

Abstract
Phosphoinositides are an important class of anionic, low abundance signaling lipids distributed throughout intracellular membranes. The plasma membrane contains three phosphoinositides: PI(4)P, PI(4,5)P2, and PI(3,4,5)P3. Of these, PI(4)P has remained the most mysterious, despite its characterization in this membrane more than a half-century ago. Fortunately, recent methodological innovations at the chemistry–biology interface have spurred a renaissance of interest in PI(4)P. Here, we describe these new toolsets and how they have revealed novel functions for the plasma membrane PI(4)P pool. We examine high-resolution structural characterization of the plasma membrane PI 4-kinase complex that produces PI(4)P, tools for modulating PI(4)P levels including isoform-selective PI 4-kinase inhibitors, and fluorescent probes for visualizing PI(4)P. Collectively, these chemical and biochemical approaches have revealed insights into how cells regulate synthesis of PI(4)P and its downstream metabolites as well as new roles for plasma membrane PI(4)P in non-vesicular lipid transport, membrane homeostasis and trafficking, and cell signaling pathways.
Keywords: genetically encoded biosensors, GPCR signaling, lipid kinase inhibitors, phospholipid transport, phosphoinositides, PI4KIIIα, PI(4)P
1. Introduction
Cellular membranes are made of myriad types of lipids, with different structural and functional properties[1]. Phosphoinositides (PIPs) are low abundance phospholipids found on the cytoplasmic leaflet of eukaryotic cell membranes[2]. They derive from phosphatidylinositol (PI), which consists of a glycerol core linked to two hydrophobic acyl chains and a phospho myo-inositol head group (Figure 1A). The inositol headgroup is the defining characteristic of PIPs, which feature its further modification by combinatorial phosphorylation at positions 3, 4, and/or 5 for a total of seven different head group-defined species. PIPs are synthesized by a network of highly regulated lipid kinases and phosphatases that can interconvert these lipids from one species to another, and whose cellular functions and regulation are extensively reviewed elsewhere[2,3] (Figure 1B). The different PIPs are present at different levels in the cell, and their relative amounts can vary in a tissue specific manner. Broadly speaking, PI(4)P and PI(4,5)P2 are the most abundant PIPs in mammalian cells, consisting of about 0.5–1.5% of total phospholipids[4–6]. The next-most abundant PIP is PI(3)P, whose levels are ~10–25% of the levels of these PI(4)P, while PI(5)P and PI(3,4,5)P3 represent much smaller fractions (0.1–1%) of the amount of PI(4)P and PI(4,5)P2, under resting conditions[6].
Figure 1. Structures and subcellular localization of the phosphoinositides.
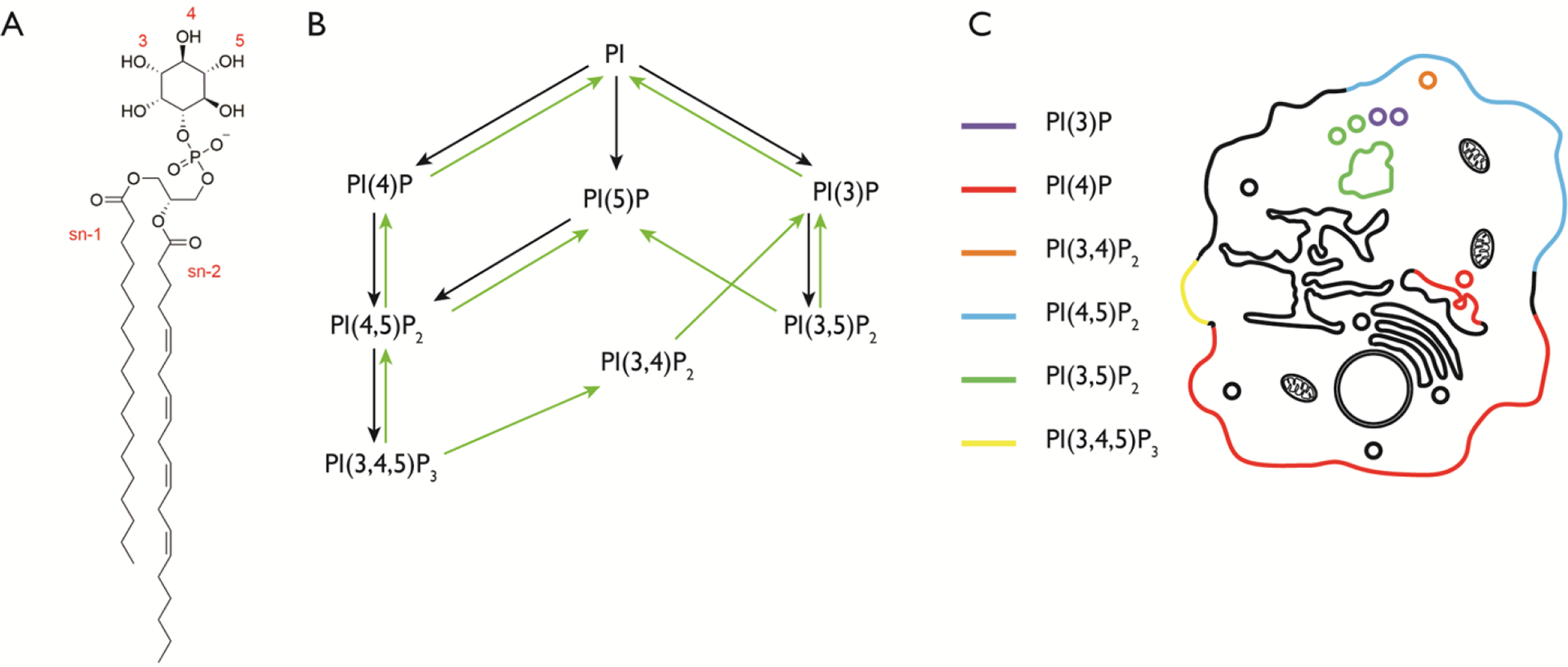
(A) The structure of phosphatidylinositol (PI). The hydroxyl groups of the inositol ring are phosphorylated at the indicated positions (3, 4, and/or 5) to generate phosphoinositides (PIPs). The illustrated PI structure is the most abundant in mammalian cells, containing stearoyl and arachidonyl groups at the sn-1 and sn-2 positions respectively. (B) The seven different PIPs are shown, with known pathways of conversion from one species to another. Black arrows represent phosphorylation reactions and green arrows represent dephosphorylation reactions. (C) Cartoon illustrating the localization of the major pools of the seven different PIP species.
Each PIP species has a distinct subcellular localization. For example, PI(4,5)P2 is predominantly localized at the plasma membrane (PM) and PI(3)P is found primarily on early endosomes[7,8]. This seemingly different localization for each PIP species, as well as their ability to engage specific protein binding partners, has led to the idea that PIPs act as organelle membrane identity markers. The picture is, however, much more complex, as certain PIPs, such as PI(4)P, are present on several organelle membranes[9–11], and certain membranes have more than one PIP species (Figure 1C).
The PM is perhaps the richest and most storied membrane in the study of PIP biology. It is home to three PIPs with precursor–product relationships: PI(4)P, PI(4,5)P2, and PI(3,4,5)P3, which are generated by sequential phosphorylation of PI at the 4, 5, and 3 positions by dedicated lipid kinases (Figure 2). PI in the PM is phosphorylated by PI4KIIIα to PI(4)P[9,12]. Further phosphorylation by PI4P 5-kinases and PI 3-kinases results in the synthesis of PI(4,5)P2[13] and PI(3,4,5)P3[14]. Whereas the functions of these lipids in the PM have been thoroughly investigated, the many of the roles of PI(4)P have only recently come to light. The lion’s share of attention has been devoted to PI(4,5)P2 and PI(3,4,5)P3, which play critical roles in regulation of physiological processes occurring in the PM, including endocytosis[15], ion channel regulation[13], actin dynamics[15], chemotaxis[16–18], mTORC2 activation and insulin signaling[19,20] and mitogenic signaling downstream of several growth factor receptors[3]. The functions of PI(4,5)P2 and PI(3,4,5)P3, the other major PM PIPs are reviewed in depth elsewhere[2,21,22]. By contrast, PI(4)P at the PM has, until recently, received far less attention. Part of this gap is due to a dearth of tools for visualizing and perturbing PM pools of this lipid and a lack of understanding for how production of PI(4)P is regulated. Fortunately, in recent years there has been a flurry of advances regarding PI(4)P at the PM, many of them at the chemistry–biology interface. Importantly, improved toolsets have spurred mechanistic biological investigations that have uncovered exciting and unexpected roles for PM PI(4)P, independent of roles as a biosynthetic precursor to PI(4,5)P2 and PI(3,4,5)P3.
Figure 2. Sequential phosphorylation reactions of PI to generate multiple phosphoinositides at the PM.
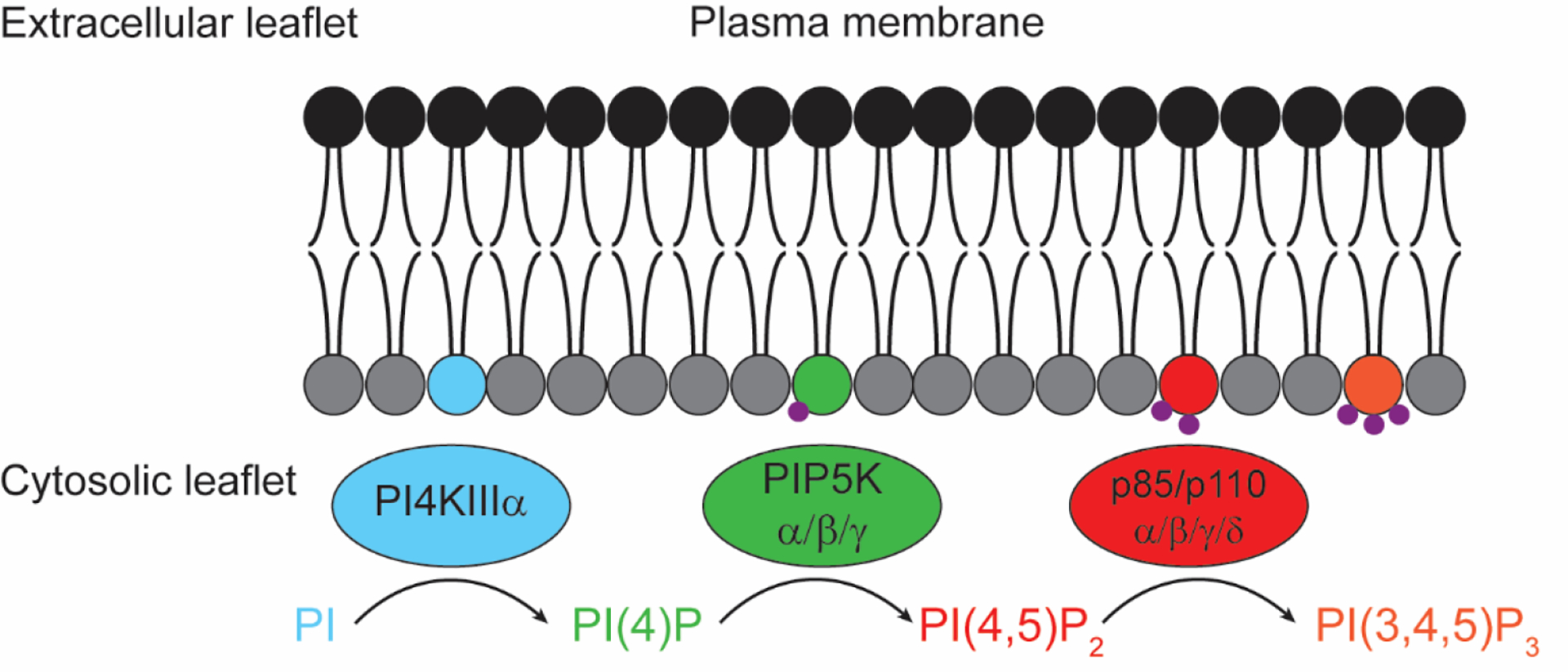
Shown are the principal lipid kinase isoforms responsible for conversion, at the PM, of PI to PI(4)P (PI4KIIIα), PI(4,5)P2 (PIP5Kα/β/γ), and PI(3,4,5)P3 (p110α/β/γ/δ).
