

Abstract
The cardinal pathophysiological finding of Parkinson’s disease (PD) is a chronic, progressive degeneration of dopamine (DA) neurons in the substantia nigra, which is responsible for the motor and some of the non-motor symptomatology. While the primary causes of nigrostriatal degeneration are hotly debated, considerable evidence supports a central role for impaired mitochondrial function. Postmortem analysis of PD patients reveals impaired respiratory chains and increased mutations of mitochondrial DNA (mtDNA), in addition to increased markers of oxidative stress indicative of mitochondrial impairment. Most animal models of PD, both genetic and toxin-based, target some component of mitochondrial function to reproduce aspects of the human disease. One model that continues to gain attention is the MitoPark mouse, created through a cell type-specific knockout of mitochondrial transcription factor A specifically in midbrain DA neurons. This model effectively recapitulates the slowly developing, adult onset motor decline seen in PD due to mass loss of DA neurons. MitoPark mice therefore represent an effective tool for studying the sequence of events that occurs in the early stages of DA neuron degeneration following mitochondrial impairment, as well as for testing the efficacy of potential disease-modifying therapies in a progressive model of neurodegeneration. A targeted review of key findings from MitoPark mice has not been published since the early years following the initial report of the model in 2007. The current review synthesizes findings from several groups that are exploring MitoPark mice and discusses implications for the future identification of disease-modifying treatments for PD.
Keywords: Parkinson’s disease, dopamine neurons, substantia nigra, striatum, mitochondria, MitoPark, model, mitochondrial transcription factor A, electrophysiology
Parkinson’s disease (PD) is the second most diagnosed neurodegenerative disorder with an economic burden in the U.S. estimated at over $23 billion per year (Huse et al., 2005). While degeneration of the pars compacta of the substantia nigra (SNc) is well established in PD, pathophysiological abnormalities are found in several other areas of the brain including the locus coeruleus, dorsal raphe, nucleus basalis of Meynert, dorsal vagal and medullary nuclei, amygdala, and limbic thalamus (Braak et al., 2003; Jellinger, 1999). The motor symptoms of the disease, as well as some non-motor symptoms, are produced by massive degeneration of the nigrostriatal pathway, which extends from the dopamine (DA) cell bodies in the SNc, projecting through the medial forebrain bundle and terminating in the caudate/putamen (or striatum). While DA neurons in the SNc are central to disease pathology, most nearby cell types are more resilient. GABA neurons of the pars reticulata of the substantia nigra are relatively protected, as are DA neurons in the adjacent ventral tegmental area (VTA; Surmeier et al., 2012). SNc DA neurons exhibit several key characteristics that likely predispose them to oxidative damage and death. Even in the absence of synaptic input, SNc DA neurons are autonomous pacemakers that fire spontaneously at 1–8 Hz and exhibit large intracellular calcium oscillations (Wilson & Callaway, 2000; Puopolo et al., 2007). This creates a high energetic demand that likely predisposes them to damage when exposed to additional metabolic “hits.” As metabolic stress can generate high basal levels of reactive oxygen species (ROS), additional oxidative stress from mitochondrial dysfunction could be especially damaging (Subramanian & Chesselet, 2013; Chan et al., 2007; Surmeier et al., 2012). This, combined with the fact that most SNc DA neurons have only a modest ability to buffer calcium (Foehring et al., 2009), suggests that any insult that increases the cytoplasmic calcium burden could expose the cells to calcium-dependent pathology. Increased calcium handling could therefore predispose SNc DA neurons to oxidative damage, a rationale that has previously justified investigation of the L-type calcium channel negative allosteric modulator isradipine as a therapeutic for PD (Guzman et al., 2010; Ilijic et al., 2011). Unfortunately, a multi-site, double-blind, placebo-controlled clinical trial for isradipine recently failed to show benefits for early stage PD patients (Parkinson Study Group, 2020). This is an example of a recurring pattern of promising preclinical findings not translating to disease modifying treatments in PD patients, a theme that we will revisit later in this review. Other factors that may predispose DA neurons to degeneration include their vast, highly arborized axonal network, and possibly the intracellular metabolism of DA itself (Duda et al. 2016; Surmeier et al., 2011).
PD pathogenesis progresses slowly over years or even decades, as cellular and circuit adaptations are apparently able to mask the presentation of symptoms for an extended time (de la Fuente-Fernandez et al., 2011). Diagnosis often only occurs following the development of the classic motor symptoms of the disease that include bradykinesia, rigidity, and resting tremor, at a point that perhaps 50–70% of DA neurons have already died (Hornykiewicz, 1975; Marsden, 1994; Damier et al., 1999; Dauer & Przedborski, 2003). The gold standard treatment for PD is the DA precursor L-DOPA, which is highly effective in relieving motor symptoms for a time (Hauser, 2009). This treatment enhances dopaminergic neurotransmission by replacing DA levels that are declining due to progressive nigrostriatal degeneration (Beckstead et al., 2004; Rodriguez et al., 2007). Unfortunately, L-DOPA treatment becomes less effective over time (often segregating into “on” and “off” oscillations) and eventually leads to adverse effects such as dyskinesias and other sensitized behavioral responses. The risk of developing complications of L-DOPA therapy are estimated at 10% per year (Stocchi, 2003) with some patients showing them as early as six months after starting treatment (Parkinson Study Group, 2004). Other commonly used therapeutics include DA receptor agonists, inhibitors of DA metabolism, and anti-cholinergic drugs. While useful for treating symptoms, the consensus is that none of these treatments arrests or slows the progression of the disease itself in any meaningful way. However, while no disease modifying treatments currently exist for PD, the slow presentation of symptoms does present a substantial temporal window to potentially target and arrest cellular pathology in the early stages of neurodegeneration. This would preserve cells that are viable but impaired at a time close to the onset of motor impairment. A challenge currently facing PD researchers is identifying the pathways and processes to be targeted, but this information remains elusive despite a wide variety of animal models currently under investigation.
Mitochondrial dysfunction in Parkinson’s disease
The processes that occur in DA neurons in the early stages of nigrostriatal degeneration prior to frank cell death remain a topic of intense research. Several theories of pathogenesis center around mitochondrial dysfunction (specifically complex I deficiency) and oxidative damage (Gomez et al., 2011; Subramanian & Chesselet, 2013), which is a consequence of mitochondrial dysfunction. Mitochondria play a critical role in cellular function by providing ATP from metabolic byproducts and by serving as key sites for calcium buffering and storage (Sheng & Cai, 2012). Mitochondria are found at elevated levels in the brain compared to other tissues due to high energetic demand. Additionally, mitochondria possess multiple copies of small extra-nuclear genomes. Most mitochondrial genes code for proteins used in the electron transport chain, and half of those participate in the complex I enzyme (Taylor & Turnbull, 2005). Not only does electron transport during oxidative phosphorylation produce ROS as a byproduct, but mitochondrial DNA (mtDNA) itself is a target of damage produced by elevated levels of ROS (Richter et al., 1988). This dual role as both the source and target of ROS could be a central factor for the role of mitochondrial dysregulation in PD and other neurodegenerative disorders.
