

Abstract
Background & aims:
Genome-wide association studies (GWAS) in primary biliary cholangitis (PBC) have failed to find X chromosome (chrX) variants associated with the disease. Here, we specifically explore the chrX contribution to PBC, a sexually-dimorphic complex autoimmune disease.
Methods:
We performed a chrX-wide association study (XWAS), including genotype data from five GWAS (from Italy, UK, Canada, China, Japan; 5,244 cases, 11,875 controls).
Results:
Single-marker association analyses found ~100 loci displaying P<5*10−4, with the most significant being a signal within the OTUD5 gene (rs3027490, P=4.80*10−6; OR=1.39 CI=1.028–1.88; Japanese cohort). While the transethnic meta-analysis evidenced only a suggestive signal (rs2239452, mapping within the PIM2 gene; OR=1.17, 95%CI=1.09–1.26; P=9.93*10−8), the population-specific meta-analysis showed a genome-wide significant locus in East Asians pointing to the same region (rs7059064, mapping within the GRIPAP1 gene; P=6.2*10−9, OR=1.33, CI=1.21–1.46). Indeed, rs7059064 tags a unique LD block including seven genes: TIMM17B, PQBP1, PIM2, SLC35A2, OTUD5, KCND1, and GRIPAP1, as well as a super-enhancer (GH0XJ048933 within OTUD5) targeting all these genes. GH0XJ048933 is predicted to target also FOXP3, the main T regulatory cell lineage-specification factor. Consistently, OTUD5 and FOXP3 RNA levels were upregulated in PBC cases (1.75- and 1.64-fold, respectively).
Conclusion:
This work represents the first comprehensive study of the chrX contribution to the genetics of an autoimmune liver disease and revealed a novel PBC-related genome-wide significant locus.
Keywords: X-wide association study, meta-analysis, superenhancer
SHORT SUMMARY
Primary biliary cholangitis (PBC) is an autoimmune liver disease showing a strong female preponderance. Here, by performing the first PBC genetic study focused on chromosome X, we identified a novel locus characterized by the presence of a control element that may regulate several PBC and autoimmune-relevant genes.
Graphical Abstract

INTRODUCTION
Primary biliary cholangitis (PBC) is a complex disease in which an inappropriately activated immune response, characterized by high-titer serum antimitochondrial autoantibodies (AMA) as well as disease-specific antinuclear autoantibodies, leads to a progressive damage of intrahepatic bile ducts that may eventually cause liver failure.1,2 The disease is characterized by a striking female predominance (female:male prevalence ratio up to 8:1), with evidence of a significant contribution of X chromosome (chrX) defects to PBC pathogenesis: in fact, women with PBC show a significantly higher frequency of X monosomy in peripheral leukocytes compared to age-matched healthy women.3,4 However, there is a substantial lack of explanation for female predominance that is also emphasized by the absence of risk loci mapping on chrX.5
PBC is characterized by a strong genetic predisposition, with the major histocompatibility complex (MHC) class-II haplotypes (primarily HLA-DRB1, DQB1, and DPB1) showing the strongest association with the disease.6–10 In addition, genome-wide association studies (GWAS) have identified more than 40 non-MHC loci contributing to the disease risk. Most of these non-MHC loci implicate genes that contribute to cell-mediated immune mechanisms.10–17 These GWAS studies show an overlap in susceptibility loci between European and East-Asian populations, albeit with some degree of locus heterogeneity.10–17
Notwithstanding these efforts, only a modest fraction of PBC heritability (~15%) has been explained.18 Of note, the role of chrX in PBC still remains largely unknown, with no association signal reported at a genome-wide threshold of significance. This could also be explained by the fact that chrX polymorphisms have not been included in GWAS analysis, and, especially in the past, also by the lack of chrX-specific bioinformatics pipelines to be used in the analytic steps.19 These limitations have indeed a more general impact on genetics of complex diseases: chrX constitutes 5% of the nuclear genome and mutations in genes mapping on this chromosome account for ~10% of Mendelian disorders;20 nevertheless, only 114 chrX susceptibility loci (0.8%) at P≤5*10−8 have been described, on a total of ~15,000 signals identified by GWAS studies for more than 300 traits.21
Here, we examine the chrX contribution to the genetic architecture of PBC by applying an analysis pipeline accounting for X-specific quality check (QC), imputation, and association tests.22,23
MATERIALS AND METHODS
Study design and participants
This study included genotype data on chrX principally derived from five previously performed GWAS (Supplementary Table 1).7,11,12,15–17 All participants gave written informed consent for genetic studies. Local Institutional Review Boards approved the respective study protocols. All cases met internationally accepted criteria for PBC24. Most individuals were positive for serum AMA. Nevertheless, AMA positivity was not used as an inclusion criterion, considering previous data suggesting no effect of AMA status on the profile of disease-associated loci.7
QC of genotype data
QC steps were applied with a stepwise procedure separately for each dataset. First, we removed individuals: i) showing cryptic relatedness based on identity-by-state status (PI_Hat>0.10), ii) having >10% missing genotypes, iii) with reported sex not matching the heterozygosity rates observed on chrX; and iv) with significant differences in call rate between cases and controls.25 Next, we excluded nucleotide polymorphisms (SNPs) having: i) >10% missingness throughout the dataset, ii) a minor allele frequency (MAF) <0.005, iii) a departure from the Hardy-Weinberg equilibrium in control females (P<1*10−4), iv) significant differences in MAF between males and females in control individuals (P<0.05/number of SNPs), and v) a location in pseudoautosomal regions. Finally, we also removed SNPs exhibiting differential missingness between males and females (P<1*10−4).
Correction for population stratification
We corrected for possible population stratification using chrX-derived principal components (PCs), which have been demonstrated to provide a more accurate population stratification correction for XWAS in admixed populations.22 This procedure was performed using the principal component analysis method implemented in the EIGENSOFT program (https://genetics.med.harvard.edu/reich/Reich_Lab/Software.html),26,27 after pruning for linkage disequilibrium (LD) and removing large LD blocks.28 For assessment and correction for population stratification we used the first 10 PCs of each dataset,27 and excluded all individuals inferred to be of an ancestry different from that of the specific dataset.
Imputation
Prephasing was performed using the SHAPEIT software, v2.17 (https://mathgen.stats.ox.ac.uk/shapeit),29 using the parameters suggested for chrX. Datasets were imputed using the IMPUTE2 software, v2.3.2 (https://mathgen.stats.ox.ac.uk/impute/impute_v2.html#reference_5),30 based on 1000 Genomes Project whole-genome and whole-exome haplotype data (reference panel: 1000Genome Phase3).31 IMPUTE2 has improved the imputation accuracy on chrX by taking into account the reduced effective population size available for this chromosome, by assuming that it is 25% less than that of the autosomes. As recommended by IMPUTE2 authors,30 the effective population size was set to 20,000, and k value to 1,000. Variants with MAF <0.005 or with informativeness <0.7 were considered of low confidence, and hence not considered in further analyses. Imputed datasets were finally submitted to QC steps, using the PLINK-XWAS v1.1 software (http://keinanlab.cb.bscb.cornell.edu/content/xwas),23 using the above-described criteria.
