

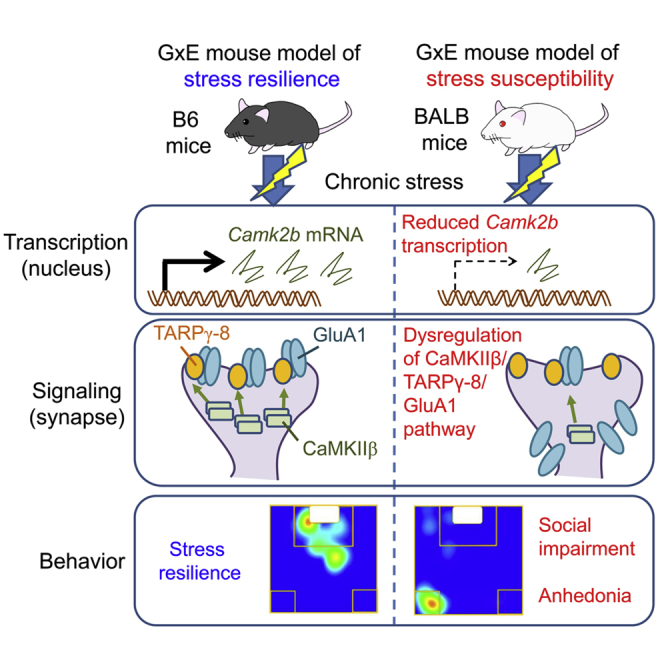
Summary
Although stressful events predispose individuals to psychiatric disorders, such as depression, not all people who undergo a stressful life experience become depressed, suggesting that gene-environment interactions (GxE) determine depression risk. The ventral hippocampus (vHPC) plays key roles in motivation, sociability, anhedonia, despair-like behaviors, anxiety, sleep, and feeding, pointing to the involvement of this brain region in depression. However, the molecular mechanisms underlying the cross talk between the vHPC and GxE in shaping behavioral susceptibility and resilience to chronic stress remain elusive. Here, we show that Ca2+/calmodulin-dependent protein kinase IIβ (CaMKIIβ) activity in the vHPC is differentially modulated in GxE mouse models of depression susceptibility and resilience, and that CaMKIIβ-mediated TARPγ-8 phosphorylation enhances the expression of AMPA receptor subunit GluA1 in the postsynaptic sites to enable stress resilience. We present previously missing molecular mechanisms underlying chronic stress-elicited behavioral changes, providing strategies for preventing and treating stress-related psychiatric disorders.
Subject areas: Behavioral neuroscience, Molecular neuroscience, Cellular neuroscience
Graphical abstract
Highlights
-
•
CaMKIIβ activity is differentially modulated in a depression/resilience GxE model
-
•
CaMKIIβ activity is critical for determining stress susceptibility and resilience
-
•
CaMKIIβ-mediated TARPγ-8 activation exerts pro-resilience effects
-
•
TARPγ-8-mediated synaptic expression of GluA1 confers chronic stress resiliency
Behavioral neuroscience; Molecular neuroscience; Cellular neuroscience.
Introduction
Major depressive disorder is a major health concern worldwide. Core symptoms include depressed mood, loss of interest or pleasure, suicidality, and reduced motivation or hopelessness (Krishnan and Nestler, 2008; McIntosh et al., 2019). Although chronic stress can increase the risk of depression in humans (Caspi et al., 2003; Hariri and Holmes, 2015; Kendler et al., 1999; Tost et al., 2015), it remains unclear why some people become depressed but others do not. Epidemiological studies indicate that both genetic and environmental factors underlie the risk for psychiatric disorders (Chen, 2020; Russo et al., 2012; Sharma et al., 2016; Southwick and Charney, 2012). Deciphering how genes and the environment interact at the molecular level to shape stress vulnerability and resilience will allow for a better understanding of the pathogenic mechanisms underlying depression.
The hippocampus (HPC) is structurally plastic and vulnerable to damage from a variety of psychological stressors (Duman et al., 2019; McEwen, 2001). Indeed, aberrant structural and functional changes in this brain structure have been implicated in depression (Akil et al., 2018; Anacker and Hen, 2017; Nestler et al., 2002; Sheline et al., 2019). Studies in rodents have shown that chronic stress often leads not only to core depression symptoms, such as anhedonia and loss of interest, but also to aberrant synaptic and structural plasticity in the HPC (Airan et al., 2007; Chattarji et al., 2015; Kheirbek et al., 2012; McEwen et al., 2015; Nestler et al., 2002; Nestler and Hyman, 2010; Uchida et al., 2018).
The HPC is functionally heterogeneous along its longitudinal axis (Fanselow and Dong, 2010; Strange et al., 2014), the dorsal region predominantly regulating episodic memory and spatial navigation, and the ventral region being associated with emotion, motivation, anhedonia, sociability, and antidepressant activity (Anacker and Hen, 2017; Bagot et al., 2016; Bigio et al., 2016; Jimenez et al., 2018; Kheirbek et al., 2013; LeGates et al., 2018; Okuyama et al., 2016; Padilla-Coreano et al., 2016; Yoshida et al., 2019). Despite accumulating evidence supporting a role of the ventral HPC (vHPC) in multiple behaviors associated with depressed symptoms, the molecular mechanisms within this brain structure underlying susceptibility and resilience to chronic stress, and how these mechanisms are affected by gene-environment interactions (GxE), are yet to be determined.
We aimed to clarify the molecular mechanisms underlying the GxE interactions shaping risk and resilience to stress-linked psychiatric disorders, such as depression. We established an animal model of depression, meeting the three primary criteria: face validity, construct validity, and predictive validity (Dzirasa and Covington, 2012; Slattery and Cryan, 2017; Willner, 1984). We then focused on the molecular mechanisms underlying resilience and susceptibility to stress through GxE factors. Our results point to potential strategies for the treatment of depression.
Results
GxE animal models of stress resilience and susceptibility
Mouse strains displaying substantial differences in emotional behavior and stress reactivity have often been used as models of the influence of GxE factors on brain functions and behaviors (Hovatta et al., 2005; Mozhui et al., 2010; Uchida et al., 2011b). Indeed, we found that BALB mice subjected to a 6-week period of chronic ultra-mild stress (CUMS; Figure 1A), a procedure based solely on environmental and social stressors (Lanfumey et al., 1999; Rangon et al., 2007; Uchida et al., 2011b), showed decreased social interaction time in a social interaction test (SIT), reduced sucrose preference in a sucrose preference test (SPT), and increased immobility time in a forced swim test (FST); whereas B6 mice did not show such behavioral abnormalities (Figures 1B–1D). These abnormal CUMS-elicited behaviors in BALB mice were prevented by chronic (21 consecutive days), but not subchronic (5 consecutive days), imipramine (IMI) treatment (Figures 1E–1H), suggesting that BALB mice can be used as an animal model of depression.
Figure 1.

Differential regulation of CaMKIIβ activity in the vCA1 of stress-susceptible and stress-resilient mouse strains
(A) Experimental timeline for chronic ultra-mild stress (CUMS) and behavioral testing. SIT, social interaction test; SPT, sucrose preference test; FST, forced swim test; NS, non-stress control.
(B–D) BALB mice show significantly reduced social interaction time in the SIT (B), reduced sucrose preference in the SPT (C), and increased immobility time in the FST (D) following CUMS. n = 24 mice per group.
(E) Experimental timeline for CUMS, imipramine (IMI) treatment, and behavioral testing. IMI was dissolved in the drinking water and administered during the last 3 weeks (IMI-21d) or 5 days (IMI-5d) of the CUMS session and during behavioral assays.
(F–H) Chronic treatment with IMI prevents CUMS-elicited behavioral abnormalities in the SIT (F), SPT (G), and FST (H). n = 16 mice per group.
(I) Experimental timeline for subchronic and mild social defeat stress (smSDS) and behavioral testing.
(J–L) BALB mice show significantly reduced time spent in the target zone (J) and increased time spent in the corner zone (K) in the SIT, and decreased sucrose preference in the SPT (L), following smSDS. n = 39–48 mice per group.
(M and N) Representative coronal slices showing Fos-positive cells in the ventral CA1 (vCA1) region (M). CUMS reduces Fos expression in the vCA1 region of BALB, but not B6 mice (N). n = 8 mice per group. Scale bar, 100 μm.
(O) Specific brain regions were isolated from brain tissue sections using tissue punch (dCA1 and vCA1). Scale bar, 1 mm.
(P) Gene expression profiling for CaMK family members within the vCA1 showing reduced Camk2b expression in BALB mice following CUMS (n = 6 mice per group).
(Q) Increased Camk2b expression following chronic treatment with IMI (n = 8 mice per group).
(R and S) Downregulation of CaMKIIβ expression in BALB mice and increased phosphorylation of CaMKIIβ (T287) in B6 mice following CUMS (n = 8 mice per group).
(T) Q-PCR data showing reduced Camk2b expression in BALB mice following smSDS. n = 8 mice per group.
(U and V) Downregulation of CaMKIIβ expression in BALB mice and increased phosphorylation of CaMKIIβ (T287) in B6 mice following smSDS (n = 6–8 mice per group).
two-way ANOVA followed by a Tukey's post hoc test (in B, D, J, K, N, S, T, P and V), Kruskal-Wallis test followed by a Dunn's multiple comparison (in C and L), one-way ANOVA followed by a Tukey's post hoc test (in F-H, and Q), and multiple comparisons (in P) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM.
See also Figure S1. Complete statistical summaries are provided in Table S2.
To further validate BALB and B6 mice as GxE models of depression and resilience, we subjected them to subthreshold social defeat stress for 5 consecutive days (5 min per defeat session, one session per day), referred to in the following as subchronic and mild social defeat stress (smSDS), and examined their behaviors using SIT and SPT (Figure 1I). BALB but not B6 mice subjected to smSDS spent less time in the interaction zone and showed reduced sucrose preference (Figures 1J-L) than the non-stressed controls. Taken together, these data showed differential responses to stress by the two strains, suggesting that BALB and B6 mice are, respectively, susceptible and resilient to stress.
Differential regulation of CaMKIIβ activity in animal models of stress resilience and susceptibility
We next examined the expression pattern of Fos, an immediate-early gene, as an indicator of neuronal activity and presynaptic neurotransmitter release to assess the basal activation (Bagot et al., 2015) of hippocampal subregions 48 hr after the end of a 6-week CUMS exposure. Immunofluorescent staining revealed that BALB mice after CUMS had significantly fewer Fos-positive cells in the vCA1 region of HPC (Figures 1M and 1N), but normal Fos expression in the dorsal region (dCA1) (Figure S1A). Fos-positive cells in BALB mice were increased in the ventral part of the dentate gyrus (vDG) but reduced in the dorsal part (dDG; Figures S1B–S1C). In contrast, B6 mice did not show differences in the number of Fos-positive cells after CUMS in either the ventral or dorsal parts of CA1 and dentate gyrus (Figures 1M, 1N, and S1A–S1C). These data suggest that decreased neuronal activity in the vCA1 of BALB mice may be associated with the development of depression-like behaviors after chronic stress.
