

INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a leading cause of liver disease worldwide. Animal models are widely used to investigate the mechanisms of fatty liver disease, but they do not faithfully represent NAFLD in humans.1 Thus, there is strong interest in studying NAFLD pathogenesis directly in humans whenever possible. Some researchers have utilized embryonic or induced pluripotent stem cells (iPSCs) derived from individual human subjects to generate complex liver platforms that model a NAFLD-like state in culture.2–4 Our group has taken a different approach, investigating iPSC-derived hepatocytes (iPSC-Heps) from a population of NAFLD patients in the hope of gaining insight into the human disease. In this study we generated iPSCs and iPSC-Heps from a well-defined cohort of NAFLD patients. Our objective was to determine whether as a group, in the absence of any metabolic challenge, they exhibit common disease-specific signatures that are distinct from healthy controls.
METHODS
RESULTS
iPSCs were generated from peripheral blood mononuclear cells collected from 21 NAFLD patients and 16 healthy controls. Clinical and genotyping data for all 37 subjects are reported in Table S1. Three NAFLD patients had isolated hepatic steatosis and 18 had histologic nonalcoholic steatohepatitis (NASH); 14/18 (78%) had advanced fibrosis. Fifteen NAFLD patients (71%) and 3 controls (19%) were homozygous for the PNPLA3 G allele (P < 0.001).
iPSCs from both control and NAFLD subjects differentiated efficiently into iPSC-Heps (Figure S1A, B). NAFLD iPSC-Heps had higher lipid content than control iPSC-Heps, although there was some variability (Figure 1A). Transcriptomic profiling of iPSCs before differentiation could not distinguish subjects by disease status (Figure S1C); unsupervised clustering of iPSC-Heps, however, yielded disease-specific segregation of the cohort into NAFLD and control clusters (Figure 1B). The NAFLD cluster comprised two sub-groups, one clearly distinct from controls and another with intermediate status. The NAFLD cluster in the center of the heatmap was dominated by subjects with a PNPLA3 GG genotype. Subjects in the other clusters displayed mixed expression of NAFLD-related SNPs, with no individual harboring more than two disease-promoting or disease-mitigating SNPs.
Figure 1. Comparison of iPSC-Heps from NAFLD and control subjects.