Single-SNP association analyses
Single-SNP association tests were performed using PLINK-XWAS v1.1.23
We assumed uniform and complete X-inactivation in females and a similar effect size between males and females. Hence, females are considered to have 0, 1, or 2 copies of an allele (like in autosomal analyses), whereas males are considered to have 0 or 2 copies of the same allele (i.e. male hemizygotes are considered equivalent to female homozygotes). This test is implemented in PLINK under the Model-2 option.
We then performed a second test by analyzing separately each sex (cases vs controls), with males coded as either having 0 or 2 copies of an allele as above. The female-only and male-only P values were then combined using the weighted Stouffer’s method,32 which allows combining P values not only accounting for potential effect size and direction between males and females, but also weighting the two test statistics (by using the square-root of the male/female sample size).
All the samples recruited in China were processed and analyzed as described above in a Chinese server to comply with the Regulation of the People’s Republic of China on the Administration of Human Genetic Resources. The summary statistics, with no individual-level data were used for all subsequent analyses (e.g., meta-analysis with other panels).
Quantile-quantile (QQ) plots, genomic inflation factors (λ) calculations, and Manhattan plots were obtained using the R program (https://www.r-project.org/).33 Single-SNP association results were clumped by the PLINK 1.9 software (https://www.cog-genomics.org/plink/1.9/), adopting P<0.001, r2>0.5, and 250kb as parameters.
Meta-analysis
We filtered the SNP lists to include only those polymorphisms for which the association result was available from all cohorts (110,370 SNPs). Meta-analysis was performed both by combining data of all analyzed populations (transethnic meta-analysis) and by separately considering Caucasian and East-Asians populations.
The transethnic meta-analysis was carried out by using the MR-MEGA (Meta-Regression of Multi-Ethnic Genetic Association) software, which models allelic effects of a variant across datasets, weighted by their corresponding standard errors, in a linear regression framework, including the axes of genetic variation as covariates.34
The Caucasian- and East-Asian-specific meta-analyses were performed using the Stouffer’s method, taking into account weights and effect directions, as implemented in the Metainter software.35 This software uses a modified version of the meta-analytic approach based on multivariate generalized least squares estimation suggested by Becker and Wu,36 and is equivalent to the fixed effect model. Meta-analysis results were clumped together using the SECA software,37 extracting subsets of independent SNPs via LD. The procedure was “P-value informed”, using r2>0.1 and 1Mb (in LD with the index SNP) as parameters.
Finally, the genome-wide associated PBC risk locus in Asians was closely examined, by considering SNPs in the region surrounding the top hit (i.e., rs7059064; ±200kb). Pairwise LD among the SNPs was calculated to detect potential independent signals. SNPs showing Pmeta<0.01 and low LD with the rs7059064 SNP (r2<0.5) were selected for conditional analysis.
In all our analyses, we considered loci with P<5*10−8 (genome-wide level) as significant, and loci with P<5*10−5 as suggestive of association. Although, P<1*10−5 is the threshold at which, under the null hypothesis, one false positive result is expected per X-wide scan of ~100K SNPs, we chose the less stringent threshold of P<5*10−5, based on the high level of LD characterizing chrX.38
Measurements of mRNA levels
Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation on a Lympholyte Cell separation medium (Cederlane Laboratories Limited, Hornby, Canada) gradient. Total RNA was isolated using the EuroGold Trifast kit (Euroclone, Wetherby, UK).
Random examers (Promega, Madison, USA) and the Superscript-III Reverse Transcriptase (Thermo Fisher Scientific, Waltham, USA) were used to perform first-strand cDNA synthesis, following the manufacturer’s instructions. Semi-quantitative real-time reverse transcriptase-polymerase chain (RT-PCR) reactions were accomplished by using 1μl of the RT reaction, the SYBR Premix Ex Taq II (TaKaRa, Japan), and a touchdown thermal protocol on a LightCycler 480 (Roche, Basel, Switzerland). HMBS (hydroxymethylbilane synthase) was used as housekeeping gene. Reactions were performed in triplicate, and expression data analyzed using the GeNorm software.39 Primer sequences will be provided upon request.
RESULTS
For evaluating the contribution of chrX to the genetic architecture of PBC, we extended the chrX marker sets from five GWAS cohorts by imputing non-pseudoautosomal regions in a total of ~17,000 individuals. For the analyses, we obtained up to 240,385 high-quality SNPs (Supplementary Table 1).
Single-SNP association analysis within individual cohorts
We performed two different tests: within each cohort, the associations were studied considering males and females together (Test 1), or separately. For the separate analysis males and females were combined using the Stouffer method (Test 2). QQ plots for each test, along with the corresponding λ calculations, showed well-calibrated test statistic distributions (Supplementary Figure 1).
Association analyses did not reveal any genome-wide significant signal (Figure 1A), with the most significant being a signal within the OTUD5 gene (rs3027490, P=4.80*10−6; OR=1.39 CI=1.028–1.88; Japanese cohort, Test 1; Supplementary Table 2). The association signals were consistent between the two used association methods within each population (Supplementary Tables 2, 3). In particular, a total of 115 and 104 SNPs in the five cohorts displayed a nominal P<0.0005 for Test 1 and Test 2 analysis, respectively, with 79 overlapping signals; >40% of signals were within “gene-desert” regions (Supplementary Table 4). Genes pinpointed by these signals showed few overlaps among populations (Figure 1B).
Figure 1. Single-SNP association analysis results.

A) Manhattan plots showing the associations of chrX SNPs with PBC in the analyzed cohorts (CA, Canadians; ITA, Italians; UK, British; JP, Japanese; CH, Chinese) for the Stouffer analysis (Test 2). The blue line represents the P=1*10−5 significance level. SNPs showing lowest P values are indicated by an arrow.
B) Venn diagrams show the number of genes mapping in correspondence/proximity of SNPs at P<0.0005 for each population. Chinese and Japanese show the major number of overlapping signals (genes are listed); the only gene shared by three populations is also highlighted.
Transethnic meta-analysis
Based on single-SNP association results, we performed transethnic meta-analyses including all the five cohorts by using two approaches: 1) results from the Test-1 analysis were directly combined; 2) results from the Test-2 analysis were used in a sex-differentiated meta-analysis. The genomic inflation factors for these meta-analyses were between 0.979 and 1.114 (Supplementary Figure 2), indicating only a minimal residual bias.