Postmortem analysis of DA neurons from PD patients shows deficient respiratory chains and increased markers of oxidative stress (Bender et al., 2006). Mitochondrial dysfunction is therefore believed to be a mechanism central to idiopathic PD (Pickrell & Youle, 2015). While genetic mutations account for only a minority of PD cases, most of the genes that have been implicated in familial forms of PD (such as SNCA [α-synuclein], leucine rich repeat kinase 2 [LRRK2], DJ-1, parkin, and phosphatase and tensin homologue [PTEN]-induced kinase 1 [PINK1]) also have roles in the regulation of mitochondrial function (Shen & Cookson, 2004; Park et al., 2006; Clark et al., 2006). Additionally, mutations in mtDNA have been implicated in sporadic PD (Dauer & Przedborski, 2003). Finally, mitochondrial respiratory chain dysfunction in DA neurons appears to be a convergent mechanism central to most rodent models of PD (Subramanian & Chesselet, 2013; Betarbet et al., 2002; Schober, 2004) and is likely responsible for the mass death of DA neurons in these models (Dawson et al., 2010; Shen & Cookson, 2004; Chen et al., 2019b). Taken together, these factors strongly implicate mitochondrial dysfunction as an early and central event in PD-related nigrostriatal degeneration.
Modeling Parkinson’s disease in rodents
Despite decades of work, there are currently no disease modifying agents approved for the treatment of PD in humans. Progress has unfortunately been hindered by the multifactorial nature of the disease and limitations in animal models available to study the early stages of degeneration. In order to move the field forward we must make use of the best available animal models in order to understand the cellular changes that occur, and also work toward developing new models that are faithful to the disease process. Generally speaking, mouse genetic models of PD based on human inheritance studies have been ineffective at mimicking profound DA neuron loss and motor impairment (Dawson et al., 2010). While many of the best tools currently available to biomedical researchers are genetic manipulations in model systems, only 10–20% of PD cases have a cause that can be directly attributed to genetics (Schulte & Gasser, 2011). The fact that the vast majority of cases occur in patients with no family history is likely a contributing factor to the slow development of therapeutics based on human genetics.
Historically, most preclinical rodent studies have instead made use of neurotoxins that selectively destroy DA neurons, most notably 6-hydroxydopamine and the opioid analogue N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP), but also rotenone and paraquat which are relevant to agrichemical-induced PD (Nisticò et al., 2011). These toxins either poison mitochondria directly or cause damage indirectly through reactive oxygen and nitrogen species (Przedborski & Ischiropoulos, 2005). They are selectively taken up by DA neurons and are generally more effective than genetic models at producing targeted degeneration of the nigrostriatal pathway. Perhaps the greatest strength of these models is the ability to study downstream events following the mass neuronal lesion. The vital information that continues to be obtained from toxin models is beyond the scope of the present review. However, while the dose of the neurotoxin can be titrated for effect, toxin models often exhibit highly variable results and suffer from systemic toxicities (Ekstrand & Galter, 2009). Neurodegeneration subsequent to toxin administration also occurs on a relatively quicker time scale than the progression of PD in humans. Therefore, while toxin models are useful for studying downstream events following degeneration, they are not well suited for studying disease genesis and progression. Furthermore, the compensatory processes that occur following toxin treatment are likely distinct from those that occur during the progressive loss of DA neurons that may themselves become part of the disease. While challenges remain, the disease-modifying therapeutics that arise from studies on animal models of PD will likely emanate from a combination of experimental approaches.
Progressive nigrostriatal degeneration in the MitoPark model
First described in 2007, the MitoPark mouse model was engineered as a midbrain DA neuron-specific knockout of the gene coding for mitochondrial transcription factor A (Tfam), a mitochondrial protein of nuclear origin (Ekstrand et al., 2007). Cell type specificity is conveyed by using mice that express Cre-recombinase specifically in cells with the plasmalemmal DA transporter (DAT), i.e. DAT-Cre mice. Tfam is necessary for normal transcription initiation at mtDNA promoters, the viability of mtDNA, and regulation of mtDNA copy number (Ekstrand et al., 2004; Falkenberg et al., 2002). Consistent with this, Tfam elimination in DA neurons reduces expression of mtDNA and produces a deficiency in the function of the respiratory chain (Ekstrand et al., 2007). However, despite the central role for mitochondria in cellular metabolism, MitoPark mice maintain outwardly normal phenotypes into adulthood. Beginning around 12 weeks and extending to 45 weeks of age, these mice experience a massive degeneration of the nigrostriatal pathway as the direct result of mitochondrial dysfunction, similar to what has been hypothesized in PD (Ekstrand et al., 2007). During this process, the MitoPark model faithfully recapitulates most of the key characteristics of nigrostriatal degeneration in clinical PD including adult onset, age-dependent loss of DA neurons in the SNc, progressive motor impairment, a positive response to L-DOPA treatment, and a loss of L-DOPA efficacy with time (Ekstrand et al., 2007; Galter et al., 2010; Li et al., 2013). Intracellular inclusion bodies also develop and exhibit a double-membrane structure consistent with aberrant mitochondria. However, unlike Lewy body formation in PD, these aggregates do not stain positive for α-synuclein (Ekstrand et al., 2007). Also similar to PD, the DA neurons in the adjacent VTA appear to be relatively spared despite lacking Tfam (Ekstrand et al., 2015), or more accurately stated, the VTA degenerates at a later time point than the SNc (Ricke et al., 2020). Importantly, the loss of neurons in the SNc is widespread and terminal, meaning that living DA neurons studied in the intervening period can be safely assumed to be actively undergoing the degenerative process. Currently, MitoPark mice represent the only rodent model that exhibits this combination of key features associated with motor loss in humans with PD.
The MitoPark model has been touted by some as the most phenomenologically faithful rodent model of nigrostriatal degeneration (Beal, 2010; Figure 1). Anatomical findings in MitoPark DA neurons begin around 12 weeks of age with a decrease of striatal innervation following aggregation of intracellular inclusion bodies (Ekstrand et al., 2007). Our group has discovered that decrements in somatodendritic morphology in DA neurons begin at around 16 weeks and become severe by around 27 weeks (Lynch et al., 2018). Mass DA neuron death occurs in the 20–30 week range (Ekstrand et al., 2007). Perhaps the greatest strength of the MitoPark model is that it allows for an analysis of the sequence of events that occur in the early stages of nigrostriatal degeneration following mitochondrial impairment. DA is necessary for the initiation of voluntary movement and an array of reward-related behaviors, which can be assayed in a longitudinal manner to accompany findings obtained later in single cells. Furthermore, the principles learned from studying MitoPark mice could apply to other neuronal types or other neurodegenerative diseases in which mitochondria play a role, including Alzheimer’s disease and amyotrophic lateral sclerosis (Islam, 2017).
Figure 1. Modeling the progressive neurodegeneration of Parkinson’s disease.
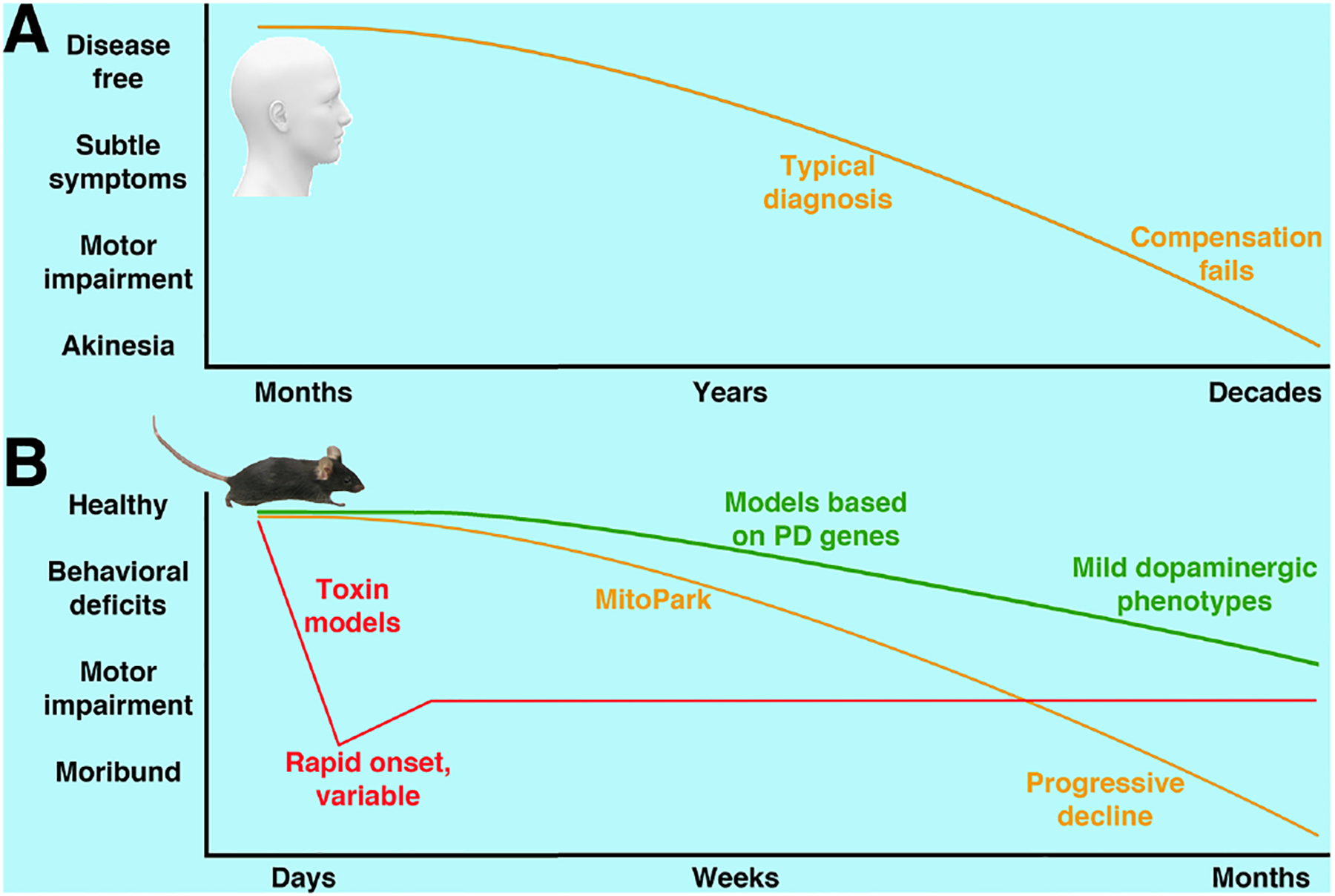
A) PD pathogenesis occurs years before onset of symptoms and typical diagnosis. Compensatory processes can mask or limit symptoms until death of dopaminergic neurons is widespread. While some compensations such as upregulation of postsynaptic receptors have been identified, they are not well understood and may contribute to the disease process itself. B) Like humans with PD, MitoPark mice exhibit a decline in nigrostriatal function that is adult onset, steadily progressive, and ultimately terminal. Toxin models can mimic neuronal death and can be titrated for effect, but are notoriously variable and produce degeneration on a rapid time scale that does not mimic PD. Models based on human genetic studies unfortunately have until now shown only mild behavioral and neurodegenerative phenotypes.
An additional strength of the MitoPark model is that the time course of decline lends itself to testing the role of therapeutics on the progressive degeneration of the pathway, whether by pharmacological agents or modulation of gene expression. The model is unique in its combination of characteristics that both model symptomatology in PD and provide predictive validity, evidenced by the efficacy and loss of L-DOPA response over time. One caveat of the model is that its reliance on cells that express DAT makes it DA-centric. Therefore, while it can effectively model progressive nigrostriatal degeneration and some downstream events, the full array of PD pathologies is not observed throughout the brain. Selectively altering a single system can however be a strength when the goal is to study degeneration of a single pathway in an otherwise healthy animal (Chen et al., 2019a). Another limitation is that MitoPark mice do not lend themselves to studies exploring the role of individual genes that contribute to familial forms of PD. While Tfam expression is known to decline in the normally aging mouse brain as well as in single DA neurons in idiopathic PD (Thomas et al., 2012; Grunewald et al., 2016; Kang et al., 2018), Tfam itself has not been directly implicated in PD pathogenesis (Belin et al., 2007). Therefore, although knocking out Tfam can reliably model mitochondrial impairment in DA neurons, it’s unclear if it models a specific component of the deficit in mitochondrial function commonly observed in PD. As with any animal model of human disease, an understanding of the strengths and weaknesses of the MitoPark model is imperative for proper study design and interpretation of results.
Early deficits in physiology drive initial behavioral decline
Over the last several years, we and others have identified deficits in DA neuron physiology, anatomy, gene expression, and the related behavioral output across the full age range of nigrostriatal degeneration in the MitoPark model. In this section we describe the progression of the appearance of pathophysiological phenotypes in MitoPark mice (Figure 2) based on approximations from published findings.
Figure 2. Earliest reported decrements observed in MitoPark mice.

Published findings were used to approximate the age that declining parameters first reach statistical significance in MitoPark mice, in most cases when compared to littermate controls. Reports are fairly consistent across labs, and some preference in this figure is given to studies that rigorously quantified events at ages prior to onset. Decrements in DA neuron (DA-N) function and neurotransmission tend to coincide with initial behavioral deficits and, with the exception of the appearance of intracellular inclusion bodies, precede significant anatomical findings. Nuclear gene expression changes tend to occur at later time points, but have not been broadly explored. HVA: homovanillic acid, NAc: nucleus accumbens, TH: tyrosine hydroxylase. Sources cited in the figure: 1) Branch et al., 2016; 2) Good et al., 2011; 3) Ekstrand et al., 2007; 4) Lynch et al., 2018; 5) Li et al., 2013; 6) Grauer et al., 2014; 7) Gordon et al., 2016; 8) Chen et al., 2019a; 9) Ricke et al., 2020; 10) Ghaisas et al., 2019; 11) Chen et al., 2019b; 12) Galter et al., 2010.
One of the first deficits noted at 6 weeks of age is decreased mitochondrial DNA gene expression, a direct result of the absence of Tfam (Ekstrand et al., 2007). Beyond that, many of the initial deficits observed at a very young age in MitoPark mice are indicative of a robust decline of DA neuron function (Branch et al., 2016; Good et al., 2011). Patch clamp electrophysiology has been a useful tool for assessing decrements of the individual ion channel currents that drive the basal firing properties of these cells. DA neurons from young MitoPark mice exhibit reduced amplitudes of multiple intrinsic ion conductances, including HCN channel-mediated H-current and afterhyperpolarization (Branch et al., 2016; Good et al., 2011), which in the SNc is largely dependent on “small” conductance calcium-dependent potassium (SK) channels (Wolfart et al., 2001). Basal electrophysiological properties are also affected in single DA neurons including input resistance, which is elevated due to a reduction in ion channel activity, and cell capacitance, which is reduced (Branch et al., 2016; Good et al., 2011). As the action potential initiation and propagation are affected by these ion channels and basal parameters, it is not surprising that alterations in the rate and pattern of impulse activity are also observed at a young age. DA neurons from MitoPark mice exhibit unusually variable firing rates across cells when compared to wild type controls (Good et al., 2011), as well as enhanced variability of inter-spike intervals within cells (Branch et al., 2016).
Decrements in the pre- and postsynaptic components of dopaminergic neurotransmission have also been identified at early time points. In addition to axonal release of DA in terminal fields such as the striatum, DA neurons also communicate with each other in the midbrain through a unique form of dendritic neurotransmission (Beckstead et al., 2004). The inhibitory postsynaptic currents (IPSCs) that comprise this form of neurotransmission are dependent on DA D2 autoreceptors and G protein coupled inwardly rectifying potassium (GIRK) channels (Beckstead et al., 2004). They can be measured directly by patch clamping DA neurons in freshly isolated brain slices and by applying local stimulation, either electrical or optogenetic (Beckstead et al., 2004; Tschumi et al., 2019). In MitoPark mice, these D2-IPSCs are reduced in amplitude versus littermate controls from the youngest ages tested (Branch et al., 2016). This is primarily a consequence of strongly reduced presynaptic vesicular content of DA. Currents produced by amphetamine, which causes efflux of vesicular DA, are reduced in young MitoPark mice, while D2 receptor-mediated currents elicited by application of exogenous DA are not as severely reduced and not until a later timepoint (Branch et al., 2016). A reduction in striatal DA release has also been observed at a very young age in MitoPark mice using fast scan cyclic voltammetry (Good et al., 2011; Chen et al., 2019a and 2019b).
The numerous deficits in DA neuron physiology appear to be coincident with the initial behavioral deficits observed at 8 weeks of age, which are impaired novel object recognition and spatial learning (Li et al., 2013). This is soon followed by a decline in horizontal locomotion and vertical movements such as rearing (Li et al., 2013; Chen et al., 2019a, Galter et al., 2010). Decreased performance on a rotarod subsequently follows (Gordon et al., 2016). The fact that observable rotarod deficits occur later than those on spontaneous locomotion could suggest, consistent with PD, that forced behaviors are preserved for longer than spontaneous ones during dopaminergic degeneration (Chen et al., 2019a). Interestingly, the behaviors that show decline at the earliest age are not typically associated with dopaminergic areas. Spatial learning is thought to be mostly hippocampal, while novel object recognition relies on cortical regions (Li et al., 2013). Both of these areas are direct targets of projections from VTA DA neurons. However, the behavioral deficits can be observed in MitoPark mice at an age prior to decreased DA levels in at least two VTA terminal fields, the cortex and the nucleus accumbens (Galter et al., 2010; Chen et al., 2019b). Li et al. (2013) go on to speculate that these cognitive deficits may develop indirectly from the SNc through other brain areas such as the thalamus. No electrophysiological studies have been performed on VTA DA neurons from MitoPark mice, therefore it is not known if their physiological properties are affected from a very early age, as has been observed in in the SNc.
Perhaps surprisingly, the decline in anatomical parameters that comprise the next wave of deficits in MitoPark mice are not coincident with deficits in physiology and the initial behavioral decline, but rather become evident at later ages. In clinical PD, it has been hypothesized that DA neurons may “die back” from the caudate/putamen, beginning with their axonal arbor and proceeding in a retrograde direction that ultimately results in death of the neuron (Bernheimer et al., 1973). Although challenging to ascertain from postmortem tissue, considerable evidence has been identified to support this notion (Kordower et al., 2013; Grosch et al., 2016;). Findings in MitoPark mice are also generally consistent with this hypothesis; a reduction in tyrosine hydroxylase positive fibers can be observed in the striatum at around 14 weeks, dendritic morphology is affected at 16 weeks, and DA neuron death begins to take hold around 20 weeks (Lynch et al., 2018; Ricke et al., 2020). This pattern of terminal damage preceding effects at the soma has also been observed in partial toxin models of PD, including hemiparkinsonian primates following unilateral infusion of MPTP into the internal carotid artery (Betarbet et al., 2000). The anatomical decline in MitoPark mice is coincident with more grave behavioral signs, including the loss of body weight as the mice become unable to consume a sufficient amount of food to maintain homeostasis. The only anatomical finding that is observed at an early age is the initial presence of intracellular inclusion bodies (Ekstrand et al., 2007). It’s unclear to what extent the inclusion bodies contribute to early cellular and behavioral pathology.
Published reports of significant alterations in nuclear gene expression in the MitoPark model are relatively few in number. Using quantitative (q)PCR of mRNA isolated from the substantia nigra, we discovered an increase in expression of genes associated with excitability of DA neurons in MitoPark mice versus littermate controls (Branch et al., 2016). However, with the exception of the L-type calcium channel subunit CACN1AC (Cav1.2), the changes in sodium and calcium channel subunit gene expression occurred at a late timepoint more consistent with compensation following denervation rather than any causative process. Gordon and colleagues (2016) reported an intriguing increase in the expression of prokineticin-2 (PK-2), a member of the AVIT protein family, at 8 weeks of age in MitoPark mice. Their findings suggest that PK-2 could be neuroprotective, and could be useful either as an early marker of degeneration or as a potential target for therapeutics through the downstream effector and transcriptional activator peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α. Hong et al. (2020) used a high fat diet to induce insulin resistance and observed that α-synuclein expression was elevated over the low levels normally observed in MitoPark mice (Ekstrand & Galter, 2009). In vitro follow-up experiments in differentiated human DA neurons suggested that reduced PGC-1α signaling may mechanistically link insulin resistance to dysregulation of mitochondrial biogenesis (Hong et al., 2020). As much of the gene expression work done thus far on the MitoPark model has used bulk tissue and has been narrow in scope, future work is needed to illuminate novel pathways both in a cell type-specific manner and using transcriptomic and epigenomic approaches. A more complete understanding of the structural, functional, and genetic adaptations produced by mitochondrial impairment could lay the groundwork for therapeutics that halt or slow PD-related neurodegeneration.
DA neuron physiology in other PD models
MitoPark mice exhibit a slowly progressive decline in health of DA neurons ultimately resulting in mass cell death. This unique characteristic among PD models makes them suitable for timed patch clamp experiments on individual DA neurons that have begun the process of neurodegeneration but are not yet dead. DA neuron electrophysiology and neurotransmission has also been studied to a limited extent in other PD models, both genetic and toxin-based.
A wide range of effects on firing activity in SNc DA neurons are reported in rodent models of familial PD. Mice overexpressing human α-synuclein (SNCA-OVX) show age dependent reductions in DA neuron firing rates by 18 months and altered firing patterns during movement despite minimal abnormalities in motor activity (Janezic et al., 2013; Dodson et al., 2016). In a model expressing mutant α-synuclein (SNCA-A53T), mice aged 7–8 months show dramatic increases in firing frequency in vivo and in brain slices due to oxidization of Kv4.3 channels causing a decrease in function (Subramaniam et al., 2014). Mice deficient in the kinase PINK1 and the mitochondrial serine protease HtrA2/Omi show increased variability in firing in brain slices and in vivo by 3–4 months and 1–2 months respectively, both due to reduced SK channel functioning as a result of impaired mitochondrial regulation of intracellular calcium (Bishop et al., 2010). PINK1 mutations in humans are autosomal recessive and concordantly PINK1 heterozygous mutant mice do not show any changes in SNc DA neuron function and quantity (Madeo et al., 2013). No clear differences in basal firing activity have been found in mice with mutations in PD-associated proteins LRRK2 (Qin et al., 2017; Sloan et al., 2016; Tong et al., 2009) or DJ-1 (Goldberg et al., 2005; Pisani et al., 2006). However, reduced D2 autoreceptor function has been observed in both of these models (Goldberg et al. 2005; Tong et al., 2009). Further, despite a limited phenotype, SNc DA neurons from DJ-1 deficient mice are more susceptible to the effects of oxygen-glucose deprivation, rotenone, and Na+/K+ ATPase inhibition compared with controls (Pisani et al., 2006). While similarities exist in the electrophysiological effects observed in MitoPark versus other genetic models of PD, MitoPark mice have not been broadly tested for susceptibility to additional metabolic challenges.