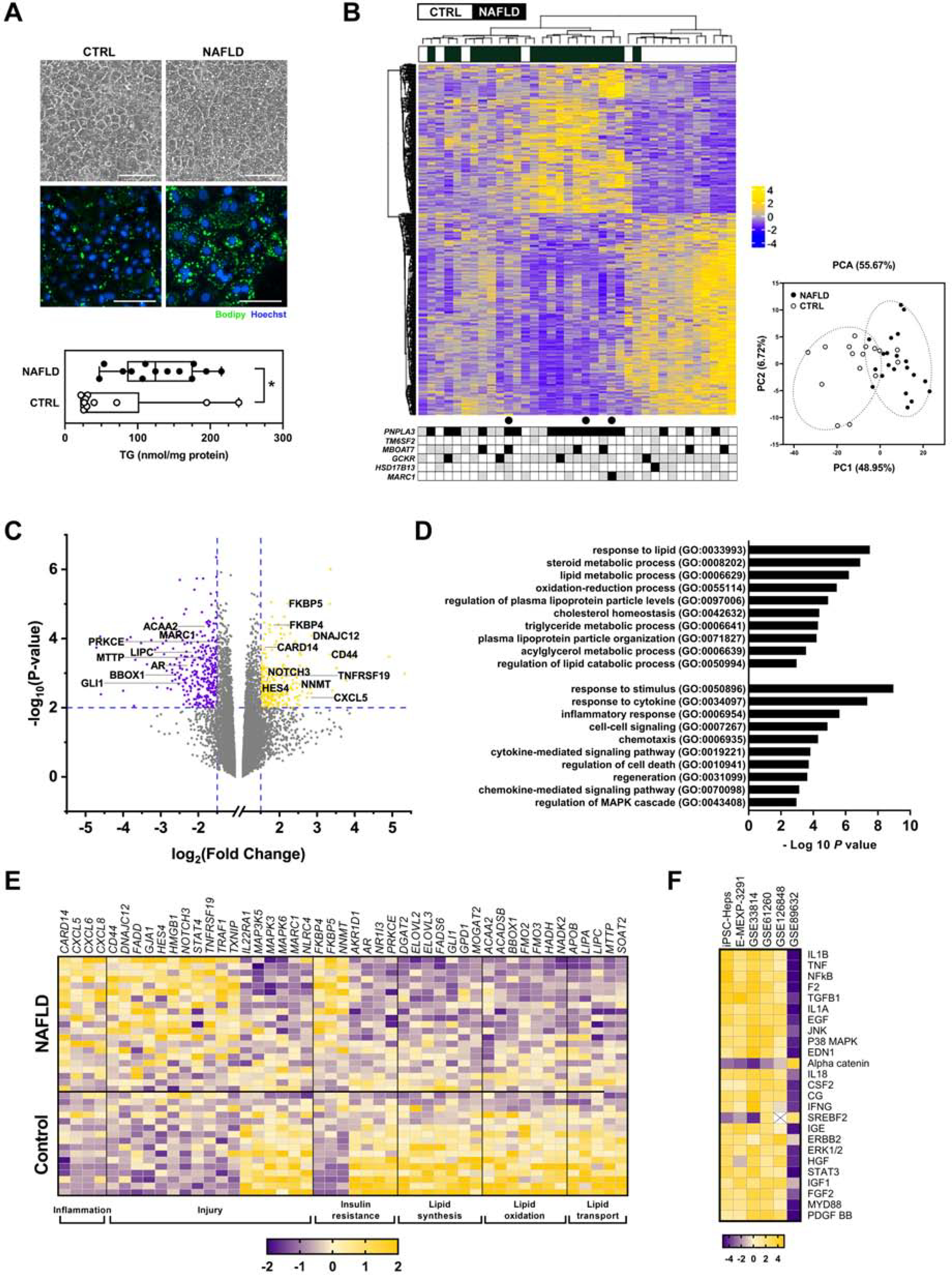
(A) Photomicrographs illustrate control and NAFLD iPSC-Heps on day 22 of differentiation. Panels depict phase contrast morphology and fluorescent images documenting lipid content (Bodipy 493/503). Graph demonstrates the triglyceride content of iPSC-Heps at day 22. * P < 0.05. (B) Heatmap illustrates unsupervised clustering of RNA sequencing data and the accompanying principal component analysis from control and NAFLD iPSC-Heps (367 genes, |FC| > 1.5, FDR < 0.05). Beneath the heatmap is the SNP genotyping profile of each subject for PNPLA3 (rs738409), TM6SF2 (rs58542926), MBOAT7 (rs641738), GCKR (rs780094), HSD17B13 (rs72613567) and MARC1 (rs2642438). Black = homozygous for risk or protective allele; gray = heterozygous; white = no risk or protective allele. ● denotes subjects with histologic hepatic steatosis but not NASH. (C) Volcano plot illustrates 235 upregulated and 331 downregulated genes in NAFLD iPSC-Heps vs. controls (|FC| > 1.5, P < 0.01). Select genes are noted by name. (D) Histogram illustrates select Gene Ontology biological processes that were significantly dysregulated in NAFLD vs. control iPSC-Heps (|FC| > 1.5, FDR < 0.25). (E) Heatmap illustrates differential regulation of select candidate genes in NAFLD vs. control iPSC-Heps, organized by function. (F) Heatmap demonstrates the top 25 differentially expressed upstream regulators in NAFLD vs. control iPSC-Heps, compared to data from 5 publicly available datasets of liver tissue from humans with NAFLD as noted by accession number (via Ingenuity Pathway Analysis; see Methods).
Among 15,730 evaluated genes, 235 were upregulated and 331 were downregulated in NAFLD vs. control iPSC-Heps (|FC| > 1.5, P < 0.01) (Figure 1C). The biological processes highlighted by these genes pertained to lipid metabolism and cell injury (Figure 1D). Injury-related genes upregulated in NAFLD iPSC-Heps included chemokines and molecules associated with cell death and transformation (Figure 1E). Prominent among these was CD44 (Figure S1D), whose expression is increased in hepatocytes in liver injury and carcinogenesis.5 Also upregulated were select genes in the NOTCH signaling pathway (NOTCH3, HES4), which is relevant to NAFLD and fibrosis.6 Interestingly, MAP3K5 (apoptosis signal-regulating kinase 1; ASK1), a purported driver of NAFLD, was downregulated in NAFLD iPSC-Heps.
