

Abstract
Purpose of review
Splicing mutations are among the most recurrent genetic perturbations in hematological malignancies, highlighting an important impact of splicing regulation in hematopoietic development. However, compared to our understanding of splicing factor mutations in hematological malignancies, studies of splicing components and alternative splicing in normal hematopoiesis have been less well investigated. Here, we outline the most recent findings on splicing regulation in normal hematopoiesis and discuss the important questions in the field.
Recent findings
Recent studies have highlighted critical role of splicing regulation in hematopoiesis, including characterization of splicing components in normal hematopoiesis, investigation of transcriptional alterations on splicing, and identification of stage-specific alternative splicing events during hematopoietic development.
Summary
These interesting findings provide insights on hematopoietic regulation at a co-transcriptional level. More high-throughput RNA sequencing and functional genomic screens are needed to advance our knowledge of critical alternative splicing patterns in shaping hematopoiesis.
Keywords: SF3B1, SRSF2, U2AF1, ZRSR2, lineage-specific alternative splicing
INTRODUCTION
Hematopoiesis is the process of blood cell production by a rare population of hematopoietic stem cells (HSCs). Self-renewal and differentiation of HSCs are orchestrated by a series of transcriptional and gene regulatory events[1,2]. Our understanding of molecular regulation of normal hematopoiesis to date mostly stems from the study of transcription factors, post-translational modifications, and cell extrinsic factors which ultimately modify gene expression.
RNA splicing, the process by which non-coding sequences are removed from premature RNA to form mature messenger RNA, is a key regulator of gene expression and mediator of gene expression. This complex and dynamic process is executed by spliceosome machineries, large ribonucleoprotein complexes consisting of small nuclear RNAs (snRNAs) and splicing factor proteins. There are two types of spliceosome machineries in most eukaryotic cells: the major and minor spliceosomes. The majority of introns (>99.5%), which typically have GT-AC at their termini and variable sequences at their 5’ ends, are recognized and removed by the major spliceosome (Figure 1)[3]. The remaining class of introns (known as “U12-dependent introns”), present in <0.5% of human genes, are defined by highly conserved 5’ and 3’ oligonucleotides which define their termini (Figure 1)[3–7]. This rare class of minor introns are recognized and excised by the minor spliceosome (also known as the “U12 spliceosome”)[6,8]. The two spliceosome machineries are distinct in their snRNA composition and a portion of their associated splicing factor proteins (reviewed recently[9,10]).
Figure 1. Sequence features defining major and minor introns.
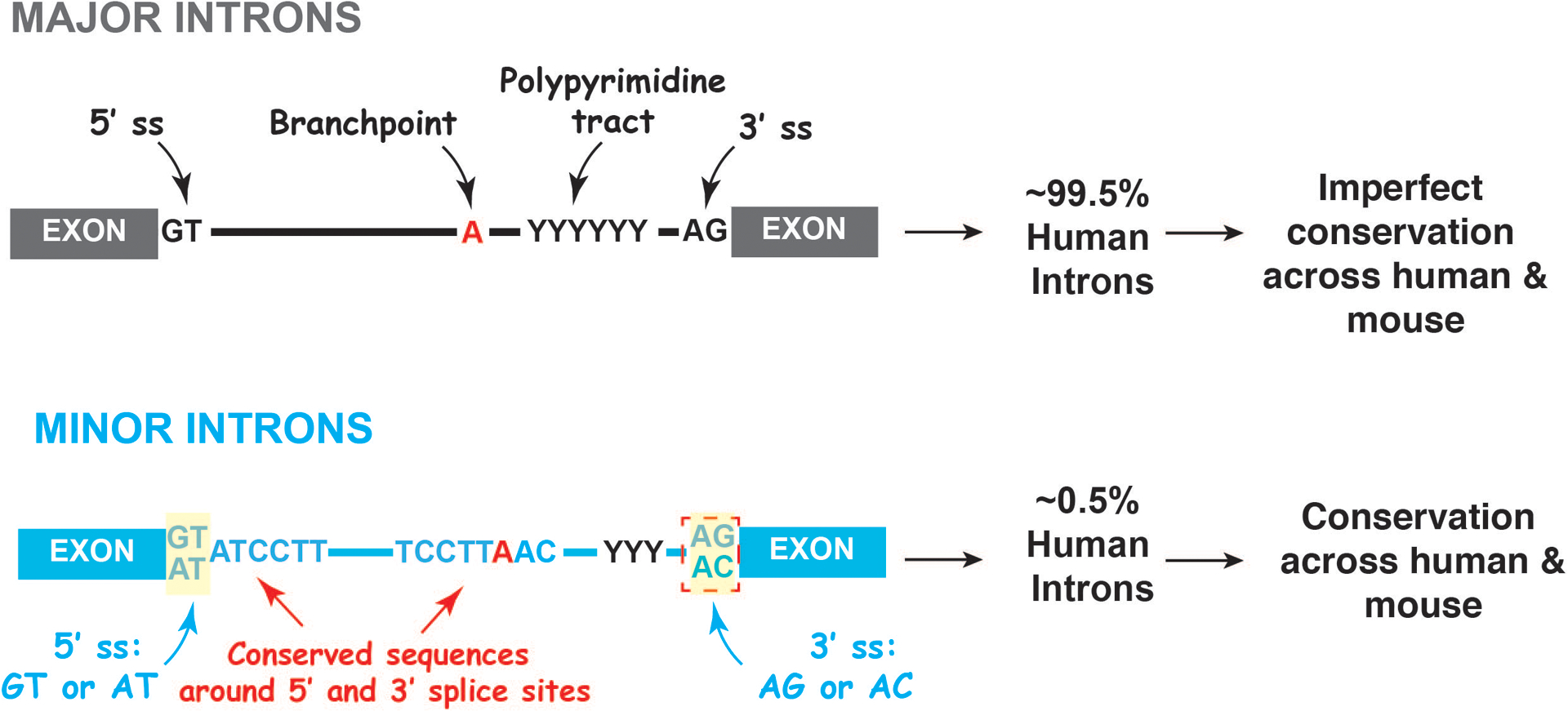
The majority of introns (>99.5%) have GT-AC dinucleotides at their termini and are not well conserved across species (upper panel). These introns are recognized and excised by the major spliceosome. The remaining class of introns (known as “U12-type minor introns”), present in <0.5% of human genes, have highly conserved 5’ and 3’ sequences at their termini (lower panel). This rare class of minor introns are recognized and excised by the minor spliceosome.
Splicing factor mutations are common to all forms of myeloid malignancies including acute myeloid leukemia (AML) and myeloid proliferative neoplasms (MPN)[11–18]. In particular, more than 50% of patients with myelodysplastic syndromes (MDS), clonal blood disorders that are characterized by impaired hematopoiesis, carry a mutation affecting an RNA splicing factor gene[11–13,18]. The molecular effects of mutations in RNA splicing factors have been described in previous reviews[19–21]. In this review, we focus on recent insights on the regulation of splicing during normal hematopoietic development, including the biological role of splicing factors and other RNA regulators in normal hematopoiesis, and stage-specific alternative splicing patterns during hematopoietic development.
Splicing factors altered in hematopoiesis
RNA splicing factor mutations in leukemia are concentrated in four genes (SF3B1, SRSF2, U2AF1, and ZRSR2)[11–13,18,22]. The discoveries that mutations affecting splicing factors are amongst the most recurrent genetic alterations in hematological malignancies underscore the importance of fine-tuned splicing regulation of hematopoiesis. Extensive efforts have been devoted to determining the biological and molecular impacts of splicing factor mutations in hematological malignancies. Here, we summarize recent research on the role of splicing factors in normal hematopoiesis (Table 1).
Table 1-.
Summary of splicing regulators and their roles in normal hematopoiesis from recent publications.
| Splicing regulators | Description | Animal model and hematopoietic phenotypes |
|---|---|---|
| SF3B1 | SF3B1 is a component of the U2 snRNP that recognizes branch point, and promotes the binding of U2 snRNA to the branchpoint [11,12,23,24]. | Germline Sf3b1+/− murine model; Sf3b1 haploinsufficiency decreases HSC repopulating potential but does not increase ring sideroblast formation [27,28]. |
| U2AF1 | U2AF1 is a member of the U2AF heterodimer involved in the recognition of AG-dinucleotide at the 3’ splice site during pre-mRNA splicing [29–31]. |
U2af1fl/fl;Mx1-Cre murine model; Loss of U2af1 causes detrimental effects on HSC function and normal hematopoiesis [32*]. |
| SRSF2 | SRSF2 is a member of the serine/arginine-rich (SR) protein family involved in exon inclusion by binding to specific exonic splicing enhances (ESE) sequences [34–39]. |
Srsf2fl/fl;Mx1-Cre murine model; Srsf2 deletion leads to leukopenia, anemia, and bone marrow aplasia and compromised HSC self-renewal [39]. |
| ZRSR2 | ZRSR2 is a component of the minor spliceosome and primarily responsible for the U12 type minor intron excision [6,8]. |
Zrsr2fl/fl;Mx1-Cre murine model; Loss of Zrsr2 increases HSC number and self-renewal capability [40**]. |
| Breast carcinoma amplified sequence (BCAS2) | BCAS2 is a component of Prp19 complex involved in spliceosome assembly [44]. |
bcas2−/− zebrafish model; Bcas2 deletion causes severe defects in HSC function and definitive hematopoiesis [47]. |
| Protein arginine methyltransferase 5 (PRMT5) | PRMT5 mediates symmetric demethylation of arginines (SDMA) on Sm (D1, B/B, D3) proteins, a modification required for spliceosome assembly [51]. |
Prmt5fl/fl;Mx1-Cre murine model; Loss of Prmt5 decreases HSC quiescence leading to HSC exhaustion [54*]. |
| DEAD-box Helicase 41 (DDX41) | DDX41 is an RNA helicase that interacts with spliceosome components and is implicated in regulating pre-mRNA splicing [55]. |
ddx41sa14887 zebrafish model; Loss-of-function Ddx41 results in increased rate of endothelial-to-hematopoietic transition (ETH) and HSPC expansion and suppresses the expansion and differentiation of erythroid progenitors [57**,58]. |
Evaluation of the role of splicing factors in hematopoiesis in vivo
There are limited studies on the role of splicing components in normal hematopoiesis. SF3B1 is a member of the U2 small nuclear ribonucleoprotein complex and the most frequently mutated splicing component in MDS[11,12,23,24]. SF3B1 mutations in MDS are strongly associated with refractory anemia with ring sideroblasts (RARS)[11,12,25]. Consistent with SF3B1’s role in the core spliceosome, Sf3b1 plays an essential role during embryonic development and germline Sf3b1 knockout mice are embryonic lethal [26]. Moreover, studies of Sf3b1+/− mice showed that heterozygous deficiency of Sf3b1 decreases competitive advantage of HSCs in vivo suggesting that Sf3b1 function in a haploinsufficient manner in the hematopoietic system [27,28]. Further depletion of Sf3b1 by shRNA in Sf3b1+/− HSCs resulted in a greater defect in HSC repopulating capacity [27], revealing a critical role of Sf3b1 in HSC function. However, Sf3b1 haploinsufficiency does not result in formation of ring sideroblasts [27,28], which is consistent with the concept that MDS-associated mutations in SF3B1 confer a change of RNA splicing activity (and not simply loss of function).
U2AF1 is a member of the U2af heterodimer involved in the recognition of the 3’ splice site during pre-mRNA splicing[29–31]. Mutations in U2AF1 (mainly at the S34 and Q157 residues) are recurrent in MDS[11,13]. In a conditional U2af1 knockout mouse model, U2af1 deletion (via the Mx1-cre system) leads to early death with impaired hematopoietic stem cell (HSC) repopulation capacity and defective hematopoiesis[32*]. Hematopoietic stem and progenitor cell (HSPC) gene signatures were profoundly downregulated, and cell death and DNA damage were increased upon U2af1 deficiency[32*]. Clearly, U2af1 is essential in the maintenance of HSPC function. Loss of U2af1 in hematopoietic cells mostly caused exon skipping events[32*], consistent with a requirement of U2af1 in normal splicing catalysis. Clearly different from the molecular phenotype of U2af1 S34F mutant mouse models, sequence-specific changes at 3’ splice site were not observed in U2af1 null hematopoietic cells[32*,33].
SRSF2 is a member of the serine/arginine-rich (SR) protein family that facilitates exon recognition by binding to exonic splicing enhances (ESE) sequences within pre-mRNAs through its RNA recognition motif (RRM) domain[34–37]. SRSF2 recognizes consensus CCNG and GGNG motif sequence in mRNA, thereby promoting exon inclusion[38,39]. Loss of SRSF2 decreases cassette exons inclusion bearing either ESE. Previous work from our group showed that homozygous deletion of Srsf2 (through the Mx1-cre system) causes leukopenia, anemia, and bone marrow aplasia in the mice, and leads to severe compromise in HSC self-renewal in competitive transplantation[39]. We therefore demonstrated an indispensable role of Srsf2 in hematopoiesis. Noteworthy, unlike mouse model with conditional expression of heterozygous Srsf2P95H mutant (a hotspot SRSF2 mutant in human MDS), deletion of Srsf2 does not induce myeloid dysplastic phenotypes. These results indicate that the SRSF2 mutant exerts a change-of-function effect in the pathogenesis of MDS.
As noted above, “minor” or “U12-type” introns are present in only 700–800 genes in humans[5]. However, in contrast to the majority of introns, sequences at the 5’ and 3’ ends of minor introns are highly evolutionarily conserved[4,6], suggesting important functional regulatory properties. However, the biological role of the minor spliceosome function is largely unexplored. ZRSR2 is a minor spliceosome component (encodes by X-linked Zrsr2 gene), and mutations in ZRSR2 are commonly mutated in myeloid malignancies. Our group recently reported that aberrant splicing of U12-type introns driven by ZRSR2 loss contributes to profound expression changes of genes with important biological functions (Figure 2), such as the tumor suppressor gene LZTR1[40**]. Using a murine model for conditional deletion of Zrsr2 in hematopoietic cells, we discovered that loss of ZRSR2 increases the number as well as self-renewal capacity of HSCs in vivo[40**]. Further functional screens mimic RNA splicing events inducing nonsense mediated mRNA decay identified that mis-splicing of the RAS ubiquitination regulator Lztr1[41–43] contributes to clonal advantage in Zrsr2-deficient hematopoietic cells[40**]. This study demonstrates intriguing and unique role of minor splicing factor and U12-type introns in regulating HSC function.
Figure 2. Molecular and biological consequence of ZRSR2 loss in hematopoietic cells.
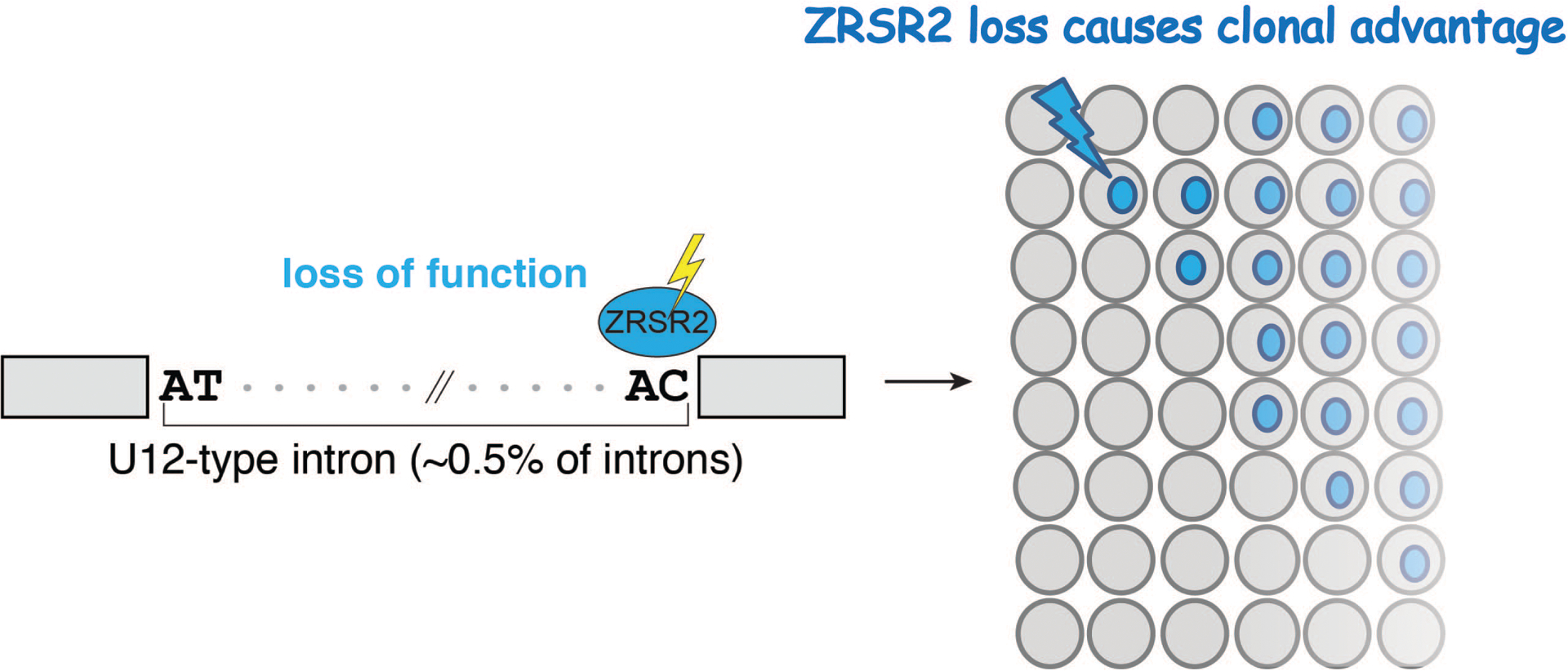
Loss of ZRSR2 induces aberrant splicing on U12 type minor introns and promotes hematopoietic clonal expansion.
BCAS2 (breast carcinoma amplified sequence) is a splicing related Prp19 Complex component which may be involved in spliceosome assembly[44]. Previous work has revealed important roles of BCAS2 in splicing regulation during developmental processes[45,46]. Recently, Yu, et al reported a novel role of BCAS2 in developmental hematopoiesis. The authors observed severe defects in HSPCs and definitive hematopoiesis in bcas2−/− zebrafish model, suggesting that bcas2 is required for HSPC development[47]. Mechanistically, bcas2 deletion induces exon 6 exclusion in Mdm4 and increases Mdm4 short isoform mRNA levels, which results in the production of truncated Mdm4 protein[47]. As a consequence, the alternative splicing of Mdm4 in bcas2−/− zebrafish embryos activate p53 pathway and trigger p53-mediated apoptosis in HSPCs, leading to impaired HSPC maintenance[47].
Other RNA regulators involved in regulation of hematopoiesis
Protein arginine methyltransferase 5 (PRMT5) regulates hematopoietic differentiation and plays important role in the context of AML[48–50]. Interestingly, PRMT5 mediates symmetric demethylation of arginines (SDMA) on Sm (D1, B/B, D3) proteins, a modification required for spliceosome assembly[51]. PRMT5 inhibitors which preferentially kill splicing factor mutant cells over their wild-type counterparts are currently in clinal trials for spliceosomal mutant myeloid neoplasms[52,53]. However, a recent study highlighted the importance of maintaining PRMT5 protein levels in the preservation of homeostatic hematopoiesis. PRMT5 deficiency causes decreased quiescence and subsequent exhaustion of the HSC compartment, leading to a detrimental impact on HSC function[54*]. The severe effect of PRMT5 reduction to HSCs was due to disruption of the splicing landscape, mostly affecting genes involved in the DNA damage repair pathway. The altered splicing events mostly consisted of intron retention and exon skipping. Importantly, majority of these splicing perturbations generate premature termination codons (PTCs), and are therefore predicted to lead to downregulation of gene expression[54*].
The gene encoding the DEAD-box Helicase 41 (DDX41) was recently been found mutated in hematological malignancies[55,56]. Ddx41 interacts with spliceosome components and is implicated in regulating pre-mRNA splicing[55]. A recent study established a critical role of DDX41 in regulating hematopoietic homeostasis. Zebrafish expressing a loss-of-function Ddx41 mutant uncovered that decreased Ddx41 resulted in increased rate of endothelial-to-hematopoietic transition (EHT) and HSPC expansion due to R-loop accumulation induced cGAS-STING inflammation pathway[57**]. At the same time, loss of Ddx41 suppressed the expansion and differentiation of erythroid progenitors, revealing an important role of Ddx41 in regulating erythroid differentiation[58]. In Ddx41 mutant HSPC and erythrocytes, pre-mRNA splicing pathway was one of the top downregulated gene sets when compared to their WT counterparts[57**,58]. The detailed mechanism of DDX41 in regulating RNA splicing is not yet clear.
Stage-specific splicing switch in hematopoietic development
Different stages of hematopoietic development are associated with distinct changes in cellular morphology as well as the transcriptome and proteome. High-throughput RNA sequencing (RNA-seq) has massively improved our understanding of alternative splicing throughout normal hematopoiesis for the past decade. As detailed reviewed by Inoue, et al.[20], the stage-specific switches in mRNA splicing have been studied using bulk RNA-seq of hematopoiesis. These include studies of normal human HSCs and downstream progenitors[59], murine granulopoiesis[60], murine and human erythropoiesis[61,62], and murine megakaryocyte differentiation[61]. These studies have illustrated that each stage of hematopoiesis is defined by lineage-specific alternative splicing, resulting in isoform specificity and stability control of the encoded proteins in each cell identity. However, few stage-specific splicing events have been functionally defined in normal hematopoiesis. Here, we summarize recent studies on the regulation and functional roles of stage-specific splicing switch in hematopoiesis.
Stage-specific, annotated alternative RNA isoforms
More than 90% of human genes undergo alternative splicing to generate multiple mRNA isoforms to subsequently give rise to distinct protein isoforms and functional diversity[63]. A study mapping stage-specific splicing isoform in human HSC development from fetal liver to cord blood and to bone marrow revealed isoform diversity along development[64]. This identified key HSC regulators displaying splicing alterations without affecting differential gene expression level[64]. For example, exon skipping of gene encoding high-mobility group AT hook 2 (HMGA2) was induced by splicing kinase CLK3[64], which phosphorylates serine/arginine-rich domains on splicing factors[65]. Functional experiments further validated that modulation of splicing of HMGA2 transcripts affects human HSCs function[64]. This comprehensive study characterized isoform diversity along human HSC development and highlighted the contribution of alternative splicing to developmental identity.
Another recent study revealed differential function of splicing variants in erythroid differentiation. The gene encoding BMP2K (bone morphogenetic protein 2 (BMP-2)-inducible kinase) is abundant in erythroid lineage cells[66,67]. Interestingly, this work identified that the longer isoform of BMP2K promotes, while the shorter isoform represses, erythropoiesis[67]. The antagonistic functions of each BMP2K isoform resulted from their distinct roles in autophagic degradation[67]. Importantly, this study proposed a model where not only splicing variant expression level, but also splicing variant ratio, are critical during erythroid differentiation.
Stage-specific intron retention
Physiologic regulation of intron retention has been posited to be an important mediator of normal hematopoietic lineage differentiation processes. For example, increasing abundance of intron-retained transcripts have been identified in maturation of normal erythroid development [61,62], where the intron retained transcripts are mainly sequestered in the nucleus [62]. Interestingly during granulopoiesis, intron-retained transcripts were reported in one study to be exported to the cytoplasm and undergo nonsense-mediated decay (NMD)[60]. While both fates of retained introns lead to decreased level of encoded protein, the mechanistic basis for distinct localization fates of retained introns in different hematopoiesis is not clear. Moreover, how intron retention patterns are changed during hematopoietic differentiation was not clarified by these studies. A recent study by Ullrich & Guigó performed a comprehensive characterization of intron retention events during hematopoietic differentiation[68*]. In line with previous reports[60], global intron retention levels were found to be highest in neutrophils and monocytes[60,68*]. Interestingly, intron retention events increase during the differentiation process from B-cell precursors towards mature cells in lymphoid organs but decrease in the late B cell affinity maturation stage[68*]. Importantly, based on RNA sequencing and eCLIP-sequencing analysis, the authors proposed that inefficient splicing due to lower expression levels of several non-core splicing factors may explain the increased global intron retention level during B cell differentiation[68*]. This finding implicates that lineage-specific regulation of splicing factors may also affect lineage commitment. More efforts are needed to characterize stage-specific regulation of splicing machineries in normal hematopoiesis. Apart from downregulation of encoded proteins by intron retention, a number of intron-retained mRNAs may proceed to translation of novel proteins. Ribosome profiling and functional genomics may help to define the potential role of novel intron-retained transcripts in hematopoietic development.
CONCLUSION
Recent findings have expanded our knowledge of normal hematopoiesis regulated at the pre-mRNA splicing level. However, there is a lack of investigation on transcriptional regulation of RNA splicing machinery during hematopoietic development and alternative splicing pattern during hematopoietic aging and lymphoid-to-myeloid bias. Additionally, it will be interesting to explore the potential crosstalk between splicing and extrinsic regulators on hematopoiesis, such as cytotoxic stresses, pro-inflammation induced cytokine storm, and metabolic changes. Further, high-throughput RNA sequencing and functional screen applications are in critical need to comprehensively address the above important questions in the field.
Key points:
Splicing factors function in the major (or U2) spliceosome are essential for hematopoietic stem cell (HSC) function and hematopoietic differentiation.
In contrast to major spliceosome components, which are essential for HSC survival, loss of certain minor spliceosome factors results in both increased numbers of HSCs in mice as well as increased HSC self-renewal.
Fine-tuning regulation of alternative splicing is critical for hematopoietic development.
Financial support and sponsorship
This work was supported by Lady Tata Memorial International Awards for Research in Leukaemia and ASH Research Restart Award to S.C. as well as R01 HL128239, R01 CA251138, and the Edward P. Evans MDS Foundation to O.A.-W.
Footnotes
Conflicts of interest
O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, Prelude Therapeutics, and Janssen, and is on the Scientific Advisory Board of Envisagenics Inc., AIChemy, and Pfizer Boulder; O.A.-W. has received prior research funding from H3B Biomedicine and LOXO Oncology unrelated to the current manuscript. The remaining authors declare no competing interests.
REFERENCES
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008, 132:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet 2000, 1:57–64. [DOI] [PubMed] [Google Scholar]
- 3.Sheth N, Roca X, Hastings ML, et al. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res 2006, 34:3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall SL, Padgett RA. Conserved sequences in a class of rare eukaryotic nuclear introns with non-consensus splice sites. J Mol Biol 1994, 239:357–365. [DOI] [PubMed] [Google Scholar]
- 5.Alioto TS. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 2007, 35:D110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell AG, Charette JM, Spencer DF, Gray MW. An early evolutionary origin for the minor spliceosome. Nature 2006, 443:863–866. [DOI] [PubMed] [Google Scholar]
- 7.Tarn WY, Yario TA, Steitz JA. U12 snRNA in vertebrates: evolutionary conservation of 5’ sequences implicated in splicing of pre-mRNAs containing a minor class of introns. RNA 1995, 1:644–656. [PMC free article] [PubMed] [Google Scholar]
- 8.Tarn WY, Steitz JA. A novel spliceosome containing U11, U12, and U5 snRNPs excises a minor class (AT-AC) intron in vitro. Cell 1996, 84:801–811. [DOI] [PubMed] [Google Scholar]
- 9.Wilkinson ME, Charenton C, Nagai K. RNA Splicing by the Spliceosome. Annu Rev Biochem 2020, 89:359–388. [DOI] [PubMed] [Google Scholar]
- 10.Rahman MA, Krainer AR, Abdel-Wahab O. SnapShot: Splicing Alterations in Cancer. Cell 2020, 180:208–208 e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478:64–69. [DOI] [PubMed] [Google Scholar]
- 12.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011, 365:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 2012, 44:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014, 371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014, 371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559:400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med 2018, 24:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28:241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 2016, 16:413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue D, Bradley RK, Abdel-Wahab O. Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev 2016, 30:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obeng EA, Stewart C, Abdel-Wahab O. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov 2019, 9:1493–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thol F, Kade S, Schlarmann C, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119:3578–3584. [DOI] [PubMed] [Google Scholar]
- 23.Cretu C, Schmitzova J, Ponce-Salvatierra A, et al. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol Cell 2016, 64:307–319. [DOI] [PubMed] [Google Scholar]
- 24.Teng T, Tsai JH, Puyang X, et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun 2017, 8:15522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malcovati L, Stevenson K, Papaemmanuil E, et al. SF3B1-mutant MDS as a distinct disease subtype: a proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isono K, Mizutani-Koseki Y, Komori T, et al. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev 2005, 19:536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, Sashida G, Saraya A, et al. Depletion of Sf3b1 impairs proliferative capacity of hematopoietic stem cells but is not sufficient to induce myelodysplasia. Blood 2014, 123:3336–3343. [DOI] [PubMed] [Google Scholar]
- 28.Matsunawa M, Yamamoto R, Sanada M, et al. Haploinsufficiency of Sf3b1 leads to compromised stem cell function but not to myelodysplasia. Leukemia 2014, 28:1844–1850. [DOI] [PubMed] [Google Scholar]
- 29.Merendino L, Guth S, Bilbao D, et al. Inhibition of msl-2 splicing by Sex-lethal reveals interaction between U2AF35 and the 3’ splice site AG. Nature 1999, 402:838–841. [DOI] [PubMed] [Google Scholar]
- 30.Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3’ splice site AG by the splicing factor U2AF35. Nature 1999, 402:832–835. [DOI] [PubMed] [Google Scholar]
- 31.Gozani O, Potashkin J, Reed R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 1998, 18:4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutta A, Yang Y, Le BT, et al. U2af1 is required for survival and function of hematopoietic stem/progenitor cells. Leukemia 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This report describes the role of U2af1 in murine hematopoietic stem and progentor cells.
- 33.Ilagan J, Ramakrishnan A, Hayes B, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. BioRxiv 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graveley BR, Maniatis T. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell 1998, 1:765–771. [DOI] [PubMed] [Google Scholar]
- 35.Liu HX, Chew SL, Cartegni L, et al. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol 2000, 20:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaal TD, Maniatis T. Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol 1999, 19:261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zahler AM, Damgaard CK, Kjems J, Caputi M. SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing. J Biol Chem 2004, 279:10077–10084. [DOI] [PubMed] [Google Scholar]
- 38.Daubner GM, Clery A, Jayne S, et al. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 2012, 31:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim E, Ilagan JO, Liang Y, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue D, Polaski JT, Taylor J, et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat Genet 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This is the first study to delineate the biological and molecluar impact of a minor spliceosome component in hematopoeisis, which are disctinct from splicing factors involved in major spliceosome. It identifies unique roles for minor spliceosome excision in HSC self-renewal.
- 41.Bigenzahn JW, Collu GM, Kartnig F, et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362:1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steklov M, Pandolfi S, Baietti MF, et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362:1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castel P, Cheng A, Cuevas-Navarro A, et al. RIT1 oncoproteins escape LZTR1-mediated proteolysis. Science 2019, 363:1226–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ajuh P, Kuster B, Panov K, et al. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J 2000, 19:6569–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen PH, Lee CI, Weng YT, et al. BCAS2 is essential for Drosophila viability and functions in pre-mRNA splicing. RNA 2013, 19:208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou MH, Hsieh YC, Huang CW, et al. BCAS2 Regulates Delta-Notch Signaling Activity through Delta Pre-mRNA Splicing in Drosophila Wing Development. PLoS One 2015, 10:e0130706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu S, Jiang T, Jia D, et al. BCAS2 is essential for hematopoietic stem and progenitor cell maintenance during zebrafish embryogenesis. Blood 2019, 133:805–815. [DOI] [PubMed] [Google Scholar]
- 48.Liu F, Cheng G, Hamard PJ, et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest 2015, 125:3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013, 13:37–50. [DOI] [PubMed] [Google Scholar]
- 50.He X, Zhu Y, Lin YC, et al. PRMT1-mediated FLT3 arginine methylation promotes maintenance of FLT3-ITD(+) acute myeloid leukemia. Blood 2019, 134:548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matera AG, Wang Z. A day in the life of the spliceosome. Nature reviews. Molecular cell biology 2014, 15:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eram MS, Shen Y, Szewczyk M, et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem Biol 2016, 11:772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fong JY, Pignata L, Goy PA, et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36:194–209 e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan DQ, Li Y, Yang C, et al. PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep 2019, 26:2316–2328 e2316. [DOI] [PubMed] [Google Scholar]; * This study describes the consequence of loss of PRMT5 on biological function and splicing pattern changes in hematopoietic stem cells.
- 55.Polprasert C, Schulze I, Sekeres MA, et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27:658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017, 17:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weinreb JT, Ghazale N, Pradhan K, et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev Cell 2021, 56:627–640 e625. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This is the first report of the role of Ddx41 in hematopoietic stem and progenitor cells during embryonic development.
- 58.Weinreb JT, Gupta V, Sharvit E, et al. Ddx41 inhibition of DNA damage signaling permits erythroid progenitor expansion in zebrafish. Haematologica 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen L, Kostadima M, Martens JH, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science 2014, 345:1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong JJ, Ritchie W, Ebner OA, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 2013, 154:583–595. [DOI] [PubMed] [Google Scholar]
- 61.Edwards CR, Ritchie W, Wong JJ, et al. A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood 2016, 127:e24–e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pimentel H, Parra M, Gee SL, et al. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cesana M, Guo MH, Cacchiarelli D, et al. A CLK3-HMGA2 Alternative Splicing Axis Impacts Human Hematopoietic Stem Cell Molecular Identity throughout Development. Cell Stem Cell 2018, 22:575–588 e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Colwill K, Pawson T, Andrews B, et al. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. The EMBO journal 1996, 15:265–275. [PMC free article] [PubMed] [Google Scholar]
- 66.Potts MB, Kim HS, Fisher KW, et al. Using functional signature ontology (FUSION) to identify mechanisms of action for natural products. Sci Signal 2013, 6:ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cendrowski J, Kaczmarek M, Mazur M, et al. Splicing variation of BMP2K balances abundance of COPII assemblies and autophagic degradation in erythroid cells. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ullrich S, Guigo R. Dynamic changes in intron retention are tightly associated with regulation of splicing factors and proliferative activity during B-cell development. Nucleic Acids Res 2020, 48:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This is the first study to analyze intron retention in B cells and explains the potential mechanism of intron retention level change during B cell differentiation.