Toxins commonly used to induce a parkinsonian state and nigrostriatal degeneration include 6-hydroxydopamine, MPTP, and rotenone (Konnova et al., 2018). These toxins all induce rapid SNc DA neuron death through inhibition of mitochondrial complex I and the generation of oxidative stress. Few abnormalities in SNc DA neuron firing activity have been found in the neurons remaining after treatment with 6-hydroxydopamine and rotenone (Bilbao et al., 2006; Hollerman & Grace, 1990; Martella et al., 2016). However, many studies have investigated the acute effects of these toxins to gain insights into how SNc DA neuron electrophysiological activity is affected before neuronal death. In brain slices, acute application of 6-hydroxydopamine, MPP+ (the active form of MPTP), and rotenone cause membrane hyperpolarization and a reduction in spontaneous firing rate (Berretta et al., 2005; Freestone et al., 2009; Liss et al., 2005; Yee et al., 2014). This is due to K-ATP channel activation that occurs in response to the stress on the cell, and this hyperpolarization can be reduced by pretreatment with antioxidants or blocked by K-ATP antagonists. MitoPark mice have not been tested for the pathological contribution of K-ATP channels. However, the dearth of reports of electrophysiological deficits following toxin treatment suggests that a prolonged progressive phase, with or without cellular and circuit compensation, could itself play a central role in PD pathology.
Non-dopaminergic findings and assessment of potential therapies
While DA neuron degeneration is central to the motor deficits associated with PD, non-dopaminergic processes also contribute to disease presentation. Indeed, constipation, hyposmia, and sleep disturbances are among the earliest indicators of future PD (Rodríguez-Violante et al., 2017) and are not typically associated with DA. Although MitoPark mice are produced by selectively knocking out a single gene in DA neurons, non-dopaminergic systems are also affected (Chen et al., 2019a). Similar to PD, L-DOPA administration in MitoPark mice induces sensitization to the drug in neural circuits downstream of DA neurons themselves, possibly making it suitable for studies of L-DOPA-induced dyskinesias (Shan et al., 2015). Consistent with this, KCl-induced glutamate release in the dorsal striatum is increased at 28 weeks in MitoPark mice, and expression of striatal astrocyte genes GLT-1, GFAP, and mGluR5 is also affected (Farrand et al., 2016). Non-motor systems are also altered. Consistent with effects observed in PD, circadian rhythms are progressively disrupted in MitoPark mice, ultimately resulting in complete circadian arrhythmia by 26 weeks (Fifel & Cooper, 2004). Findings from MitoPark mice are also consistent with the evolving notion that gut-brain interactions could play a central role in PD pathogenesis. MitoPark mice exhibit decreased gastrointestinal motility at 8 weeks (Ghaisas et al., 2019), which is one of the earliest non-motor symptoms observed in PD and is consistent with constipation in humans being a predictor of future diagnosis. Other gastrointestinal effects in MitoPark mice include decreased colon length (12 weeks), increased transit time (16 weeks), altered fecal metabolomes (20 weeks), and decreased water content and constipation (24 weeks; Ghiasas et al., 2019). Further, treatment of MitoPark mice with probiotics starting at 8 weeks improves motor abilities starting at 14–22 weeks including beam walking, rotarod performance, and several stride and gait parameters (Hsieh et al., 2020). These non-dopaminergic findings are surprising, given that the Tfam knockout in MitoPark mice is confined to DAT-expressing cells. If these abnormalities are conveyed through circuit mechanisms and are downstream from DA neurons, then correcting early deficits in dopaminergic function could have broad benefits in PD. Alternatively, these findings could be consequences of extra-dopaminergic DAT expression, perhaps in peripheral monocytes (Gopinath et al., 2020) or during development. These possibilities could be experimentally addressed using viral approaches instead of a genetic cross to induce the Tfam deletion.
The slow development of degeneration in MitoPark mice has also made the model a useful tool to explore non-dopaminergic neurotransmitter receptor systems for their potential in treating symptoms and/or modulating progression. Multiple studies have explored the efficacy of adenosine 2A (A2A) antagonists in MitoPark mice, which likely target DA D2 receptor-expressing indirect pathway striatal medium-sized spiny projection neurons. Both acute and chronic administration of the A2A antagonist MSX-3 starting at 12 weeks ameliorates the decrease of locomotor behaviors in MitoPark mice, however the progression of neurodegeneration is not affected (Marcellino et al., 2010). Similarly, administration of the A2A receptor antagonist SCH 412348 restores hindlimb bradykinesia and increases GABA in the globus pallidus of MitoPark mice, as well as partially restoring rotarod performance (Smith et al., 2014). The findings are consistent with the notion that treatment with A2A antagonists could be an effective strategy to treat early motor symptoms in PD and could delay the necessity for L-DOPA treatment. Multiple clinical trials over the last two decades have explored A2A ligands as potential PD treatments (LeWitt et al., 2020), and the antagonist istradefylline has now been approved in both Japan and the United States as an add-on to L-DOPA in patients experiencing “OFF” episodes (Chen & Cunha, 2020). Metabotropic glutamate receptor ligands have been explored as well; the mGluR4 positive allosteric modulator ADX88178 works synergistically with L-DOPA to enhance locomotion in 17-week-old MitoPark mice (Le Poul et al., 2012).
MitoPark mice have also been used to explore mechanisms responsible for the predisposition of neurons to degeneration. Mitochondria play an essential role in cell health not only through cellular respiration but also as an essential source of calcium buffering. DA neurons from MitoPark mice exhibit increased markers of oxidative stress, and SNc microglia exhibit altered morphology indicative of neuroinflammation (Langley et al., 2017). Recent work suggests that mitochondrial dysfunction produces an imbalance in the GSH/GSSG redox ratio in DA neurons from MitoPark mice, which can be ameliorated by blocking voltage gated calcium channels with isradipine or by inhibiting mitochondrial calcium influx with RU360 (Ricke et al., 2020). The authors of that study conclude that mitochondrial dysfunction interacts with increased intracellular calcium to produce toxicity in DA neurons (Ricke et al., 2020). That study also noted a higher number of neurons expressing the calcium binding protein calbindin-D28K in 20-week-old MitoPark mice compared to controls, consistent with the notion that calbindin negative neurons may be more sensitive to degeneration. Calbindin expression has been previously identified as a key feature of DA neurons in the VTA and SNc that could help explain the relative protection of neuron subpopulations, both in clinical PD and in animal models such as MPTP (Yamada et al., 1990; Lavoie & Parent, 1991; Iacopino et al., 1992; Zampese & Surmeier, 2020). Electrophysiological work conducted thus far in MitoPark mice has exclusively focused on the SNc and has not been accompanied by staining for calbindin. It would therefore be instructive to determine the role of calbindin expression on the physiology of single DA neurons in the SNc and VTA at ages prior to degeneration in these mice. The Ricke et al. study is a recent example of preclinical work supporting isradipine as a potential treatment for PD, seemingly at odds with the result of the recent clinical trial (Parkinson Study Group, 2020). While the reasons for this discrepancy are not clear, the effects of normal aging could themselves be an underappreciated factor. By far the bulk of preclinical work in PD models is performed in young animals, despite the fact that advanced age is the leading risk factor for clinical PD. Our work has identified disrupted pacemaker firing and a dramatic decline in nimodipine-sensitive (presumably L-type calcium) currents in SNc DA neurons from healthy aged mice (Branch et al., 2014; Howell et al., 2020). This could indicate a restricted temporal window for the neuroprotective effects of isradipine, possibly limited to ages prior to the typical onset of motor symptoms in PD (Branch et al., 2014).