We then aimed to clarify the molecular mechanisms underlying the decreased basal neuronal activity within the vCA1 and increased depression-like behaviors in BALB mice, as well as those underlying the stress-resilient phenotype of B6 mice. Since Fos expression is controlled by a calcium-activated signaling pathway (Sheng et al., 1990; Takemoto-Kimura et al., 2017; Uchida and Shumyatsky, 2018), we focused on calcium signaling and measured the mRNA expression in the HPC (Figure 1O) of genes encoding calcium/calmodulin-dependent protein kinases (CaMKs), a major calcium-dependent signaling protein family (Bayer and Schulman, 2019), in the vCA1 48 hr after the end of CUMS exposure. BALB mice had reduced Camk1g expression than the B6 mice; however, in BALB mice, only the expression level of Camk2b mRNA was reduced upon CUMS (Figure 1P), and this effect was reversed by chronic (21 consecutive days), but not subchronic (5 consecutive days), treatment with IMI (Figure 1Q) or fluoxetine (Figure S1D). In contrast, Camk2b mRNA expression in the vCA1 of B6 mice was not affected by CUMS (Figure 1P), nor was Camk2b mRNA expression in the dCA1 region of BALB mice (Figure S1E). CaMKIIβ protein levels in the vCA1 (Figures 1R and 1S) confirmed these results. Interestingly, autophosphorylation at threonine 287 of CaMKIIβ (pCaMKIIβ) was upregulated in the vCA1 of B6 mice following CUMS (Figures 1R and 1S). Similar results were observed by applying a smSDS stressor (Figures 1T–1V and S1F). These data suggest that vCA1 CaMKIIβ expression and activity are differentially regulated in the two strains and may contribute to their different responses to stress.
CaMKIIβ activation in the vCA1 leads to pro-resilient and antidepressant-like responses
To determine whether upregulation of CaMKIIβ in the vCA1 could lead to pro-resilient phenotypes in BALB mice, we used adeno-associated virus (AAV)-mediated gene transfer to overexpress wild-type CaMKIIβ, a constitutively active form of CaMKIIβ (CaMKIIβ-CA; T287D CaMKIIβ, in which the autophosphorylation site is replaced with the phosphomimetic residue aspartate), or green fluorescent protein (GFP) as a control, in the vCA1 of BALB mice (Figures 2A–2C). The overexpression of CaMKIIβ or CaMKIIβ-CA prevented social impairments in the SIT, increased sucrose preference in the SPT, and reduced immobility time in the FST under the stressful condition (Figures 2D–2F). In addition, CaMKIIβ activation prevented the CUMS-induced reduction of Fos expression in the vCA1 of BALB mice (Figures 2G and S2), suggesting an important role of CaMKIIβ in chronic stress-mediated changes in neural plasticity within the vCA1.
Figure 2.

CaMKIIβ activation in the vCA1 drives stress resilience and has antidepressant-like effect
(A) AAV vectors engineered to overexpress a control construct (AAV-GFP), CaMKIIβ (AAV-CaMKIIβ), or a constitutively active form of CaMKIIβ (AAV-CaMKIIβ-CA). ITR, inverted terminal repeats; PCamk2a, Camk2a promoter; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element.
(B) Experimental timeline for chronic stress and behavioral testing. CUMS, chronic ultra-mild stress; SIT, social interaction test; SPT, sucrose preference test; FST, forced swim test.
(C) Schematic representation showing the microinjection of AAV into the vCA1 region of mice. Region-specific expression of GFP in vCA1 but not dCA1 is shown. Scale bar, 500 μm.
(D–F) BALB mice overexpressing CaMKIIβ or CaMKIIβ-CA show significantly increased social interaction time in the SIT (D), increased sucrose preference in the SPT (E), and decreased immobility time in the FST (F) under stress. n = 12–13 mice per group.
(G) CaMKIIβ-CA overexpression prevents CUMS-induced reduction of Fos expression in the vCA1 of BALB mice. n = 6 mice per group.
(H) AAV-mediated spatiotemporal gene expression strategy using a cocktail of AAV-Camk2a-tTA and AAV-TRE-CaMKIIβ-CA or AAV-TRE-GFP.
(I) Experimental timeline for CaMKIIβ-CA overexpression after the termination of CUMS episodes. BALB mice were treated with doxycycline (dox) after AAV injection and exposed 21 days later to CUMS for 6 weeks; they were then taken off dox, and 4 days later behavioral tests were performed.
(J) AAV microinjection into the vCA1 region. Region-specific and dox-regulated expression of GFP in the vCA1 is shown. Scale bar, 200 μm.
(K–M) CaMKIIβ activation after CUMS leads to a significant increase in social interaction time in the SIT (K), a tendency for increased sucrose preference in the SPT (L), and reduced immobility time in the FST (M). n = 15–16 mice per group.
One-way ANOVA followed by a Tukey's post hoc test (in D-G) and two-tailed Student's t-test (in K-M) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM.
See also Figure S2. Complete statistical summaries are provided in Table S2.
Given that the activation of CaMKIIβ before and during stress episodes prevented stress-induced depression-like behaviors, we next tested whether activation of CaMKIIβ could also reverse depression-like behaviors after stress induction. We used a tetracycline system to overexpress CaMKIIβ-CA after the termination of CUMS episodes and injected AAVs expressing a tetracycline-dependent transcription activator (tTA) under the control of the Camk2a promoter (AAV-Camk2a-tTA) together with AAV-TRE-CaMKIIβ-CA or control AAV-TRE-GFP into the vCA1 of BALB mice (Figure 2H). A doxycycline (Dox) solution was used to turn off transgene expression until the termination of the CUMS episodes. At day 42, mice were switched to normal water to induce transgene expression (Figures 2I and 2J). CaMKIIβ-CA overexpression after CUMS rescued social impairments in the SIT, decreased immobility time in the FST, and tended to rescue anhedonia in the SPT (Figures 2K–2M).
CaMKIIβ inhibition in the vCA1 increases stress susceptibility in B6 mice
To determine whether CaMKIIβ downregulation induced depression-like behaviors in B6 mice, we used AAV-mediated gene transfer of short hairpin RNA (shRNA) to knockdown Camk2b vCA1 expression (Figures 3A–3C). The shRNA reduced CaMKIIβ but not CaMKIIα expression in the vCA1 (Figures 3D, S3A, and S3B). Unexpectedly, behavioral assays in mice injected with AAV-CaMKIIβ-shRNA did not show behavioral abnormalities in the SIT, SPT, or FST under the non-stressed condition (Figures 3E–3G). However, after CUMS, shRNA-injected mice showed reduced social interaction time in the SIT, decreased sucrose preference in the SPT, and increased immobility time in the FST (Figures 3E–3G). Potential off-target shRNA effects were ruled out by injecting AAV-CaMKIIβ-shRNA together with a shRNA-resistant CaMKIIβ-CA (AAV-CaMKIIβ-CARes), which rescued the social impairments in the SIT and showed a tendency to increase sucrose preference in the SPT (Figures 3H–3L). In addition to the CUMS model, we found that B6 mice injected with AAV-CaMKIIβ-shRNA showed reduced time in the interaction zone in the SIT following subthreshold smSDS exposure, which was rescued by the overexpression of CaMKIIβ-CARes (Figures 3M and 3N).
Figure 3.

CaMKIIβ inhibition in the vCA1 increases stress susceptibility
(A) AAV vectors used for the control construct (AAV-shControl) and CaMKIIβ knockdown (AAV-shCaMKIIβ). ITR, inverted terminal repeats; PCMV, cytomegalovirus promoter; PCamk2a, Camk2a promoter; U6, U6 promoter; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element.
(B) Experimental paradigm for behavioral testing. CUMS, chronic ultra-mild stress; SIT, social interaction test; SPT, sucrose preference test; FST, forced swim test.
(C) AAV microinjection into the vCA1. Region-specific expression of GFP in vCA1 is shown. Scale bar, 100 μm.
(D) Specific knockdown of CaMKIIβ but not CaMKIIα by the AAV-shCaMKIIβ.
(E–G) CaMKIIβ knockdown in the vCA1 of B6 mice leads to significantly decreased social interaction time in the SIT (E), decreased sucrose preference in SPT (F), and increased immobility time in FST (G) following CUMS exposure. n = 12–15 mice per group.
(H) AAV vectors engineered to overexpress a constitutively active form of CaMKIIβ (CaMKIIβ-CA) with resistance against shCaMKIIβ (AAV-CaMKIIβ-CARes) or a control construct (AAV-mCherry).
(I) Experimental paradigm for behavioral testing.
(J) AAV microinjection into the vCA1. Region-specific expression of GFP and mCherry in the vCA1 is shown. Scale bar, 100 μm.
(K and L) Overexpression of CaMKIIβ-CARes together with shCaMKIIβ increases time in the interaction zone in the SIT (K) and tends to increase sucrose preference in the SPT (L) in B6 mice, when compared to the mice overexpressing mCherry together with shCaMKIIβ. n = 14–15 mice per group.
(M) Experimental paradigm for behavioral testing. smSDS, subchronic and mild social defeat stress.
(N) Decreased time in the interaction zone in B6 mice given AAV-shCaMKIIβ following smSDS exposure is prevented by overexpression of CaMKIIβ-CARes. n = 15–16 mice per group.
(O) AAV vectors engineered to overexpress a dominant-negative form of CaMKIIβ (AAV-CaMKIIβ-DN) or a control construct (AAV-GFP).
(P) Experimental paradigm for behavioral testing.
(Q) AAV microinjection into the vCA1. Region-specific expression of GFP in vCA1 is shown. Scale bar, 100 μm.
(R–S) Overexpression of CaMKIIβ-DN decreases time in the interaction zone in the SIT (R) and sucrose preference in the SPT (S) in B6 mice following smSDS. n = 13–15 mice per group.
(T) Overexpression of CaMKIIβ-DN decreases Fos levels in the vCA1 of B6 mice following smSDS exposure. n = 6 for all groups.
Two-way ANOVA followed by a Tukey's post hoc test (in E-G, N, R, and S), one-way ANOVA followed by a Tukey's post hoc test (in N), and two-tailed Student's t-test (in K, L, and T) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM.
See also Figure S3. Complete statistical summaries are provided in Table S2.
In addition, we tested the behaviors of B6 mice injected with AAV vectors expressing a dominant-negative form of CaMKIIβ (CaMKIIβ-DN; CaMKIIβ-K43R), which lacks kinase activity (Fink et al., 2003), into the vCA1 (Figures 3O–3Q). We found that mice injected with AAV-CaMKIIβ-DN did not show any behavioral abnormality, as assessed by SIT and SPT in non-stressed condition. However, mice that underwent smSDS episodes had decreased time in the interaction zone in the SIT and reduced sucrose preference in the SPT (Figures 3R and 3S), thus confirming that downregulation of vCA1 CaMKIIβ function leads to enhanced stress susceptibility. We found that CaMKIIβ inhibition led to the reduction of Fos expression in the vCA1 of B6 mice following smSDS exposure (Figures 3T and S3C). Together, these results suggest that CaMKIIβ function in the vCA1 determines resilience or susceptibility to stress.
CaMKIIβ function is associated with stress-induced alterations of GluA1 expression in postsynaptic sites
How does CaMKIIβ downstream signaling modulate stress resilience or susceptibility? Previous reports have shown that the AMPAR subunit GluA1 is a target of CaMKIIs: (1) CaMKIIs phosphorylate GluA1 subunits at S831, increasing the average conductance of the channels, and (2) CaMKIIs phosphorylate the transmembrane AMPAR regulatory proteins (TARPs), increasing the number of synaptic AMPARs (Lisman et al., 2012). Therefore, to explore the potential mechanisms by which CaMKIIβ drives stress resilience or susceptibility, we first investigated the subcellular localization of GluA1 by the synaptosome fractionation of mouse vHPC. Further digestion of synaptosomes yields an insoluble ‘PSD-enriched’ (synaptic) membrane fraction and a ‘non-PSD-enriched’ (extrasynaptic) membrane fraction (Milnerwood et al., 2010; Pacchioni et al., 2009; Uchida et al., 2014, 2017; Wang et al., 2018). We confirmed the successful separation of PSD-enriched and non-PSD-enriched membranes by analyzing the distribution of the postsynaptic markers (PSD-95, GluA1, GluA2, and GluA3) in the PSD fraction and that of the presynaptic markers (synaptophysin and synapsin II) in the non-PSD fraction (Figures S4A and S4B). We found that in BALB mice, GluA1 levels in the synaptosomal fraction and whole cell extracts were not affected by CUMS (Figures 4A and 4B). However, we found reduced GluA1 levels in the PSD fraction and increased GluA1 levels in the non-PSD fraction after CUMS exposure (Figures 4C and 4D). Such CUMS-induced changes in GluA1 localization were prevented by chronic, but not subchronic, treatment with IMI (Figures 4C and 4D). In contrast, GluA1 levels in the PSD fraction of B6 mice were not affected by CUMS (Figure 4E). A similar alteration of GluA1 localization was also observed in BALB mice subjected to smSDS, in which GluA1 levels in the PSD fraction were decreased, without affecting GluA1 levels in the synaptosomal fraction; whereas GluA1 levels in the PSD fraction were normal in B6 mice exposed to the same stressor (Figures 4F–4H). We did not observe any significant differences in PSD or non-PSD GluA2 and GluA3 levels in BALB mice subjected to smSDS (Figures S4C–S4E).
Figure 4.

Disruption of GluA1 expression in PSD fraction in susceptible mice is mediated by CaMKIIβ dysfunction
(A–D) Immunoblot estimation of GluA1 level in the whole cell extract (A), synaptosomal fraction (B), postsynaptic membrane (PSD) fraction (C), and non-PSD fraction (D) of the vCA1 in BALB mice exposed to chronic ultra-mild stress (CUMS) or non-stress (NS), treated with imipramine (IMI) or water. PSD95 and synaptophysin (Syn) were used as PSD and non-PSD markers, respectively. n = 9–10 mice per group.
(E) Immunoblot estimation of GluA1 levels in the PSD fraction of the vCA1 in B6 mice exposed to CUMS. n = 7–8 mice per group.
(F–H) Immunoblot estimation of GluA1 levels in the synaptosomal fraction (F), PSD fraction (G), and non-PSD fraction (H) of the vCA1 in BALB and B6 mice exposed to subchronic and mild social defeat stress (smSDS) or NS. n = 8–10 mice per group.
(I) Experimental timeline. BALB mice were injected with AAV-CaMKIIβ-CA or AAV-GFP into the vCA1, subjected to smSDS, and euthanized to measure GluA1 levels.
(J and K) Immunoblot estimation of GluA1 levels in the PSD fraction (J) and synaptosomal fraction (K). n = 8–10 mice per group.
L. Experimental paradigm. B6 mice were injected with AAV-CaMKIIβ-DN or AAV-GFP into the vCA1, subjected to smSDS, and euthanized to measure GluA1 levels.
(M and N) Immunoblot estimation of GluA1 levels in the PSD (M) and synaptosomal (N) fractions. n = 10 mice per group.
One-way ANOVA followed by a Tukey's post hoc test (in A-D), two-way ANOVA followed by a Tukey's post hoc test (in F-H), and two-tailed Student's t-test (in E, J, K, M, and N) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM.
See also Figure S4. Complete statistical summaries are provided in Table S2.
To assess whether CaMKIIβ function in the vCA1 is critical for GluA1 synaptic localization, we injected AAV-CaMKIIβ-CA into the vCA1 of BALB mice, subjected them to subthreshold smSDS, and measured GluA1 levels in the synaptosomal and PSD fractions (Figure 4I). We found that CaMKIIβ activation enhanced GluA1 levels in the PSD but not in the synaptosomal fraction (Figures 4J and 4K). We also investigated the effect of CaMKIIβ downregulation on GluA1 synaptic localization. GluA1 levels were measured in the synaptosomal and PSD fractions of B6 mice injected with AAV-CaMKIIβ-DN and exposed to smSDS (Figure 4L). CaMKIIβ inhibition reduced GluA1 levels in the PSD fraction but not in the synaptosomal fraction (Figures 4M and 4N). These data suggest a key role for CaMKIIβ function in stress-induced dysregulation of GluA1 synaptic localization.
To test whether the alteration of GluA1 localization is associated with its phosphorylation, we measured the levels of GluA1 phosphorylation (GluA1 pS831) in the vCA1 of BALB mice after CUMS, and found them to be comparable to those of non-stressed controls (Figure S4F). In addition, AAV-mediated overexpression of CaMKIIβ-CA into the vCA1 of BALB mice did not affect GluA1 pS831 levels (Figure S4G). These results suggest that CaMKIIβ-mediated synaptic localization of GluA1 is independent of its phosphorylation.
GluA1 function is necessary for stress resilience
To directly address the role of AMPARs in the behavioral response to stress, we used the AMPAR potentiator CX614. BALB mice received CX614 infusions into the vCA1 during a 5-day period of smSDS and were tested by SIT and SPT (Figure 5A). Mice infused with CX614 did not show behavioral changes in the no-stress condition, whereas, under the stress condition, treatment with CX614 increased the time in the interaction zone in the SIT, when compared to the vehicle-treated controls, and prevented the development of anhedonia in the SPT (Figures 5B and 5C). To investigate whether the structural plasticity is influenced by smSDS exposure and AMPAR function, we performed Golgi staining to measure the dendritic spine density within the vCA1. We found that smSDS exposure reduced dendritic spine density in BALB mice, whereas this reduction was prevented by treatment with an AMPAR potentiator (Figures S5A and S5B). These data suggest that AMPAR activation is associated with neural plasticity and pro-resilient behavioral effects.
Figure 5.

GluA1 function is crucial for behavioral responses to chronic stress
(A) Experimental paradigm. BALB mice were subjected to 5-day subchronic and mild social defeat stress (smSDS). An AMPA receptor potentiator was infused into their vCA1 1 hr before smSDS exposure (day 1 to day 5, once per day). SIT, social interaction test; SPT, sucrose preference test.
(B and C) Treatment with AMPAR potentiator rescues smSDS-elicited reduction of the time in the interaction zone in the SIT (B) and prevents smSDS-induced anhedonia in the SPT (C). n = 13–14 mice per group.
(D) AAV vectors engineered to overexpress a GluA1CT (AAV-GluA1CT-T2A-mCherry) or a control construct (AAV-mCherry). PCamk2a, Camk2a promoter.
(E) Experimental paradigm. B6 mice were injected with AAV-GluA1CT-T2A-mCherry or AAV-mCherry and subjected to 5-day exposure to smSDS.
(F) AAV microinjection into the vCA1. Region-specific expression of mCherry in the vCA1 is shown. Scale bar, 100 μm.
(G) Overexpression of GluA1CT decreases Fos levels in the vCA1 of B6 mice following smSDS exposure. n = 6 mice per group.
(H) Representative examples of behavior traces of stressed mice given either AAV-mCherry or AAV-GluA1CT-T2A-mCherry during the SIT (target session).
(I and J) GluA1CT overexpression reduces time in the interaction zone in the SIT (I) and induces anhedonia in the SPT (J) after smSDS. n = 14–15 mice per group.
two-way ANOVA followed by a Tukey's post hoc test (in B, C, I, and J) and two-tailed Student's t-test (in G) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM. See also Figure S5. Complete statistical summaries are provided in Table S2.
We next investigated the effect of AAV-mediated overexpression of a dominant-negative form of GluA1 (GluA1CT), which blocks GluA1 synaptic incorporation (Rumpel et al., 2005), on the stress-resilient phenotypes of B6 mice. B6 mice were injected with AAV overexpressing GluA1CT into the vCA1 and subjected to smSDS (Figures 5D–5F). We found that GluA1CT overexpression led to the reduction of Fos expression following smSDS exposure (Figures 5G and S5C). Behavioral tests revealed social impairments in the SIT and anhedonia in the SPT (Figures 5H–5J) in both no-stress and stress conditions, thereby suggesting that GluA1 is critical for the development of behavioral stress susceptibility and resilience.
TARPγ-8 activation increases PSD GluA1 and promotes stress resilience
Given that modulating CaMKIIβ function in the vCA1 did not affect GluA1 phosphorylation in BALB mice (Figures S4G), we focused on the role of stargazin/TARPγ-8, which has key roles in GluA1 synaptic insertion and AMPAR-mediated synaptic transmission (Rouach et al., 2005), and has been reported to be regulated by CaMKIIβ phosphorylation (Park et al., 2016). These findings led us to speculate that TARPγ-8 could be involved in the stress-induced alteration of GluA1 localization and behavioral response to stress. This idea was supported in part by findings showing that TARPγ-8 mRNA expression is enriched and co-expressed with Camk2b in the vCA1 (Figure 6A), and that TARPγ-8 phosphorylation at S277 (pTARPγ-8), a major CaMKII phosphorylation site (Park et al., 2016), was differentially regulated by stress in the two strains. While B6 mice showed increased pTARPγ-8 levels after smSDS, the same stressor reduced pTARPγ-8 levels in BALB mice (Figures 6B and 6C). However, total TARPγ-8 protein levels were left unchanged by smSDS in both strains (Figures 6B and 6C). We next investigated the correlation between pTARPγ-8 expression and PSD GluA1 levels in the vCA1 and found a positive correlation in both strains (Figure 6D). We also observed positive correlation between pTARPγ-8 levels and time spent in the interaction zone in the SIT, in both strains (Figure 6E). These data suggest a potential contribution of TARPγ-8 in determining stress resilience and susceptibility.
Figure 6.

TARPγ-8-mediated GluA1 expression in the PSD fraction is associated with behavioral resilience to chronic stress
(A) Expression of the Cacng8 gene (encoding TARPγ-8) by RNA FISH (RNAscope) in a coronal section of the mouse brain. vCA1, ventral part of CA1 in the hippocampus. Scale bar, 50 μm.
(B and C) Immunoblot estimation of phosphorylated TARPγ-8 (pTARPγ-8) and total TARPγ-8 levels in the vCA1 of BALB and B6 mice exposed to subchronic and mild social defeat stress (smSDS) or no stress (NS). n = 8–10 mice per group.
(D) Linear correlation between pTARPγ-8 levels and postsynaptic membrane (PSD) GluA1 levels in the vCA1 of BALB and B6 mice.
(E) Linear correlation between pTARPγ-8 levels in the vCA1 and social interaction in BALB and B6 mice.
(F) Schematic representation of the AAV vectors engineered to overexpress a constitutively active form of TARPγ-8 (AAV-TARPγ-8D/D-T2A-mCherry) or a control construct (AAV-mCherry).
(G) Experimental paradigm for behavioral testing. BALB mice were injected with AAV-TARPγ-8D/D-T2A-mCherry or AAV-mCherry and subjected to 5-day smSDS exposure. SIT, social interaction test; SPT, sucrose preference test.
(H) Coronal view of mCherry signals in the vCA1 of BALB mice injected with AAV-TARPγ-8D/D-T2A-mCherry. Scale bar, 100 μm.
(I and J) No change in synaptosomal GluA1 levels was observed between mice injected with AAV-TARPγ-8D/D-T2A-mCherry and AAV-mCherry (I). BALB mice overexpressing TARPγ-8D/D show increased PSD GluA1 levels in the PSD fraction under stress condition (J). n = 8 mice per group.
(K and L) TARPγ-8D/D overexpression leads to a significant increase of time in the interaction zone in the SIT (K) and sucrose preference in the SPT (L). n = 14–16 mice per group.
(M) Overexpression of TARPγ-8D/D increases Fos levels in the vCA1 of BALB mice following smSDS exposure. n = 6 mice per group.
(N) Experimental paradigm for stress exposure and behavioral testing.
(O) SIT occupancy heat maps during target session obtained by averaging the location of testing animals in each group.
(P) GluA1CT overexpression prevents the increase in social interaction time induced by the activation of TARPγ-8 under the smSDS condition. n = 12 mice per group.
(Q) Experimental paradigm for stress exposure, drug treatment, and behavioral testing.
(R) B6 mice given vehicle control showed normal social interaction following smSDS exposure, but intra-vCA1 JNJ55511118 injection decreased time in the interaction zone.
One-way ANOVA followed by a Tukey's post hoc test (in P), two-way ANOVA followed by a Tukey's post hoc test (in C and R), two-tailed Student's t-test (in I-M), and Pearson's correlation (in D and E) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM.
See also Figure S6. Complete statistical summaries are provided in Table S2.
To investigate the direct role of pTARPγ-8 in GluA1 synaptic localization and behavioral response, we injected AAVs expressing TARPγ-8D/D, a phosphomimic form of TARPγ-8 with the CaMKII phosphorylation sites at S277 and S281 mutated to aspartic acid (D/D) (Park et al., 2016), into the vCA1 of BALB mice, examined their behaviors, and measured PSD GluA1 levels after smSDS (Figures 6F–6H). Overexpression of TARPγ-8D/D increased GluA1 levels in the PSD, but not in the synaptosomal fractions (Figures 6I and 6J). BALB mice overexpressing TARPγ-8D/D showed increased time in the interaction zone in the SIT and sucrose preference in the SPT (Figures 6K and 6L), and enhanced Fos levels (Figures 6M and S6A). Moreover, we found that overexpressing TARPγ-8D/D prevented the reduction of vCA1 dendritic spine density by smSDS exposure (Figures S6B and S6C). These behavioral, cellular, and morphological data suggest a pro-resilient effect of TARPγ-8 activation.
We next examined whether chronic stress influenced the formation of the TARPγ-8-GluA1 complex. Immunoprecipitation experiments showed that smSDS exposure reduced the binding of GluA1 with TARPγ-8 in BALB but not in B6 mice (Figures S6D and S6E), suggesting an important role of the TARPγ-8-GluA1 interaction in the behavioral response to chronic stress.
To test whether increased PSD GluA1 would be required for the TARPγ-8-mediated behavioral resilience observed in Figure 6K, GluA1CT was co-overexpressed with TARPγ-8D/D to inhibit GluA1 synaptic insertion in the vCA1 of BALB mice and an SIT was performed following smSDS exposure (Figure 6N). We found that the TARPγ-8-mediated increase in social interaction in stressed mice was completely blocked by GluA1CT overexpression (Figures 6O and 6P), suggesting that increased synaptic GluA1 might be necessary for TARPγ-8-mediated behavioral resilience.
We further investigated the effect of pharmacological inhibition of AMPARs containing TARPγ-8 on the stress-resilient phenotypes of B6 mice, using JNJ55511118, a negative modulator of AMPARs containing TARPγ-8, with minimal activity against other TARP-less AMPARs (Maher et al., 2016, 2017). We infused it bilaterally into the vCA1 area of B6 mice during a period of smSDS episodes and performed the SIT (Figure 6Q). Mice injected with JNJ55511118 showed reduced time in the interaction zone following smSDS exposure (Figure 6R), indicating a disruption of stress resilience by TARPγ-8 inhibition in B6 mice.
Taken together, these data suggest that the phosphorylation of TARPγ-8 plays important roles in the synaptic localization of GluA1 and in the behavioral response to chronic stress. Increased CaMKIIβ-GluA1 pathway activation in the lateral habenula (LHb) has been implicated in depression (Li et al., 2013). Given that mRNA expression of TARPγ-8 was not observed in the LHb (Figure S6F), the molecular mechanisms underlying GluA1 synaptic localization and controlling AMPAR function may differ between the HPC and LHb (see also Discussion).
TARPγ-8 activation is required for CaMKIIβ-mediated enhancement of PSD GluA1 expression and stress resilience
To better understand the role of TARPγ-8 in CaMKIIβ-mediated GluA1 synaptic localization and behavioral responses to stress, we examined the effects of modulating CaMKIIβ activity on pTARPγ-8 levels, by injecting AAVs expressing CaMKIIβ-CA into the vCA1 of BALB mice and measuring pTARPγ-8 levels after smSDS (Figure 7A). We found increased levels of pTARPγ-8, but not of total TARPγ-8, upon CaMKIIβ activation (Figure 7B). Conversely, B6 mice injected with AAVs expressing CaMKIIβ-DN into the vCA1 showed reduced levels of pTARPγ-8, but not of total TARPγ-8, after smSDS (Figures 7C and 7D).
Figure 7.

TARPγ-8 function is required for CaMKIIβ-mediated enhancement of GluA1 expression in the PSD and behavioral response to chronic stress
(A) Experimental paradigm for AAV injection, stress exposure, and behavioral testing. BALB mice were injected with AAV- CaMKIIβ-CA and subjected to 5-day exposure to subchronic and mild social defeat stress (smSDS).
(B) Immunoblot estimation of phosphorylated TARPγ-8 (pTARPγ-8) and total TARPγ-8 levels in the vCA1 of BALB mice injected with AAV-CaMKIIβ-CA or AAV-GFP. n = 8 mice per group.
(C) Experimental paradigm for AAV injection, stress exposure, and behavioral testing. B6 mice were injected with AAV-CaMKIIβ-DN and subjected to 5-day smSDS exposure.
(D) Immunoblot estimation of pTARPγ-8 and total TARPγ-8 levels in the vCA1 of B6 mice injected with AAV-CaMKIIβ-DN or AAV-GFP. n = 8 mice per group.
(E) Experimental paradigm for AAV injection, stress exposure, drug treatment, and behavioral testing.
(F–H) Immunoblot estimation of the PSD (G) and synaptosomal (H) GluA1 levels in the vCA1 of BALB mice exposed to smSDS. JNJ55511118 or vehicle was infused into the vCA1 region during the 5-day smSDS session. n = 8 mice per group.
(I) Increased time in the interaction zone after CaMKIIβ-CA overexpression is disrupted by treatment with JNJ55511118. n = 14–15 mice per group.
(J) Experimental paradigm for AAV injection, stress exposure, drug treatment, and behavioral testing.
(K–M) Immunoblot estimation of the PSD (L) and synaptosomal (M) GluA1 levels in the vCA1 of smSDS-exposed B6 mice. Mice were injected with AAV-CaMKIIβ-DN or AAV-GFP and AAV-TARPγ-8D/D-T2A-mCherry or AAV-mCherry. n = 10 mice per group.
(N) Decreased time in the interaction zone after CaMKIIβ-DN overexpression is prevented by TARPγ-8 activation. n = 14–15 mice per group.
(O) Proposed model of stress-induced depression based on vCA1 CaMKIIβ: In stress-resilient animals, the Camk2b transcript is normal but CaMKIIβ activity is enhanced following stress episodes. Increased CaMKIIβ activity stimulates the phosphorylation of TARPγ-8, which promotes GluA1 expression in synaptic sites, leading to stress adaptation. In stress-susceptible animals, the Camk2b transcript and CaMKIIβ function are downregulated, leading to reduced TARPγ-8 activity and GluA1 insertion. As a result, vCA1 dysfunction may cause depression-like behavioral alterations such as social impairment and anhedonia.
One-way ANOVA followed by a Tukey's post hoc test (in I and N) and two-tailed Student's t-test (in B, D, G, H, L, and M) were used for statistical analyses. ∗p < 0.05. Bar graphs show mean ± SEM. Complete statistical summaries are provided in Table S2.
Finally, we examined the direct role of TARPγ-8 in CaMKIIβ-mediated changes in GluA1 localization and behavioral changes following stress, by overexpressing CaMKIIβ-CA in the vCA1 of BALB mice using AAV-mediated gene transfer: The mice were infused with JNJ55511118 during a period of smSDS episodes and then subjected to SIT and PSD GluA1 level measurement (Figure 7E). Treatment with JNJ55511118 reduced GluA1 levels in the PSD but not the synaptosomal fractions (Figures 7F–7H). Behaviorally, intra-vCA1 injection of JNJ55511118 in mice overexpressing CaMKIIβ-CA reduced the time in the interaction zone in the SIT, when compared to the vehicle-treated mice overexpressing CaMKIIβ-CA (Figure 7I), suggesting that TARPγ-8 mediates the pro-resilient phenotypes induced by CaMKIIβ activation.
To examine whether the stress-susceptible behavior induced by CaMKIIβ downregulation in B6 mice (Figure 3) could be rescued by TARPγ-8 activation, we co-expressed CaMKIIβ-DN and TARPγ-8D/D in the vCA1 of B6 mice using AAV-mediated gene transfer, subjected the mice to smSDS, performed the SIT, and measured GluA1 levels (Figure 7J). Mice overexpressing TARPγ-8D/D had higher GluA1 levels in the PSD but not the synaptosomal fractions (Figures 7K–7M). Accordingly, overexpression of TARPγ-8D/D prevented the social impairment caused by CaMKIIβ downregulation under stress (Figure 7N), suggesting that TARPγ-8 activation rescues the increased stress susceptibility incurred by CaMKIIβ dysfunction.
Taken together, our data demonstrate that activation of CaMKIIβ, leading to enhanced TARPγ-8 phosphorylation, is required for GluA1 synaptic localization and stress resilience and that, conversely, dysfunction of this molecular pathway may be associated with depression (Figure 7O).
Discussion
How genes and the environment interact to shape the risk and resilience to depression is a critical question. In particular, the vHPC molecular network perturbed by chronic stress exposure remains to be clarified. To this end, we first developed a GxE animal model of depression and resilience, which indicated that CaMKIIβ, TARPγ-8, GluA1, and their interactions in the vCA1 are differentially modulated by GxE factors, thus determining susceptibility or resilience to stressful life events. In particular, TARPγ-8 is required for CaMKIIβ-mediated GluA1 synaptic localization and the subsequent stress resilience. Given that postmortem studies have demonstrated the downregulation of these three molecules in patients with major depression (Duric et al., 2013; Labonte et al., 2017; Nagy et al., 2020; Seney et al., 2018), this study highlights the importance of molecular data in shedding light on the pathophysiology and treatment of depression.
Although stressful life events increase the risk for depression, there is significant individual variability in such vulnerability, likely mediated by GxE interactions (Krishnan and Nestler, 2008), the mechanisms of which remain to be elucidated. We hypothesized that different mechanisms are involved in different mouse strains, just as different mechanisms may be involved in genetically heterogeneous human populations, so that results obtained from different mouse strains might be translatable to humans. We demonstrated that BALB and B6 mice exhibited differential behavioral responses to stress. BALB mice showed social impairment and anhedonia after CUMS or smSDS, and these abnormalities were prevented by chronic, but not acute, treatment with antidepressants, but in contrast, B6 mice did not. These GxE models thus enabled us to examine the molecular mechanisms underlying how genes and the environment interact to shape depression risk and resilience.
Recent optogenetic studies showed that acute activation of vCA1 cells projecting to the nucleus accumbens, lateral hypothalamus, or medial prefrontal cortex enhances pro-depressive and anxiety behaviors (Bagot et al., 2015; Ciocchi et al., 2015; Jimenez et al., 2018; Padilla-Coreano et al., 2016), in contrast with our results showing decreased neuronal activity in the vCA1 of stress-susceptible BALB mice. However, the inhibition of lateral septum-projecting vHPC cells was shown to have anxiogenic effects (Parfitt et al., 2017) and the activity of the vCA1-medial prefrontal cortex pathway was found necessary for antidepressant-like behavior (Carreno et al., 2016). Furthermore, recent reports have shown that pyramidal neurons in the deep and superficial layers of the hippocampal CA1 region differ in their physiological and behavioral properties (Danielson et al., 2016; Pi et al., 2020), suggesting opposite roles for the vCA1 superficial and deep layers in modulating anxiety-related behaviors (Pi et al., 2020). Thus, the vCA1 is a physiologically and functionally heterogeneous structure and distinct subpopulations of neurons within it may respond to positive or negative valence quality. Hence, vCA1 neurons may route distinct behavior-contingent information selectively to different target areas according to information context (Ciocchi et al., 2015). A single-cell gene expression profile of the HPC revealed that the dCA1 region is composed of relatively homogeneous cells, while many cell types are observed in the vCA1 (Shah et al., 2016), corresponding to the diverse roles of the vCA1 in modulating multiple behaviors. Importantly, CaMKIIβ loss-of-function did not alter social interaction, sucrose preference, or immobility time in the non-stressed condition, but induced behavioral abnormalities after chronic stress, demonstrating the role of vCA1 cell populations in the response to long-term environmental stimulation. To better understand the role of the vCA1 in stress-induced depression-like behaviors, further investigations will have to clarify the effect of chronic stimulation of the neuronal activity of specific vCA1 cell subpopulations on physiological and behavioral properties.
Although numerous studies have focused on acute stimulation and cell type-specific control of the activity of vCA1 pyramidal neurons projecting from/to other brain areas with optogenetic approaches, less attention was paid to the dynamic molecular events in the vCA1 under chronic stress (Eagle et al., 2020). We demonstrated a critical role of CaMKIIβ-mediated expression of AMPA receptor GluA1 in the PSD fraction of vCA1 pyramidal neurons in the behavioral response under stress. Decreasing and increasing CaMKIIβ function in the vCA1 leads to stress susceptibility and resilience, respectively. Previous studies have shown that stress-induced CaMKIIβ upregulation in the LHb leads to more GluA1 insertion into synapses, resulting in increased synaptic efficacy and behavioral depression (Li et al., 2013). Thus, CaMKIIβ expression/function is differentially modulated in a brain region-specific manner by chronic stress. This model is similar to that described in the ventral tegmental area and HPC, where BDNF levels are positively and negatively regulated by chronic social defeat stress (Berton et al., 2006; Tsankova et al., 2006). Given that TARPγ-8 is enriched in the HPC, but not in the LHb, we suggest an HPC-specific mechanism by which chronic stress modulates the expression of PSD GluA1 via the CaMKIIβ/TARPγ-8 pathway. Whether and how GxE interaction influences CaMKIIβ phosphorylation and expression in the vCA1 and LHb warrants further investigation.
How CaMKIIs influence GluA1 expression in the PSD sites in response to stress remains an important open question. The glutaminergic system has been implicated in the pathophysiology and treatment of depression (Duman and Aghajanian, 2012; Popoli et al., 2011; Thompson et al., 2015). To explore the mechanism underlying depression susceptibility and resilience, we investigated the potential involvement of AMPARs, implicated in depression pathophysiology and treatment (Autry et al., 2011; Duric et al., 2013; Li et al., 2010), as downstream substrates of CaMKIIβ. Although a link between GluA1 phosphorylation and depression-like and/or antidepressant-like behavior has been reported (Cai et al., 2013; Popoli et al., 2011; Zhang et al., 2016), we did not find changes in GluA1 phosphorylation (at S831) either in the GxE animal model of depression or in mice with activated CaMKIIβ. However, it should be noted that the levels of phosphorylated GluA1 at S831 are nearly negligible (less than 1%), and most synapses do not contain any phosphorylated AMPAR (Hosokawa et al., 2015). Indeed, GluA1 phosphorylation at S831 and S845 did not affect the total and surface expression or the subcellular localization of GluA1 (Makino et al., 2011) and basal synaptic transmission and synaptic plasticity were quite normal in mutant mice lacking S831 phosphorylation (Lee et al., 2010). In addition, CaMKII-mediated delivery of GluA1 to synapses is independent of phosphorylation at S831 (Hayashi et al., 2000), confirming our results and also suggesting that other molecules could be involved in CaMKIIβ-mediated changes in the expression of PSD GluA1 in response to stress. We provide direct evidence demonstrating that TARPγ-8 inhibition alone is sufficient to decrease the levels of PSD GluA1, and that TARPγ-8 inhibition disrupts CaMKIIβ-mediated enhancement of PSD GluA1 expression and stress resilience, suggesting that TARPγ-8 is required for the pro-resilience action of CaMKIIβ. Indeed, a previous report has shown that long-term potentiation and memory formation are substantially impaired in mice lacking TARPγ-8 CaMKII phosphorylation sites (Park et al., 2016), supporting an important role for the CaMKIIβ-TARPγ-8 pathway in synaptic transmission. Hence, we identified TARPγ-8 as a key molecular determinant of behavioral depression, using a combination of molecular, biochemical, genetic, behavioral, and pharmacological approaches.
We thus propose that TARPγ-8 may have potential therapeutic utility for depression. As noted above, CaMKIIβ-mediated GluA1 insertion was respectively up- and downregulated in the LHb and vCA1 regions by chronic stress. Given that TARPγ-8 is not expressed in the LHb, it would be more beneficial if GluA1-associated TARPγ-8 could be selectively activated by small molecule compounds to ameliorate depression symptoms. Thus, the development of a TARPγ-8 activator may enable more precise brain region targeting and higher effectiveness. The targeting of receptor-associated proteins has recently garnered great interest in neuropsychiatry, as it may provide more versatile and selective pharmacotherapy, as distinct brain regions are differentially and sometimes oppositely perturbed in neurobiological processes (Maher et al., 2017).
In conclusion, our data suggest that downregulation of the CaMKIIβ/TARPγ-8/GluA1 pathway in the vCA1 region mediates chronic stress susceptibility, whereas enhancement of this pathway contributes to behavioral resilience. The elucidation of the mechanisms underlying individual differences in the regulation of the CaMKIIβ/TARPγ-8/GluA1 pathway caused by GxE interacting factors is a critical step toward the development of mechanism-based treatments for depression.
Limitations of the study
Although we used a subcellular fractionation assay to measure GluA1 expression in PSD and non-PSD fractions of mice in this study, we did not provide direct evidence indicating the altered synaptic trafficking of GluA1 as consequences of the exposure of chronic stress and/or the CaMKIIβ/TARPγ-8 activation/inhibition. Thus, because of the limitations of the subcellular fractionation assay, there is still a need for further experimental validations in follow-up studies to gain further mechanistic insight into GluA1 trafficking in vivo with higher sensitivity and precision.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| c-Fos (9F6) Rabbit mAb | Cell Signaling Technology | Cat# 2250, RRID:AB_2247211 |
| GFP Polyclonal Antibody | Thermo Fisher Scientific | Cat# A-11122, RRID:AB_221569 |
| Living Colors® mCherry Monoclonal Antibody | Takara Bio | Cat# 632543, RRID:AB_2307319 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody | Thermo Fisher Scientific | Cat# A-21206, RRID:AB_2535792 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-21203, RRID:AB_141633 |
| CaMKII beta Monoclonal Antibody (CB-beta-1) | Thermo Fisher Scientific | Cat# 13-9800, RRID:AB_2533045 |
| Phospho-CaMKII (Thr286) (D21E4) Rabbit mAb antibody | Cell Signaling Technology | Cat# 12716, RRID:AB_2713889 |
| CaMKIIalpha (6G9) antibody | Santa Cruz Biotechnology | Cat# sc-32288, RRID:AB_626787 |
| Anti-GluR1-NT (NT) Antibody, clone RH95 | Millipore | Cat# MAB2263, RRID:AB_11212678 |
| Anti-Glutamate Receptor 2, extracellular, clone 6C4 antibody | Millipore | Cat# MAB397, RRID:AB_2113875 |
| AMPA Receptor (GluR 3) (D47E3) Rabbit mAb antibody | Cell Signaling Technology | Cat# 4676, RRID:AB_10547136 |
| Anti-phospho-GluR1 (Ser831) Antibody, clone N453, rabbit monoclonal | Millipore | Cat# 04-823, RRID:AB_1977218 |
| Anti-α-Tubulin antibody | Sigma-Aldrich | Cat# T9026, RRID:AB_477593 |
| Rabbit Anti-PSD95 Polyclonal Antibody | Cell Signaling Technology | Cat# 2507, RRID:AB_561221 |
| Mouse Anti-Synaptophysin Monoclonal antibody | Millipore | Cat# MAB5258-50UG, RRID:AB_95187 |
| Rabbit Anti-Synapsin II Monoclonal Antibody | Abcam | Cat# ab76494, RRID:AB_2200420 |
| Anti-Histone H4 antibody | Millipore | Cat# 07-108, RRID:AB_2279758 |
| Anti-GM130 (C-terminal) antibody produced in rabbit | Sigma-Aldrich | Cat# G7295, RRID:AB_532244 |
| Anti-β-Actin Antibody | Sigma-Aldrich | Cat# A5441, RRID:AB_476744 |
| Anti-TARPγ-8 antibodt |
Park et al. (2016) Sumioka et al. (2011) |
N/A |
| Anti-phospho-TARPγ8 S277 antibody |
Park et al. (2016) Sumioka et al. (2011) |
N/A |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076, RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| AMPA Receptor 1 (GluA1) (D4N9V) antibody | Cell Signaling Technology | Cat# 13185, RRID:AB_2732897 |
| Anti-rabbit IgG (Conformation Specific) (L27A9) mAb (HRP Conjugate) antibody | Cell Signaling Technology | Cat# 5127, RRID:AB_10892860 |
| Bacterial and virus strains | ||
| AAV8-Camk2a-GFP | Higuchi et al. (2016) | N/A |
| AAV8-Camk2a-CaMKIIβ | This paper | N/A |
| AAV8-Camk2a-CaMKIIβ-CA | This paper | N/A |
| AAV8-Camk2a-CaMKIIβ-DN | This paper | N/A |
| AAV9-CMV-GFP-U6-shControl | Vector Biolabs | N/A |
| AAV9-CMV-GFP-U6-shCaMKIIβ | Vector Biolabs | N/A |
| AAV8-Camk2a-CaMKIIβ-CARes | This paper | N/A |
| AAV8-Camk2a-mCherry | This paper | N/A |
| AAV8-Camk2a-GluA1CT-T2A-mCherry | This paper | N/A |
| AAV8-Camk2a-GluA1CT-T2A-EGFP | This paper | N/A |
| AAV8-Camk2a-TARPγ-8D/D-T2A-mCherry | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Imipramine | Sigma | I7379 |
| Fluoxetine | FujiFilm Wako | 062-05681 |
| Doxycycline | Sigma | D9891 |
| CX614 | Tocris | 5149 |
| Hydroxypropyl-β-cyclodextrin | Sigma | 332607 |
| Hydroxypropyl methylcellulose | Sigma | 09963 |
| JNJ-55511118 | Tocris | 5666 |
| DAPI | Sigma | D9542 |
| TRIzol Reagent | Thermo Fisher Scientific | 15596026 |
| Protein G Dynabeads | Thermo Fisher Scientific | 10004D |
| Critical commercial assays | ||
| SYBR green PCR Master Mix | Thermo Fisher Scientific | 4334973 |
| Direct-zol RNA Miniprep Plus | Zymo research | R2072 |
| RNAscope Fluorescent Multiplex 2.0 assay | Advanced Cell Diagnostics | 320850 |
| Pierce 660nm Protein Assay Reagent | Thermo Fisher Scientific | 22662 |
| KOD Plus Mutagenesis kit | TOYOBO | SDMK-101 |
| FD Rapid Golgi Stain kit | FD NeuroTechnologies | PK401 |
| Deposited data | ||
| Original images of the Western blots | This paper | Mendeley Data, https://doi.org/10.17632/m8zyb8mmtg.1 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | Charles River | RRID: IMSR_JAX:000664 |
| Mouse: BALB/c | Charles River | RRID:MGI:6323059 |
| Mouse: CD1 | Charles River | RRID:IMSR_CRL:022 |
| Mouse: C3H | Charles River | RRID:IMSR_CRL:025 |
| Oligonucleotides | ||
| Real-time PCR primers | This paper | See Table S1 |
| Recombinant DNA | ||
| pAAV-CMV-MCS | Higuchi et al. (2016) | N/A |
| pAAV-Camk2a-MCS | Higuchi et al. (2016) | N/A |
| pAAV-Camk2a-GFP | Higuchi et al. (2016) | N/A |
| pAAV-SYP1-miniSOG-T2A-mCherry vectors | Lin et al. (2013) | Addgene #50972 |
| pEGFPC1-GluR1Ctail | Rumpel et al. (2005) | Addgene #32441 |
| pAAV-Camk2a-CaMKIIβ | This paper | N/A |
| pAAV-Camk2a-CaMKIIβ-CA | This paper | N/A |
| pAAV-Camk2a-CaMKIIβ-DN | This paper | N/A |
| pAAV-CMV-GFP-U6-shControl | Vector Biolabs | N/A |
| pAAV-CMV-GFP-U6-shCaMKIIβ | Vector Biolabs | N/A |
| pAAV-Camk2a-CaMKIIβ-CARes | This paper | N/A |
| pAAV-Camk2a-mCherry | This paper | N/A |
| pAAV-Camk2a-GluA1CT-T2A-mCherry | This paper | N/A |
| pAAV-Camk2a-GluA1CT-T2A-EGFP | This paper | N/A |
| pAAV-Camk2a-TARPγ-8D/D-T2A-mCherry | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software | RRID:SCR_002798 |
| ImageJ | NIH | RRID:SCR_003073 |
| Excel | Microsoft | RRID:SCR_016137 |
| Photoshop CS6 | Adobe | RRID: SCR_014199 |
| Any-maze | Stoeling | RRID:SCR_014289 |
| BZ-X800 analyzer | Keyence | https://www.keyence.com/ |
| ImageStudio software | LI-COR Biosciences | RRID:SCR_013715 |
| Other | ||
| RNAscope probe: Cacng8 | Advanced Cell Diagnostics | cat#556161-C1 |
| RNAscope probe: Camk2b | Advanced Cell Diagnostics | cat#453601-C2 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shusaku Uchida (Uchida.shusaku.3n@kyoto-u.ac.jp).
Materials availability
All unique reagents used in this study are available from the Lead Contact upon reasonable requests.
Data and code availability
Original data for the Western blots in the paper are available Mendeley Data https://doi.org/10.17632/m8zyb8mmtg.1.
Experimental model and subject details
Animals
All of the animal procedures were approved by the Institutional Animal Care and Use Committees of Kyoto University and Yamaguchi University, and performed in accordance with the Guidelines for Animal Care and Use of Yamaguchi University and Kyoto University. Experimental animals were either male C57BL/6J or BALB/c mice (Charles River, Japan), 8 weeks old at the time of experimentation. In addition, retired breeder CD1 male mice (Charles River, Japan) were used as aggressors in the social defeat stress procedure and in the social interaction test (SIT); however, they were not included in any analysis. Four to six-week-old C3H male mice (Charles River, Japan) were used in the free-moving SIT. C57BL/6J, BALB/c, and C3H mice were housed under standard conditions in groups of 4-5 animals; CD-1 mice were housed individually until the start of the experiment on a 12-h light/dark cycle with mouse chow and water ad libitum.
Method details
Chronic ultra-mild stress (CUMS)
The CUMS procedure (Uchida et al., 2011b) was solely based on environmental and social stressors, which did not include food/water deprivation. A total of three stressors were used in this study. For the first stressor, two of the following five stressors were delivered randomly over a period of 1–2 h with a 2-h stress-free time period between the two stressors: a period of cage tilt (30°), confinement to a small cage (11 × 8 × 8 cm), paired housing, soiled cage (50 ml water per 1 l of sawdust bedding), and odor (10% acetic acid). The second stressor consisted of four nocturnal stressors, including one overnight period with difficult access to food, one overnight period with permanent light, one overnight period with a 30° cage tilt, and one overnight period in a soiled cage. For the third stressor, a reversed light/dark cycle was used from Friday evening to Monday morning. This procedure was scheduled over a 1-week period and repeated 6 times, but the reversed light/dark cycle was omitted during the weekend of the last week (sixth week) of the session. Non-stressed mice were handled every day for weighing purposes.
Social defeat stress
For chronic social defeat stress (CSDS) (Berton et al., 2006; Golden et al., 2011), retired male breeder CD1 mice were used as aggressors and housed in the social defeat cage (18.8 cm width × 29.6 cm depth × 12.8 cm height) 72 h before the start of defeats on one side of the cage separated by a clear perforated Plexiglas divider. On the first day of stress, the C57BL/6J mouse was placed in the same side of the cage as the CD1 aggressor for a period of 10 min/day. During this time, the C57BL/6J mouse was physically attacked by the unfamiliar CD1 aggressor. Following the physical bout, C57BL/6J mice were removed and housed on the opposite side of the divider, where it remained in sensory contact with the CD1 aggressor, over a 24 h period. This session was repeated for 10 consecutive days, and the C57BL/6J mouse was then exposed to a new CD1 aggressor in a different defeat cage. In the control group, C57BL/6J mice were double-housed in the same social defeat cage separated by a clear perforated Plexiglas divider for the length of the stress period and rotated daily in a similar manner without being exposed to the CD1 aggressor.
In order to assess stress susceptibility, we used an abbreviated subthreshold social defeat stress, referred to as subchronic mild social defeat stress (smSDS) in this study. In this paradigm, BALB/c mice were subjected to defeat stress for a period of 5 min over 5 consecutive days while the other procedures were the same as in the CSDS condition described above.
Social interaction test
For mice subjected to CUMS episodes, the test mouse was placed in a neural cage and allowed to explore freely for at least 60 min (habituation). At the end of the habituation, a male juvenile mouse (C3H strain) was introduced into the cage, and the amount of time spent in social interaction (e.g., grooming, licking, sniffing, or crawling over or under the other mouse) by the tested adult mouse was recorded during a 3-min session.
For mice subjected to CSDS or smSDS, the test mice were placed in an open arena (42 cm width × 42 cm depth × 42 cm height) with an empty wire cage (10 cm width × 6.5 cm depth × 42 cm height). Animals were given 3 min to explore the arena as a first stage (empty session) and then removed. A novel CD1 mouse enclosed in the wire cage was placed in the arena, and the procedure was repeated as a second stage (target session). Time spent in an area surrounding the wire cage (interaction zone, 24 cm × 14 cm) and in the corner zones (9 cm × 9 cm) opposite to the interaction zone was measured automatically with an ANY-maze tracking system (Stoeling).
Sucrose preference test
Mice were habituated to drink water from two bottles for at least 7 days. Mice were then submitted to forced exposure to 1.5% sucrose solution for 24 h to avoid neophobia. Mice were subjected to water deprivation for 16 h prior to the SPT. Then, mice were given two pre-weighed bottles (one containing tap water and another containing 1.5% sucrose solution) for 4 h. The positions of the water and sucrose bottles (left or right) were switched every 2 h. The bottles were weighed again, and the weight difference was considered to be the mouse intake from each bottle. The preference for the sucrose solution was expressed as the percentage of sucrose intake relative to the total intake (sum of the water and sucrose intakes).
Forced swim test
Mice were placed in a water tank (20 cm height × 14 cm diameter filled with 23-24°C water to a depth of 13 cm) for 5 min, and the duration of floating (i.e., the time during which the mouse only performed the small movements necessary to keep its head above water) was measured.
Drug treatment
For the treatment with imipramine (IMI, Sigma, I7379) and fluoxetine (FLX, Fuji Film Wako, 062-05681) was dissolved in tap water at a concentration of 160 mg/l (Abe-Higuchi et al., 2016; Higuchi et al., 2016; Uchida et al., 2011b). Drug solutions were protected from light in opaque water bottles and were replaced every other day. Vehicle-treated animals received regular (tap) drinking water.
For the administration of doxycycline (Dox), solutions containing 200 μg/ml Dox (Sigma, D9891) in drinking water were prepared and, due to the sensitivity of Dox to light, supplied in light-protected bottles and replaced every 3 days.
For the intra-vCA1 injection, CX614 (Tocris) was dissolved in hydroxypropyl-β-cyclodextrin vehicle (Sigma) as a stock solution and diluted with saline just before injection (final conc., 20 μM). JNJ-55511118 (Tocris) was dissolved in hydroxypropyl methylcellulose (Sigma, final conc., 1 μM) (Maher et al., 2016). Mice were anesthetized with Avertin (250 mg/kg body weight) and stainless steel guide cannulae (26 gauge, Plastics One) were implanted into the vCA1 (bregma, −3.28 mm anteroposterior (AP), ±3.3 mm mediolateral (ML), −3.0 mm dorsoventral (DV)). Seven days after surgery, mice were injected the compound using a microliter syringe (10 μl, Hamilton, USA) connected by polyethylene tubing to an internal cannula (33 gauge, Plastics One) that projected 1 mm below the guide cannula. The compounds were infused (0.5 μl/hemisphere) at a rate of 0.1 μl/min. On completing the experimentation, we confirmed the location of the injection sites by injecting 0.5 μl of Chicago Sky Blue solution bilaterally, followed by histological analysis of coronal brain slices. Mice that did not receive symmetrical and bilateral injections in the vCA1 were excluded from the study.
Immunohistochemistry
Mice were deeply anesthetized via intraperitoneal injection of Avertin (250 mg/kg body weight) and transcardially perfused with a fixative solution containing 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.4). Brains were post-fixed for 24 h in 4% PFA at 4°C and cryoprotected for 3 days in 30% sucrose in phosphate buffered saline (PBS) at 4°C. The brains were then frozen and 20-μm coronal sections were cut with a cryostat (Leica) and stored at −20°C in a solution containing 30% ethylene glycol (v/v), 30% glycerol (v/v), and 0.1 M phosphate buffer (pH 7.4). Free-floating sections were incubated with blocking solution (3% normal goat serum, 3% bovine serum albumin, and 0.3% TritonX-100 in PBS) for 1 h. Primary antibodies against Fos (1:500, Cell Signaling Technology, #2250), GFP (1:1000, Thermo Fisher Scientific, #A11122), and mCherry (1:1000, Takara Bio Clontech, #632543) were diluted in blocking solution and sections were incubated overnight at 4°C with gentle shaking. The sections were then washed three times in PBS and incubated with the secondary antibodies (Alexa Fluor 488 donkey anti-rabbit IgG, 1:1000, Thermo Fisher Scientific, #A21206; Alexa Fluor 594-conjugated anti-mouse IgG, 1:1000, Thermo Fisher Scientific, #21203) in blocking solution for 2 h at room temperature. The sections were then washed and stained with DAPI (Sigma, D9542) for 10 min. After the final washes in PBS, the sections were mounted in Vectashield (Vector Labs). Images were acquired using a BZ-X800 fluorescence microscope (Keyence) and analyzed using BZ-X800 analyzer software (Keyence).
RNA extraction, cDNA synthesis, and real-time polymerase chain reaction (PCR)
Mice were sacrificed by rapid decapitation; tissues were dissected, flash-frozen, and stored at −80°C until processing. Specific brain regions were isolated from brain tissue sections using disposable punches (dCA1, Bregma −1.34 mm to −2.30 mm and vCA1, Bregma −2.92 mm to −3.52 mm). Total RNA from dissected tissues was extracted using the TRIzol Reagent (Thermo Fisher Scientific) and Direct-zol RNA Miniprep Plus (Zymo research), according to the manufacturers' instructions. The quality of RNA was determined using a spectrophotometer (NanoDrop, Thermo Fisher Scientific), based on the A260/A280 ratio, which was 1.90–2.05 for all RNA preparations. One microgram of total RNA was used for cDNA synthesis using the QuantiTect Reverse Transcription Kit (Qiagen). The cDNA was stored at −80°C until use. Real-time PCR amplification was performed using a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) with SYBR green PCR Master Mix (Thermo Fisher Scientific) according to the manufacturer's protocol. PCR conditions were 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 30 s at 60°C. The sequences of all primers are listed in Table S1. Amplification of a single PCR product was confirmed by monitoring the dissociation curve and electrophoresis on 1.2% agarose gels stained with ethidium bromide. Amplification curves were visually inspected to set a suitable baseline range and threshold level. The relative quantification method was employed for the quantification of target molecules according to the manufacturer's protocol, where the ratio between the amount of each target molecule and a reference molecule within the same sample was calculated. All measurements were performed in triplicate. Gapdh and β-actin were used to normalize the relative expression levels of each target mRNA. All data shown are normalized to Gapdh. β-actin-normalized data were very similar to Gapdh-normalized data.
RNAscope fluorescent in situ hybridization (FISH)
Brains were rapidly dissected, flash-frozen with isopentane chilled with dry ice, and stored at −80°C until sectioning for FISH. Coronal brain slices (20 μm) containing the lateral habenula or vHPC were sectioned on a cryostat (Leica) at −20°C, mounted directly onto slides, and stored at −80°C until FISH processing. FISH for Cacng8 (TARPγ-8) and Camk2b mRNA was performed using the RNAscope Fluorescent Multiplex 2.0 assay (Advanced Cell Diagnostics) as per the manufacturer's instructions. Briefly, slides were fixed in 4% PFA for 15 min at 4°C and subsequently dehydrated for 5-10 min with 50%, 70%, and 100% ethanol at room temperature. Sections were then incubated with protease-IV solution for 30 min at room temperature. Slides were washed with PBS, before being incubated with appropriate probes for 2 h at 40°C in the HybEZ oven (Advanced Cell Diagnostics). All probes used were commercially available from Advanced Cell Diagnostics: Cacng8 (cat#556161-C1) and Camk2b (cat#453601-C2). After washing with wash buffer, the signal was amplified by incubating the sections in amplification buffers at 40°C in the HybEZ oven. After the final rinse, DAPI solution was applied to the sections at room temperature. Finally, slides were coverslipped and visualized under a fluorescence microscope (Keyence).
Preparation of total, synaptosomal, PSD, and non-PSD fractions
Mice were sacrificed by rapid decapitation; tissues (dorsal or ventral hippocampi) were dissected, flash-frozen, and stored at −80°C until processing. For whole cell extracts from the dorsal or ventral hippocampi, the tissues were homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.4), 0.1% SDS, 1% TritonX-100, 0.5% sodium deoxycholate, 1 mM EDTA, 150 mM NaCl, supplemented with Halt Protease and Phosphatase inhibitor cocktail (Thermo Fisher Scientific), sonicated, and centrifuged at 10,000 x g for 10 min at 4°C. The protein concentration of the resulting supernatants was determined using a Pierce 660nm Protein Assay Reagent (Thermo Fisher Scientific).
For the subcellular fractionation (Uchida et al., 2014, 2017), the ventral and dorsal HPC were isolated and nuclear, S2, non-PSD, and PSD fractions were isolated according to a standard protocol (Milnerwood et al., 2010; Pacchioni et al., 2009; Wang et al., 2018) with minor modifications. The tissues were homogenized in cold buffer containing 0.32 M sucrose, 50 mM NaF, and 10 mM HEPES (pH 7.4). The homogenates were centrifuged twice at 1000 x g for 10 min to yield the nuclear enriched pellet and the S1 fraction. To obtain the S2 fraction, the S1 fraction was centrifuged at 12,000 x g for 20 min at 4°C, and the supernatants (microsomes and cytosol) were collected. To obtain the synaptosomal fraction, the S1 fraction was filtered using a nitrocellulose filter (5 μm, Millipore) and centrifuged at 1000 x g for 20 min. The synaptosomal pellet was rinsed twice (4 mM HEPES, 1 mM EDTA, pH 7.4; centrifugation for 20 min at 12,000 x g). The resulting pellet was incubated (20 mM HEPES, 100 mM NaCl, 0.5% Triton X-100, pH 7.2) for 15 min and then centrifuged at 12,000 x g for 20 min. The supernatant (Triton-soluble NP fraction) containing non-PSD membranes was retained. The pellet was resuspended in buffer containing 20 mM HEPES, 0.15 mM NaCl, 1% Triton X-100, 1% deoxycholic acid, and 1% SDS, pH 7.5, for 1 h and was then centrifuged at 10,000 x g for 15 min. The supernatant contained the PSD, or Triton-insoluble, fraction. The protein concentrations were determined using a Pierce 660nm Protein Assay Reagent (Thermo Fisher Scientific).
Protein expression analysis
Equal amounts of proteins were separated on 10% or 12% Bis-Tris gels (Thermo Fisher Scientific), and transblotted onto polyvinylidene difluoride membranes (GE Healthcare Bio-Sciences). After blocking the membrane with 5% skim milk or 5% BSA in TBS-T (1% Tween20 in Tris-buffered saline (TBS)), the membranes were incubated with primary antibodies diluted in blocking buffer at 4°C overnight. The following primary antibodies were used for Western blotting: mouse anti-CaMKIIβ (Thermo Fisher Scientific, 13-9800, 1:2000), rabbit anti-phospho-CaMKII T286 (Cell Signaling Technology, 12716, 1:1000), mouse anti-CaMKIIα (Santa Cruz Biotechnology, sc-32288, 1:2000), mouse anti-GluA1 (Millipore, MAB2263, 1:1000), mouse monoclonal anti-GluA2 (Millipore, MAB397, 1:1000), rabbit polyclonal anti-GluA3 (Cell Signaling Technology, 4676, 1:1000), rabbit anti-phospho-GluA1 S831 (Millipore, 04-823, 1:1000), mouse anti-α-Tubulin (Sigma, T9026, 1:10000), rabbit anti-PSD95 (Cell Signaling Technology, 2507, 1:1000), mouse anti-synaptophysin (Millipore, MAB5258, 1:1000), rabbit monoclonal synapsin II (Abcam, ab76494, 1:5000), rabbit anti-histone H4 (Millipore, 07-108, 1:1000), rabbit anti-GM130 (Sigma, G7295, 1:1000), and mouse anti-β-actin (Sigma, A5441, 1:10000). The specificity of primary antibodies against TARPγ-8 (1:1000) and phospho-TARPγ-8 S277 (1:1000) was validated as previously reported (Park et al., 2016; Sumioka et al., 2011). After rinsing three times, membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies diluted in TBS-T (HRP-linked anti-mouse IgG, Cell Signaling Technology, 7076, 1:2000; or HRP-linked anti-rabbit IgG antibody, Cell Signaling Technology, 7074, 1:2000) for 1 h at room temperature. Then the washed blots were developed with an Amersham Imager system (GE Healthcare) using a Pierce ECL-Plus Western Blotting Substrate (Thermo Fisher Scientific). When the phospho-specific antibodies were used, all buffers included sodium fluoride (50 mM). Densitometric analysis was performed with ImageStudio software (LI-COR Biotechnology) and the protein load was normalized to that of the appropriate loading control.
Immunoprecipitation
For the Immunoprecipitation experiments (Uchida et al., 2011b), vHPC tissues were homogenized in a Dounce homogenizer with 1.0 ml ice-cold lysis buffer containing 0.32 M sucrose, 10 mM HEPES (pH 7.4), 2 mM EDTA, and Halt Protease and Phosphatase inhibitor cocktail (Thermo Fisher Scientific); the homogenate was kept at 4°C for 30 min before the debris was removed by centrifugation at 1,000 g for 15 min. The resulting supernatant was then centrifuged at 10,000 g for 30 minutes. The resulting pellet was lysed with 0.5% Triton X-100 in PBS at 4°C overnight and then stored at -80°C until use. Anti-GluA1 antibodies (mixture of rabbit polyclonal anti-GluA1, Millipore, MAB2263 and rabbit polyclonal anti-GluA1, Cell Signaling Technology, #13185) were bound and crosslinked to Protein G Dynabeads (Thermo Fisher Scientific) according to the manufacturer's instructions. The resulting supernatant was incubated with antibody conjugated Protein G Dynabeads in lysate solution. Beads were washed five times with 0.2% Tween-20 in PBS, and coimmunoprecipitated proteins were recovered in 4x Bolt LDS Sample Buffer (Thermo Fisher Scientific) and heated at 90°C for 5 minutes. Western blot analyses of the immunoprecipitated anti-GluA1, input, and negative control (rabbit IgG) samples were performed with anti-TARPγ-8 or anti-GluA1 antibody as described above. An HRP-linked anti-rabbit IgG (Conformation specific, Cell Signaling Technology, 5127, 1:1000) was used as the secondary antibody to avoid the detection of both light and heavy chains.
Viral vector construction
The plasmids for generating AAV vectors were constructed using standard molecular cloning methods. The plasmids were constructed via PCR amplification of the open reading frame of the gene of interest (CaMKIIβ, GluA1CT, EGFP, mCherry) and the genomic region of promoters (mouse Camk2a, human U6 promoter, CMV promoter). Codon-optimized full-length TARPγ-8 DNAs were synthesized due to high GC content. These DNA fragments were inserted into the pAAV-CMV-MCS (Agilent), pAAV-Camk2a-MCS, pAAV-Camk2a-GFP (Higuchi et al., 2016) or pAAV-SYP1-miniSOG-T2A-mCherry vectors (a gift from Roger Tsien; Addgene plasmid #50972, (Lin et al., 2013). GluA1CT and mCherry and T2A-mCherry DNA fragments were obtained from pEGFPC1-GluR1Ctail (a gift from Roberto Malinow; Addgene plasmid #32441, (Rumpel et al., 2005)) and pAAV-SYP1-miniSOG-T2A-mCherry. The mutations (CaMKIIβ-CA, CaMKIIβ-DN, CaMKIIβ-CARes, and TARPγ-8D/D) were induced using a KOD Plus Mutagenesis kit (TOYOBO, Japan). The pAAV-cis plasmid encoding a shRNA specific to Camk2b (GAAGCATTCCAACATTGTA) was generated by Vector Biolabs, as were recombinant viruses (AAV serotype 8 or 9). The genomic titer of each virus was determined using Q-PCR: AAV8-Camk2a-GFP (6.7 × 1013 vector genomes (vg)/ml), AAV8-Camk2a-CaMKIIβ (3.4 × 1013 vg/ml), AAV8-Camk2a-CaMKIIβ-CA (2.8 × 1013 vg/ml), AAV8-Camk2a-CaMKIIβ-DN (6.1 × 1013 vg/ml), AAV9-CMV-GFP-U6-shControl (1.8 × 1013 vg/ml), AAV9-CMV-GFP-U6-shCaMKIIβ (1.8 × 1013 vg/ml), AAV8-Camk2a-CaMKIIβ-CARes (5.2 × 1013 vg/ml), AAV8-Camk2a-mCherry (7.9 × 1013 vg/ml), AAV8-Camk2a-GluA1CT-T2A-mCherry (5.2 × 1013 vg/ml), AAV8-Camk2a-GluA1CT-T2A-EGFP (1.1 × 1013 vg/ml), and AAV8-Camk2a-TARPγ-8D/D-T2A-mCherry (7.5 × 1013 vg/ml).
AAV-mediated gene transfer
For the Stereotaxic surgeries (Uchida et al., 2011b, 2017), mice were anesthetized with Avertin (250 mg/kg body weight) and placed in a stereotaxic frame (Kopf). The skull was exposed, and a small portion of the skull above the vHPC was removed bilaterally using a dental drill. The AAV vector was dissolved in physiological saline (0.2 μl for behavioral experiments and 1.0 μl for biochemical experiments) and injected bilaterally into the CA1 area (bregma, −3.28 mm anteroposterior (AP), ±3.3 mm mediolateral (ML), −4.0 mm dorsoventral (DV)) at a rate of 0.1 μl/min followed by a 5-min rest period to prevent backflow. The mice were allowed to recover for at least 3 weeks after surgery. Successful transduction of GFP, mCherry, HA tag, or flag tag in the hippocampal region was confirmed by immunohistochemistry and/or Western blotting.
Golgi staining
Mice were deeply anesthetized via intraperitoneal injection of Avertin (250 mg/kg body weight) and transcardially perfused with a fixative solution containing 4% PFA in 0.1 M phosphate buffer (pH 7.4). Golgi staining was performed using the FD Rapid Golgi Stain kit according to the manufacturer's protocol (FD NeuroTechnologies) (Uchida et al., 2011a). The brains were then sectioned to a thickness of 60 μm using a cryostat. Bright-field microscopic (Keyence) images (at 60× magnification) of vCA1 pyramidal neurons (20 dendrites per mouse) were obtained. Only fully impregnated neurons displaying dendritic trees without obvious truncations and isolated from neighboring impregnated neurons were retained for analysis. Quantification of spine density was limited to dendrites 100–150 μm from the soma. The dendritic spine density is expressed as the number of spines per micrometer of dendritic length.
Quantification and statistical analysis
All statistical parameters are reported in Table S2 and the sample sizes for specific analyses are shown in the figure legends. For all experiments, mice were randomly assigned to the groups. Student's t tests, Kruskal-Wallis tests, and one- or two-way analysis of variance tests were used as appropriate to determine statistical differences using GraphPad Prism v.7.0 and SPSS Statistics v.25. Kolmogorov-Smirnov and/or Shapiro-Wilk tests were used to assess data normality. For ANOVA tests, significant effects were followed by Tukey's post-hoc tests. For multiple comparisons, adjusted p-values were obtained by applying the Benjamini-Hochberg procedure (for Figure 1P) or Dunn's comparison (for behavioral tests). For correlation analysis, the Pearson's correlation coefficient was determined, and the associated linear regression was performed. In all cases, the comparisons were considered statistically significant when p < 0.05. All data are presented as mean ± SEM.
Acknowledgments
We thank Mr. Naoto Yasuda, Ms. Kaede Kuroda, Ms. Ayumi Kobayashi, and Ms. Kumiko Hara for technical assistance. S.U. was supported by Grant-in-Aids for Scientific Research (B) (JP18H02750, JP15H04895), Grant-in-Aid for Scientific Research (C) (JP15K09807), Grant-in-Aids for Scientific Research on Innovation Areas “Integrative research toward elucidation of generative brain systems for individuality” (JP19H04905) and “Constructive understanding of multi-scale dynamism of neuropsychiatric disorders” (JP19H05214 and JP21H00198) from MEXT of Japan, CREST (14530640) from Japan Science and Technology Agency, Grant for Research on Development of New Drugs (JP20ak0101136h001) from AMED, and by research grants from the Takeda Science Foundation, the Uehara Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the Naito Foundation. H.Y. was supported by a grant from the Strategic Research Program for Brain Sciences (Integrated Research on Neuropsychiatric Disorders). H.L. and Y.S. were supported by Grant-in-Aids for Research Activity Start-up (JP19K23776 and JP19K23775) and young Scientists (JP21K13749 and JP21K15711). S.T. was supported by NIH/MH077939 and the NIDA center core (P30DA018343).
Author contributions
Y.S., H.L., and S.U. designed the study. Y.S., H.L., N.I., Y.F., E.I., A. K., H.Y., T.S., T.H., and S.U. conducted the experiments. S.N. and S.T. provided critical reagents. Y.S., H.L., and S.U. wrote the manuscript with input from Y.W. and T.M. All authors have read and approved the final version of this manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102504.
Supplemental information
References
- Abe-Higuchi N., Uchida S., Yamagata H., Higuchi F., Hobara T., Hara K., Kobayashi A., Watanabe Y. Hippocampal sirtuin 1 signaling mediates depression-like behavior. Biol. Psychiatry. 2016;80:815–826. doi: 10.1016/j.biopsych.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Airan R.D., Meltzer L.A., Roy M., Gong Y., Chen H., Deisseroth K. High-speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007;317:819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- Akil H., Gordon J., Hen R., Javitch J., Mayberg H., McEwen B., Meaney M.J., Nestler E.J. Treatment resistant depression: a multi-scale, systems biology approach. Neurosci. Biobehav Rev. 2018;84:272–288. doi: 10.1016/j.neubiorev.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anacker C., Hen R. Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat. Rev. Neurosci. 2017;18:335–346. doi: 10.1038/nrn.2017.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry A.E., Adachi M., Nosyreva E., Na E.S., Los M.F., Cheng P.F., Kavalali E.T., Monteggia L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot R.C., Cates H.M., Purushothaman I., Lorsch Z.S., Walker D.M., Wang J., Huang X., Schluter O.M., Maze I., Pena C.J. Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron. 2016;90:969–983. doi: 10.1016/j.neuron.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot R.C., Parise E.M., Pena C.J., Zhang H.X., Maze I., Chaudhury D., Persaud B., Cachope R., Bolanos-Guzman C.A., Cheer J.F. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat. Commun. 2015;6:7062. doi: 10.1038/ncomms8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer K.U., Schulman H. CaM kinase: still inspiring at 40. Neuron. 2019;103:380–394. doi: 10.1016/j.neuron.2019.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O., McClung C.A., Dileone R.J., Krishnan V., Renthal W., Russo S.J., Graham D., Tsankova N.M., Bolanos C.A., Rios M. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bigio B., Mathe A.A., Sousa V.C., Zelli D., Svenningsson P., McEwen B.S., Nasca C. Epigenetics and energetics in ventral hippocampus mediate rapid antidepressant action: implications for treatment resistance. Proc. Natl. Acad. Sci. U S A. 2016;113:7906–7911. doi: 10.1073/pnas.1603111113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X., Kallarackal A.J., Kvarta M.D., Goluskin S., Gaylor K., Bailey A.M., Lee H.K., Huganir R.L., Thompson S.M. Local potentiation of excitatory synapses by serotonin and its alteration in rodent models of depression. Nat. Neurosci. 2013;16:464–472. doi: 10.1038/nn.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreno F.R., Donegan J.J., Boley A.M., Shah A., DeGuzman M., Frazer A., Lodge D.J. Activation of a ventral hippocampus-medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol. Psychiatry. 2016;21:1298–1308. doi: 10.1038/mp.2015.176. [DOI] [PubMed] [Google Scholar]
- Caspi A., Sugden K., Moffitt T.E., Taylor A., Craig I.W., Harrington H., McClay J., Mill J., Martin J., Braithwaite A., Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chattarji S., Tomar A., Suvrathan A., Ghosh S., Rahman M.M. Neighborhood matters: divergent patterns of stress-induced plasticity across the brain. Nat. Neurosci. 2015;18:1364–1375. doi: 10.1038/nn.4115. [DOI] [PubMed] [Google Scholar]
- Chen A. Academic Press; 2020. Stress Resilience : Molecular and Behavioral Aspects. [Google Scholar]
- Ciocchi S., Passecker J., Malagon-Vina H., Mikus N., Klausberger T. Brain computation. Selective information routing by ventral hippocampal CA1 projection neurons. Science. 2015;348:560–563. doi: 10.1126/science.aaa3245. [DOI] [PubMed] [Google Scholar]
- Danielson N.B., Zaremba J.D., Kaifosh P., Bowler J., Ladow M., Losonczy A. Sublayer-specific coding dynamics during spatial navigation and learning in hippocampal area CA1. Neuron. 2016;91:652–665. doi: 10.1016/j.neuron.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R.S., Aghajanian G.K. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman R.S., Sanacora G., Krystal J.H. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102:75–90. doi: 10.1016/j.neuron.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duric V., Banasr M., Stockmeier C.A., Simen A.A., Newton S.S., Overholser J.C., Jurjus G.J., Dieter L., Duman R.S. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int. J. Neuropsychopharmacol. 2013;16:69–82. doi: 10.1017/S1461145712000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzirasa K., Covington H.E., 3rd Increasing the validity of experimental models for depression. Ann. N. