the stimulation of TREM-1 and its functions by binding to human TREM-1 as well as other species of primates. These antibodies attenuate TREM-1 signaling by preventing the formation of functional complex between PGLYRP-1 and TREM-1 or by preventing the individual TREM-1 molecules to form dimers or multimers. Thus, these antibodies can block PGLYRP-1-induced activation of TREM-1 and can ultimately regulate pro-inflammatory cytokine release from myeloid cells. Among the investigated monoclonal antibodies, mAb0170 efficiently can bind to human and cynomolgus TREM-1 with greater affinity as compared to commercially available TREM-1 antibodies and made it advantageous over other commercially available antibodies. The specific binding was further confirmed through humanization of human and cynomolgus TREM-1 which hindered the binding affinity with these antibodies. Further studies indicated that mAb0170 blocked the cytokine release from synovial tissue cells from rheumatoid arthritis patients stimulated by PGLYRP-1 which was further confirmed in mouse arthritis model by inducing single paw arthritis in female C57BL/6 mice. A significant reduction in inflammation was observed in the paw with monoclonal antibodies over control mice, suggesting that these antibodies show true TREM-1 blocking features and find utility in the treatment of inflammatory conditions [62].
Further, Griffin and colleagues [63] found the viscosity issue with the antibody discovered by Stennicke and colleagues [62]. They resolved the viscosity issue by applying specific negatively charged amino acid mutations of uncharged amino acids that are mainly involved in the TREM-1 antibodies self-interactions to reduce the viscosity and retain the original binding affinity towards TREM-1.
High mobility group box 1 (HMGB1) is a nuclear protein that interacts with nucleosomes, histones, and transcription factors to regulate transcriptional activity. During inflammatory conditions, HMGB1 is automatically released by damaged tissue and activated myeloid cells, and functions as a damage associated molecular protein (DAMP) molecule to induce inflammatory response. Wu et al. [64] first observed the direct interactions between HMGB1 and TREM-1 by employing cross-linking and immunoprecipitation assays [64]. HMGB1 needs co-activating molecules to trigger TREM-1 stimulation and thus reflected as an inducer of inflammation via activation of various pattern recognition receptors (PRR) [65]. The expression of HMGB1 is observed in inflammatory conditions like colitis, ischemia, fibrosis, arthritis, cancer, and pancreatitis.
Among potential DAMPs, heat shock protein 70 (HSP70) has also been considered as a TREM-1 ligand. HSP70 is present in the necrotic cell lysates of myeloid cells and are responsible for the induction of proinflammatory cytokine production. The HSP70 expression was reduced by blocking the TREM-1, thus confirming the role of HSP70 as a potential endogenous ligand for TREM-1 [66,67].
Interestingly, all endogenous ligands of TREM-1 identified to date are associated with inflammatory conditions. Various TREM-1 modulators and ligands are summarized in Table 2.
Table 2:
TREM-1 modulators and ligands in various disease conditions
| Modulators/phenotype/dose | Ligands | Disease condition | Animal Model | References |
|---|---|---|---|---|
| LP17/protective/1.0 mg | HMGB1, HSP70, CD177, PGLYTRP1 | Hemorrhagic shock | Rat | [68] |
| LR12/protective/5 mg/kg | HMGB1, HSP70, CD177, PGLYTRP1 | Atherosclerosis | Mice | [12,13] |
| LR12/protective/5 mg/kg | HMGB1, HSP70, CD177, PGLYTRP1 | Myocardial Infarction | Mice | [15] |
| LP17/protective/0.2 mg, rat 1.0 mg | HMGB1, HSP70, CD177, PGLYTRP1 | Colitis | Mice | [69] |
| LP17/ protective/3.5 mg/kg | HMGB1 | Mesenteric Ischemia-Reperfusion Injury | Rat | [70] |
| LP17/protective/0.2 mg TREM-1 adenovirus/protective |
HMGB1, HSP70, CD177, PGLYTRP1 | Collagen Induced Arthritis | Mice Mice |
[71] [72] |
| TREM-1 knock out/protective | HMGB1, HSP70, CD177, PGLYTRP1 | Renal fibrosis | [68] | |
| LP17/no effect/0.05–0.2 mg LR12/no effect/0.2 mg |
HMGB1 HSP70 |
Renal Ischemia-Reperfusion Injury | Mice Mice |
[18] [18] |
| TREM-1 knock out/protective | HMGB1, HSP70, CD177, PGLYTRP1 | Hepatocellular Carcinoma | [64] | |
| LP17/protective/1.0 mg | HMGB1, HSP70, CD177, PGLYTRP1 | Pancreatitis | Rat | [73] |
| TREM-1 knock out/no effect | HMGB1, HSP70, CD177, PGLYTRP1 | Psoriasis | [12] | |
| M3/protective/10 mg/kg | Extracellular CIRP | Acute lung injury | Mice | [74] |
CIRP, cold-inducible RNA-binding protein; HMGB1, high mobility group box 1; HSP70, heat shock protein 70; M3, a 7-amino acid peptide derived from human extracellular CIRP
3.2.4. eCIRP
Extracellular cold-inducible RNA-binding protein (eCIRP) is a recently discovered DAMP, mainly expressed on macrophages, lymphocytes, and neutrophils to amplify the production of cytokines [75]. Administration of recombinant murine CIRP developed acute lung injury (ALI) in healthy mice through the activation of macrophages, neutrophils, and endothelial cells, while CIRP−/− mice were protected from ALI [76,77]. The treatment of infected animals with eCIRP antibody attenuated ALI and prolonged the survival [75,78]. Consistent with these findings, eCIRP is considered as a novel and prominent contributing factor in the pathogenesis of sepsis. Denning et al. identified a strong binding affinity between eCIRP and TREM-1 which was confirmed via fluorescence resonance energy transfer assay in macrophages. These investigators also confirmed that LP17 peptide can block these interactions and eCIRP-induced inflammation is attenuated. They also produced a new 7-amino acid peptide M3 which also significantly inhibited eCIRP-induced systemic inflammation and tissue injury by blocking the interactions between CIRP and TREM-1 [74].
3.2.5. Copper labeled probe
Michelle and colleagues [79] reported for the first-time labeled probe (agonist antibody) by targeting TREM-1 for the detection of inflammatory disease conditions. The labeled probe was developed by using copper-64 (64Cu-DOTA TREM-1 probe) that can selectively bind to TREM-1 with high affinity in vitro and in vivo and can be detected by Positron Emission Tomography imaging. Particularly, it was revealed that the labeled probe can link directly to the extracellular domain of TREM-1 through lysine amino acid [79].
3.2.6. Non-endogenous ligands
Pathogen associated molecular patterns (PAMPs) are also considered as possible TREM-1 ligands. One well known PAMP is lipopolysaccharide (LPS) which is involved in TREM-1 activation and thus can be considered as its non-endogenous ligand [6,39], but it is still a quest whether LPS activates TREM-1 directly or indirectly through other receptors [80]. Other possible ligands of TREM-1 are microbe associated molecular patterns (MAMP), including zymosan, HIV gp41, Marburg virus glycoprotein and Schistosoma mansoni egg antigens. Administration of schistosoma egg homogenate antigen to a mouse macrophage cell line downregulated the expression of TREM-1 and lowered the macrophage-mediated inflammatory response of infected hosts [81]. HIV gp41-induced modulation of cytokines triggered the expression of MMP-1 and TREM-1 which also led to the conversion of surface TREM-1 into soluble TREM-1 [82]. The modulation was so specific that a single mutation within the conserved immunosuppressive domain of gp41 abrogated the cytokine response [83]. Stimulation of Kupffer cells with HIV or HCV induces the upregulation of TREM1 through NF-кB signaling and enhanced activation of the ERK1/2 signaling cascade. Moreover, silencing of TREM1 dampened the inflammatory immune responses elicited by HIV or HCV stimulation [84]. Marburg virus (MARV) and Ebola virus (EBOV), members of the viral family Filoviridae which causes fatal hemorrhagic fevers, activate TREM-1 on human neutrophils, leading to various post activation sequelae including DAP12 phosphorylation, TREM-1 shedding, mobilization of intracellular calcium and secretion of proinflammatory cytokines [85]. Zymosan is a potent ligand found on the surface of fungi like yeast. Transgene expression in the liver of soluble form of extracellular domain of TREM-1 to antagonize DAP12 signaling remarkably inhibited zymosan A-induced granuloma formation [86].
3.2.7. Targeting TREM-1 other than ligand binding domain
Besides targeting ligand-binding domain of TREM-1, Sigalov and colleagues [87] in 2006 made successive measures to inhibit TREM-1 function directing transmembrane domain. Therefore, ligand-independent TREM-1 inhibition using signaling chain homo-oligomerization (SCHOOL) is a promising strategy to develop clinical therapeutics. In this strategy, ligand binding initiates receptor clustering of signaling oligomers that leads to the recruitment of tyrosine kinase to ITAM-bearing adaptors [87, 88]. Therefore, it is feasible to develop peptides and other compounds that can interfere with the interactions of TREM-1 and TREM-1 homodimers or TREM-1 and DAP12 complexes.
Various research groups are actively involved to design peptides or other molecules by employing SCHOOL approach. GF9, a small peptide sequence developed by Sigalov and colleagues in 2014 [53] was selected from the transmembrane core region of human (GFLSKSLVF) [53] and mouse (GLLSKSLVF) TREM-1 [88]. The significant reports on the use of this small peptide in various inflammatory conditions have been summarized previously [89]. Recent studies on mouse collagen-induced arthritis (CIA) model also provide the evidence of therapeutic use of GF9 in rheumatoid arthritis by blocking the TREM-1 pathway. Mice treated with GF9 in the dose of 25 mg/kg i.p. daily for 21 days showed significant delay in arthritis with reduced severity [90]. Similarly, in 2017, Shen and colleagues [91] reported anti-pancreatic cancer ability of GF9 via suppressing the specific inflammatory response through silencing the TREM-1-mediated signaling pathway when tested in three pancreatic cancer xenograft mouse model. Blocking TREM-1 pathway by injecting the peptide significantly halted the production of IL-1α, IL-6, and M-CSF. This anti-cancer effect was persisting even after stopping the peptide treatment. The peptide was also found well-tolerated in both free and in the formulated form with lipopeptide given to enhance the shelf-life [91]. Besides these studies, therapeutic efficacy of this peptide has also been tested in several in vitro and in vivo models of oxygen-induced retinopathy [92], alcohol-induced liver cirrhosis [93], and liver cancer [94], and confirmed its high efficacy. Furthermore, to enhance its stability, half-life, and target-specific delivery, Tornai and colleagues [93] modified the sequence of peptide chemically to produce two 31-amino acid long chain peptides, GA31 and GE31, which further demonstrated significant TREM-1 inhibitory effect in vivo. These studies indicate that the inhibition of TREM-1 pathway using short synthetic peptides that employ a novel ligand-independent mechanism of action (SCHOOL peptides) may represent a steppingstone in the development of future drug candidates for various inflammatory conditions.
In addition to the peptides as modulators of TREM-1, Wang and colleagues [95] evaluated the TREM-1/DAP12 inhibitory effect of organically synthesized drug Pravastatin, which is a clinically used lipid-lowering drug [95] (Figure 1). The investigators found that pravastatin inhibits the complex formation between TREM-1 and DAP12 which further attenuates the release of inflammatory cytokines (TNF-α and IL-1) and can reduce the area of atherosclerotic plaques, improve plaque formation by reducing lipid deposits and alleviate plaque inflammatory responses.
4. TREM-2 signaling and its potential therapeutic applications
TREM-2 is expressed on a wide range of cells, including myeloid cells [4,7,8], microglia [95], osteoclasts [96], and tissues including liver, kidney, heart, lungs, and brain [95]. In the recent past, researchers around the globe have focused their efforts to investigate the role of TREM-2 on various myeloid cells. Recent single-cell RNA sequencing (scRNA-seq) studies [97] suggested that TREM-2 is also expressed in a minor subset of tissue-specific macrophages. In addition to microglia, TREM-2 is expressed in macrophages of adrenal gland, adipose tissue and placenta [98]. TREM-2 interacts with a wide range of ligands, majority of which occur as the hallmarks of tissue damage and so far, involved in a variety of functions including cell survival, maturation, proliferation, phagocytic activation, and regulation of inflammation [99,100]. The importance of TREM-2 is generally highlighted after genetic studies that confirmed its association with neurodegenerative disorders such as Nasu-Hakola disease (NHD), also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, and Alzheimer’s disease (AD). Individuals with rare and different sets of mutations in TREM-2 encoding genes such as arginine to histidine at position 47 (R47H) are at higher risk of developing NHD and AD at a much younger age. The mutations that can cause AD and NHD produced mutant TREM-2 protein that differs from the normal TREM-2 protein by only a single amino acid. In order to find out how different mutations affecting the same protein develops two different disorders, Kober et al. employed X-ray crystallography and confirmed that mutations found in NHD are buried deep inside the folded TREM-2 while for AD, mutations lie on the surface of TREM-2 [101]. Further studies revealed that in NHD, the ability of TREM-2 to fold correctly is hindered and results in fewer TREM-2 proteins being present on the surface of immune cells. TREM-2 binds to glycosaminoglycans in NHD, while in AD, TREM-2 is unable to bind to glycosaminoglycans. Therefore, these findings suggest that TREM-2 binding to glycosaminoglycans is important for preventing AD. Recently, it has also been revealed that the superimposition of diabetes mellitus enhanced TREM2-MAPK signaling in cognitive deficits, neuroinflammation, and neuronal cell death. Knockout of TREM-2 intensified while TREM-2 enhancement suppressed the MAPK signaling and pro-inflammatory production under high glucose and hypoxic conditions [102]. These studies indirectly indicate that TREM-2 expression should be enhanced to treat the neurodegenerative disorders.
In addition to neuroprotection, increasing the expression of TREM-2 also helps to ameliorate the inflammatory infections like sepsis. It has been confirmed that the administration of TREM-2 overexpressing macrophages significantly prolonged survival in aged septic mice by lowering the level of IL-23 and IL-17 [103]. Similarly, TREM-2 knockout mice were found more prone to develop hepatocellular carcinoma, and conditioned media from human hepatic stellate cells overexpressed TREM-2 and attenuated the hepatocyte proliferation also confirms the protective role of this receptor in protection against liver damage and inflammation [104].
TREM-2 signaling associates with adaptor proteins such as DAP10 and DAP12 via oppositely charged amino acid residues in their transmembrane domains and mediates activation of Syk and PI3K [105] (Figure 2). The in vivo picture of TREM-2 signaling is highly complex. In AD brain, TREM-2 could bind directly with pathological β-amyloid (Aβ) oligomers, anionic and zwitterionic lipids [106,107], and lipo- and apo-lipoproteins (LPL, APOE) [108] which together make Aβ-plaques, a pathological hallmark of AD.
Figure 2:
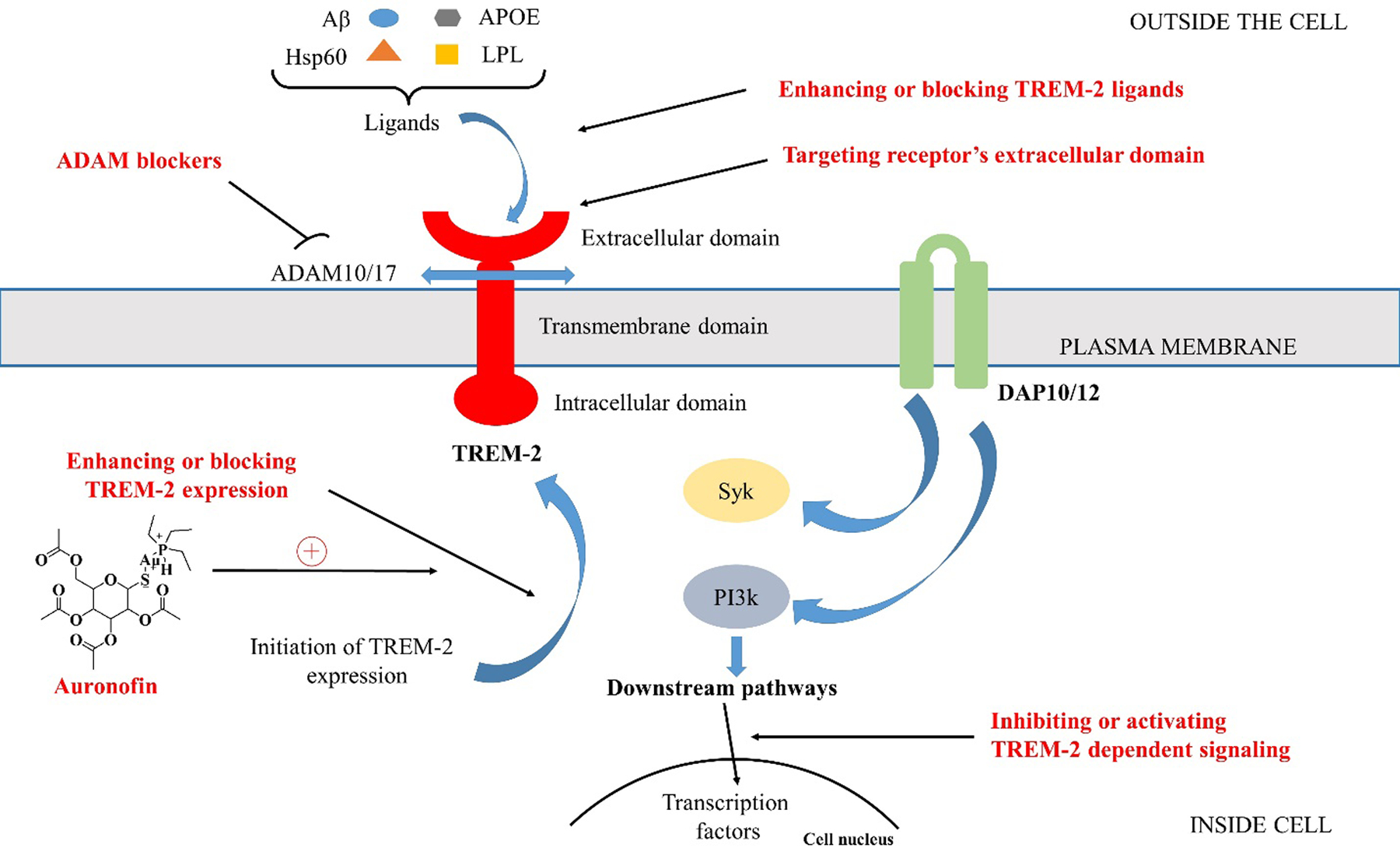
TREM-2 pathway: TREM-2 interacts with one of its ligands by extracellular binding domain, and signal is propagated by DAP10 and DAP12, that activates PI3K and Syk, respectively. Splitting of TREM-2 from its stalk region by ADAMs results in the production of sTREM-2 which further inhibits TREM-2 signaling. Various possible approaches to target TREM-2 are also shown.
In mouse model of AD, microglia, the myeloid cells of the brain, respond to the pathology of AD by amplifying apolipoprotein E (APOE). In addition, TREM-2 signaling results in enhanced production of LPL [109]. It was confirmed that in AD mouse model, TREM-2-deficient myeloid cells in the brain show limited expression of APOE [110]. Jaitin and colleagues in 2019 confirmed that LPL and APOE are amplified by the TREM-2 pathway in adipose tissue macrophages because of high-fat diet-induced obesity in mice [111].
5. Possible therapeutics targeting TREM-2
There are a number of possible approaches to control TREM-2 pathway and to treat various pathological conditions (Figure 2).
5.1. Targeting extracellular domain of the receptor:
The most prominent strategy is to directly target the extracellular domain of the TREM-2 receptor with specific antibody or small molecule peptide that could block or activate downstream signaling. Many biologists are investigating the effect of TREM2 receptor to activate microglia to phagocytose or clear the Aβ deposition. Alector, LLC company used an agonistic monoclonal antibody for TREM-2, i.e., AL002 that was under clinical intervention Phase I trial to assess its safety and tolerability and outcomes are recently published in December 2020 which revealed that AL002 is safe and well tolerated and is a promising candidate for AD therapy [112,113].
5.2. Targeting ADAMs
A Disintegrin and metalloproteinase domain-containing protein (ADAM10/17) is a cellular surface protein. These ADAMs cleave human TREM-2 at His157-Ser158 peptide bond and thus release sTREM-2. Further cleavage of transmembrane region of sTREM-2 by other enzymes (ℽ-secretase) release DAP12 and blocks sTREM-2 signaling. Inhibiting or initiating the shedding of TREM-2 is another approach of intervention [114]. However, due to the vast range of substrates of ADAM10/17, it is unable to inhibit specific protease or substrate activity because this may lead to various unwanted side effects. So, there is a pressing need for specific and fine-tuned modulation to block TREM-2 shedding. In the German center for neurodegenerative diseases, scientists targeted the cleavage site of TREM-2 to interrupt receptor shedding by ADAMs that increased the membrane concentration of receptor and amplify the signaling. They developed antibody 4D9 that could bind to the stalk region of TREM-2 and disrupt the proteolytic shedding [115]. Additionally, this antibody was also found to activate TREM-2 signaling and increase Aβ uptake thus cause microglia survival. Furthermore, in 2020, they also tested the antibody 4D9 in mouse model of AD and found that antibody significantly reduced the level of Aβ plaques but did not improve cognitive ability [116].
5.3. Targeting signaling cascade
To inhibit or activate the specific intracellular signaling cascade, modulation of TREM-2 is another approach. In 2017, Kang and colleagues [117] reported gold-containing agents for the treatment of liver cirrhosis or liver fibrosis via increasing the TREM-2 expression. Using Western blot analysis, they confirmed that especially auranofin (gold containing agent) (Figure 2) can induce transformation of macrophages to M2-type mouse macrophage cell line (RAW264.7) by increasing the expression of TREM-2 which can further effectively inhibit liver fibrosis. Recently in 2020, Mohamed et al. [118] treated Bisphenol-A and ℽ-radiation induced neuroinflammation with methylsulfonylmethane (MSM; organosulfur compound) in rats. They found that this treatment significantly improved the histopathological defects and neuroinflammation in animals by modulating TREM-2/DAP-12/Syk pathway. One major concern with this approach is the non-specificity due to the involvement of DAP10/12, Syk, and PI3K in many other signal transduction pathways. Thus, they are unlikely to become effective target in the context of TREM-2 specific therapy.
These strategies which have been employed in the treatment of neurodegenerative disorders can also apply to other obesity-related comorbidities like atherosclerosis [110].
5.4. Therapeutics targeting TREM-2 for the treatment of Alzheimer’s disease
Shedding of TREM-2 by regulated intramembrane proteolysis results in decreased cell surface TREM-1 and reduced phagocytic activity of microglia leading to increased accumulation of fuzzy amyloid plaques and the disease pathology. Thus, targeting TREM-2 cleavage to prevent TREM-2 shedding and increase its surface expression is an emerging therapeutical strategy in neurological diseases. Various research groups around the globe focused on to target TREM-2 using antibody compositions to treat this disease condition. Antibody clone 14D8 has very high affinity to inhibit TREM-2 cleavage [119]. Antibody clone 14D8 has a binding site within the ectodomain of the TREM-2 and inhibits TREM-2 cleavage. Inhibition of TREM-2 cleavage will stabilize TREM-2 protein on cell surface to decrease risk of the disease. Recently, Brand et al. [120] proposed an antibody targeting stalk (IgSF) region to stabilize TREM-2. Inhibiting TREM-2 cleavage will be useful in the treatment or prevention of neurological disorders including Alzheimer’s disease, frontotemporal lobar degeneration, FTLD-like syndrome, Parkinson’s disease, Nasu-Hakola disease, multiple sclerosis, Huntington disease, immune-mediated neuropathies, and amyotrophic lateral sclerosis. Other than inhibiting TREM-2 cleavage, activating TREM-2 and the downstream signaling via TREM-2/DAP12 with antigen binding proteins as agonists [121] in the myeloid cells is an alternative strategy to treat the disease associated with TREM-2 loss. These antibodies specifically bind to an extracellular domain of human TREM-2 and modulates its activities in microglia, macrophages, dendritic cells, and osteoclasts. Activation of TREM-2 suppresses expression and secretion of inflammatory cytokines in macrophages and microglia [122] resulting in decreased inflammation and progression of neurological diseases. However, the pathological role of heterozygous and homozygous TREM-2 variants in late onset Alzheimer’s disease and Nasu-Hakola disease has also been documented. In such conditions, increased number of microglia and increased expression of TREM-2 on these microglia play an important role in the pathogenesis. Fassler et al. proposed antibodies and antigen-binding fragments binding to extracellular domain of TREM-2 to attenuate TREM-2 expression [123]. Contrary to this, in case of tumor biology, increased recruitment of immune cells including macrophages, neutrophils, and dendritic cells results in adverse outcome. It has been proposed that targeting increased TREM-2 in tumor microenvironment might be useful and anti-TREM-2 antibodies seem to be useful to attenuate tumor growth [124]. Additionally, use of anti-TREM-2 antibody with anti-PD1 antibody has synergistic effect on tumor growth.
The TREM-2 variant, TREM-2R47H increases the AD risk by impairing ligand binding and accumulate β-amyloids. Recently, it was reported that anti-TREM-2 antibody (hT2AB) stimulates microglia activation in TREM-2R47H mice by acting as a surrogate ligand of TREM-2 which suggests that anti-TREM-2 can treat AD by strengthening microglial response [125]. Similarly, Fassler et al. also contributed in the development of monoclonal TREM-2 antibody that induced the microglial activation by binding to its extracellular domain and treated the cognitive function in animal model of AD [126]. In vitro experiments showed that antibody treatment enhances the cellular proliferation, uptake of β-amyloids and apoptotic neurons. During in vivo treatment, the antibody was found to improve cognitive function in amyloidopathy models and to facilitate plaque-associated microglial coverage and activation [126]. Similarly, Denali Therapeutics Inc. [127] and Alector LLC [128] also published patents on monoclonal antibodies that can induce the activation of microglial cells in a Syk- and Akt-dependent manner and could be used as therapeutic treatments for AD and cognitive impairment.
6. TREM-3, TREM-4 and TREM-5 receptors
TREM-3 receptors could be detected at low levels in T cell lines, but are not found in NK, mast cells and B cell lines. Similar to TREM-1 and TREM-2, these receptors also associate with the adaptor protein DAP12 and form receptor signaling complex thus activate myeloid cells [129]. TREM-4 is a transmembrane glycoprotein mainly found in the endothelium of capillaries, cardiac tissue, and testis [130]. It has been documented that the modulation of TREM-4 could be of therapeutic use to treat heart-related complications, inflammatory diseases, and male infertility [130]. TREM-5 is also a transmembrane glycoprotein selectively expressed in bone marrow-derived population of leukocytes and dendritic cells. TREM-5 is upregulated in conditions like inflammation, cell activation, or unusual dendritic cell function. Inhibition of TREM-5 with monoclonal antibodies can cure skin diseases or disorders associated with dendritic cells [130].
7. CONCLUSION
At the end of the literature survey for the period of 2015–2020, it is interesting to underline that major efforts have been made on the development of the modulators of TREM-1 and TREM-2 receptors. Most of the available TREM-1 modulators are peptide-based, rationally designed from TREM-1 ligand-binding domain. LR12 is the characteristic example of TREM-1 inhibitory peptide that is currently under clinical trials in the treatment of septic shock. On the other hand, targeting transmembrane domain (SCHOOL approach) could also be a lead strategy in the coming future, in which blocking peptides (eg. GF9) prevent TREM-1/DAP12 interactions. Modulation of TREM-2 expression by various peptides and antibodies developed so far should be the focus of further research. Instability of peptide-based compounds is the major concern which could limit their clinical use. Various pharmaceutical approaches should be employed to enhance their relationship between dose, bioavailability and therapeutic effect. By understanding the biochemical nature of their molecular skeleton, group modifications could also improve their stability profile, which could be a novel approach to target TREM. Further conceptual understanding associated with these receptors is essential to develop novel therapeutic strategies for the control of various diseases regulated by them.
8. EXPERT OPINION
Following the discovery of relatively novel TREM family, our understanding of innate immunity has significantly increased. Intriguingly, the main biological interventions based on these receptors mainly associate with TREM-1 and TREM-2 function. A number of ongoing clinical trials using TREM-1 and TREM-2 as biomarkers in the biological samples of patients suffering from various disease conditions show the importance of these two receptors (Table 3). Indeed, the role of TREM-1 is not limited to infectious diseases but it has also been found to be involved in several non-infectious diseases. TREM-1 generally amplifies the inflammatory response initiated by DAMPs upon tissue injury. Evidence suggest that the association of TREM-1 in the pathophysiology can be organ- or disease-oriented. But its precise mechanism of action is still a quest for the scientific community which needs to be addressed for the future use of TREM-1 as a potential target.
Table 3:
List of clinical trials and their current status during the years 2015–2020 using TREM-1 and TREM-2 as pathological biomarkers in biological samples of patients with various diseases.
| TREM-1 | |||||||
|---|---|---|---|---|---|---|---|
| Condition | Sample type | Sponsors | Starting date | Status | Number of enrolled subjects | Outcome measures | ClinicalTrials.gov Identifier |
| COVID-19 | Blood | Central Hospital, Nancy, France | Sept-10–2020 | Not yet recruiting | 1000 | Prognostic value of TREM-1 pathway activation on clinical worsening | NCT04544891 |
| Periodontitis | Crevicular fluid | Central Hospital, Nancy, France | April-2016 | Completed | 60 | Average concentration of sTREM-1 in crevicular fluid | NCT02873949 |
| Fever of unknown origin | Blood | Beijing Friendship Hospital,China | Mar-01–2016 | Unknown | 80 | Inflammatory markers TREM-1, IL-1 and IL-6 in patients with fever of unknown origin | NCT02695914 |
| Neonatal Sepsis | Blood | Assiut University, Egypt | Jan-07–2019 | Not yet recruiting | 50 | Mean difference of neutrophil CD64 expression and sTREM-1 before and after treatment | NCT03795285 |
| Prosthetic infections | Blood | Instituto Ortopedico Galeazzi, Italy | Sept-01–2018 | Recruiting | 130 | Differences in serum concentration of IL-6 and TREM-1 before and after treatment | NCT03769337 |
| Acute kidney injury | Urine | Chinese People’s Liberation Army General Hospital | Jan-2016 | Unknown | 500 | Urine sTREM-1 | NCT02920736 |
| TREM-2 | |||||||
| Periodontitis | Crevicular fluid | Central Hospital, Nancy, France | April-2016 | Completed | 60 | Average concentration of TREM-2 in crevicular fluid | NCT02873949 |
So far, LP17, LR12 and M3 are classic examples of TREM-1 inhibitory protein peptides that can directly bind to ligand-binding domain and further inhibit inflammatory responses during infectious or non-infectious disorders. Among these peptide blockers of TREM-1, LR12 is the first clinical-phase agent being evaluated as a prominent novel therapy to treat patients with inflammatory conditions such as septic shock [47]. The peptide exhibited favorable safety and pharmacokinetic profile at three different doses of 1, 5 and 10 mg/kg and acts as a hit lead for further clinical development.
Targeting transmembrane domain by employing SCHOOL approach is also a promising strategy to inhibit TREM-1 signaling and to develop novel therapeutic compounds. GF9, a peptide sequence discovered by Sigalov et al. [53], is an excellent example of TREM-1 inhibitors developed through this approach. Various pre-clinical experiments in animal models have justified its pre-clinical success against various disease conditions such as rheumatoid arthritis, pancreatic cancer, retinopathy and liver cirrhosis [91–94]. These results indicate that SCHOOL approach could open new panoramas in the near future to develop novel therapeutics.
TREM-2 receptor has been established as a major pathology-induced immune signaling hub mainly associated with neurodegenerative disorders. TREM-2 activating ligands have been proven to be highly beneficial in the treatment of neurodegenerative diseases like AD [131]. To date, various approaches have been employed by the scientific community to develop therapeutics to modulate TREM-2 functions. Modulation by targeting the active domain of TREM-2, Alector and LLC Company developed antibody AL002 for TREM-2, the prolonged administration of which can reduce filamentous plaques and impacted behavior, neurite dystrophy and tempered microglial inflammatory response [112]. Recently completed Phase I clinical trials on this antibody also confirmed that it is a safe and well tolerated in first-in-human clinical experiments which suggests it as a promising candidate for the treatment of AD [112,113]. Similarly, scientists at the German center of neurodegenerative diseases developed antibody 4D9, which can bind to the stalk region of TREM-2 (ADAMs targeting) and can increase the Aβ uptake and significantly reduce the level of plaques [115,116]. A direct targeting signaling pathway is also an approach by which TREM-2 expression can be modulated. Kang and colleagues [117] postulated the approach by developing a gold-containing agent ‘Auronofin’ which can effectively inhibit liver fibrosis. But due to the involvement of various transduction pathways in the pathology of disease condition associated with TREM-2, this approach needs a careful attention to make it more target specific to become effective.
According to our knowledge with TREM receptors, this is just the beginning of the drug development, but considerable efforts are still needed in this area of research prior to clinical trials. By keeping in mind, the stability issues with the peptides to modulate TREM expression, there is a pressing need to develop stable peptides or their mimetic compounds which should be stable at physiological pH. Chemical modifications can be performed within the molecular structure of the existing peptides to enhance their stability as well as binding affinity towards TREM.
Molecular dynamic simulation is the computer-based approach towards the designing of novel compounds or to perform modifications to change the existing molecular behavior of the compounds. From the pharmacological point of view, molecular dynamic simulations can predict various possible deep interactions between TREM and their so far unknown ligands which could further expedite the research findings in a short time period without performing the experimental procedures and can thus provide possible lead TREM modulators. There is also a need to introduce artificial intelligence in this field which can perform the experimental tasks automatically with the use of its own datasets and automatically might suggest new possible target specific strategies as well as modulators to treat disease associated with the modulation of TREM expression.
After gaining deeper understanding of the physiopathology of TREM receptors, we can expect a number of novel peptides as well as small molecule TREM modulators in the imminent future with potential clinical application.
Article highlights.
Various TREM ligands, which can modulate TREM expression have been identified within last five years (2015–2020).
Several TREM-1 and TREM-2 modulators are currently under evaluation in pre-clinical and clinical studies in many inflammatory diseases.
Multiple strategies employed in the modulation of TREM-1 and TREM-2 expression have been presented.
TREM-1 and TREM-2 and their soluble receptors in patients’ samples could be used as biomarkers to monitor clinical progression in several diseases.
Funding
The research work of DK Agrawal is supported by research grants R01 HL144125 and R01HL147662 from the National Institutes of Health, USA. The content of this review article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of interests
The authors have no other relevant affiliations or financial or non-financial involvement with any organization or entity with financial or non-financial interest or conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
Reviewer disclosures
A reviewer on this manuscript has disclosed that they are an employee of Denali therapeutics and they are developing antibodies targeting Trem2. All other peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
REFERENCES
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- [1].Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Radaev S, Kattah M, Rostro B, et al. Crystal Structure of the Human Myeloid Cell Activating Receptor TREM-1. Structure. 2003;11:1527–1535. [DOI] [PubMed] [Google Scholar]
- [3].Lanier LL, Corliss BC, Wu J, et al. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–707. [DOI] [PubMed] [Google Scholar]
- [4].Kelker MS, Foss TR, Peti W, et al. Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47Å. J Mol Biol. 2004b;342:1237–1248. [DOI] [PubMed] [Google Scholar]
- [5].Allcock RJN, Barrow AD, Forbes S, et al. The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur J Immunol. 2003;33:567–577. [DOI] [PubMed] [Google Scholar]
- [6].Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–4995. [DOI] [PubMed] [Google Scholar]
- [7].Bouchon A, Facchetti F, Weigand MA, et al. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. [DOI] [PubMed] [Google Scholar]
- [8].Colonna M, Facchetti F. TREM-1 (triggering receptor expressed on myeloid cells): a new player in acute inflammatory responses. J Infect Dis. 2003;187:S397–S401. [DOI] [PubMed] [Google Scholar]
- [9].Sharif O, Knapp S. From expression to signaling: Roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology. 2008;213:701–713. [DOI] [PubMed] [Google Scholar]
- [10].Chen LC, Laskin JD, Gordon MK, et al. Regulation of TREM expression in hepatic macrophages and endothelial cells during acute endotoxemia. Exp Mol Pathol. 2008;84:145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rigo I, McMahon L, Dhawan P, et al. Induction of triggering receptor expressed on myeloid cells (TREM-1) in airway epithelial cells by 1,25(OH)(2) vitamin D(3). Innate Immun. 2012;18:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Joffre J, Hau E, Zeboudj L, et al. Trem-1 is not crucial in psoriasiform imiquimod-induced skin inflammation in mice. Exp Dermatol. 2016a:25:400–402. [DOI] [PubMed] [Google Scholar]
- [13].Joffre J, Potteaux S, Zeboudj L, et al. Genetic and pharmacological inhibition of TREM-1 limits the development of experimental atherosclerosis. J Am Coll Cardiol. 2016b;68:2776–2793. [DOI] [PubMed] [Google Scholar]
- [14].Zysset D, Weber B, Rihs S, et al. TREM-1 links dyslipidemia to inflammation and lipid deposition in atherosclerosis. Nat Commun. 2016;7:13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Boufenzer A, Lemarie J, Simon T, et al. TREM-1 mediates inflammatory injury and cardiac remodeling following myocardial infarction. Circ Res 2015;116:1772–1782. [DOI] [PubMed] [Google Scholar]
- [16].Liu M, Wu W, Zhao Q, et al. High expression levels of trigger receptor expressed on myeloid cells-1 on neutrophils associated with increased severity of acute pancreatitis in mice. Biol Pharm Bull. 2015;38:1450–1457. [DOI] [PubMed] [Google Scholar]
- [17].Lee J, Lee SY, Lee J, et al. Monosodium urate crystal-induced triggering receptor expressed on myeloid cells 1 is associated with acute gouty inflammation. Rheumatology (Oxford). 2016;55:156–161. [DOI] [PubMed] [Google Scholar]
- [18].Tammaro A, Kers J, Emal D, et al. Effect of TREM-1 blockade and single nucleotide variants in experimental renal injury and kidney transplantation. Sci Rep. 2016;6:38275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Joffre J, Hau E, Zeboudj L, et al. Trem-1 is not crucial in psoriasis form imiquimod-induced skin inflammation in mice. Exp Dermatol. 2016;25:400–402. [DOI] [PubMed] [Google Scholar]
- [20].Matesanz-Isabel J, Sintes J, Llinas L, et al. New B-cell CD molecules. Immunol Lett. 2011;134:104–112. [DOI] [PubMed] [Google Scholar]
- [21].Rigo I, McMahon L, Dhawan P, et al. Induction of triggering receptor expressed on myeloid cells (TREM-1) in airway epithelial cells by 1,25(OH)2 vitamin D3. Innate Immun. 2011;18:250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol. 2006;7:1266–1273. [DOI] [PubMed] [Google Scholar]
- [23].Gomez-Pina V, Soares-Schanoski A, Rodríguez-Rojas A, et al. Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J Immunol. 2007;179:4065–4073 [DOI] [PubMed] [Google Scholar]
- [24].Gingras M-C, Lapillonne H, Margolin JF. TREM-1, MDL-1, and DAP12 expression is associated with a mature stage of myeloid development. Mol Immunol. 2002;38:817–824. [DOI] [PubMed] [Google Scholar]
- [25].Gibot S, Cravoisy A, Levy B, et al. Soluble triggering receptor expressed on myeloid cells and the diagnosis of pneumonia. N Engl J Med. 2004;350:451–458. [DOI] [PubMed] [Google Scholar]
- [26].Gibot S, Kolopp-sarda M, Rene MC, et al. Plasma level of a triggering receptor expressed on myeloid cells-1: its diagnostic accuracy in patients with suspected sepsis. Ann Intern Med. 2004;141:9–15. [DOI] [PubMed] [Google Scholar]
- [27].Barraud D, Gibot S. Triggering receptor expressed on myeloid cell 1. Crit Care Clin. 2011;27:265–279. [DOI] [PubMed] [Google Scholar]
- [28].Kluckova K, Kozak J, Szaboova K, et al. TREM-1 and TREM-2 Expression on Blood Monocytes Could Help Predict Survival in High-Grade Glioma Patients. Mediators Inflamm. 2020;1798147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Determann RM, Weisfelt M, deGans J, et al. Soluble triggering receptor expressed on myeloid cells 1: a biomarker for bacterial meningitis. Intensive Care Med. 2006;32:1243–1247. [DOI] [PubMed] [Google Scholar]
- [30].Bishara J, Hadari N, Shalita-Chesner M, et al. Soluble triggering receptor expressed on myeloid cells-1 for distinguishing bacterial from aseptic meningitis in adults. Eur J Clin Microbiol Infect Dis. 2007;26:647–650. [DOI] [PubMed] [Google Scholar]
- [31].Nguyen AH, Lim VM, Fleegel JP, et al. Cutaneous expression of TREM, vitamin D receptor and HMGB1 in vitamin D deficiency. Int J Clin Exp Pathol. 2016; 9(8): 8506–8512. [PMC free article] [PubMed] [Google Scholar]
- [32].Rao VH, Rai V, Stoupa S, et al. Tumor Necrosis Factor-α Regulates Triggering Receptor Expressed on Myeloid Cells-1-dependent Matrix Metalloproteinases in the Carotid Plaques of Symptomatic Patients with Carotid Stenosis. Atherosclerosis. 2016;248:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Subramanian S, Pallati PK, Sharma P, et al. Significant association of TREM-1 with HMGB1, TLRs and RAGE in the pathogenesis of insulin resistance in obese diabetic populations. Am J Transl Res. 2017;9:3224–3244. [PMC free article] [PubMed] [Google Scholar]
- [34].Tessarz AS, Crwenka A. The TREM-1/DAP12 pathway. Immunol Lett. 2008;116:111–116. [DOI] [PubMed] [Google Scholar]
- [35].Lemarie J, Boufenzer A, Popovic B, et al. Pharmacological inhibition of the triggering receptor expressed on myeloid cells-1 limits reperfusion injury in a porcine model of myocardial infarction. ESC Heart Fail. 2015;2:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dower K, Ellis DK, Saraf K, et al. Innate immune responses to TREM-1 activation: Overlap, divergence, and positive and negative cross-talk with bacterial lipopolysaccharide. J Immunol. 2008;180:3520–3534. [DOI] [PubMed] [Google Scholar]
- [37].Arts RJW, Joosten LAB, vanderMeer JWM, et al. TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol. 2013;93:209–215. [DOI] [PubMed] [Google Scholar]
- [38].Haselmayer P, Grosse-Hovest L, vonLandenberg P, et al. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood. 2007;110:1029–1035. [DOI] [PubMed] [Google Scholar]
- [39].Kevin C, Amir B, Lucie J, et al. TREM-1 multimerization is essential for its activation on monocytes and neutrophils. Cell Mol Immunol. 2019;5:460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mariani M, Sinigaglia F, Panina P. Diagnostic and prognostic compounds and method. US20060183125A1.
- [41].Wong-Baeza I, Gonzalez-Roldan N, Ferat-Osorio E, et al. Triggering receptor expressed on myeloid cells (TREM-1) is regulated post-transcriptionally and its ligand is present in the sera of some septic patients. Clin Exp Immunol. 2006;145:448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gibot S, Buonsanti C, Massin F, et al. Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect Immun. 2006;74:2823–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wong-Baeza I, Gonzalez-Roldan N, Ferat-Osorio E, et al. Triggering receptor expressed on myeloid cells (TREM-1) is regulated post-transcriptionally and its ligand is present in the sera of some septic patients. Clin Exp Immunol. 2006;145:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cioni B, Zaalberg A, Beijnum JRV, et al. Androgen receptor signaling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat Commun. 2020;11:4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dantas PHS, Matos AO, Filho ES et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) as a therapeutic target in infectious and noninfectious disease: a critical review. Int Rev Immunol. 2020;39:188–202. [DOI] [PubMed] [Google Scholar]
- [46].Henri Poincare University. Method of diagnosing infectious disease by measuring the level of soluble TREM-1 in a sample. 2007. US20070281319A1.
- [47].Clinicaltrials.govNCT03463044. Safety, Tolerability and Pharmacokinetic Profiles of MOTREM (LR12) in Healthy Male Subjects. Available from: https://clinicaltrials.gov/ct2/show/NCT03463044?term=NCT03463044&draw=2&rank=1 (Retrieved on 10-Oct-2020)
- [48].Clinicaltrials.govNCT03158948. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of 3 Doses of MOTREM in Patients With Septic Shock. Available from: https://clinicaltrials.gov/ct2/show/NCT03158948?term=NCT03158948&draw=2&rank=1. (Retrieved on 10-Oct-2020)
- [49].Derive M, Bouazza Y, Sennoun N, et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J Immunol. 2012;188:5585–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Weber B, Schuster S, Zysset D, et al. TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance. PLoS Pathog. 2014;10:e1003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Derive M, Massin F, Gibot S. Effects of a TREM-like Transcript-1-derived peptide during hypodynamic shock in pigs. Shock. 2013;39:176–82. [DOI] [PubMed] [Google Scholar]
- [52].Derive M, Boufenzer A, Gibot S. Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate. Anesthesiology 2014;120:9350–42. [DOI] [PubMed] [Google Scholar]
- [53].Sigalov AB. A novel ligand-independent peptide inhibitor of TREM-1 suppresses tumor growth in human lung cancer xenografts and prolongs survival of mice with lipopolysaccharide-induced septic shock. Int Immunopharmacol. 2014;21:208–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Amatngalim GD, Nijnik A, Hiemstra AN, et al. Cathelicidin Peptide LL-37 Modulates TREM-1 Expression and Inflammatory Responses to Microbial Compounds. Inflammation. 2011;34:412–425. [DOI] [PubMed] [Google Scholar]
- [55].Lalezari P, Murphy GB, Allen FH Jr. NB1, a new neutrophil specific antigen involved in the pathogenesis of neonatal neutropenia. J Clin Invest. 1971;50:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Colonna M, Klesney-Tait J, Panina P. Screening, therapy and diagnosis. US20100306863A1. 2010.; ** Study identified CD177 as a ligand of TREM-1
- [57].Fu L, Han L, Xie C, et al. Identification of extracellular actin as a ligand for triggering receptor expressed on myeloid cells 1, signaling. Front Immunol. 2017;8:917. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Study identified extracellular actin as TREM-1 ligand
- [58].Mohamadzadeh M, Coberley SS, Olinger GG, et al. Activation of triggering receptor expressed on myeloid cells-1 on human neutrophils by Marburg and Ebola viruses. J Virol. 2006;80:7235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Read CB, Kuijper JL, Hjorth SA. Cutting edge: Identification of Neutrophil PGLYRP1as a Ligand of TREM-1. J Immunol. 2015;194:1417–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Saha S, Jing X, Park S, et al. Peptidoglycan recognition proteins protect mice from experimental colitis by promoting normal gut flora and preventing induction of interferon-gamma. Cell Host Microbe. 2010;8:147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zulfiqar F, Hozo I, Rangarajan S, et al. Genetic association of peptidoglycan recognition protein variants with inflammatory bowel disease. PloS One. 2013;8:e67393. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Developed antibodies that can attenuate TREM-1 signaling by preventing the complex formation between PGLYRP-1 and TREM-1 or by inhibiting the TREM-1 to form dimer or multimer.
- [62].Stennicke VW, Read CB, Grell SN, et al. Antibodies that bind and block triggering receptor expressed on myeloid cells-1 (TREM-1). US20150274825A1.
- [63].Griffin et al. Novo Nordisk: Asignee. Site directed mutagenesis of TREM-1 antibodies for decreasing viscosity. EP2975056A1.
- [64].Wu B, Brooks JD. Gene expression changes induced by unilateral ureteral obstruction in mice. The Journal of Urology. 2012;188:1033–1041. [DOI] [PubMed] [Google Scholar]
- [65].Lo TH, Tseng KY, Tsao WS, et al. TREM-1 regulates macrophage polarization in ureteral obstruction. Kidney Int. 2014;86:1174–1186. [DOI] [PubMed] [Google Scholar]
- [66].Mezayen RE, Gazzar ME, Seeds MC, et al. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. 2007;111:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wu J, Salcedo JL, Mivechi NF et al. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72:3977–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gibot S, Massin F, Alauzet C, et al. Effects of the TREM 1 pathway modulation during hemorrhagic shock in rats, Targeting HMGB1 in inflammation. Shock. 2009;32:633–637. [DOI] [PubMed] [Google Scholar]
- [69].Saurer L, Zysset D, Rihs S, et al. TREM-1 inhibition attenuates inflammation and tumor within the colon. Sci Rep. 2017;7:14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gibot S, Massin F, Alauzet C, et al. Effects of the TREM-1 pathway modulation during mesenteric ischemia-reperfusion in rats. Crit Care Med. 2008;36:504–510. [DOI] [PubMed] [Google Scholar]
- [71].Murakami Y, Akahoshi T, Aoki N, et al. Intervention of an inflammation amplifier, triggering receptor expressed on myeloid cells 1, for treatment of autoimmune arthritis. Arthritis Rheum. 2009;60:1615–1623. [DOI] [PubMed] [Google Scholar]
- [72].Denninger KCM, Litman T, Marstrand T, et al. Kinetics of gene expression and bone remodeling in the clinical phase of collagen-induced arthritis. Arthritis Res Ther. 2015; 17:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Dang S, Shen Y, Yin K, et al. TREM-1 promotes pancreatitis-associated intestinal barrier dysfunction. Gastroenterol Res Pract. 2012;2012:720865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Denning NL, Aziz M, Murao A, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight. 2020;5:e134172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Qiang X, Yang WL, Wu R, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19:1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Yang WL, Sharma A, Wang Z, et al. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep. 2016;6:26571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ode Y, Aziz M, Wang P. CIRP increases ICAM-1+ phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol. 2018;103:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Aziz M, Brenner M, Wang P. Extracellular CIRP (eCIRP) and inflammation. J Leukoc Biol. 2019;106:133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Study identified extracellular CIRP as a novel TREM-1 ligand and developed new peptide (M3) to block the interaction between TREM-1 and eCIRP
- [79].James ML, Andreasson KI. Labeled probe and methods of use. WO2017083682A1; https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017083682
- [80].Arts RJ, Joosten LA, Dinarello CA, et al. TREM-1 interaction with the LPS/TLR4 receptor complex. European Cytokine Netw. 2011;22:11–14. [DOI] [PubMed] [Google Scholar]
- [81].Cheng PC, Lin CN, Chen YJ, et al. Triggering receptor expressed on myeloid cells (TREM)-1 participates in Schistosoma mansoni inflammatory responses. Parasite Immunol. 2011;33:276–286. [DOI] [PubMed] [Google Scholar]
- [82].Denner J, Eschricht M, Lauck M, et al. Modulation of cytokine release and gene expression by the immunosuppressive domain of gp41 of HIV-1. PLoS One. 2013;8:e55199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Morozov VA, Morozov AV, Semaan M et al. Single mutations in the transmembrane envelope protein abrogate the immunosuppressive property of HIV-1. Retrovirology. 2012;9;67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hyun J, McMahon RS, Lang AL, et al. HIV and HCV augments inflammatory responses through increased TREM-1 expression and signaling in Kupffer and Myeloid cells. PLoS Pathog. 2019;15: e1007883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mohamadzadeh M, Coberley SS, Olinger GG, et al. Activation of triggering receptor expressed on myeloid cells-1 on human neutrophils by marburg and ebola viruses. J Virol. 2006;80:7235–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nochi H, Aoki N, Oikawa K, et al. Modulation of hepatic granulomatous responses by transgene expression of DAP12 or TREM-1-Ig molecules. Am J Pathol. 2003;162:1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sigalov AB. Immune cell signaling: A novel mechanistic model reveals new therapeutic targets. Trends Pharmacol Sci. 2006;27:518–24. [DOI] [PubMed] [Google Scholar]
- [88].Sigalov A Signablok Inc, assignee. Inhibition of TREM receptor signaling with peptide variants. US Patent 20120202733A1; https://patents.google.com/patent/US20120202733A1/en; ** Developed SCHOOL approach to design peptides that block TREM-1/DAP12 interaction at the transmembrane domain
- [89].Pelham CJ, Pandya AN, Agrawal DK. Triggering receptor expressed on myeloid cells (TREM) receptor family modulators: a patent review. Expert Opin Ther Pat. 2014;24:1383–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shen ZT, Sigalov AB. Rationally designed ligand-independent peptide inhibitors of TREM-1 ameliorate collagen-induced arthritis. J Cell Mol Med. 2017; 21:2524–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Shen ZT, Sigalov AB. Novel TREM-1 Inhibitors Attenuate Tumor Growth and Prolong Survival in Experimental Pancreatic Cancer. Mol Pharm. 2017;14:4572–4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rojas MA, Shen ZT, Caldwell RB et al. Blockade of TREM-1 prevents vitreoretinal neovascularization in mice with oxygen-induced retinopathy. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2761–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tornai D, Furi I, Shen ZT, et al. Inhibition of Triggering Receptor Expressed on Myeloid Cells 1 Ameliorates Inflammation and Macrophage and Neutrophil Activation in Alcoholic Liver Disease in Mice. Hepatol Commun. 2019;3:99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Wu Q, Zhou W, Yin S, et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology. 2019;70:198–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang HM, Gao JH, Lu JL. Pravastatin improves atherosclerosis in mice with hyperlipidemia by inhibiting TREM-1/DAP12, Eur Rev Med Pharmacol Sci. 2018;22: 4995–5003. [DOI] [PubMed] [Google Scholar]; * Provide evidence to inhibit interactions between TREM-1 and DAP12 by synthetic drug pravastatin
- [96].Schmid CD, Sautkulis LN, Danielson PE, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83:1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Han X, Zhou Z, Fei L, et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303–309. [DOI] [PubMed] [Google Scholar]; ** Study identified the expression of TREM-2 in a small set of tissue-specific macrophages
- [98].Jaitin DA, Adlung L, Thaiss CA, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2- Dependent Manner. Cell 2019;178:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. 2016;17:201–207. [DOI] [PubMed] [Google Scholar]
- [100].Hall SC, Agrawal DK. Increased TREM-2 expression on the subsets of CD11c+ cells in the lungs and lymph nodes during allergic airway inflammation, Sci Rep. 2017;7:11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kober DL, Brett TJ. TREM2-ligand interactions in health and disease. J Mol Biol. 2017;429:1607–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhang J, Liu Y, Zheng Y, et al. TREM-2-p38 MAPK signaling regulates neuroinflammation during chronic cerebral hypoperfusion combined with diabetes mellitus. J Neuroinflammation. 2020;17:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chen QX, Yang Y, Wu XL, et al. Triggering Receptor Expressed On Myeloid Cells-2 Protects Aged Mice Against Sepsis By Mitigating The IL-23/IL-17A Response. Shock, 2020, doi: 10.1097/SHK.0000000000001668 [DOI] [PubMed] [Google Scholar]
- [104].Esparza-Baquer A, Labiano I, Sharif O, et al. TREM-2 defends the liver against hepatocellular carcinoma through multifactorial protective mechanisms. Gut 2020;319227:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Peng Q, Malhotra S, Torchia JA, et al. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3:ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zhong L, Wang Z, Wang D, et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol Neurodegener. 2018;13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yeh FL, Wang Y, Tom I, et al. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–340. [DOI] [PubMed] [Google Scholar]
- [109].Keren-Shaul H, Spinrad A, Weiner A, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169:1276–1290. [DOI] [PubMed] [Google Scholar]
- [110].Parhizkar S, Arzberger T, Brendel M, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019;22:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Jaitin DA, Adlung L, Thaiss CA, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell. 2019;178:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Clinicaltrials.govNCT03635047. A Phase I Study for Safety and Tolerability of AL002. Available from https://clinicaltrials.gov/ct2/show/NCT03635047?term=NCT03635047&draw=2&rank=1 (Retrieved on 10-Oct-2020).
- [113].Wang A, Mustafa M, Yuede CM, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. 2020;21:e20200785. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Study developed monoclonal antibody AL002 for TREM-2
- [114].Voytyuk I, De Strooper B, Chavez-Gutierrez L. Modulation of gand b-Secretases as Early Prevention Against Alzheimer’s Disease. Biol Psychiatry. 2018;83:320–327. [DOI] [PubMed] [Google Scholar]
- [115].Schlepckow K, Kleinberger G, Fukumori A, et al. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. 2017;9:1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Schlepckow K, Monroe KM, Kleinberger G, et al. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020;12:e11227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kang KW, Kim N, Yoon KR. Pharmaceutical composition, comprising gold compound, for preventing or treating liver fibrosis or liver cirrhosis. EP3184113A2.
- [118].Abdel-Rafei M, Thabet NM. Modulatory effect of methylsulfonylmethane against BPA/γ-radiation induced neurodegenerative alterations in rats: Influence of TREM-2/DAP-12/Syk pathway. Life Sci. 2020;260:118410. [DOI] [PubMed] [Google Scholar]
- [119].Haass C, Kleinberger G, Schlepckow K. TREM2 cleavage modulators and uses thereof. WO2018015573A2.
- [120].Brand V, Feuerbach D, Gasparini F, et al. TREM2 stabilizing antibodies. WO20200079580A1.
- [121].Foltz I, Sambashivan S, Chen I, et al. TREM2 antigen binding proteins and uses thereof. WO2018195506A1.
- [122].Holtzman D, Ulrich J, Jiang H. Anti-TREM-2 agonist antibodies. WO2020055975A1.
- [123].Fassler M, George J. TREM2 antibodies and uses thereof. WO2020121195A1.
- [124].Streuli M, Sriram V, Pal A, et al. Anti-TREM-2 antibodies and related methods. WO2019118513A1.
- [125].Ellwanger DC, Wang S, Brioschi S, et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. PNAS. 2021;118:e2017742118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Fassler M, Rappaport MS, Benaim C, et al. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. J Neuroinflammation. 2021;18:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Dennis M, Duncan S, Lisaingo K, et al. Anti-TREM2 antibodies and methods of use thereof. WO2020172457A1.
- [128].Tina S, Eric B, Philip K, et al. Anti-TREM2 antibodies and methods of use thereof. US2020317776A1.
- [129].Chung DH, Seaman WE, Daws MR. Characterization of TREM-3, an activating receptor on mouse macrophages: Definition of a family of single ig domain receptors on mouse chromosome 17. Eur J Immunol. 2002;32:59–66. [DOI] [PubMed] [Google Scholar]
- [130].Colonna M, Panina P. Bioxell SpA, assignee. Novel receptor TREM (triggering receptor expressed on myeloid cells) and uses thereof. US20060263770A1.
- [131].Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of alzheimer’s disease. N Engl J Med. 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]