Growth factor ligands such as glial cell-derived neurotrophic factor (GDNF) and neurturin have also been explored as potential treatments for clinical PD, with limited success (Pascual et al., 2011; Warren Olanow et al., 2015). This strategy presents several challenges, such as determining the most efficacious method to target the growth factors to the SNc or the striatum (Sidorova & Saarma, 2020). In this regard, MitoPark mice have been used with some success to test cell graft approaches. Whole body irradiation followed by peripheral injection of modified macrophages can be used as a strategy to deliver GDNF to the brain. When the injection is performed at 18 weeks in MitoPark mice it protects against further neurodegeneration and improves behaviors such as locomotion and nest building (Chen et al., 2018; Chen et al., 2019). Direct implantation of DA neurons has also been tested in MitoPark mice. When injected at 20 weeks of age, DA neurons lacking the PTEN gene increase locomotion by 36 weeks. These mice also exhibit improved striatal optical density, increased DA release as measured with fast scan cyclic voltammetry, and an increase in the number of tyrosine hydroxylase positive cells (Zhang et al., 2012).
Other mechanisms and treatments have also been explored in MitoPark mice. Consistent with PD in humans, voluntary exercise delays onset of motor impairment in MitoPark mice (Lai et al., 2019). Chronic administration of the natural product quercetin improves locomotion and increases the number of tyrosine hydroxylase positive neurons in 20-week-old MitoPark mice, possibly though activation of neuroprotective protein kinase D1 and Akt pathways (Ay et al., 2017). Finally, one study found that administration of the LRRK2 inhibitor MLi-2 fails to improve markers of degeneration and symptomatology in MitoPark mice (Fell et al., 2015). Future work will continue to identify mechanisms responsible for progressive neurodegeneration in MitoPark mice and the associated cellular and behavioral pathologies.
It must be restated that all of the targets identified using animal models have thus far failed to produce disease modifying treatments for PD. While it is tempting to lay the blame for this on the animal models themselves, study design (both in trials and in preclinical work) represents an ongoing challenge to all researchers. There are currently no reliable biomarkers of prodromal PD that would allow for earlier initiation of prospective trials. Given the advanced stage of neuronal death that occurs prior to typical diagnosis, preclinical studies that aim to explore potential mid- to late stage treatments must carefully consider when to begin treatment of their animal subjects. Clinical trials themselves are constrained by patient and resource availability that does not allow for comprehensive exploration of dose amounts and timing of those doses, in addition to issues involving pharmaco-kinetics and dynamics. These challenges must be solved if we are to slow or halt the decline of brain function in afflicted individuals.
Conclusion
MitoPark mice currently hold a unique place as the only animal model of PD that is both based on modulation of a cellular process that plays a major role in the etiology of the disease and produces a robust, adult onset, progressive nigrostriatal degeneration and development of a terminal motor symptomatology. While much has already been learned, further work from this and other models will be necessary to achieve the first disease-modifying treatment for the millions of patients currently afflicted with PD.
Acknowledgements
We would like to thank Drs. Amanda Sharpe, Chris Tschumi, and Tommy Lewis for critical reads of the manuscript. This work was supported by National Institutes of Health grant R01 AG052606, funding from the Presbyterian Health Foundation (PHF), and the Oklahoma Center for Adult Stem Cell Research (OCASCR).
Declaration of competing interest
Michael Beckstead is a current recipient of contract funding from Nitrome Biosciences to perform a Parkinson’s disease research study. This study does not involve MitoPark mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ay M, Luo J, Langley M, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG (2017) Molecular mechanisms underlying protective effects of quercetin against mitochondrial dysfunction and progressive dopaminergic neurodegeneration in cell culture and MitoPark transgenic mouse models of Parkinson’s Disease. J Neurochem 141: 766–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF (2010) Parkinson’s disease: a model dilemma. Nature 466: S8–10. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT (2004) Vesicular DA release elicits an inhibitory postsynaptic current in midbrain DA neurons. Neuron 42: 939–946. [DOI] [PubMed] [Google Scholar]
- Belin AC, Bjork BF, Westerlund M, Galter D, Sydow O, et al. (2007) Association study of two genetic variants in mitochondrial transcription factor A (TFAM) in Alzheimer’s and Parkinson’s disease. Neurosci Lett 420: 257–262. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E et al. (2006) High levels of mitochondrial DNA deletions in Substantia nigra neurons in aging and Parkinson disease. Nat Genet 38: 515–517 [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 20: 415–455. [DOI] [PubMed] [Google Scholar]
- Berretta N, Freestone PS, Guatteo E, de Castro D, Geracitano R, Bernardi G, Mercuri NB, Lipski J (2005) Acute effects of 6-hydroxydopamine on dopaminergic neurons of the rat substantia nigra pars compacta in vitro. Neurotoxicology 26: 869–881. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Porter RH, Greenamyre JT (2000) GluR1 glutamate receptor subunit is regulated differentially in the primate basal ganglia following nigrostriatal dopamine denervation. J Neurochem 74: 1166–1174. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT (2002) Animal models of Parkinson’s disease. Bioessays 24: 308–318. [DOI] [PubMed] [Google Scholar]
- Bilbao G, Ruiz-Ortega JA, Miguens N, Ulibarri I, Linazasoro G, Gomez-Urquijo S, Garibi J, Ugedo L (2006) Electrophysiological characterization of substantia nigra dopaminergic neurons in partially lesioned rats: effects of subthalamotomy and levodopa treatment. Brain Res 1084: 175–184. [DOI] [PubMed] [Google Scholar]
- Bishop MW, Chakraborty S, Matthews GA, Dougalis A, Wood NW, Festenstein R, Ungless MA (2010) Hyperexcitable substantia nigra dopamine neurons in PINK1- and HtrA2/Omi-deficient mice. J Neurophysiol 104: 3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: 197–211. [DOI] [PubMed] [Google Scholar]
- Branch SY, Chen C, Sharma R, Lechleiter JD, Li S, Beckstead MJ (2016) Dopaminergic neurons exhibit an age-dependent decline in electrophysiological parameters in the MitoPark mouse model of Parkinson’s disease. J Neurosci 36: 4026–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch SY, Sharma R, Beckstead MJ (2014) Aging decreases L-type calcium currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J Neurosci. 34: 9310–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ (2007) ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447: 1081–1086. [DOI] [PubMed] [Google Scholar]
- Chen JF, Cunha RA (2020) The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal 16: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Guderyon MJ, Li Y, Ge G, Bhattacharjee A, Ballard C, He Z, Masliah E, Clark RA, O’Connor JC, Li S (2019) Non-toxic HSC Transplantation-Based Macrophage/Microglia-Mediated GDNF Delivery for Parkinson’s Disease. Mol Ther Methods Clin Dev 17: 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Li X, Ge G, Liu J, Biju KC, Laing SD, Qian Y, Ballard C, He Z, Masliah E, Clark RA, O’Connor JC, Li S (2018) GDNF-expressing macrophages mitigate loss of dopamine neurons and improve Parkinsonian symptoms in MitoPark mice. Sci Rep 8:5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Hsieh TH, Kuo TT, Kao JH, Ma KH, Huang EY, Chou YC, Olson L, Hoffer BJ (2019a) Release parameters during progressive degeneration of dopamine neurons in a mouse model reveal earlier impairment of spontaneous than forced behaviors. J Neurochem 150: 56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Wang V, Huang EY, Chou YC, Kuo TT, Olson L, Hoffer BJ (2019b) Delayed Dopamine Dysfunction and Motor Deficits in Female Parkinson Model Mice. Int J Mol Sci 20(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ et al. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166. [DOI] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. Brain 122: 1437–1408. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39: 889–909. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson’s disease. Neuron 66: 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Fernández R, Schulzer M, Kuramoto L, Cragg J, Ramachandiran N, Au WL, Mak E et al. (2011) Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol 69: 803–810. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Dreyer JK, Jennings KA, Syed EC, Wade-Martins R, Cragg SJ, Bolam JP, Magill PJ (2016) Representation of spontaneous movement by dopaminergic neurons is cell-type selective and disrupted in parkinsonism. Proc Natl Acad Sci U S A 113(15): E2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda J, Potschke C, Liss B (2016) Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J Neurochem 139 Suppl 1: 156–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13: 935–944. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Galter D (2009) The MitoPark Mouse—An animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat Disord 15: S185. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S et al. (2007) Progressive parkinsonism in mice with respiratory-chain-deficient DA neurons. Proc Natl Acad Sci U S A 104: 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM (2002) Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 31: 289–294. [DOI] [PubMed] [Google Scholar]
- Farrand AQ, Gregory RA, Bäckman CM, Helke KL, Boger HA (2016) Altered glutamate release in the dorsal striatum of the MitoPark mouse model of Parkinson’s disease. Brain Res 1651: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell MJ, Mirescu C, Basu K, Cheewatrakoolpong B, DeMong DE, Ellis JM, Hyde LA, Lin Y, Markgraf CG, Mei H, Miller M, Poulet FM, Scott JD, Smith MD, Yin Z, Zhou X, Parker EM, Kennedy ME, Morrow JA (2015) MLi-2, a Potent, Selective, and Centrally Active Compound for Exploring the Therapeutic Potential and Safety of LRRK2 Kinase Inhibition. J Pharmacol Exp Ther 355: 397–409. [DOI] [PubMed] [Google Scholar]
- Fifel K, Cooper HM (2014) Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol Dis 71: 359–369. [DOI] [PubMed] [Google Scholar]
- Foehring RC, Zhang XF, Lee JC, Callaway JC (2009) Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J Neurophysiol 102: 2326–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freestone PS, Chung KK, Guatteo E, Mercuri NB, Nicholson LF, Lipski J (2009) Acute action of rotenone on nigral dopaminergic neurons--involvement of reactive oxygen species and disruption of Ca2+ homeostasis. Eur J Neurosci 30: 1849–1859. [DOI] [PubMed] [Google Scholar]
- Galter D, Pernold K, Yoshitake T, Lindqvist E, Hoffer B, Kehr J, Larsson NG, Olson L (2010) MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson’s disease. Genes Brain Behav 9: 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaisas S, Langley MR, Palanisamy BN, Dutta S, Narayanaswamy K, Plummer PJ, Sarkar S, et al. (2019) MitoPark transgenic mouse model recapitulates the gastrointestinal dysfunction and gut-microbiome changes of Parkinson’s disease. Neurotoxicology 75: 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, et al. (2005) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45: 489–496. [DOI] [PubMed] [Google Scholar]
- Gómez FJ, Aguirre P, Gonzalez-Billault C, Núñez MT (2011) Iron mediates neuritic tree collapse in mesencephalic neurons treated with 1-methyl-4-phenylpyridinium (MPP+). J Neural Transm 118: 421–431. [DOI] [PubMed] [Google Scholar]
- Good CH, Hoffman AF, Hoffer BJ, Chefer VI, Shippenberg TS, Backman CM, Larsson NG, et al. (2011) Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson’s disease. FASEB J 25: 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath A, Doty A, Mackie PM, Hashimi B, Francis M, Saadatpour L, Saha K, Shaw G, Ramirez-Zamora A, Okun MS, Streit WJ, Khoshbouei H (2020) A novel approach to study markers of dopamine signaling in peripheral immune cells. J Immunol Methods 476: 112686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Neal ML, Luo J, Langley MR, Harischandra DS, Panicker N, Charli A et al. (2016) Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nat Commun 7: 12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauer SM, Hodgson R, Hyde LA (2014) MitoPark mice, an animal model of Parkinson’s disease, show enhanced prepulse inhibition of acoustic startle and no loss of gating in response to the adenosine A(2A) antagonist SCH 412348. Psychopharmacology (Berl) 231: 1325–1337. [DOI] [PubMed] [Google Scholar]
- Grosch J, Winkler J, Kohl Z (2016) Early Degeneration of Both Dopaminergic and Serotonergic Axons - A Common Mechanism in Parkinson’s Disease. Front Cell Neurosci 10: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM (2016) Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann Neurol 79: 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468: 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser RA (2009) Levodopa: past, present, and future. Eur Neurol 62: 1–8. [DOI] [PubMed] [Google Scholar]
- Hollerman JR, Grace AA (1990) The effects of dopamine-depleting brain lesions on the electrophysiological activity of rat substantia nigra dopamine neurons. Brain Res 533(2): 203–212. [DOI] [PubMed] [Google Scholar]
- Hong CT, Chen KY, Wang W, Chiu JY, Wu D, Chao TY, Hu CJ, Chau KD, Bamodu OA (2020) Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornykiewicz O (1975) Parkinson’s disease and its chemotherapy. Biochem Pharmacol 24: 1061–1065. [DOI] [PubMed] [Google Scholar]
- Howell RD, Dominguez-Lopez S, Ocañas SR, Freeman WM, Beckstead MJ (2020) Female mice are resilient to age-related decline of substantia nigra dopamine neuron firing parameters. Neurobiology of Aging 95: 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Kuo CW, Hsieh KH, Shieh MJ, Peng CW, Chen YC, Chang YL, et al. (2020) Probiotics Alleviate the Progressive Deterioration of Motor Functions in a Mouse Model of Parkinson’s Disease. Brain Sci 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse DM, Schulman K, Orsini L, Castelli-Haley J, Kennedy S, Lenhart G (2005) Burden of illness in Parkinson’s disease. Mov Disord 20: 1449–1454. [DOI] [PubMed] [Google Scholar]
- Iacopino A, Christakos S, German D, Sonsalla PK, Altar CA (1992) Calbindin-D28K-containing neurons in animal models of neurodegeneration: possible protection from excitotoxicity. Brain Res Mol Brain Res 13: 251–261. [DOI] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, Surmeier DJ (2011) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis 43: 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MT (2017) Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res 39: 73–82. [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, et al. (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A 110(42), E4016–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA (1999) Post mortem studies in Parkinson’s disease--is it possible to detect brain areas for specific symptoms? J Neural Transm Suppl 56: 1–29. [DOI] [PubMed] [Google Scholar]
- Kang I, Chu CT, Kaufman BA (2018) The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett 592: 793–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnova EA, Swanberg M (2018) Animal Models of Parkinson’s Disease, in: Stoker TB, Greenland JC (Eds.), Parkinson’s Disease: Pathogenesis and Clinical Aspects. Brisbane (AU). [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JH, Chen KY, Wu JC, Olson L, Brené S, Huang CZ, Chen YH, et al. (2019) Voluntary exercise delays progressive deterioration of markers of metabolism and behavior in a mouse model of Parkinson’s disease. Brain Res 1720: 146301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley M, Ghosh A, Charli A, Sarkar S, Ay M, Luo J, Zielonka J, Brenza T, Bennett B, Jin H, Ghaisas S, Schlichtmann B, Kim D, Anantharam V, Kanthasamy A, Narasimhan B, Kalyanaraman B, Kanthasamy AG (2017) Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxid Redox Signal 27: 1048–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie B, Parent A (1991) Dopaminergic neurons expressing calbindin in normal and parkinsonian monkeys. Neuroreport 2: 601–604. [DOI] [PubMed] [Google Scholar]
- Le Poul E, Boléa C, Girard F, Poli S, Charvin D, Campo B, Bortoli J, et al. (2012) A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson’s disease. J Pharmacol Exp Ther 343: 167–177. [DOI] [PubMed] [Google Scholar]
- LeWitt PA, Aradi SD, Hauser RA, Rascol O (2020) The challenge of developing adenosine A2A antagonists for Parkinson disease: Istradefylline, preladenant, and tozadenant. Parkinsonism Relat Disord 80 Suppl 1: S54–S63. [DOI] [PubMed] [Google Scholar]
- Li X, Redus L, Chen C, Martinez PA, Strong R, Li S, O’Connor JC (2013) Cognitive dysfunction precedes the onset of motor symptoms in the MitoPark mouse model of Parkinson’s disease. PLoS One 8:e71341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Haeckel O, Wildmann J, Miki T, Seino S, Roeper J (2005) K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci 8: 1742–1751. [DOI] [PubMed] [Google Scholar]
- Lynch WB, Sharpe AL, Tschumi CW, Branch SY, Chen C, Ge G, Li S, Beckstead MJ (2018) Progressively disrupted somatodendritic morphology in dopamine neurons in a mouse Parkinson’s model. Mov Disord 33: 1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo G, Schirinzi T, Martella G, Latagliata EC, Puglisi F, Shen J, Valente EM, et al. (2014) PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov Disord 29: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino D, Lindqvist E, Schneider M, Müller CE, Fuxe K, Olson L, Galter D (2010) Chronic A2A antagonist treatment alleviates parkinsonian locomotor deficiency in MitoPark mice. Neurobiol Dis 40: 460–466. [DOI] [PubMed] [Google Scholar]
- Marsden CD (1994) Parkinson’s disease. J Neurol Neurosurg Psychiatr 57: 672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella G, Madeo G, Maltese M, Vanni V, Puglisi F, Ferraro E, Schirinzi T, et al. (2016) Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol Dis 91: 21–36. [DOI] [PubMed] [Google Scholar]
- Nisticò R, Mehdawy B, Piccirilli S, Mercuri N (2011) Paraquat- and rotenone-induced models of Parkinson’s disease. Int J Immunopathol Pharmacol 24: 313–322. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E et al. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group (2004) Levodopa and the progression of Parkinson’s disease. N Engl J Med 351: 2498–2508. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group STEADY-PD III Investigators (2020) Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann Intern Med 172: 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A, Hidalgo-Figueroa M, Gómez-Díaz R, López-Barneo J (2011) GDNF and protection of adult central catecholaminergic neurons. J Mol Endocrinol 46: R83–92. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85: 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Costa C, Mercuri NB, Bernardi G, Shen J, Calabresi P (2006) Enhanced sensitivity of DJ-1-deficient dopaminergic neurons to energy metabolism impairment: role of Na+/K+ ATPase. Neurobiol Dis 23: 54–60. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Ischiropoulos H (2005) Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal 7: 685–693. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27: 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Zhi LT, Li XT, Yue ZY, Li GZ, Zhang H (2017) Effects of LRRK2 Inhibitors on Nigrostriatal Dopaminergic Neurotransmission. CNS Neurosci Ther 23: 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C, Park JW, Ames BN (1988) Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 85: 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke KM, Paß T, Kimoloi S, Fährmann K, Jüngst C, Schauss A, Baris OR et al. (2020) Mitochondrial Dysfunction Combined with High Calcium Load Leads to Impaired Antioxidant Defense Underlying the Selective Loss of Nigral Dopaminergic Neurons. J Neurosci 40: 1975–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez M, Morales I, González-Mora JL, Gómez I, Sabaté M, Dopico JG, Rodríguez-Oroz MC, Obeso JA (2007) Different levodopa actions on the extracellular dopamine pools in the rat striatum. Synapse 61: 61–71. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Violante M, Zerón-Martínez R, Cervantes-Arriaga A, Corona T (2017) Who Can Diagnose Parkinson’s Disease First? Role of Pre-motor Symptoms. Arch Med Res 48: 221–227. [DOI] [PubMed] [Google Scholar]
- Schober A Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP (2004) Cell Tissue Res 318: 215–224. [DOI] [PubMed] [Google Scholar]
- Schulte C, Gasser T (2011) Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl Clin Genet 4: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Diaz O, Zhang Y, Ladenheim B, Cadet JL, Chiang YH, Olson L, Hoffer BJ, Bäckman CM (2015) L-Dopa induced dyskinesias in Parkinsonian mice: Disease severity or L-Dopa history. Brain Res 1618: 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Cookson MR (2004) Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron 43: 301–304. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Cai Q (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13: 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova YA, Saarma M (2020) Can Growth Factors Cure Parkinson’s Disease? Trends Pharmacol Sci 41: 909–922. [DOI] [PubMed] [Google Scholar]
- Sloan M, Alegre-Abarrategui J, Potgieter D, Kaufmann AK, Exley R, Deltheil T, Threlfell S, et al. (2016) LRRK2 BAC transgenic rats develop progressive, L-DOPA-responsive motor impairment, and deficits in dopamine circuit function. Hum Mol Genet 25: 951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Browne SE, Jayaraman S, Bleickardt CJ, Hodge LM, Lis E, Yao L et al. (2014) Effects of the selective adenosine A2A receptor antagonist, SCH 412348, on the parkinsonian phenotype of MitoPark mice. Eur J Pharmacol 728: 31–38. [DOI] [PubMed] [Google Scholar]
- Stocchi F (2003) Prevention and treatment of motor complications. Parkinsonism Relat Disord 9: S73–S81. [DOI] [PubMed] [Google Scholar]
- Subramaniam M, Althof D, Gispert S, Schwenk J, Auburger G, Kulik A, Fakler B, Roeper J (2014) Mutant alpha-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J Neurosci 34: 13586–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam SR, Chesselet MF (2013) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 106–107: 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT (2012) Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb Perspect Med 2: a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA (2011) The origins of oxidant stress in Parkinson’s disease and therapeutic strategies. Antioxid Redox Signal 14: 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6: 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RR, Khan SM, Smigrodzki RM, Onyango IG, Dennis J, Khan OM, Portelli FR, Bennett JP Jr (2012) RhTFAM treatment stimulates mitochondrial oxidative metabolism and improves memory in aged mice. Aging (Albany NY) 4: 620–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J (2009) R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A 106: 14622–14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschumi CW, Sharma R, Lynch WB, Beckstead MJ (2019) Retrograde neurotensin release from dopamine neurons drives long-term depression of substantia nigra dopamine signaling. [Pre-print] bioRxiv, doi: 10.1101/717843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren Olanow C, Bartus RT, Baumann TL, Factor S, Boulis N, Stacy M, Turner DA, et al. (2015) Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: A double-blind, randomized, controlled trial. Ann Neurol 78: 248–257. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Callaway JC (2000) A coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol 83: 3084–3100. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Neuhoff H, Franz O, Roeper J (2001) Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci 15: 3443–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, McGeer PL, Baimbridge KG, McGeer EG (1990) Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res 526: 303–307. [DOI] [PubMed] [Google Scholar]
- Yee AG, Lee SM, Hunter MR, Glass M, Freestone PS, Lipski J (2014) Effects of the Parkinsonian toxin MPP+ on electrophysiological properties of nigral dopaminergic neurons. Neurotoxicology 45: 1–11. [DOI] [PubMed] [Google Scholar]
- Zampese E, Surmeier DJ (2020) Calcium, Bioenergetics, and Parkinson’s Disease. Cells 9: 2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Granholm AC, Huh K, Shan L, Diaz-Ruiz O, Malik N, Olson L et al. (2012) PTEN deletion enhances survival, neurite outgrowth and function of dopamine neuron grafts to MitoPark mice. Brain 135(Pt 9): 2736–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]