This review will focus on the latest advances in our understanding of molecular roles for the pool of PI(4)P at the PM. First, we will describe recent findings concerning the biosynthetic machinery responsible for producing and regulating PI(4)P production at the PM, including structural biology breakthroughs to characterize this machinery at high resolution. Second, we will survey recently reported chemical and biochemical toolsets for visualizing, measuring, and perturbing PI(4)P at the PM. These tools include fluorescent biosensors for visualizing PI(4)P, advanced mass spectrometry methods for profiling PI(4)P, highly selective inhibitors for the PI(4)P biosynthetic machinery, and genetically encoded enzymes for perturbing PM PIP content. Third, we will summarize selected examples of newly discovered biological roles for the PM pool of PI(4)P, including functions in endocytosis, non-vesicular lipid transport, and G protein-coupled receptor signaling. Finally, we will conclude by providing an outlook focusing on remaining outstanding questions and opportunities for further methodology development.
2. The plasma membrane PI(4)P biosynthetic machinery
2.1. PI 4-kinase Type IIIα (PI4KIIIα) forms complexes that synthesize PI(4)P at the PM
PI(4)P is found in independent pools at the PM, where it is one of the most abundant PIPs[23], in the Golgi complex, and on certain populations of endosomes and vesicles. The pools of PI(4)P on different organelles are largely independent of each other, as each is synthesized by phosphorylation of PI by a different PI 4-kinase isoform[24], and loss of one isoform is not effectively compensated by the activities of the remaining isoforms[25,26]. The bulk of the PM pool of PI(4)P is synthesized by PI 4-kinase Type IIIα, a 237 kDa globular protein (PI4KIIIα, encoded by PI4KA in humans and known as Stt4 in yeast)[9]. Although it is believed to phosphorylate PI in the PM, PI4KIIIα is not irreversibly anchored in this membrane. In fact, it was initially reported to localize to several intracellular organelle membranes[27,28]. However, studies of its yeast ortholog Stt4, later confirmed in mammalian cells, have established that PI4KIIIα is recruited to the PM in a complex with at least two accessory proteins, EFR3A/B (Efr3 in yeast) and TTC7A/B (Ypp1 in yeast)[9,29].
EFR3 serves as a membrane anchor for the PI4KIIIα complex. The mammalian EFR3s are ~90 kDa peripheral membrane proteins bearing posttranslational S-acylation (i.e., palmitoylation) within a Cys-rich N-terminal motif[9,30]. This palmitoylation, which is absent from yeast Efr3, anchors EFR3 to the membrane, and a polybasic patch on its N-terminal folded domain, which based on x-ray crystallography studies of yeast Efr3 is an all-α-helical rod, restricts EFR3 to the PM, preventing its endocytosis[9,31].
EFR3 does not bind directly to PI4KIIIα; instead, both proteins interact with a ~95 kDa scaffold protein, TTC7A/B (Ypp1 in yeast). TTC7 is a mostly globular, α-helical protein that is functionally conserved from yeast to humans. A third non-catalytic component of the PM PI4KIIIα complex, FAM126 (a ~57 kD protein also encoded by two genes, FAM126A/B, in mammals), is specific to the metazoan lineage, and orthologs are absent from yeast. FAM126 forms a tight heterodimer with TTC7, stabilizing TTC7 and therefore further enhancing PM recruitment of PI4KIIIα[32].
Though complete loss of PI4KIIIα is lethal in all tested organisms from yeast to mice, mutations to certain accessory proteins can permit life but cause disease. Mutations to TTC7A and FAM126A cause congenital human diseases affecting immunity and gastrointestinal function (TTC7A) and a hypomyelinating leukodystrophy (FAM126A, also known as hyccin).[27,33–36] Patient-derived cells exhibit defects in PM PI(4)P synthesis, suggesting that tissue-specific defects in this lipid synthetic pathway underlies disease pathogenesis[32,37]. Even inactivating mutations to PI4KA, whose expression is highest in the brain, can cause a neurological disease, though presumably there is residual PI4KIIIα activity in these patients[38]. In addition to its developmental functions, PI4KIIIα is required for the successful replication of several types of viruses[39], the most extensively studied being the Hepatitis C virus (HCV)[40–43]. Reduction of PI4KIIIα activity using siRNA or PI4KIIIα-selective inhibitors resulted in drastically reduced viral replication[41,43–45]. Understanding the regulatory networks governing PI4KIIIα and PI(4)P synthesis at the PM, therefore, has strong implications for human health.
Beyond EFR3, TTC7, and FAM126, a fifth protein is reported to take part in PI4KIIIα recruitment and activation at the PM. Sfk1, or suppressor of four-kinase, is a small (29 kDa), six-pass transmembrane protein identified in yeast that aids in Stt4 recruitment to the plasma membrane[46]. Of five potential mammalian orthologs of Sfk1 (TMEM150A–C and DRAM1/2), only TMEM150A can recapitulate the function of Sfk1[47]. TMEM150A promotes the recovery of PM levels of PI(4,5)P2 only after acute, PLC-mediated depletion of both PI(4,5)P2 and PI(4)P[47]. Interestingly, the cytosolic tail of TMEM150A facilitates its interaction with PI4KIIIα and EFR3 but prevents interaction of these proteins with TTC7. This observation supports a model wherein PI4KIIIα can exist in two different complexes, both with EFR3 but one containing TTC7 (and presumably FAM126) and a second containing TMEM150A. However, this model has yet to be tested rigorously, and mechanisms by which PI4KIIIα switches between the two complexes, as well as their functional differences, remain unexplored.
2.2. Cryo-EM and HDX-MS have revealed structure and dynamics of the PI4KIIIα complex
Whereas the x-ray crystallographic characterization of various non-catalytic components of the PI4KIIIα complex discussed above, e.g., yeast Efr3 and Ypp1[31], and mammalian TTC7/FAM126[32], revealed important information about how these molecules interact with the membrane and with each other, there was no structural information about PI4KIIIα itself or how it assembles with its non-catalytic partners. In 2017, the Reinisch lab reported a cryo-EM structure for what could be considered the core PI4KIIIα complex (PI4KIIIα, TTC7B, and the folded, globular portion of FAM126A[37,48] At a maximum resolution of 3–4 Å at the core of the complex, this structure revealed several previously unknown features of the kinase and its non-catalytic subunits. Interactions between PI4KIIIα, TTC7 and FAM126 bury an extensive surface area of the proteins, supporting the idea that these proteins form a complex in the cytosol and are recruited en masse to the membrane by EFR3 via interactions with its long, unstructured C-terminal tail (Figure 3).
Figure 3. Structure of the PI4KIIIα complex at the PM.
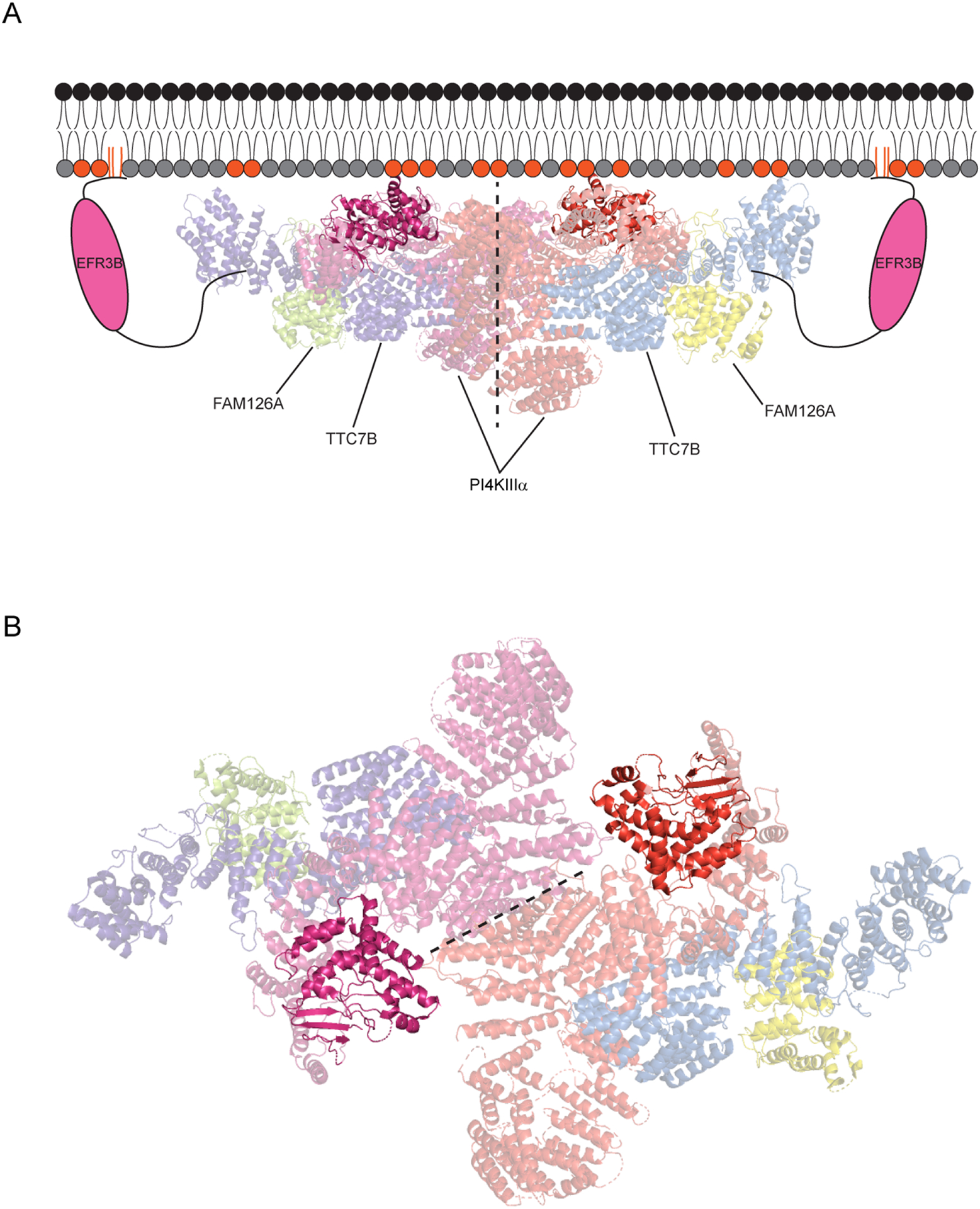
(A) EFR3B anchors the complex at the PM and recruits an ~800 kDa, hexameric complex involving a dimer of TTC7B/FAM126A/PI4KIIIα heterotrimers (~1 MDa with two molecules of EFR3B included). The kinase and TTC7B engage in stabilizing electrostatic interactions with negatively charged lipids in the inner leaflet of the PM (indicated with colored head groups). The dimerization interface between the kinases is shown with a dotted line, and the active site of each PI4KIIIα molecule is highlighted, to show its proximity to the membrane. (B) A view of the PI4KIIIα complex looking down from the plasma membrane with the kinase domains highlighted. The kinase dimerization interface is represented by a dotted line and the PI4KIIIα active sites are highlighted.
This model is consistent with the observation that forced overexpression of PI4KIIIα results in elevated protein levels (and minimal degradation) in the presence of forced co-expression with TTC7B and FAM126A, which facilitates their co-purification as a complex, as well as imaging studies showing colocalization of fluorescently tagged versions of these three proteins in the same compartment[32,37]. Additionally, this structure revealed the PI4KIIIα–TTC7–FAM126 complex as a hexamer, i.e., a dimer of heterotrimers, suggesting that, at the PM, the kinase complex likely assembles in such a form, containing two molecules of each protein component and constituting roughly 800 kDa total. Incorporating two molecules of EFR3, which are not present in the cryo-EM structure but are necessary for PI4KIIIα localization to the PM via direct interactions with TTC7, leads to a model for a rather hefty PM PI4KIIIα complex, weighing in at approximately 1 MDa (Figure 3).
Interestingly, this cryo-EM structure revealed that TTC7B and PI4KIIIα engage in electrostatic interactions with the PM through conserved basic residues on the protein surface. These interactions are not sufficient to localize these proteins to the PM in the absence of EFR3 but are likely to stabilize the assembled complex once recruited to the membrane and could in principle aid in partitioning it within the bilayer to regions enriched in certain lipids or bring the catalytic domain closer to the membrane to facilitate interaction with its lipid substrate, PI. This finding is consistent with an earlier study of PI4KIIIα that showed that overexpressing the kinase alone resulted in cytoplasmic localization of the protein, with transient interactions with the PM observable by TIRF microscopy[9].
Subsequently, Burke and coworkers reported a similar overall structure of the PI4KIIIα–TTC7–FAM126 complex by fitting the high-resolution structural data from the Reinisch lab’s structure into a lower resolution electron density map generated from cryo-EM. Here, the authors used complexes with either the N-terminal FAM126 globular domain (as in the previous work) or full-length FAM126, incorporating its uncharacterized long, disordered C-terminal tail into the structure[48]. Interestingly, kinase activity assays revealed that inclusion of the C-terminal FAM126 tail substantially lowered PI4KIIIα activity relative to the complex with only the N-terminal, globular FAM126 domain, suggesting a regulatory role for this previously mysterious tail.
The dynamics of PI4KIIIα complex assembly was examined using hydrogen-deuterium exchange mass spectrometry (HDX-MS). This method measures the kinetics of exchange of polypeptide backbone hydrogen for deuterium in the presence of D2O, a property that is heavily influenced by participation in stabilizing interactions (e.g., secondary structure, protein-protein interactions, protein-membrane interactions, protein-small molecule interactions, etc.)[48,49]. HDX-MS analysis of the PI4KIIIα complex revealed several insights. First, the FAM126 C-terminal tail engaged in no discernable interactions with the rest of the complex, suggesting its mechanism for attenuating kinase activity might lie in altered membrane binding of the kinase. As well, the mechanisms of two PI4KIIIα inhibitors (to be discussed more below) were investigated: the highly specific GSK-A1 and the non-specific Pik93. Both inhibitors reduced hydrogen-deuterium exchange in the ATP binding pocket and the ligand-binding site of the kinase. However, GSK-A1 also induced large-scale changes in deuterium exchange, suggesting that its binding causes allosteric structural changes in the kinase. Other regions showed an increase in hydrogen-deuterium exchange upon GSK-A1 binding, such as the N-terminal lobe of the kinase. Such changes were not observed in presence of Pik93 and may be a characteristic of specific, highly potent PI4KIIIα inhibition and could be useful in future inhibitor design efforts.
Collectively, these structural studies mark major advances in our understanding of PI4KIIIα assembly and regulation at the PM and targeting by small molecules. They lay the groundwork to address the many questions that remain unanswered about this lipid kinase complex: (1) How is assembly and disassembly of the kinase complexes regulated? (2) What factors govern handoff of PI4KIIIα between the non-TMEM150A-containing and TMEM150A-containing complexes? (3) What roles do dynamic posttranslational modifications (e.g., phosphorylation, palmitoylation) play in regulation of the kinase? (4) Can specific interactions between viral non-structural proteins and PI4KIIIα be harnessed to develop drugs that block this interaction but still permit physiological PI(4)P production at the PM?
The roles of the PI4KIIIα complex subunits in diverse human diseases are also not fully understood. The phenotypic manifestation of mutations in EFR3A, TTC7A, and FAM126A occur in a tissue-specific manner[32–35,50–52], raising the possibility of specialized functions or tissue-specific PI(4)P requirements fulfilled by these subunits. Mutations in TTC7A have been linked to severe combined immunodeficiency and early onset irritable bowel disease[51] and disruption of the epithelial barrier and cell polarity inversion in the gastrointestinal tract[52]. Most of these mutations result in complete loss of function, though one patient variant of TTC7A carries a mutation resulting in loss of only a small portion of its C terminus. This TTC7A variant is expressed at wild-type levels and permits PI4KIIIα complex assembly but not function, indicating that multiple interactions within the PI4KIIIα complex may be necessary for its full physiological function[37]. Expression data from the Human Protein Atlas show much higher expression of TTC7A relative to its functional paralog, TTC7B, in the gastrointestinal tract, suggesting a tissue-specific function for TTC7A. The significantly lower expression level of TTC7B in this tissue may explain why it cannot compensate for the loss of TTC7A function.
By contrast, EFR3A exhibits ubiquitous expression, whereas EFR3B is selectively expressed in the brain. Therefore, the link of EFR3A to autism spectrum disorders is harder to rationalize mechanistically, though in this instance the genetics is more complex, i.e., it is an increased risk factor identified by an elevated burden of de novo mutations in ASD probands compared to control[50], as opposed to a more clear-cut situation like TTC7A, where recessive inheritance of two loss-of-function alleles is causative for the disease.
Regarding FAM126A, the genetics are similar to TTC7A (i.e., autosomal recessive, disease-causing gene), but in a different disease affecting a different tissue: Hypomyelination and Congenital Cataract (HCC), a leukoencephalopathy that perturbs formation of the myelin sheath during childhood development. Based on expression data in the Human Protein Atlas, both FAM126A and its functional paralog, FAM126B, are expressed ubiquitously, though, in the nervous system, FAM126B is specifically abundant in photoreceptor cells whereas FAM126A mRNA is more ubiquitous, including in certain neuronal cell lines. It is important to note that, because of the complex cell–cell interactions involved in the formation of myelin, other more well-characterized heritable leukoencephalopathies can occur due to dysfunction not only in myelin-producing oligodendrocytes and Schwann cells but also in other cell types in the brain, including neurons and microglia.
Moreover, considering the well-established roles for PI(4)P, PI(4,5)P2[53] and PI(3,4,5)P3[54–56] for the formation and maintenance of the myelin sheath and the concomitant substantial membrane expansion, it is understandable that proper function and regulation of PI4KIIIα is required. FAM126A has been shown to increase the activity of PI4KIIIα, likely through a stabilizing mechanism[32], which may help oligodendrocytes and Schwann cells meet the high demands for phosphoinositides in membrane expansion and myelination. The loss of stability of the PI4KIIIα complex may explain the important role of FAM126A in myelin formation and maintenance and its connection to HCC, although the exact cell type(s) implicated in this dysfunction remain to be characterized.
3. Tools to visualize and perturb PI(4)P localization, abundance, and dynamics
The complex regulatory network controlling levels of PIPs is notoriously difficult to study. Visualizing and measuring different PIP pools poses significant challenges. Assigning functions to these specialized pools of lipids is also difficult because they are synthesized by enzymes that can generate potentially distinct lipid pools and play other regulatory roles. The design of isoform-selective lipid kinase inhibitors must surmount the conservation in active site architectures between different kinases. Yet, lipid kinase inhibitors are both powerful tool compounds for basic cell biology and, in several instances, have been translated for use in treating diseases caused by aberrant PIP metabolism[47,57–61]. As new functions for PI(4)P have been discovered, so to have the tools to study this lipid become more advanced; indeed, there has been a virtuous cycle between chemical and biochemical tool development and biological discovery in this arena. In this section, we will discuss tools to study PI(4)P and present their strengths and pitfalls to aid researchers in selecting the appropriate methods for their experiments.
3.1. Genetically encoded fluorescent probes for visualizing PM PI(4)P
Many proteins contain phosphoinositide-binding domains, the largest group being those bearing pleckstrin homology (PH) domains. These domains can be repurposed, when fused to fluorescent proteins, to act as genetically encoded probes, providing spatial information on the location of their target PIP in live cells (Figure 4). However, use of such probes comes with several caveats. Early probes such as the PH domains of FAPP1/2, Osh1, and OSBP bind PI(4)P in conjunction with ADP-ribosylation factor 1 (Arf1)[62–65], a Golgi-resident protein. Thus, these probes provide a Golgi-biased localization of PI(4)P and are unable to accurately report on PM PI(4)P (Figure 4A). Though the PH domain of OSBP is also capable of binding PI(4,5)P2, the PI(4)P/Arf1 coincidence detection mechanism restricts it to the Golgi[62].
Figure 4. Genetically encoded probes for visualizing PI(4)P involving fluorescent protein fusions to PI(4)P-binding domains.
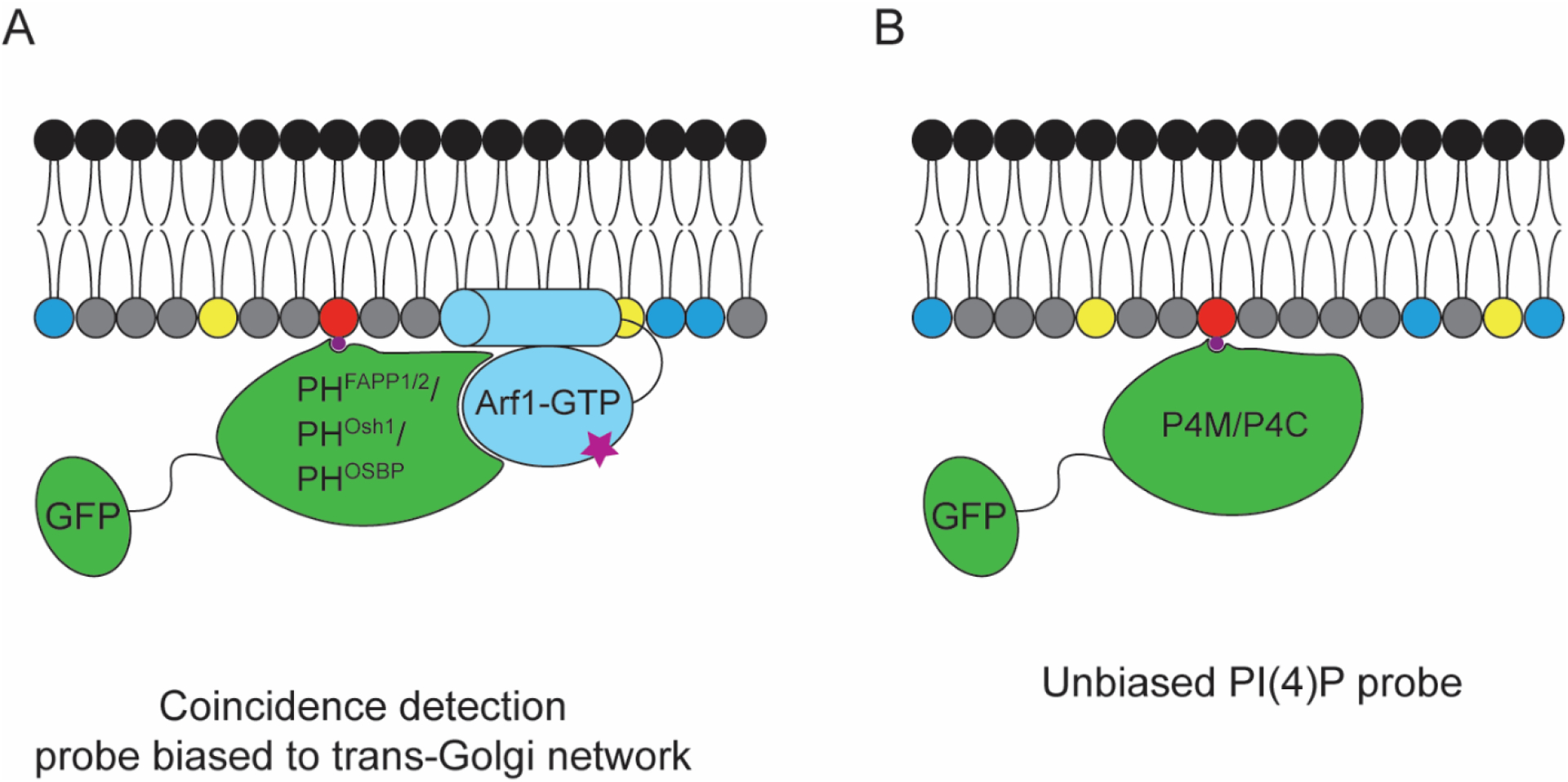
(A) PH domain-based probes from FAPP1, Osh1, and OSBP, capable of recognizing PI(4)P, but which are typically biased toward Golgi localization due to a coincidence detection mechanisms via secondary interactions with Arf1. (B) Unbiased PI(4)P probes take advantage of PI(4)P-binding motifs from Legionella pneumophila proteins SidM and SidC, termed P4M and P4C respectively, that do not engage in additional interactions that might affect probe localization.