In the category of metabolism, NAFLD iPSC-Heps exhibited dysregulation of genes associated with insulin resistance and cellular oxygen consumption (e.g., FKBP5, NNMT) as well as several genes impacting hepatic lipid synthesis/elongation, oxidation and transport (ACAA2 and others) (Figure 1C, E, S1D). Notable in NAFLD iPSC-Heps was downregulation of the Hedgehog target GLI1, modeling reports that GLI1 suppression portends hepatic lipid accumulation.7 Also noteworthy in NAFLD iPSC-Heps was downregulation of MARC1 (mitochondrial amidoxime-reducing component 1) because a SNP in MARC1 (rs2642438) was recently linked to protection from NAFLD.8
We identified several upstream regulators dysregulated in NAFLD vs. control iPSC-Heps (Figure 1F), many of which control pro-inflammatory pathways. Importantly, these genes were similarly dysregulated in liver tissue from four of five examined human NAFLD cohorts. Finally, we verified the reliability of the iPSC-Hep platform by documenting that the transcriptomic profile of individual iPSC lines is reproducible across multiple differentiations (Figure S1E, F).
DISCUSSION
Our results show that iPSC-Heps from NAFLD patients display several features indicative of NAFLD in vivo and distinct from healthy control subjects. NAFLD iPSC-Heps spontaneously accumulated more fat than control iPSC-Heps; they also displayed a distinct transcriptomic profile correlating with the NAFLD diagnosis. Regulatory pathways activated in NAFLD iPSC-Heps were highly concordant with those activated in liver tissue from NAFLD patients. Taken together, these findings argue that NAFLD iPSC-Heps represent a useful model of NAFLD in vivo.
Distinctions between NAFLD and control subjects were not detectable in undifferentiated iPSCs. They emerged only upon differentiation to hepatocyte-like cells, which indicates that in vivo, NAFLD likely arises at least in part through aberrations specific to hepatocytes. Genes differentially expressed in NAFLD iPSC-Heps pointed to a potential for these cells to accumulate lipid through impairments in fatty acid oxidation and export, as well as a predisposition to injury and immune activation.
Gene expression profiling suggested that the NAFLD phenotype in our cohort is strongly influenced by the PNPLA3 GG genotype (90% of the central NAFLD cluster were GG). Other NAFLD-related genotypes did not correlate with disease in our dataset, but the cohort is small. Indeed, despite our NAFLD iPSC-Hep collection being the largest of its type studied to date, the size of the cohort constitutes an important limitation. A larger and more diverse population will be needed to assess other genetic and non-genetic factors influencing NAFLD.
Our experiments are distinct from, but complementary to the work of others who have used pluripotent stem cells from individual subjects to investigate the pathogenesis of metabolic liver diseases.2–4 The promise of our cohort approach is its potential to identify novel characteristics that are diagnostic or even predictive of NAFLD in a larger population. The fact that we uncovered an intermediate cluster in the cohort in which 75% of the subjects (10/14) had NAFLD but did not have a clear relationship to PNPLA3 implies the existence of other factors capable of producing a NAFLD phenotype, which warrants further investigation.
In summary, the current experiments establish that iPSC-Heps are a tractable model for identifying hepatocyte-specific attributes that contribute to human NAFLD. The data indicate that iPSC-Heps from NAFLD patients share characteristics that portend a disease phenotype. The reproducibility of the NAFLD signature bodes well for expanded study of iPSC from larger, more diverse NAFLD populations. Further development of this cohort-based iPSC approach should provide novel insight into human NAFLD.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Eric Hoffman and Kevin Siao for subject recruitment and experimentation, and Dr. Stephen Duncan for advice and assistance.
FUNDING
This work was funded by research grants from the California Institute for Regenerative Medicine (IT1-06563), AbbVie, Inc. and NIH: R21 DK118380 (JJM), UG3 DK120004 (HW) and K08 DK098270 (ANM). Additional support was provided by core facilities within the UCSF Liver Center (P30 DK026743).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
PREPRINT SERVER PUBLICATION
A preliminary version of this work was posted on BioRxiv on April 22, 2020. doi: https://doi.org/10.1101/2020.04.20.052001.
REFERENCES
- 1.Teufel A, et al. Gastroenterology 2016;151:513–525. [DOI] [PubMed] [Google Scholar]
- 2.Collin de l’Hortet A, et al. Cell Metab 2019;30:385–401 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ouchi R, et al. Cell Metab 2019;30:374–384 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bin Ramli MN, et al. Gastroenterology 2020;159:1471–1486. [DOI] [PubMed] [Google Scholar]
- 5.Dhar D, et al. Cancer Cell 2018;33:1061–1077 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu C, et al. Sci Transl Med 2018;10:eaat0344.30463916 [Google Scholar]
- 7.Matz-Soja M, et al. Elife 2016;5:e13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emdin CA, et al. PLoS Genet 2020;16:e1008629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.