Adopting the genome-wide significance threshold, the transethnic meta-analysis revealed the presence of only one interesting signal: the region tagged by the rs2239452 variant, which maps in the PIM2 gene (suggestive Pmeta=9.93*10−8) (Figure 2A; Table 1). This signal was found considering all the five cohorts together, and seems to be sustained by the female component of the cohorts (suggestive Pmeta-females=1.34*10−5) (Figure 2B; Table 1).
Figure 2. Manhattan plots of meta-analyses.
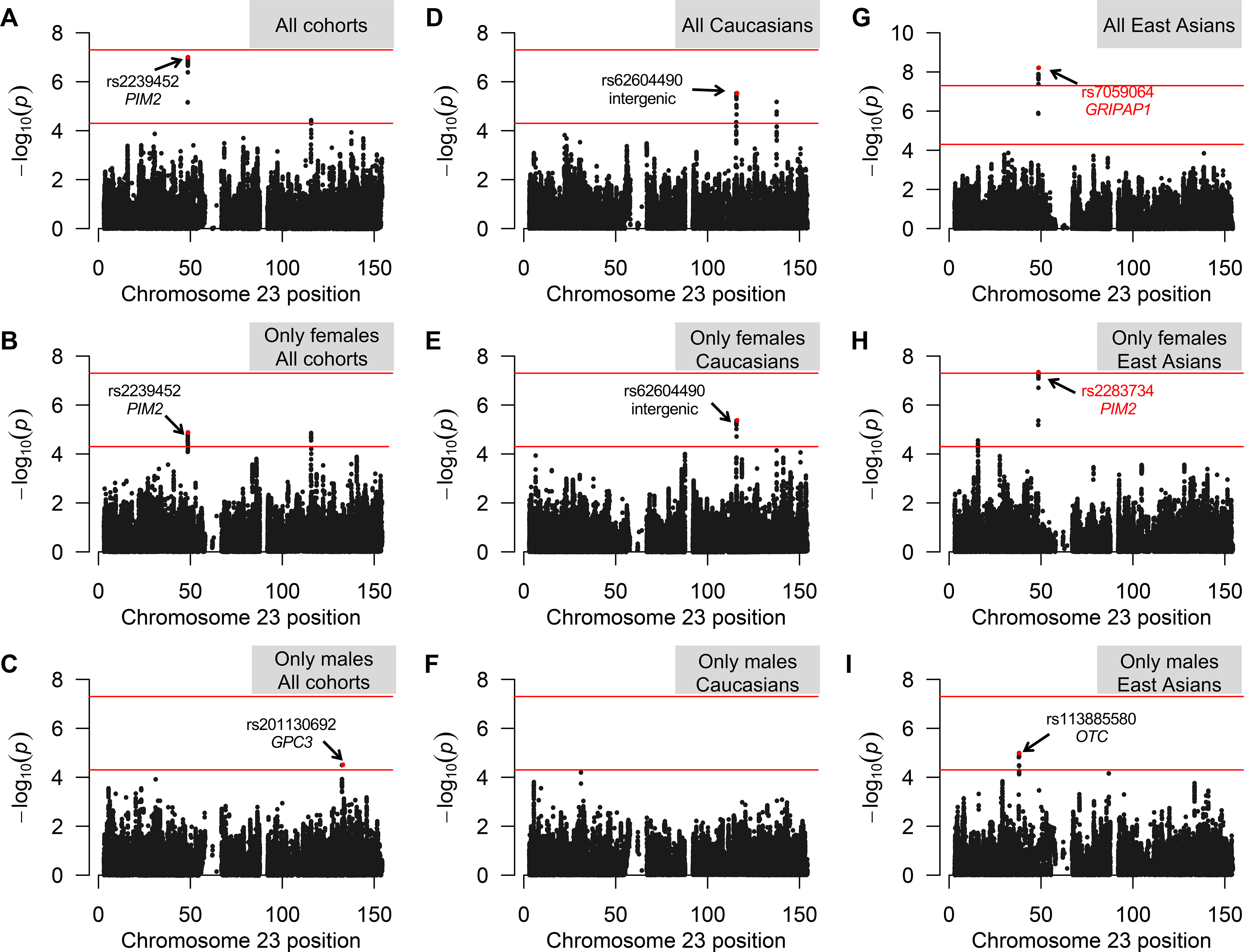
Manhattan plots summarizing the results of transethnic (A-C), population-specific (D-I), and sex-stratified meta-analyses (B,C,E,F,H,I). The horizontal lines represent the suggestive P=5*10−5 and the genome-wide Bonferroni-corrected P=5*10−8 significance levels. SNPs showing lowest P values are indicated by an arrow (if intragenic, the relevant gene is also indicated); those reported in red, survive to the Bonferroni correction for multiple testing.
Table 1:
Meta-analysis results: list of top independent suggestive signals (P<5*10−5).
| Populations | Software | SNP | ChrX Position * | A1/A2 | P_JP | P_CH | P_CAN | P_ITA | P_UK | P_meta | OR [95% CI] | Locus |
| ALL COHORTS | MR-MEGA | rs2239452 | 48775572 | G/C | 7.51e-06 | 5.41e-04 | 0.97 | 0.59 | 0.012 | 9.9e-08 | 1.17 [1.09–1.26] | PIM2 |
| FEMALES-ALL COHORTS | MR-MEGA | rs2239452 | 48775572 | G/C | 4.35e-05 | 4.83e-4 | 0.48 | 0.36 | 5.11e-3 | 1.3e-05 | 1.11 [1.02–1.21] | PIM2 |
| MALES-ALL COHORTS | MR-MEGA | rs201130692 | 132978723 | -/A | 0.015 | 0.88 | 5e-04 | 0.045 | 0.25 | 3.1e-05 | 3.16 [1.8–5.42] | GPC3 |
| P_JP | P_CH | P_meta | OR [95% CI] | Locus | ||||||||
| EAST ASIANS | META INTER | rs7059064 | 48837087 | G/A | 8.1e-06 | 1.75e-04 | 6.2e-09 | 1.33 [1.21–1.46] | GRIPAP1 | |||
| EAST-ASIAN FEMALES | META INTER | rs2283734 | 8773556 | A/G | 4.15e-05 | 2.91e-04 | 4.64e-08 | 1.38 [1.23–1.56] | PIM2 | |||
| EAST-ASIAN MALES | META INTER | rs113885580 | 38236645 | G/A | 0.0075 | 3.84e-04 | 1.06e-05 | 2.36 [1.61–3.46] | OTC | |||
| P_CAN | P_ITA | P_UK | P_meta | OR [95% CI] | Locus | |||||||
| CAUCASIANS | META INTER | rs62604490 | 116104694 | G/A | 0.36 | 0.0058 | 6.90e-05 | 2.98e-06 | 0.75 [0.66–0.85] | Intergenic | ||
| CAUCASIAN FEMALES | META INTER | rs62604490 | 116104694 | G/A | 0.47 | 7.59e-04 | 1.09e-04 | 4.24e-06 | 0.73 [0.63–0.83] | Intergenic |
Only top signals of each suggestively/genome-wide associated region is reported (see also Figure 2).
For all SNPs presented in this table, directions among cohorts were always consistent, except for rs2239452 (all cohort analysis).
A1: tested allele (MAF allele). JP, Japanese; CH, Chinese; CAN, Canadians; ITA, Italians; UK, British; OR, odds ratio; CI, confidence interval.
According to human genome release Feb. 2009, GRCh37/hg19.
Population-specific meta-analysis evidenced a novel PBC locus
Due to the evidence for locus heterogeneity in PBC susceptibility among different ethnicities,10–17 we also performed separate European and East Asian-specific meta-analyses, as well sex-specific meta-analyses, using the same strategy described above; genomic inflation factors for these meta-analyses were well calibrated (Supplementary Figure 3).
The population-specific meta-analysis evidenced one locus with an association signal at genome-wide significance, i.e. the region tagged by the rs7059064 polymorphism, which maps within the GRIPAP1 gene (Pmeta=6.17*10−9; OR=1.33, 95%CI=1.21–1.46) (Figures 2, 3; Table 1; Supplementary Table 5). This signal was found in East Asians, and corresponds to the top region evidenced by the transethnic meta-analysis (the GRIPAP1 and the PIM2 genes are only 53 kb apart).
Figure 3. The GRIPAP1/PIM2 locus.
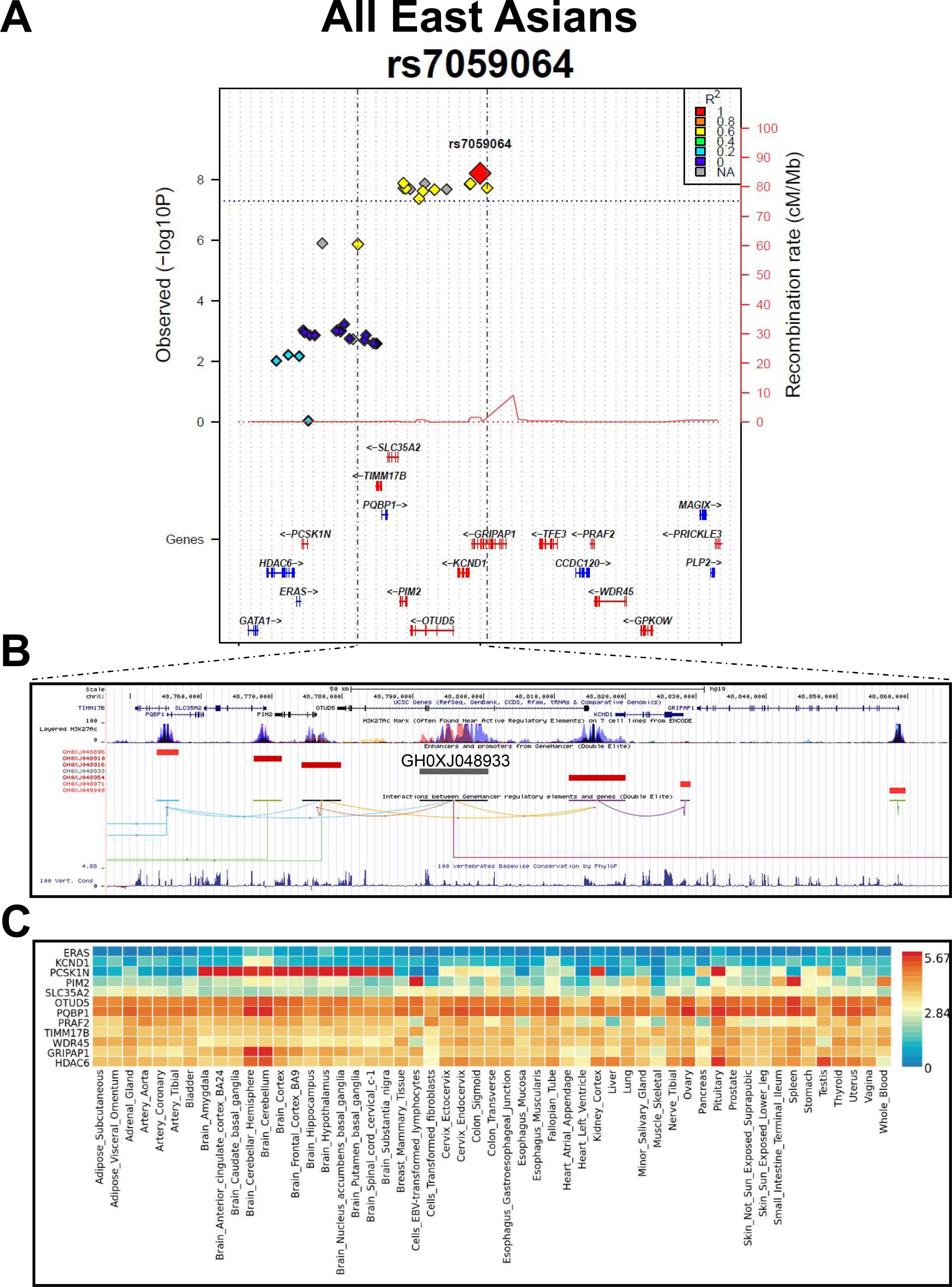
A) Plot of the regional association signals surrounding the rs7059064 top hit in East Asians. The plot was built using the LocusTrack site (https://gump.qimr.edu.au/general/gabrieC/LocusTrack/).40
B) Screenshot from the UCSC Genome browser (http://genome.ucsc.edu/; GRCh37/hg19) highlighting the PBC-associated LD region (coordinates chrX:48,750,000–48,865,000). The panel shows the following tracks: i) the ruler with the scale at the genomic level; ii) chrX nucleotide numbering; iii) the UCSC RefSeq track; iv) ENCODE data (https://www.encodeproject.org/) for H3K4Me1, H3K4Me3, H3K27Ac histone marks, derived from seven cell lines; v) enhancers (grey bars) and promoters (red bars) from GeneHancer41 with the GH0XJ048933 enhancer targets; vi) interactions (curved lines) connecting GeneHancer regulatory elements/genes; vii) basewise conservation track.
C) Expression panel across tissues of the genes depicted in panel A (GTEx data; https://gtexportal.org/home/).
In Europeans, there were only suggestive associations, one in an intergenic region (rs62604490; Pmeta=2.98*10−6) (Table 1; Supplementary Figure 4), and a second mapping within the FGF13 gene (rs73241097; Pmeta=6.77*10−6) (Table 1; Supplementary Figure 5).
The sex-stratified analysis found a novel suggestive signal among East-Asian males (i.e., rs113885580 mapping within the OTC gene, Pmeta-males=1.06*10−5) (Figure 2; Table 1; Supplementary Figure 6), and evidenced that both the signal in the GRIPAP1 region and the intergenic region pinpointed by the rs62604490 SNP are sustained by the female component (Pmeta-females=4.64*10−8 and Pmeta-females=4.24*10−6, respectively). The strongest signal among East-Asian females corresponded to rs2283734, a SNP mapping in the PIM2 gene (Figure 2), the same gene highlighted by the transethnic meta-analysis.
Dissecting the genome-wide significant GRIPAP1/PIM2 locus
The strongest signal evidenced by the meta-analysis was further investigated. Table 2 shows the association summary statistics for the lead SNP (rs7059064) in each of the analyzed cohorts, including the European ones: there are indeed differences between the frequencies of the rs7059064-G minor allele in European cases (from 9.7 to 11.4%) vs those observed in East Asian patients (16 to 16.2%), thus possibly explaining the lack of association observed among Europeans. The effect of the rs7059064-G allele among East Asians was comparable between males and females (OR=1.50, 95%CI=1.24–1.81 for females; OR=1.53, 95%CI=0.99–2.38 for males), thus indicating that the apparent major contribution of females to the association signal simply stems on the higher number of analyzed female patients (Supplementary Table 1).
Table 2:
Association data for the lead rs7059064 polymorphism in all analyzed populations.
| Position* | Minor allele/ Major allele | MAF cases | MAF controls | OR [95% CI] | P value | Population | Pmeta | Transethnic Pmeta | |
|---|---|---|---|---|---|---|---|---|---|
| rs7059064 | 48837087 | G/A | 0.114 | 0.119 | 0.99 [0.77–1.25] | 0.908 | Canadian | 0.350 | 9.93e-08 |
| 0.111 | 0.0935 | 1.21 [0.92–1.59] | 0.174 | Italian | |||||
| 0.0973 | 0.116 | 0.89 [0.78–1.02] | 0.0942 | British | |||||
| 0.162 | 0.119 | 1.38 [1.20–1.59] | 8.14e-06 | Japanese | 6.2e-09 | ||||
| 0.160 | 0.123 | 1.28 [1.13–1.46] | 1.75e-04 | Chinese |
Minor allele frequencies (MAF) and P values of association tests are given for all populations (Model-2 analysis). P values are presented for both the population-specific and transethnic meta-analyses.
According to human genome release Feb. 2009, GRCh37/hg19
Within a ±200kb window centered on the rs7059064 polymorphism, there were 25 SNPs with association signals at Pmeta<0.01. However, after conditional analysis, none of them remained significant, indicating that rs7059064 tagged a single haplotype that could account for the association signal in this region. Indeed, this region is characterized by a unique LD block including seven genes (Figure 3A, B): TIMM17B, PQBP1, PIM2, SLC35A2, OTUD5, KCND1, and GRIPAP1. Among these genes, only PQBP1 was previously associated at the genome-wide level with a phenotype (i.e., type II diabetes mellitus; https://www.ebi.ac.uk/gwas/).
With the exception of KCND1, each of these genes show expression in most tissues, including liver and whole blood (Figure 3C). While this region does not contain significant expression quantitative-trait loci (eQTLs; GTEx portal and eQTL Catalogue at EBI), it is characterized by the presence of a strong epigenetic signature (the activating H3K27Ac histone mark) within OTUD5 intron 2, associated with the presence of a superenhancer (SE, element ID: GH0XJ048933; Figure 3B, 4A). Two SNPs, in perfect LD with the top-hit rs7059064, fall within this SE (Supplementary Table 5). GH0XJ048933 is known to target 13 genes, including six out of seven mapping in the PBC-associated region (KCND1 is the only one not targeted by the enhancer; data from FANTOM5 Human Enhancers project; Figure 4B).44 Among these 13 genes, we found the immunologically relevant transcription factor forkhead box P3 (FOXP3). Hence, to further study the potential impact of the identified haplotype, we evaluated the expression levels of both OTUD5 and FOXP3 by semi-quantitative real-time RT-PCRs comparing PBMCs from 16 female PBC patients and 18 healthy female controls. Only females were examined due to the possibility of confounding sex effects (especially for FOXP3; see Discussion). We found a significant 1.75- and 1.64-fold upregulation in PBC patients of OTUD5 (P=0.0013) and FOXP3 (P=0.046), respectively (Figure 5).
Figure 4. The GH0XJ048933 SE codes for an eRNA and co-regulates the genes of the GRIPAP1/PIM2 locus.
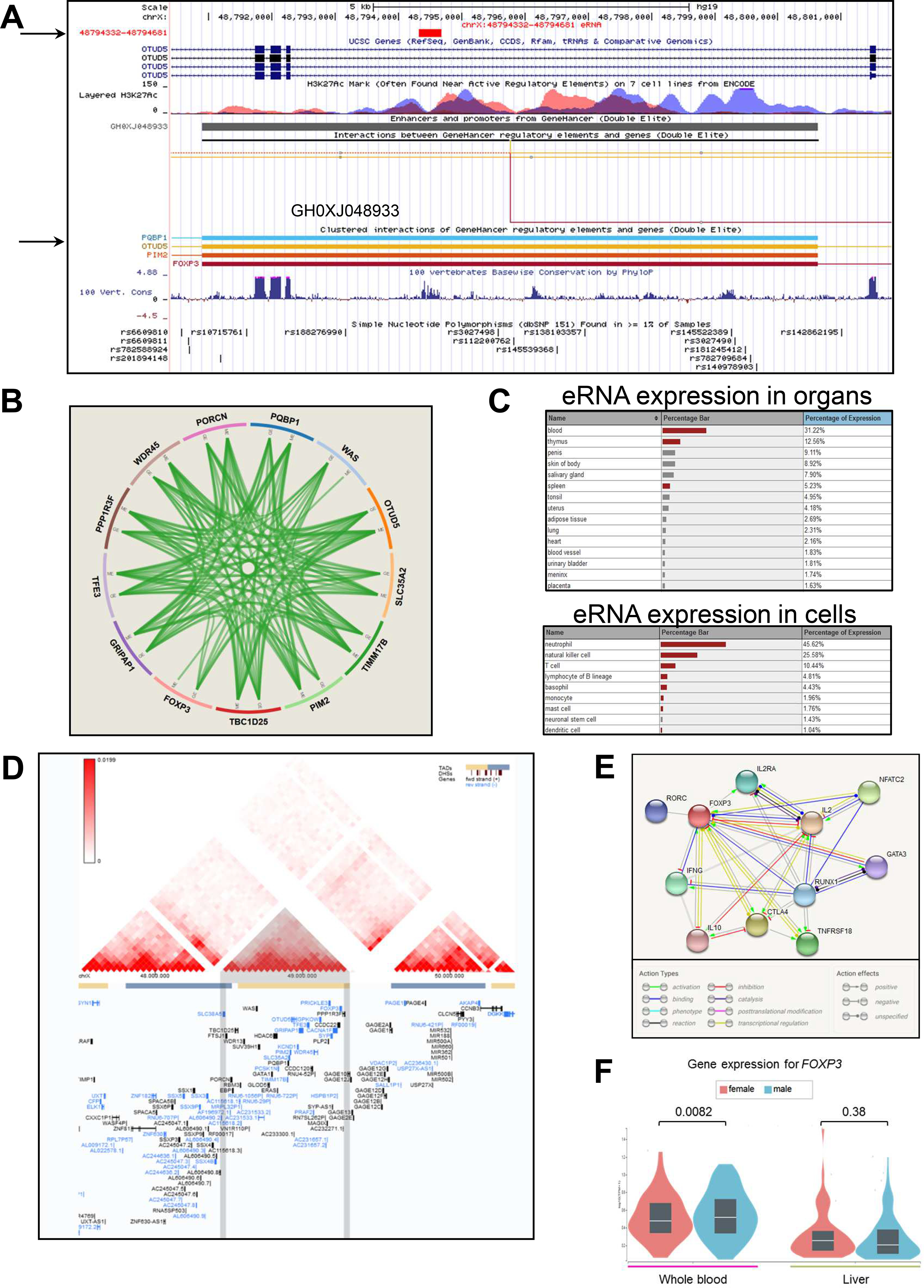
A) Screenshot from the UCSC Genome browser showing the GH0XJ048933 SE region (chrX:48,791,000–48,802,000). Listed tracks are: i) the ruler with the scale at the genomic level; ii) chrX nucleotide numbering; iii) the track for eRNAs from the FANTOM5 Human Enhancers project (http://slidebase.binf.ku.dk/human_enhancers/); iv) the UCSC RefSeq track; v) ENCODE data for H3K4Me1, H3K4Me3, H3K27Ac histone marks, derived from seven cell lines; vi) the enhancers/promoters track from GeneHancer;41 the grey bar indicates the GH0XJ048933 enhancer; vii) interactions connecting GeneHancer regulatory elements and genes (interactions with OTUD5, PIM2, PQBP1, FOXP3 are depicted); viii) basewise conservation track; ix) the dbSNP(151) track for common polymorphisms.
B) Integration of gene expression (GE), protein expression (PE), copy number (CN) and methylation (ME) relative to the 13 genes regulated by the GH0XJ048933 SE. Data come from the TCGA portal (https://tcga-data.nci.nih.gov/docs/publications/tcga/?). Circle plot was built by using the Zodiac tool (http://www.compgenome.org/zodiac/).42 Only significant intergenic interactions are shown (FDR≤0.1). Green lines indicate positive interactions.
C) The tables show expression data (>1%) in organs/cells for the eRNA gene mapping within GH0XJ048933. Red bars indicate a significant over-representation of the transcript (FANTOM5 data).
D) TAD structure of the chrX:47,480,000–50,440,000 region. The central TAD contains all genes of the PBC-associated region tagged by rs7059064. The panel was produced though the 3D-Genome Browser (http://3dgenome.org),43 using Hi-C data produced in HepG2 cells (hepatocytes) and generated by the Dekker Laboratory (resolution: 40kb).
E) FOXP3 interactome. The best 10 interactions are shown (highest confidence=90%). Evidence are based on text-mining, experiments, databases, co-expression data, gene fusions, co-occurrences. The panel was produced using the STRING tool (https://string-db.org/).
F) Violin plots show FOXP3 RNA expression levels in whole blood and liver, obtained through the GTEx portal, stratified on sex (265 males, 142 females).
Figure 5. OTUD5 and FOXP3 are overexpressed in PBC.

Boxplots show expression levels of OTUD5 (A) and FOXP3 (B) measured by semi-quantitative real-time RT-PCR in PBMCs of a PBC case-control cohort. Boxes define the interquartile range; thick lines refer to the median. Results were normalized to expression levels of the HMBS housekeeping gene and are presented as rescaled values. The number of subjects is indicated (N). Significance levels of t-tests: *: P<0.05; **: P<0.005.
For the other top loci (the intergenic rs62604490 polymorphism, the FGF13 locus, and the OTC gene), their main features are illustrated in Supplementary Figures 4–6.
DISCUSSION
GWAS have been a fruitful method to disclose genes/regions involved in the predisposition to complex diseases, however, chrX is notable for the paucity of associated loci.19 For example, the most recent meta-analysis on multiple sclerosis, in another complex disorder with an autoimmune etiology and a marked female preponderance, identified 233 loci associated with the disease at genome-wide level, but just one locus was reported on chrX.45 In our study, we adopted an analysis pipeline specifically designed for chrX, to search for novel potential contributors to PBC heritability, and, possibly, to its female preponderance.
Indeed, the best association signal observed both in the transethnic and in the population-specific meta-analyses points to a unique LD region characterized by the presence of 7 genes (TIMM17B, PQBP1, PIM2, SLC35A2, OTUD5, KCND1, and GRIPAP1) and a SE, GH0XJ048933 (within OTUD5), which presents features with a potential impact on PBC pathogenesis. First, the enhancer is site of active transcription of an enhancer RNA (eRNA), which has been described as significantly expressed in blood, thymus, and spleen, as well as in blood cells such as neutrophils, natural killer, T, and B cells (Figure 4A, C; FANTOM5 data). This type of non-coding RNAs usually contributes to the enhancer activity and to the in-cis regulation of nearby genes.46 Second, the enhancer is enriched in binding sites for immune-related nuclear factor of activated T-cells (NFAT) transcription factors (particularly, NFATC1 and NFATC3), thus stressing its possible involvement in an immune-mediated regulation of target genes. Third, the enhancer targets 13 genes that, by integrating gene expression, protein expression, and methylation data, seem to be strongly co-regulated (Figure 4C), as it could be predicted for an enhancer having its cognate promoters located in the same topologically associating domain (TAD) (Figure 4D). Forth, GH0XJ048933 targets also the FOXP3 gene. FOXP3 is a specific marker of T regulatory cells (Tregs), which are critical for the correct maintenance of immune tolerance (especially self-tolerance) and have been implicated in the pathogenesis of many autoimmune diseases.47–49 Fifth, FOXP3 interacts with important determinants of the immune response (Figure 4E), and the transcript is among the few ones mapping on chrX to show a significant differential expression between males and females (in blood, P=0.0082; Figure 4F).50 Last, but not least, different Foxp3 transgenic mouse models have been developed;51–53 particularly interesting are: i) the Foxp3−/− knockout mice, which developed an intense multiorgan inflammatory response and loss of CD4+ CD25+ Treg cells;51 ii) the Foxp3 conditional-knockout mice (Foxp3floxR26CreERT2), which showed increased levels of IgE and autoantibodies;52 and, more importantly, iii) the so called Scurfy mice (Foxp3sf mutant), i.e. animals that have a mutation in Foxp3 that results in the complete abolition of Foxp3+ Tregs, which are all characterized, at 3–4 weeks of age, by the presence of high-titer serum AMA of all isotypes, by moderate to severe lymphocytic infiltrates surrounding portal areas and by evidence of biliary duct damage.53
Together with FOXP3, at least three additional genes with potential implications in PBC - PIM2, OTUD5, and GRIPAP1 - could be regulated by the GH0XJ048933 SE (Figures 3, 4). The proviral integration site for Moloney murine leukemia virus 2 (PIM2) is a serine/threonine kinase belonging to the PIM family, playing fundamental roles in proliferation/differentiation processes, and with known implications in cancer.54 A growing number of studies have also implicated PIM2 in regulating the immune response, in particular with the description of a circuit linking the PIM2 protein with FOXP3: PIM2, induced by FOXP3, was demonstrated to be essential for the expansion of Tregs and, contrariwise, PIM2 was also described as being able to inhibit the suppressive function of Tregs by phosphorylating FOXP3.55 Concerning the OTUD5 gene, it codes for a member of the OTU (ovarian tumor) domain-containing cysteine protease superfamily. Also known as DUBA (deubiquitinating enzyme A), the OTUD5 protein was shown to suppress type-I interferon (IFN-I) dependent innate immune response, by cleaving the poly-ubiquitin chain from the IFN-I adaptor protein, thus causing the disassociation of the adaptor from the downstream signaling complex, and ultimately the interruption of the IFN-I signaling cascade.56 As for GRIPAP1 (GRIP1-associated protein 1), this gene codes for a guanine nucleotide exchange factor for the Ras family of small G proteins.57 Indeed, in a study aimed at identifying autoantibodies in PBC directed against GWBs (glycine-tryptophan-containing bodies, i.e. cytoplasmic domains that are involved in mRNA processing), Stinton and colleagues58 were able to demonstrate that GRIPAP1 is one of the most common GWB autoantigen targets, being present in 17% of analyzed patients.
Although we demonstrated that OTUD5 and FOXP3 are differentially expressed in PBC patients, a major limitation of our study is the lack of functional studies, from one hand unraveling the molecular mechanisms linking SE GH0XJ048933 and its molecular targets, from the other explaining how genetic variants in this region could influence these mechanisms.
In conclusion, from the extensive analysis of chrX it emerges a number of genes possibly contributing, each with a modest effect, to PBC. This is not trivial, especially considering that chrX can be regarded as an “immunologic” chromosome (it contains the largest number of immune-related genes compared to other chromosomes).59 Our major finding is however the identification of a genome-wide significantly associated locus, i.e. the one tagged by the rs7059064 polymorphism. This locus is characterized by presence of different genes and of a superenhancer possibly involved in their co-regulation, as well as in the regulation of FOXP3 (which located in the same TAD). Future studies are mandatory for explaining the role of SE GH0XJ048933 and its targets in PBC.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT:
Primary biliary cholangitis (PBC) is an autoimmune liver disease showing a relevant female preponderance; however, genetic studies have failed to find X-chromosome variants associated with the disease.
NEW FINDINGS:
Using a chromosome X-specific meta-analysis (>5000 cases, >11500 controls), we identified a novel genome-wide significant locus, characterized by a super-enhancer targeting all the genes of the region, including FOXP3.
LIMITATIONS:
Further studies will be necessary for replicating the identified signal in independent PBC cohorts, and for unraveling the molecular mechanisms linking the super-enhancer, FOXP3, and PBC.
IMPACT:
Considering the genetic overlap among autoimmune liver diseases, as well as other autoimmune disorders with a female preponderance, our study suggests that focused studies of the role of FOXP3 may be useful.
Acknowledgements:
Marco Carbone, Alessio Gerussi, and Pietro Invernizzi are members of the European Reference Network on Hepatological Diseases (ERN RARE LIVER). This study was partly supported by Italian Ministry of Health grants (PE-2016–02363915 and GR-2018–12367794), NIH grant R01DK091823, the National Natural Science Foundation of China grants (#81830016 and 81620108002 to XM). The authors thank AMAF Monza ONLUS and AIRCS for the unrestricted research funding. This work was also supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science to Yuki Hitomi (#15K19314, #17K15924), Minae Kawashima (#15K06908), Yoshihiro Aiba (#15K19357, #17K09449), and Minoru Nakamura (#23591006, #26293181, and 17H04169), a Grant-in-Aid for Clinical Research from the National Hospital Organization to Minoru Nakamura, a grant from the Research Program of Intractable Disease provided by the Ministry of Health, Labor, and Welfare of Japan to Minoru Nakamura, a grant from the Takeda Foundation to Yuki Hitomi, and grants from the Japan Agency for Medical Research and Development (AMED) (JP19km0405205 and JP19km0405501h0001) to Katsushi Tokunaga and Masao Nagasaki.
Abbreviations:
- AMA
Anti-mitochondrial antibodies
- ChrX
X chromosome
- CI
Confidence interval
- eQTL
Expression quantitative-trait locus
- eRNA
Enhancer RNA
- FGF13
Fibroblast Growth Factor 13
- FOXP3
Transcription factor forkhead box P3
- GRIPAP1
GRIP1-associated protein 1
- GWAS
Genome-wide association studies
- GWB
Glycine-tryptophan-containing body
- HLA
Human leukocyte antigen
- IFN-I
Type-I interferon
- λ
Genomic inflation factor
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- NFAT
Nuclear factor of activated T-cells
- OR
Odds ratio
- OTUD5
OTU Deubiquitinase 5
- PBC
Primary biliary cholangitis
- PBMCs
Peripheral blood mononuclear cells
- PC
Principal component
- PIM2
Proviral integration site for Moloney murine leukemia virus 2
- QC
quality check
Quantile-quantile
- RT
Reverse transcriptase
- SE
Superenhancer
- SNP
Single nucleotide polymorphism
- TAD
topologically associating domain
- Tregs
T regulatory cells
- XWAS
X chromosome-wide association study
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med 2005;353:1261–1273. [DOI] [PubMed] [Google Scholar]
- 2.Invernizzi P, Selmi C, Gershwin ME. Update on primary biliary cirrhosis. Dig Liver Dis 2010;42:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerussi A, Cristoferi L, Carbone M, et al. The immunobiology of female predominance in primary biliary cholangitis. J Autoimmun 2018;95:124–132. [DOI] [PubMed] [Google Scholar]
- 4.Invernizzi P, Miozzo M, Battezzati PM, Bianchi I, Grati FR, Simoni G, Selmi C, Watnik M, Gershwin ME, Podda M. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004;363:533–535. [DOI] [PubMed] [Google Scholar]
- 5.Webb GJ, Hirschfield GM. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J Autoimmun 2016;66:25–39. [DOI] [PubMed] [Google Scholar]
- 6.Invernizzi P, Selmi C, Poli F, et al. Human leukocyte antigen polymorphisms in Italian primary biliary cirrhosis: a multicenter study of 664 patients and 1992 healthy controls. Hepatology 2008;48:1906–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirschfield GM, Liu X, Xu C, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med 2009;360:2544–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Invernizzi P Human leukocyte antigen in primary biliary cirrhosis: an old story now reviving. Hepatology 2011;54:714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Invernizzi P, Ransom M, Raychaudhuri S, et al. Classical HLA-DRB1 and DPB1 alleles account for HLA associations with primary biliary cirrhosis. Genes Immun 2012;13:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joshita S, Umemura T, Tanaka E, et al. Genetics and epigenetics in the pathogenesis of primary biliary cholangitis. Clin J Gastroenterol 2018;11:11–18. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Invernizzi P, Lu Y, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet 2010;42:658–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet 2011;43:329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu JZ, Almarri MA, Gaffney DJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet 2012;44:1137–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juran BD, Hirschfield GM, Invernizzi P, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum Mol Genet 2012;21:5209–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura M, Nishida N, Kawashima M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet 2012;91:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu F, Tang R, Zuo X, et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat Commun 2017;8:14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawashima M, Hitomi Y, Aiba Y, et al. Genome-wide association studies identify PRKCB as a novel genetic susceptibility locus for primary biliary cholangitis in the Japanese population. Hum Mol Genet 2017;26:650–659. [DOI] [PubMed] [Google Scholar]
- 18.Gulamhusein AF, Juran BD, Lazaridis KN. Genome-Wide Association Studies in Primary Biliary Cirrhosis. Semin Liver Dis 2015;35:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wise AL, Gyi L, Manolio TA. eXclusion: toward integrating the X chromosome in genome-wide association analyses. Am J Hum Genet 2013;92:643–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amberger J, Bocchini CA, Scott AF, et al. McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 2009;37:D793–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017;45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang D, Gao F, Slavney A, et al. Accounting for eXentricities: analysis of the X chromosome in GWAS reveals X-linked genes implicated in autoimmune diseases. PLoS One 2014;9:e113684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao F, Chang D, Biddanda A, et al. XWAS: A Software Toolset for Genetic Data Analysis and Association Studies of the X Chromosome. J Hered 2015;106:666–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindor KD, Gershwin ME, Poupon R, et al. Primary biliary cirrhosis. Hepatology 2009;50:291–308. [DOI] [PubMed] [Google Scholar]
- 25.Laurie CC, Doheny KF, Mirel DB, et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol 2010;34:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 28.Novembre J, Johnson T, Bryc K, et al. Genes mirror geography within Europe. Nature 2008;456:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods 2011;9:179–181. [DOI] [PubMed] [Google Scholar]
- 30.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auton A, Brooks LD, Altshuler D, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stouffer SA, Suchman EA, DeVinney LC, et al. Adjustment During Army Life. Princeton University Press; Princeton, NJ, USA. 1949. [Google Scholar]
- 33.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. [Google Scholar]
- 34.Mägi R, Horikoshi M, Sofer T, et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum Mol Genet 2017;26:3639–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaitsiakhovich T, Drichel D, Herold C, et al. METAINTER: meta-analysis of multiple regression models in genome-wide association studies. Bioinformatics 2015;31:151–157. [DOI] [PubMed] [Google Scholar]
- 36.Becker BJ, Wu MJ. The synthesis of regression slopes in meta-analysis. Stat Sci 2007;22:414–429. [Google Scholar]
- 37.Nyholt DR. SECA: SNP effect concordance analysis using genome-wide association summary results. Bioinformatics 2014;30:2086–2088. [DOI] [PubMed] [Google Scholar]
- 38.Schaffner SF. The X chromosome in population genetics. Nat Rev Genet 2004;5:43–51. [DOI] [PubMed] [Google Scholar]
- 39.Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuellar-Partida G, Renteria ME, MacGregor S. LocusTrack: Integrated visualization of GWAS results and genomic annotation. Source Code Biol Med 2015;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fishilevich S, Nudel R, Rappaport N, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Y, Xu Y, Helseth DL Jr, et al. Zodiac: A Comprehensive Depiction of Genetic Interactions in Cancer by Integrating TCGA Data. J Natl Cancer Inst 2015;107(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Song F, Zhang B, et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 2018;19:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lizio M, Harshbarger J, Abugessaisa I, et al. Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res 2017;45:D737–D743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science. 2019;365(6460). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hnisz D, Shrinivas K, Young RA, et al. A Phase Separation Model for Transcriptional Control. Cell 2017;169:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell 2008;133:775–787. [DOI] [PubMed] [Google Scholar]
- 48.Mohr A, Atif M, Balderas R, et al. The role of FOXP3(+) regulatory T cells in human autoimmune and inflammatory diseases. Clin Exp Immunol 2019;197:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Attias M, Al-Aubodah TA, Piccirillo CA. Mechanisms of human Foxp3(+) T(reg) cell development and function in health and disease. Clin Exp Immunol 2019;197:36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tukiainen T, Villani AC, Yen A, et al. Landscape of X chromosome inactivation across human tissues. Nature 2017;550:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin W, Truong N, Grossman WJ, et al. Allergic dysregulation and hyperimmunoglobulinemia E in Foxp3 mutant mice. J Allergy Clin Immunol 2005;116:1106–1115. [DOI] [PubMed] [Google Scholar]
- 52.Tai Y, Sakamoto K, Takano A, Haga K, Harada Y. Dysregulation of humoral immunity in Foxp3 conditional-knockout mice. Biochem Biophys Res Commun 2019;513:787–793. [DOI] [PubMed] [Google Scholar]
- 53.Zhang W, Sharma R, Ju ST, et al. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology 2009;49:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev 2014;34:136–159. [DOI] [PubMed] [Google Scholar]
- 55.Deng G, Nagai Y, Xiao Y, et al. Pim-2 kinase influences regulatory T cell function and stability by mediating Foxp3 protein N-terminal phosphorylation. J Biol Chem 2015;290:20211–20220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kayagaki N, Phung Q, Chan S, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science 2007;318:1628–1632. [DOI] [PubMed] [Google Scholar]
- 57.Ye B, Liao D, Zhang X, et al. GRASP-1: a neuronal RasGEF associated with the AMPA receptor/GRIP complex. Neuron 2000;26:603–617. [DOI] [PubMed] [Google Scholar]
- 58.Stinton LM, Swain M, Myers RP, et al. Autoantibodies to GW bodies and other autoantigens in primary biliary cirrhosis. Clin Exp Immunol 2011;163:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bianchi I, Lleo A, Gershwin ME, et al. The X chromosome and immune associated genes. J Autoimmun 2012;38:J187–J192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.