Therefore, such probes are useful to study Golgi-resident pools of PI(4)P, such as those synthesized by the PI4KIIIβ isoform, but they are not well suited for studying global PI(4)P localization or the PM pools of this lipid. The PH domain of Osh2 localizes to the PM in mammalian cells, dependent on the presence of PI(4)P in that membrane, but this probe is biased to the PM because of its additional capability to recognize PI(4,5)P2[62]. More recently, two related PI(4)P probes were derived from effector proteins from Legionella pneumophila, the causative agent of Legionnaire’s disease: SidM and SidC[66,67]. The PI(4)P-binding motifs of these proteins, known as P4M and P4C respectively, exhibit high affinity for PI(4)P and do not rely on coincidence detection of a protein or lipid co-interactor to bind (Figure 4B). Thus, they provide an unbiased detection of PI(4)P and detect both PM and Golgi pools. Additionally, these probes are able to detect pools of PI(4)P on Rab7-positive late endosomes[66].
Probes like P4M and P4C can be used to study PI(4)P dynamics at the PM. Depletion and resynthesis of PI(4)P using tools described in sections 3.3–3.4 are monitored by imaging the loss of PM-associated signal and the increase in cytosolic fluorescence upon depletion, and return to the PM during PI(4)P recovery. Care must be taken when using such probes to study PI(4)P dynamics. Small but metabolically significant fluctuations in PI(4)P levels may remain undetected because of the transient nature of these changes and the binding affinity of the probe to its target. However, recently developed BRET-based PI(4)P and PI(4,5)P2 sensors are capable of monitoring small changes in the pools of these lipids during signaling events, allowing for finer dissection of signaling pathways and homeostatic feedback loops[68].
Because several pools of PI(4)P exist on different organelles, probes like P4M and P4C are in an equilibrium with each separate organelle pool and an unbound cytosolic pool. This scenario means that changes to PI(4)P levels in one location may have consequences on the overall probe localization. Another factor worthy of consideration is that PIP-binding probes can only recognize the labile, available pool of their target PIP and may not be sensitive to specialized pools that are tightly bound to other protein factors or otherwise inaccessible to the macromolecular probe. There are also instances when overexpression of a PIP-binding biosensor reduces the bioavailability of its target PIP, leading to phenotypic effects[69]. Though this property of using PIP-binding probes to mask PIPs can be useful, as they mimic loss-of-function, it can complicate their application as simple reporters of lipid abundance and localization.
Lipid-binding probes for the purpose of studying depletion, synthesis, and localization have led to important discoveries in the field of PIP biology. Unbiased probes have enabled precise analysis of the PI(4)P pool at the PM. These probes allow for basic quantification of PIP levels and their changes and provide spatiotemporal information about their target PIP in the context of live cells, with usually minimal disturbance to the system.
Alternative probes for visualizing PIPs in situ are PIP-specific antibodies[13]. Due to the requirement of cell fixation, antibody-based methods cannot report on dynamic changes in PIP levels in real time. Furthermore, even mild permeabilization of samples, required to permit intracellular access of antibodies while retaining PIPs within membranes, can nonetheless disrupt membrane morphology and composition, so careful controls must be applied when using these methods. Despite these caveats, such antibodies are useful tools for visualizing PIPs in samples in which genetically encoded probes are not suitable, such as in tissue sections. Even in cultured cells, their ability to reveal quantitative, spatially resolved information about PIP abundance sets them apart from localization-based, genetically encoded probes.
3.2. Liquid chromatography–mass spectrometry-based detection of PI(4)P
Chromatography methods have long been used to measure the PI(4)P content and changes after treatment. Thin layer chromatography (TLC) can be used to separate different native PIP species[70]. Cells can also be labeled with 3H-radiolabeled inositol, which is metabolically incorporated into the PIP head groups. Radiolabeled PIPs can then be extracted and detected by TLC and HPLC, increasing sensitivity. Such labeling is typically performed for one or several days to allow for steady-state incorporation of radiolabeled inositol into all PIP lipids. For probing rapid PIP synthesis, 32P-labeled phosphate can be pulsed into cells, resulting in its incorporation into ATP and subsequent phosphorylation of PI and PIPs. To circumvent safety and regulatory issues associated with using radioisotopes, liquid chromatography and mass spectrometry (LC–MS) methods have become sensitive enough to detect the minute amounts of non-labeled phosphoinositides from cellular and tissue extracts[71,72].
In particular, different mass spectrometry protocols have been described to improve ionization and detection of PIPs by LC–MS. The charged nature of PIPs (especially the three isomeric PIP2s and PI(3,4,5)P3) reduces their ionization efficiency. Methylation of the PIP head groups using TMS-diazomethane has proven effective at improving detection of low-abundance PIPs[23,73]. The addition of piperidine or triethylammonium acetate to the LC–MS mobile phases also improve PIP detection[5]. More recently, the addition of sodium formate was shown to improve the detection limit even further[74]. Another approach to detect PIPs is to deacylate the lipids prior to LC–MS[75]. This method results in the loss of the fatty acid groups, reducing sample complexity and changing solubility properties to facilitate measurement of the abundance of the resultant glycerol–PIP head groups. However, all acyl tail structural information is lost in these deacylation-based LC–MS protocols.
MS methods that analyze intact PIPs provide rich molecular information on acyl chain composition, including lengths and degrees of unsaturation. This advantage is useful for detecting the variations in PIP composition from different cell lines or tissues. For example, even different degrees of cell confluence can lead to varying degrees of unsaturation and tail length combinations[74]. One challenge associated with MS-based methods is distinguishing between regioisomeric PIP head groups (e.g., PI(4)P and PI(3)P). Fortunately, PI(4)P is much more abundant than its isomers, PI(3)P and PI(5)P, particularly at the PM, which can be biochemically enriched prior to analysis to reduce sample complexity[76,77]. Further, coupling MS with prior sample separation by liquid chromatography can resolve the PIP regioisomers[75,78]. Similar issues arise in detecting isomers based on acyl tail double bond placement. Remarkably, using specialized fragmentation methods, it is even possible to distinguish isomers of certain phospholipids with different double bond placement within acyl tails[79].
3.3. Pharmacological inhibitors of the PM PI(4)P biosynthetic machinery
A useful approach to study any system is to disrupt it and observe its return to equilibrium. This general framework can be applied to study PI(4)P at the PM by using PI4KIIIα inhibitors (Figure 5) or controllable enzymes that deplete or increase PI(4)P levels at the PM. Historically, the PI4KIIIα activity was characterized as relatively insensitive to inhibition by adenosine, as compared to its counterparts PI4KIIα/β[80], but sensitive to inhibition by wortmannin[81,82], an effective inhibitor of type III PI 4-kinases but an even more potent inhibitor of class I PI 3-kinases. These early inhibitors proved useful as tools in identifying the activity of PI4KIIIα. However, these inhibitors have significant off-target effects that make their use in more detailed study of PI(4)P biology challenging. Phenylarsine oxide (PAO), which has also been used extensively as a PI4KIIIα inhibitor, is a highly reactive small molecule that covalently tags thiol groups, especially those from vicinal cysteines[83] which appear in the active site of PI4KIIIα[84]. As such, PAO inhibits PI4KIIIα[84] but has effects on many other enzymes, including inhibition of PI 3-kinases[85], tyrosine phosphatases[83], and other uncharacterized effects. The off-target effects of these inhibitors can confound data collection and analysis and make it difficult to attribute a phenotype to specific effects on PI(4)P metabolism.
Figure 5. Chemical structures and IC50 values of PI4KIIIα inhibitors.
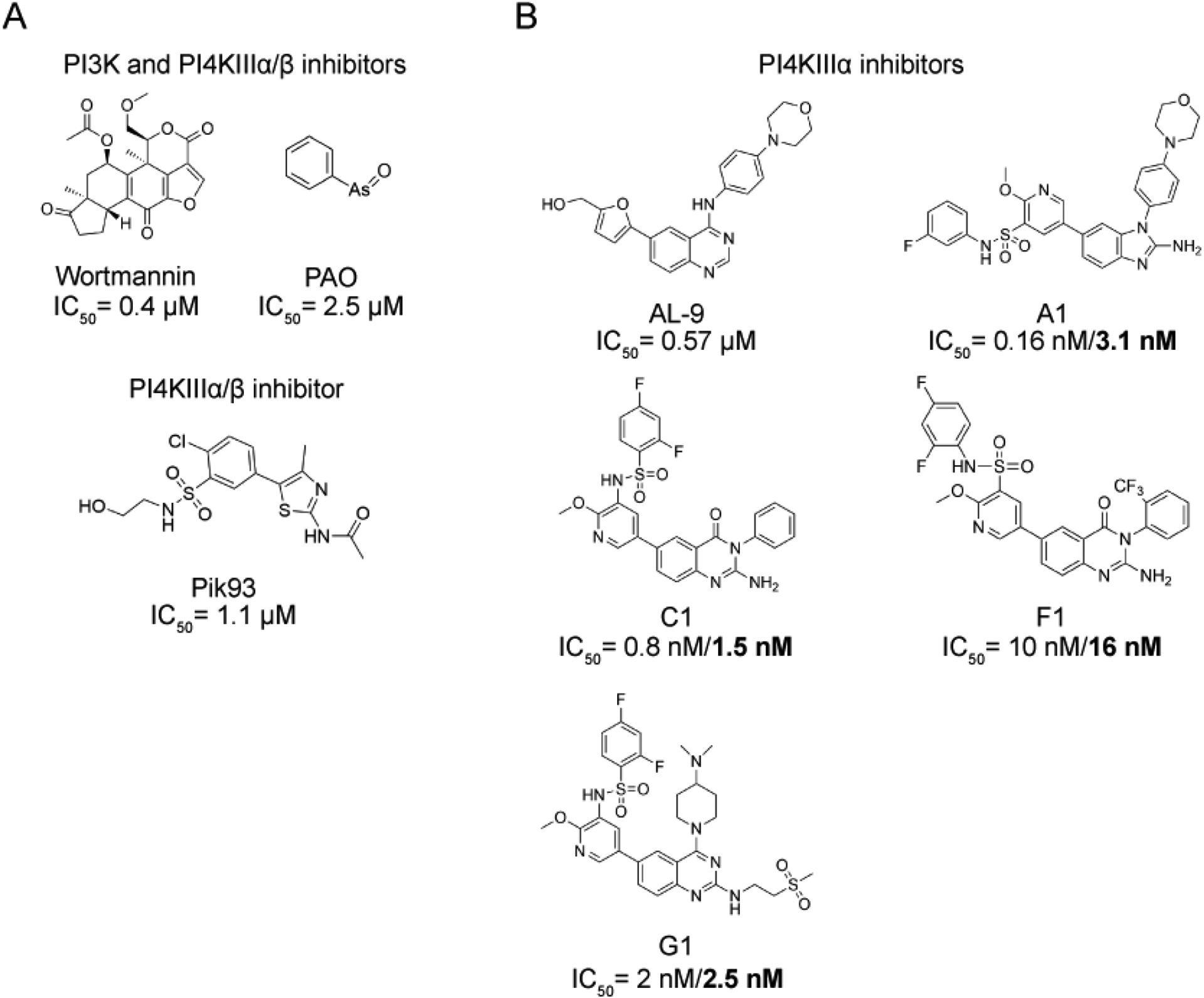
(A) Structures of relatively non-selective compounds that were, historically, used to inhibit PI4KIIIα, including phenylarsine oxide (PAO), Wortmannin, and Pik93. The IC50 values of Wortmannin, PAO and Pik93 for PI4KIIIα are referenced here, respectively[44,86,87]. PAO and wortmannin are broad-spectrum lipid kinase inhibitors, targeting PI 3-kinases and PI4KIIIα/β, whose kinase domains share homology with those of PI 3-kinases. Pik93 is more selective, inhibiting PI4KIIIα/β, with a higher affinity for the β form. (B) Structures of the recently developed, highly selective PI4KIIIα inhibitors AL-9, A1, C1, F1 and G1 with their IC50 values. Values in the standard font were measured with the ADP-Glo assay, and values in bold were measured by γ32 ATP PI-kinase assays[44].
Pik93 is an inhibitor first identified in a screen for PI 3-kinase inhibitors[87]. The authors also found that it potently inhibits PI4KIIIβ (IC50 of 19 nM) and PI4KIIIα at higher concentrations (IC50 of 1.39 μM). This inhibitor is a useful tool to inhibit the production of the Golgi-resident pool of PI(4)P synthesized by PI4KIIIβ or to completely inhibit PI(4)P synthesis when used at higher concentrations. However, it cannot be used to specifically inhibit PI4KIIIα while leaving the PI4KIIIβ-derived pool of PI(4)P intact. Additionally, Pik93 also inhibits PI 3-kinases at doses close to those that inhibit PI4KIIIα, which could have downstream effects on PIP metabolism.
More recently, AL-9, an antiviral compound that blocks HCV replication, was characterized to be a relatively selective PI4KIIIα inhibitor[88]. Shortly after the identification of AL-9, a screen for inhibitors of PI4KIIIα identified several related compounds that were highly potent and selective inhibitors of PI4KIIIα in vitro and in cells[44]. Among these, GSK-A1, often referred to as A1, was the most effective PI4KIIIα inhibitor in cultured cells. Consistent with earlier inhibitor experiments, treatment of cells with A1 resulted in loss of PM PI(4)P, with no effect seen on PM PI(4,5)P2 levels. However, after stimulation of a HEK 293 cell line stably expressing the angiotensin receptor AT1 with angiotensin II, which activates phospholipase C (PLC) and hydrolyzes PI(4,5)P2, A1-treated cells did not exhibit restoration of PI(4,5)P2 levels, confirming the role of PI4KIIIα in the resynthesis of this phosphoinositide. Interestingly, this study demonstrated that cells tolerate prolonged inhibition of PI4KIIIα for at least 24 h, an unexpected result considering the importance of PI4KIIIα for PI(4,5)P2 synthesis. This led the authors to hypothesize that there could be other sources of PI(4)P for sustaining PI(4,5)P2 at the PM, a possibility that has found support in other independent studies[11,89,90].
Importantly, A1 is a useful tool to study the PM pool of PI(4)P due to its ability to inhibit PI4KIIIα without affecting PI4KIIIβ and with minimal off-target effects. Although A1 was the most potent PI4KIIIα inhibitor in cells, other compounds identified in that study (such as G1) were more potent and more selective for PI4KIIIα than A1 in an in vitro setting, suggesting that they could be useful for in vitro kinase activity studies. The finding that inhibition of PI4KIIIα and subsequent depletion of PI(4)P have no impact on steady-state PI(4,5)P2 levels but that PI4KIIIα is required for PI(4,5)P2 recovery at the PM suggests that cells might use a specialized pool of PI(4)P dedicated to PI(4,5)P2 resynthesis[24]. This idea is consistent with the recent discoveries of independent functions of PI(4)P in the PM (discussed in section 4). Using a separate pool of PI(4)P for PI(4,5)P2 synthesis and after signaling events would ensure that sufficient PI(4)P remains in the PM to fulfill its other biological roles.
The observation that HCV requires high PI4KIIIα activity for replication within host cells but that cells can survive and resynthesize PI(4)P and PI(4,5)P2 at low PI4KIIIα activity levels suggested the potential existence of a therapeutic window in which PI4KIIIα could be inhibited enough to prevent HCV replication without impacting host cellular function. To investigate this idea, several PI4KIIIα inhibitors were tested in mice. Although cultured cells tolerated PI4KIIIα inhibition for 24 h, mice treated with compounds that were structurally related to A1 (e.g., F1, C1, M1, and J1) developed a wide range of debilitating side effects and ultimately died of cardiovascular collapse[44]. The authors hypothesized that systemic inhibition of PI4KIIIα resulted in an inability to resynthesize PI(4,5)P2, crippling Gαq signaling pathways. These results suggest that this potential therapeutic window might be very difficult to target due to different cell types’ and tissues’ PI(4)P and PI(4,5)P2 requirements. These inhibitor experiments also mirror the lethality seen in a global PI4KIIIα knockout mouse[9] and necrosis seen in tissue-specific conditional PI4KIIIα knockout mice[44].
3.4. Genetically encoded tools to disrupt PI(4)P levels in the PM
Tools to deplete PI(4)P without inhibiting PI4KIIIα are also available. Expression of a soluble form of the phosphoinositide phosphatase Sac1 results in constitutive depletion of PI(4)P from the PM[91]. To rapidly deplete PI(4)P from the PM, an artificial construct called pseudojanin[13] was designed that uses chemically-induced dimerization of FKBP and FRB fusions by rapamycin to recruit phosphoinositide phosphatase catalytic domains to desired target membranes. Inspired by the enzyme synaptojanin, which contains two distinct 5- and 4-phosphatase catalytic domains to hydrolyze PI(4,5)P2 to PI, pseudojanin recapitulates this function using fusions of the catalytic domains of Sac1, which acts on PI(4)P, and the inositol polyphosphate-5-phosphatase E (INPP5E), which removes the 5-phosphate group of PI(4,5)P2, to FKBP. When used in combination with PM-tethered FRB, this system allows rapamycin-induced degradation of PI(4)P and PI(4,5)P2. Point mutants of either catalytic domain within pseudojanin can be used to selectively dephosphorylate the 5 position of PI(4,5)P2 (and thus increase PI(4)P) or to deplete PI(4)P while leaving PI(4,5)P2 pools intact. Systems such as pseudojanin that enable temporal control of phosphoinositide depletion at specific organelle membranes are important complements to pharmacological inhibitors for probing the requirements for local pools of phosphoinositides.
Inhibitors and enzyme-based tools to deplete PI(4)P require simultaneous use of a PI(4)P-binding probe such as GFP-tagged P4M to validate proper depletion of the lipid. Although these probes are both widely used and very useful, their recruitment and dissociation from the membrane are dependent upon their affinities for the target lipid. As a result, they are most often used to provide qualitative, relative information about lipid levels (and, critically, localizations), but in some circumstances they are not able to detect minor changes in levels of PIPs. Other biochemical methods that provide highly quantitative measurements can be used to answer these questions more accurately.
4. Recently discovered functions of PI(4)P at the PM
PI(4)P is synthesized on the inner leaflet of the PM, where it resides alongside other negatively charged lipids such as phosphatidylserine (PS), phosphatidic acid (PA), PI(4,5)P2, and PI(3,4,5)P3. These anionic lipids are used to recruit proteins to the PM. For example, the PH domain of phospholipase Cδ (PLCδ) binds to PI(4,5)P2 and is used as a marker for this lipid[92,93]. More recently, the important contribution of the PM pool of PI(4)P to protein recruitment has come to light. Using a tandem Osh2 PH domain (PH-Osh2×2), which binds to PI(4)P and PI(4,5)P2, and tools for depleting one or both of these lipids,[13] it was proposed that PI(4)P can fulfill the requirement of some proteins for anionic lipid binding. This role of PI(4)P would permit stable recruitment of such proteins to the PM even during signaling-induced PI(4,5)P2 hydrolysis. This study also highlighted the requirement for PI(4)P and PI(4,5)P2 in the regulation of TRPV1, a transmembrane cation channel involved in nociception and heat sensation. These phosphoinositides can differentially regulate a wide variety of ion channels, as reviewed comprehensively elsewhere[94]. Other classes of transmembrane proteins can also be regulated by PI(4)P, including some P4-ATPases. A recent cryo-EM study of the yeast trans-Golgi network (TGN)-localized Drs2-Cdc50 flippase showed a binding site with high specificity for PI(4)P that relieved autoinhibition of the enzyme, increasing activity[95].
Recent studies have revealed roles for PM pools of PI(4)P in two different PM internalization pathways. First, a study of clathrin-mediated endocytosis in yeast, using mutants of Stt4 and Mss4, the homologs of PI4KIIIα and the sole yeast PI4P 5-kinase, respectively, suggests a role for PI(4)P, independent of PI(4,5)P2, in this process[96]. There are numerous differences in clathrin-mediated endocytosis between yeast and mammalian cells, and roles for PI(4)P in the mammalian process, other than its potential implication in uncoating following endocytosis, remain less clear[97]. Second, PI(4)P was recently characterized to play a key role in phagocytosis[98]. PI(4)P is enriched at sites of phagocytosis by specific recruitment of the PI4KIIIα complex. This specific pool of PI(4)P was proposed to ensure a sufficient supply of PI(4,5)P2 for this specialized membrane trafficking event. In this model, following phagocytosis, PI(4)P is depleted by dephosphorylation, and, interestingly, subsequently re-synthesized later on during phagosome maturation, but by the endosomal PI 4-kinase PI4KIIα. This sequence illustrates the dynamic nature of PIP metabolism and how local, targeted recruitment of specific PI 4-kinase isoforms can connect PI(4)P synthesis to membrane trafficking events.
In the past decade, a major function for PI(4)P has emerged in facilitating non-vesicular lipid transport. The production of PI(4)P on non-ER membranes allows proteins from the OSBP-related protein (ORP) family (Osh in yeast) to transport other lipids such as PS and cholesterol from their site of synthesis in the ER to non-ER target membranes. At ER–PM contact sites, a PI(4)P/PS counter-transport system was recently identified in yeast and mammalian cells (Figure 6)[59,99]. This system relies on competitive binding and differential affinities of the ORP/Osh proteins for PI(4)P and the second lipid, and localized synthesis and destruction of PI(4)P to drive a transport cycle.
Figure 6. Schematic illustrating a PI(4)P/PS counter-transport system at ER–PM contact sites involving ORP5/8.
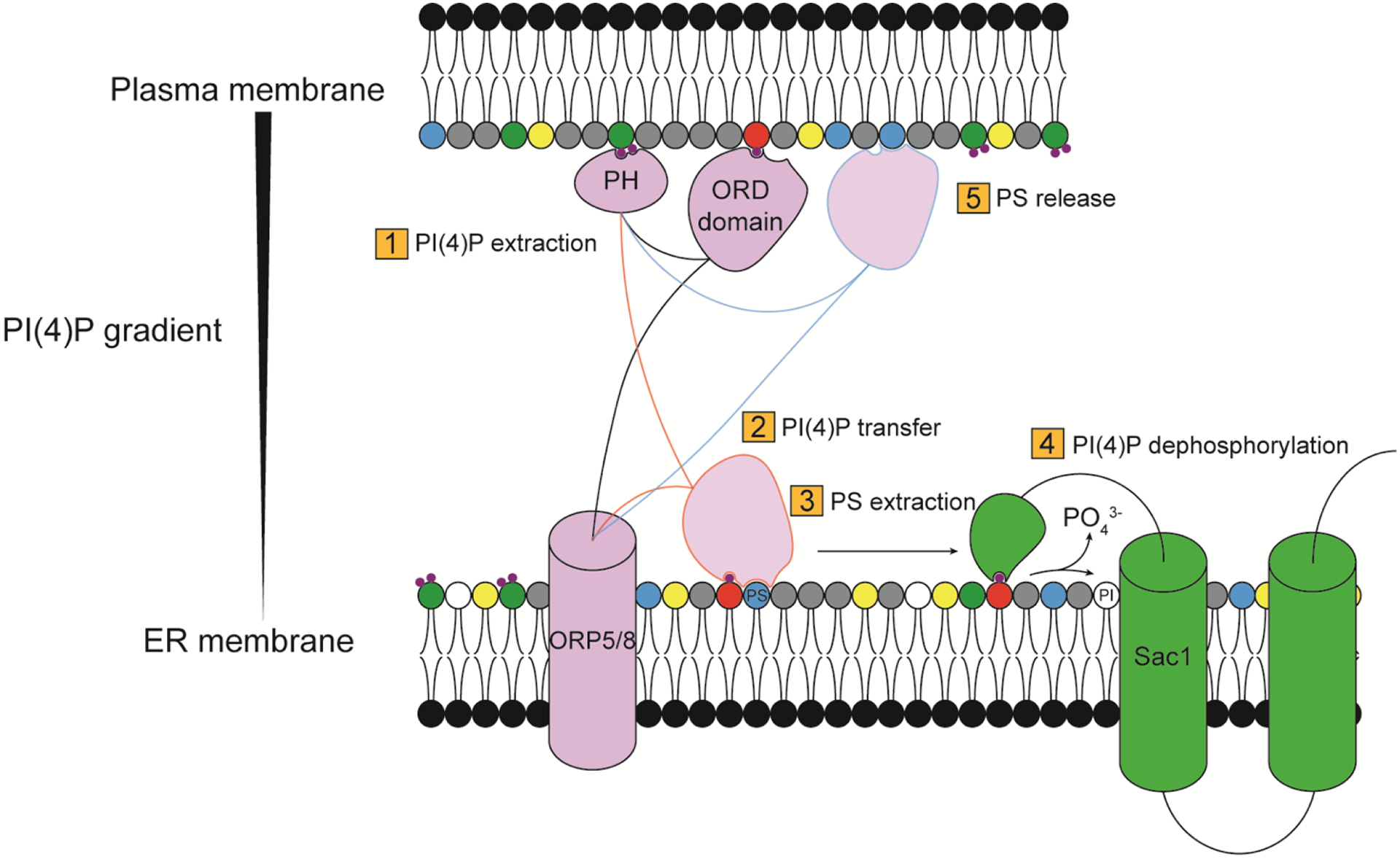
The PH domain of ORP5/8 (Osh6/7 in yeast) tethers the protein to the PM, allowing the ORD domain to capture and extract PI(4)P from the PM (1). The ORD domain transfers the PI(4)P to the ER membrane (2), where it exchanges with PS (3). PI(4)P is then dephosphorylated by the ER PI4P phosphatase Sac1 (4). The ORD domain moves back to the PM, where it releases PS and becomes free to bind and extract PI(4)P, restarting the cycle.
For example, human ORP5/8 and their yeast orthologs Osh6/Osh7 can bind to either PI(4)P or PS but interact with PI(4)P with higher affinity (Figure 6). At the ER, where PI(4)P levels are low, they extract PS. At the PM, they deposit PS and extract PI(4)P. Upon return to the ER, they release PI(4)P, which is dephosphorylated by the ER-resident PI(4)P phosphatase Sac1. Because of continued PI(4)P synthesis at the PM by PI4KIIIα/Stt4 and dephosphorylation by Sac1 at the ER, a cycle of net ATP hydrolysis drives directional PS transport from the ER to the PM[59,99]. A later study demonstrated that ORP5/8 differ in their binding affinities to PI(4,5)P2, leading to a finely tuned feedback loop ensuring stable, physiological levels of PI(4,5)P2 in the PM upon strong resynthesis of this lipid[100]. Other non-vesicular lipid transport systems between organelles also exploit PI(4)P gradients across membrane contact sites, such as counter-transport of PI(4)P and cholesterol between the TGN and the ER[101], but these rely on a different pool of PI(4)P synthesized by PI4KIIIβ.
G-protein coupled receptors (GPCRs) and their signaling pathways are iconic examples of the use of PIPs for generation of second messengers. Signaling through Gαq/11 leads to PLCβ activation and hydrolysis of PI(4,5)P2 to release soluble inositol 1,4,5-trisphosphate (IP3) and membrane-bound diacylglycerol (DAG). These second messengers activate conventional protein kinase C (PKC) enzymes and engender changes in cell physiology. Until recently, PI(4)P was not known to play a direct role in GPCR signaling, except as a metabolic precursor to replenish PI(4,5)P2. A recent study showed that GPCR activation could lead to PI(4)P depletion on internal membranes of cardiac myocytes as a result of PLCβ activation[102]. PI(4)P depletion led to protein kinase D activation and expression of hypertrophic genes. This effect was inhibited by prior depletion of PI(4)P by treatment with PAO. A subsequent study revealed that a PM pool of PI(4)P could also be depleted in several other cell types (mouse embryonic fibroblasts, rat aortic smooth muscle cells and PANC-1 pancreatic duct epithelial cells)[103] in which GPCR agonists such as endothelin-1, neurotensin, and fetal bovine serum elicited PI(4)P hydrolysis (Figure 7).
Figure 7. Diagram illustrating different GPCR signaling outcomes from PLC-mediated hydrolysis of PI(4)P and PI(4,5)P2.
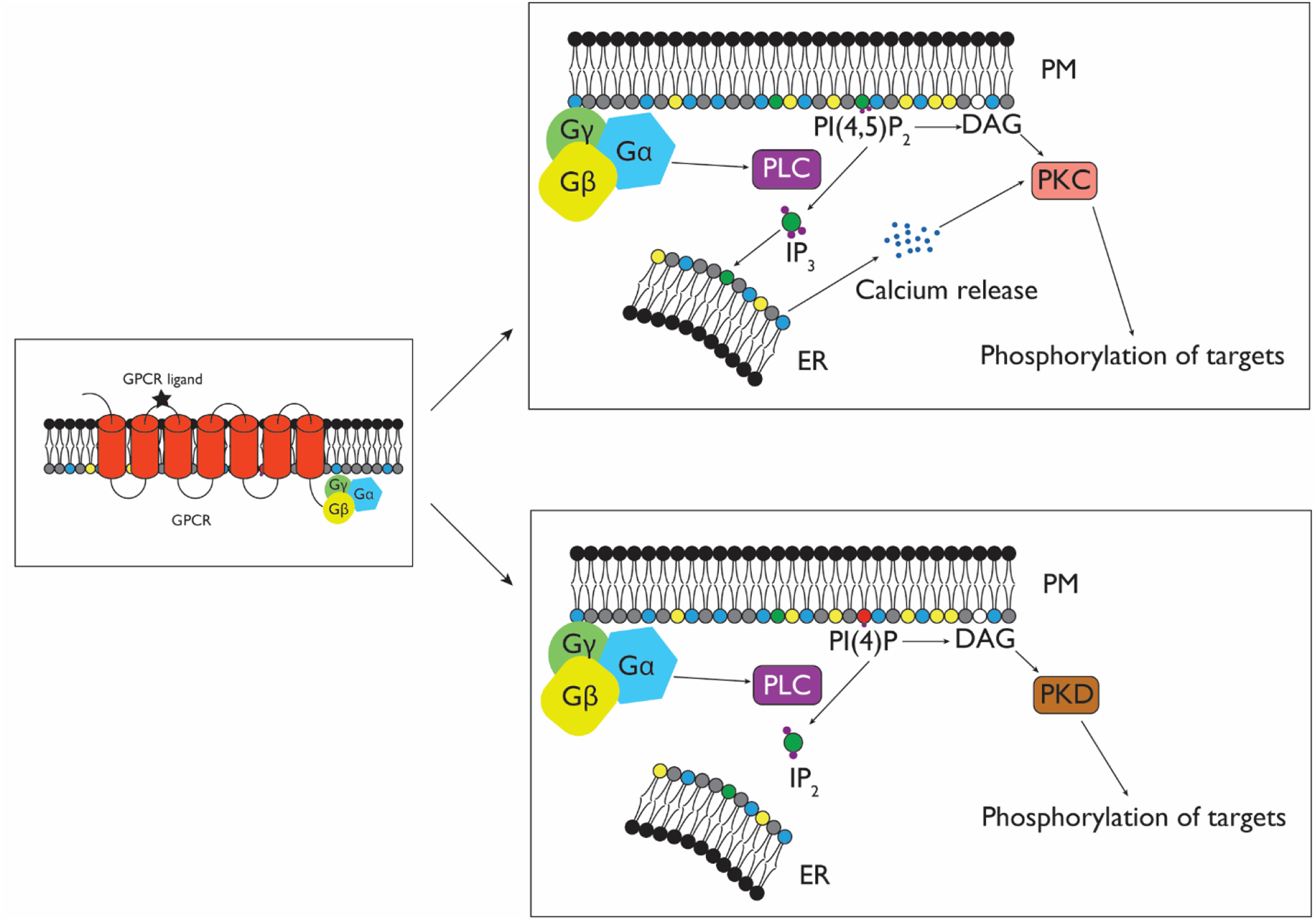
Upon GPCR activation, Gαq/11 stimulates PLC enzymes. PLC-mediated cleavage of PI(4,5)P2 (top) causes release of IP3 and DAG. IP3 activates the ER-resident IP3 receptor, triggering release of ER calcium stores into the cytosol. DAG works synergistically with Ca2+ to activate conventional PKCs, leading to phosphorylation of downstream targets. However, when PLC acts upon PI(4)P, IP2 and DAG are released. In contrast to IP3, IP2 does not stimulate Ca2+ release, resulting in a different signaling profile, namely the release of DAG, which can activate novel PKCs and PKD, leading to the phosphorylation of a different set of targets.
By using a rapamycin-inducible PI(4,5)P2 5-phosphatase, the authors showed that PI(4)P is not being phosphorylated to PI(4,5)P2 upon GPCR signaling. Instead, they proposed a model wherein GPCR activation in this context leads to PLC-mediated cleavage of PI(4)P. Whereas PI(4,5)P2 hydrolysis generates two second messengers, DAG and IP3, PI(4)P hydrolysis releases DAG and the signaling-inert IP2, resulting in different signaling outcomes. Unlike IP3, IP2 does not activate ER-resident IP3 receptors and therefore does not cause calcium release. The authors hypothesized that, whereas PI(4,5)P2 depletion is responsible for the acute response to GPCR activation, PI(4)P hydrolysis is required for the well-documented, long-term DAG production that has been observed after stimulation of GPCRs[104,105].
5. Unanswered Questions and Outlook
Though the picture of PI(4)P function of the PM has become clearer in recent years, many questions remain unanswered. PI4KIIIα, the PI 4-kinase that synthesizes the bulk of the PM PI(4)P pool, has several interaction partners responsible for its recruitment to this membrane, including EFR3A/B, TTC7A/B, FAM126A/B, and TMEM150A. Though PI4KIIIα-mediated PI(4)P synthesis is ubiquitous and can be thought of as a housekeeping function, mutation of PI4KIIIα or some of its interaction partners (e.g., TTC7A, FAM126A) can cause heritable diseases affecting different organ systems. It remains a mystery how this tissue selectivity occurs in different diseases associated with PI4KIIIα dysfunction.
At the molecular level, the regulation of PI4KIIIα complex assembly and enzymatic activity is not well understood. Studies on yeast Efr3 have identified that phosphorylation of its unstructured C-terminal domain likely reduces binding to Ypp1 (the homolog of TTC7A/B)[31]. The identity of kinases and phosphatases that regulate this phosphorylation and what agonists and signaling pathways regulate this phosphorylation, or whether such a mechanism is conserved in the mammalian complex, remain unknown. Additionally, the function of the six-pass transmembrane protein TMEM150A is not well understood. It forms an independent complex with PI4KIIIα and EFR3, but not TTC7 (and presumably not FAM126, either) that was proposed to increase the initial rate of PI(4,5)P2 resynthesis after acute depletion[47]. However, the mechanisms controlling this activation of PI4KIIIα are unknown and difficult to reconcile with the extensive stabilizing contacts between PI4KIIIα and TTC7B/FAM126A observed by cryo-EM. In yeast, overexpression of Sfk1, the homolog of TMEM150A, increases association of the kinase with the PM[46]. This scenario may not be the case in mammalian cells, as TTC7B and EFR3B are required to recruit the kinase to the PM before interaction with TMEM150A occurs, with the caveat that the studies in mammalian cells have been thus far performed with overexpressed proteins[47].
One constant in the proposed model of PI4KIIIα complexes is the presence of EFR3. Beyond issues discussed regarding regulation of its interaction with TTC7, other questions remain about how its membrane association impacts PI4KIIIα activity. There is currently no high-resolution structural information for mammalian EFR3. A structure of the soluble N-terminal domain of yeast Efr3 indicates it contains a long, all-α-helical domain[31], but the mammalian protein has some striking differences that may contribute to differential regulation of the overall complex. Yeast Efr3 is not lipidated, but mammalian EFR3A and EFR3B have, respectively, four and three Cys residues in an N-terminal palmitoylation motif. Though this palmitoylation is critical for EFR3 membrane anchoring, the dynamics of this posttranslational lipidation, and its roles in complex assembly and PI4KIIIα activity remain unknown.
Further, the stoichiometry of EFR3 relative to PI4KIIIα–TTC7–FAM126 hexamers at the PM is presently unknown, though a 1:1 relationship between EFR3 and TTC7 has been proposed[37]. Notably, in yeast, endogenously GFP-tagged Stt4 assembles in PM puncta termed PIK patches, which have been estimated to comprise approximately 30 Stt4 molecules[29]. By contrast, GFP-tagged PI4KIIIα, when recruited to the PM by EFR3 and TTC7, adopts a uniform distribution, though these studies are using overexpressed proteins[32,47]. Investigation of fluorescently tagged, endogenously expressed PI4KIIIα would address whether mammalian PI4KIIIα assembles into clusters. Such an approach would also reveal whether the localization of PI4KIIIα within the PM has a relationship to regions where its substrate, PI, which was recently discovered to be virtually absent from this membrane[106,107], may be delivered, or its product, PI(4)P, is used by PI4P 5-kinases to produce PI(4,5)P2.
Methods to visualize and perturb PI(4)P at the PM have also advanced rapidly in recent years. Here, we have focused on those tailored to understanding the biology of PI(4)P at the PM. Certain other approaches developed for PI(4,5)P2 or PI(3,4,5)P3 may be adaptable to PI(4)P down the road. These include dimerization-based or FRET-based PH domain probes that recognize these PM PIPs[108]. Compared to traditional biosensors, where lipid presence is inferred by a change in subcellular localization of the probe, these fluorescence intensity-based probes are tethered to a membrane of interest and fluoresce strongly in presence or absence of their target PIP. When a change in the concentration of the target PIP is induced, the nature of the probe fluorescence changes drastically but the probes remain anchored to their target membrane. These tools are modular and could in principle be adapted to track metabolic flux of PI(4)P in any membrane of interest. This type of probe may be ideally suited to detect transient changes in PI(4)P synthesis in response to specific stimuli. The membrane-targeted nature of these probes also enables organelle-specific study of PIPs without complications arising from bias or coincidence detection in the PIP-binding requirements of a PH domain.
Other alternative strategies for highly sensitive detection of phosphoinositides include chemical conjugation of environment-sensitive fluorophores to PIP-binding domains, followed by microinjection into cells[109,110]. A related approach uses cell-permeable peptides tagged in a similar manner[111]. In both instances, these approaches were developed for PI(4,5)P2 but in principle could be tailored to PI(4)P.
Another approach to study PIPs that would prove useful to investigating PI(4)P functions is the recent synthesis of fluorogenic analogues of PI(4,5)P2[112]. An analog termed XY-69 acts as a substrate for PLC enzymes, becoming fluorescent after hydrolysis. XY-69 was demonstrated to report on PLC activity in purified samples or in lysates, but future work could include making such a probe membrane-permeable or otherwise capable of delivery into live cells. If such a tool could be developed for PI(4)P, it would be invaluable to study events of PI(4)P hydrolysis by PLCs, for example in GPCR signaling[102,103].
Recent years have seen an explosion of innovative tool development in the field of phosphoinositide signaling. These new tools have spurred major leaps in our understanding of the localizations and functions of all PIPs. At the PM, PI(4)P is emerging from the shadows cast by the bright light that has been shown on the downstream lipids PI(4,5)P2 and PI(3,4,5)P3. Beyond its role as metabolic precursor, PI(4)P at the PM is a key player in regulating PS homeostasis, transmembrane ion channels, endocytosis, phagocytosis, GPCR signaling, and recruitment of PM-associated proteins and maintenance of the anionic character of this membrane. Future research in this area is sure to see a continuation of this virtuous cycle of tool development and biological discovery to reveal new functions for this fascinating, multifunctional lipid.
Acknowledgments
Work on phosphoinositide signaling in the Baskin Lab is supported by the National Institutes of Health (R01GM131101) and the Alfred P. Sloan Foundation (Sloan Research Fellowship to J.M.B.). The graphical abstract was created using BioRender.
Biographies

Jeremy M. Baskin was born in Montreal, Canada in 1981. He received his undergraduate education at the Massachusetts Institute of Technology, with a major in Chemistry and minors in Biology and Music. He carried out Ph.D. studies under NDSEG and NSF graduate fellowships in Carolyn Bertozzi’s group at the University of California, Berkeley, focusing on development of bioorthogonal chemistries. Jeremy received postdoctoral training in phosphoinositide signaling as a Jane Coffin Childs fellow at Yale School of Medicine with Pietro De Camilli. Since 2015, he has been Assistant Professor and Nancy and Peter Meinig Family Investigator in the Life Sciences at Cornell University, with appointments in the Department of Chemistry and Chemical Biology and the Weill Institute for Cell and Molecular Biology. Research in the Baskin laboratory centers on the chemical biology and cell biology of phospholipid signaling, with a focus both on development of tools for visualizing and manipulating phosphatidic acid signaling and elucidation of mechanisms connecting phosphatidic acid and phosphoinositide metabolism to physiological and pathological signaling events. Jeremy is recipient of numerous awards, including Beckman Young Investigator, Sloan Research Fellowship, NSF CAREER, and ASBMB Walter A. Shaw Young Investigator in Lipid Research.

Alex G. Batrouni was born in Rennes, France, in 1994. He received his undergraduate training in Biochemistry and Genetics from the University of Sheffield in the United Kingdom. He is currently a Ph.D. candidate in the Baskin laboratory in the Weill Institute for Cell and Molecular Biology at Cornell University. His research is focused on understanding the recruitment of PI4KIIIα to the plasma membrane by its interaction partners and the underlying regulatory mechanisms of PI4KIIIα complex assembly. Additionally, he is investigating the role of this kinase in PI(4,5)P2 homeostasis at the plasma membrane. Alex’s research interests include membrane biology, phosphoinositide signaling, lipid- and protein-driven membrane partitioning effects and protein clustering. Specifically, his interests lie in understanding how these biophysical phenomena impact cell signaling and how cells harness these biophysical effects to form functional, ordered membrane-bound structures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Harayama T & Riezman H Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol 19, 281–296 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Balla T Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev 93, 1019–1137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Craene JO, Bertazzi DL, Bär S & Friant S Phosphoinositides, major actors in membrane trafficking and lipid signaling pathways. Int. J. Mol. Sci 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nasuhoglu C et al. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal. Biochem 301, 243–254 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Wenk MR et al. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat. Biotechnol 21, 813–817 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Stephens LR, Jackson TR & Hawkins PT Agonist-stimulated synthesis of phosphatidylinositol(3,4,5)-trisphosphate: A new intracellular signalling system? BBA - Mol. Cell Res 1179, 27–75 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Simonsen A et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394, 494–498 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Murray JT, Panaretou C, Stenmark H, Miaczynska M & Backer JM Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic 3, 416–427 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Nakatsu F et al. Ptdins4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol 199, 1003–1016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godi A et al. ARF mediates recruitment of PtdIns-4-OH kinase-β and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol 1, 280–287 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Dickson EJ, Jensen JB & Hille B Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci 111, 2281–2290 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balla A, Tuymetova G, Tsiomenko A, Varnai P & Balla T A Plasma Membrane Pool of Phosphatidylinositol 4- Phosphate Is Generated by Phosphatidylinositol 4-Kinase Type-III Alpha: Studies with the PH Domains of the Oxysterol Binding Protein and FAPP1. Mol Biol Cell 16, 1282–1295 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammond GRV et al. PI(4)P and PI(4,5)P2 Are Essential But Independent Lipid Determinants of Membrane Identity. Science. 337, 727–730 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebner M, Lučić I, Leonard TA & Yudushkin I PI(3,4,5)P3 Engagement Restricts Akt Activity to Cellular Membranes. Mol. Cell 65, 416–431 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Senju Y & Lappalainen P Regulation of actin dynamics by PI(4,5)P2in cell migration and endocytosis. Curr. Opin. Cell Biol 56, 7–13 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Servant G et al. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 287, 1037–1040 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang F et al. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol 4, 513–518 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Ferguson GJ et al. PI(3)Kγ has an important context-dependent role in neutrophil chemokinesis. Nat. Cell Biol 9, 86–91 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Liu P et al. Ptdins(3,4,5)P3 -dependent activation of the mTORC2 kinase complex. Cancer Discov. 5, 1194–11209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foukas LC et al. Critical role for the p110α phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441, 366–370 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Lien EC, Dibble CC & Toker A PI3K signaling in cancer: beyond AKT. Curr. Opin. Cell Biol 45, 62–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammond GRV Does PtdIns(4,5)P2 concentrate so it can multi-task? Biochem. Soc. Trans 44, 228–233 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Kielkowska A et al. A new approach to measuring phosphoinositides in cells by mass spectrometry. Adv. Biol. Regul 54, 131–141 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM, T. B. Maintenance of Hormone-sensitive Phosphoinositide Pools in the Plasma Membrane Requires Phosphatidylinositol 4-Kinase III. Mol. Biol. Cell 19, 711–721 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida S, Ohya Y, Goebl M, Nakano A & Anraku Y A novel gene, STT4, encodes a phosphatidylinositol 4-kinase in the PKC1 protein kinase pathway of Saccharomyces cerevisiae. J. Biol. Chem 269, 1166–1171 (1994). [PubMed] [Google Scholar]
- 26.Flanagan CA et al. Phosphatidylinositol 4-kinase: Gene structure and requirement for yeast cell viability. Science. 262, 1444–1448 (1993). [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa T, Goto K & Kondo H Cloning, expression, and localization of 230-kDa phosphatidylinositol 4-kinase. J. Biol. Chem 271, 12088–12094 (1996). [DOI] [PubMed] [Google Scholar]
- 28.Wong K, Meyers R & Cantley LC Subcellular locations of phosphatidylinositol 4-kinase isoforms. J. Biol. Chem 272, 13236–13241 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Baird D, Stefan C, Audhya A, Weys S & Emr SD Assembly of the PtdIns 4-kinase Stt4 complex at the plasma membrane requires Ypp1 and Efr3. J. Cell Biol 183, 1061–1074 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bojjireddy N, Guzman-Hernandez ML, Reinhard NR, Jovic M & Balla T EFR3s are palmitoylated plasma membrane proteins that control responsiveness to G-protein-coupled receptors. J Cell Sci 128, 118–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X et al. Structural Insights into Assembly and Regulation of the Plasma Membrane Phosphatidylinositol 4-Kinase Complex. Dev. Cell 28, 19–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baskin JM et al. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol 18, 132–138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zara F et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat. Genet 38, 1111–1113 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Traverso M et al. Novel FAM126A mutations in hypomyelination and congenital cataract disease. Biochem. Biophys. Res. Commun 439, 369–372 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Rossi A et al. Hypomyelination and congenital cataract: Neuroimaging features of a novel inherited white matter disorder. Am. J. Neuroradiol 29, 301–305 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watt SA, Kular G, Fleming IN, Downes CP & Lucocq JM Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C δ1. Biochem. J 363, 657–666 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lees JA et al. Architecture of the human PI4KIIIα lipid kinase complex. Proc. Natl. Acad. Sci. U. S. A 114, 13720–13725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pagnamenta AT et al. Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum. Mol. Genet 24, 3732–3741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altan-Bonnet N & Balla T Phosphatidylinositol 4-kinases: Hostages harnessed to build panviral replication platforms. Trends Biochem. Sci 37, 293–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berger KL et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A 106, 7577–7582 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trotard M et al. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 23, 3780–3789 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Vaillancourt FH et al. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387, 5–10 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Tai AW et al. A Functional Genomic Screen Identifies Cellular Cofactors of Hepatitis C Virus Replication. Cell Host Microbe 5, 298–307 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bojjireddy N et al. Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J. Biol. Chem 289, 6120–6132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger KL, Kelly SM, Jordan TX, Tartell MA & Randall G Hepatitis C Virus Stimulates the Phosphatidylinositol 4-Kinase III Alpha-Dependent Phosphatidylinositol 4-Phosphate Production That Is Essential for Its Replication. J. Virol 85, 8870–8883 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Audhya A & Emr SD Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev. Cell 2, 593–605 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Chung J, Nakatsu F, Baskin JM & De Camilli P Plasticity of PI4KIIIα interactions at the plasma membrane. EMBO Rep. 16, 312–320 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dornan GL et al. Probing the Architecture, Dynamics, and Inhibition of the PI4KIIIα/TTC7/FAM126 Complex. J. Mol. Biol 430, 3129–3142 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Oganesyan I, Lento C & Wilson DJ Contemporary hydrogen deuterium exchange mass spectrometry. Methods 144, 27–42 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Gupta AR et al. Rare deleterious mutations of the gene EFR3A in autism spectrum disorders. Mol. Autism 5, 31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Avitzur Y et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 146, 1028–1039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bigorgne AE et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J. Clin. Invest 124, 328–337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nawaz S et al. Phosphatidylinositol 4,5-bisphosphate-dependent interaction of myelin basic protein with the plasma membrane in oligodendroglial cells and its rapid perturbation by elevated calcium. J. Neurosci 29, 4794–4807 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Snaidero N et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 156, 277–290 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goebbels S et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J. Neurosci 30, 8953–8964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goebbels S et al. A neuronal PI(3,4,5)P3 -dependent program of oligodendrocyte precursor recruitment and myelination. Nat. Neurosci 20, 10–15 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Boehle AS et al. Wortmannin inhibits growth of human non-small-cell lung cancer in vitro and in vivo. Langenbeck’s Arch. Surg 387, 234–239 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Juvekar A et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2, 1048–1063 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung J et al. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER - Plasma membrane contacts. Science. 349, 428–432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gayle S et al. Identification of apilimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 129, 1768–1778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitagawa M et al. Dual blockade of the lipid kinase PIP4Ks and mitotic pathways leads to cancer-selective lethality. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roy A & Levine TP Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. J. Biol. Chem 279, 44683–44689 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Godi A et al. FAPPS control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat. Cell Biol 6, 393–404 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Levine TP & Munro S Targeting of Golgi-specific pleckstrin homology domains involves both Ptdlns 4-kinase-dependent and -independent components. Curr. Biol 12, 695–704 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Jun YW, Lee JA & Jang DJ Novel GFP-fused protein probes for detecting phosphatidylinositol-4-phosphate in the plasma membrane. Animal Cells Syst. (Seoul) 23, 164–169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hammond GRV, Machner MP & Balla T A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J. Cell Biol 205, 113–126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo X et al. Structure of the Legionella Virulence Factor, SidC Reveals a Unique PI(4)P-Specific Binding Domain Essential for Its Targeting to the Bacterial Phagosome. PLoS Pathog. 11, 1–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tóth JT et al. BRET-monitoring of the dynamic changes of inositol lipid pools in living cells reveals a PKC-dependent PtdIns4P increase upon EGF and M3 receptor activation. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1861, 177–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gillooly DJ Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19, 4577–4588 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schu PV et al. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 260, 88–91 (1993). [DOI] [PubMed] [Google Scholar]
- 71.Ogiso H & Taguchi R Reversed-phase LC/MS method for polyphosphoinositide analyses: Changes in molecular species levels during epidermal growth factor activation in A431 cells. Anal. Chem 80, 9226–9232 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Milne SB, Ivanova PT, DeCamp D, Hsueh RC & Brown HA A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. J. Lipid Res 46, 1796–1802 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Clark J et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods 8, 267–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Traynor-Kaplan A et al. Fatty-acyl chain profiles of cellular phosphoinositides. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1862, 513–522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kiefer S et al. Separation and detection of all phosphoinositide isomers by ESI-MS. J. Pharm. Biomed. Anal 53, 552–558 (2010). [DOI] [PubMed] [Google Scholar]
- 76.Saheki Y et al. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat. Cell Biol 18, 504–515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Symons JL et al. Lipidomic atlas of mammalian cell membranes reveals hierarchical variation induced by culture conditions, subcellular membranes, and cell lineages. Soft Matter 17, 288–297 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bui HH et al. Direct analysis of PI(3,4,5)P 3 using liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Biochem 547, 66–76 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Castro-Perez J et al. Localization of fatty acyl and double bond positions in phosphatidylcholines using a dual stage CID fragmentation coupled with ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom 22, 1568–1569 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Endemann G, Cantley LC & Dunn SN Bovine Brain Contains Two Types of Phosphatidylinositol Kinase. Biochemistry 26, 6845–6852 (1987). [DOI] [PubMed] [Google Scholar]
- 81.Nakanishi S, Catt KJ & Balla T A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. U. S. A 92, 5317–5321 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cutler NS, Heitman J & Cardenas ME STT4 is an essential phosphatidylinositol 4-kinase that is a target of wortmannin in Saccharomyces cerevisiae. J. Biol. Chem 272, 27671–27677 (1997). [DOI] [PubMed] [Google Scholar]
- 83.Gerhard R, John H, Aktories K & Just I Thiol-modifying phenylarsine oxide inhibits guanine nucleotide binding of Rho but not of Rac GTPases. Mol. Pharmacol 63, 1349–1355 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Balla A et al. Design of drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry 47, 1599–1607 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Han YP & Kohanski RA Phenylarsine oxide inhibits insulin activation of phosphatidylinositol 3’-kinase. Biochem. Biophys. Res. Commun 239, 316–321 (1997). [DOI] [PubMed] [Google Scholar]
- 86.Dumaresq-Doiron K, Savard MF, Akam S, Costantino S & Lefrancois S The phosphatidylinositol 4-kinase PI4KIIIα is required for the recruitment of GBF1 to Golgi membranes. J. Cell Sci 123, 2273–2280 (2010). [DOI] [PubMed] [Google Scholar]
- 87.Knight ZA et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110α in Insulin Signaling. Cell 125, 733–747 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bianco A et al. Metabolism of phosphatidylinositol 4-kinase IIIα-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 8, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Falkenburger BH, Jensen JB & Hille B Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol 135, 81–97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Szentpetery Z, Várnai P & Balla T Acute manipulation of Golgi phosphoinositides to assess their importance in cellular trafficking and signaling. Proc. Natl. Acad. Sci. U. S. A 107, 8225–8230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zewe JP, Wills RC, Sangappa S, Goulden BD & Hammond GRV SAC1 degrades its lipid substrate Ptdins4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. Elife 7, 1–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lemmon MA, Ferguson KM, O’Brien R, Sigler PB & Schlessinger J Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. U. S. A 92, 10472–10476 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Várnai P & Balla T Visualization of phosphoinositides that bind pleckstrin homology domains: Calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell Biol 143, 501–510 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rohacs T Phosphoinositide regulation of TRPV1 revisited. Pflugers Arch. Eur. J. Physiol 467, 1851–1869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Timcenko M et al. Structure and autoregulation of a P4-ATPase lipid flippase. Nature 571, 366–370 (2019). [DOI] [PubMed] [Google Scholar]
- 96.Yamamoto W et al. Distinct roles for plasma membrane PtdIns(4)P and PtdIns(4,5)P2 during receptor-mediated endocytosis in yeast. J. Cell Sci 2, (2018). [DOI] [PubMed] [Google Scholar]
- 97.He K et al. Dynamics of Auxilin1 and GAK in clathrin-mediated traffic. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levin R et al. Multiphasic dynamics of phosphatidylinositol 4-phosphate during phagocytosis. Mol. Biol. Cell 28, 128–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Filseck J. M. Von, Alenka Č, Delfosse V & Vanni S Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. 349, (2015). [DOI] [PubMed] [Google Scholar]
- 100.Sohn M et al. PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER–PM contact sites. J. Cell Biol 217, 1797–1813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mesmin B et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi Tether OSBP. Cell 155, 830–843 (2013). [DOI] [PubMed] [Google Scholar]
- 102.Zhang L et al. Phospholipase Cε Hydrolyzes Perinuclear Phosphatidylinositol 4-Phosphate to Regulate Cardiac Hypertrophy. Cell 153, 216–227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rubio R. G. De et al. Phosphatidylinositol 4-phosphate is a major source of GPCR-stimulated phosphoinositide production. Science. 1210, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wright TM, Rangano LA, Shinll HS & Rabenoll DM Kinetic Analysis of 1,2-Diacylglycerol Mass Levels in Cultured Fibroblasts. J. Biol. Chem 263, 9374–9380 (1988). [PubMed] [Google Scholar]
- 105.Griendlingsb KK et al. Sustained Diacylglycerol Formation from Inositol Phospholipids in Angiotensin 11-stimulated Vascular Smooth Muscle Cells. J. Biol. Chem 261, 5901–5906 (1986). [PubMed] [Google Scholar]
- 106.Zewe JP et al. Probing the subcellular distribution of phosphatidylinositol reveals a surprising lack at the plasma membrane. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pemberton JG et al. Defining the subcellular distribution and metabolic channeling of phosphatidylinositol. J. Cell Biol 219, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hertel F et al. Fluorescent biosensors for multiplexed imaging of phosphoinositide dynamics. ACS Chem. Biol 15, 33–38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yoon Y, Lee PJ, Kurilova S & Cho W In situ quantitative imaging of cellular lipids using molecular sensors. Nat. Chem 3, 868–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu SL et al. Simultaneous in situ quantification of two cellular lipid pools using orthogonal fluorescent sensors. Angew. Chem Int. Ed 53, 14387–14391 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mondal S, Rakshit A, Pal S & Datta A Cell Permeable Ratiometric Fluorescent Sensors for Imaging Phosphoinositides. ACS Chem. Biol 11, 1834–1843 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Huang W et al. A membrane-associated, fluorogenic reporter for mammalian phospholipase C isozymes. J. Biol. Chem 293, 1728–1735 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]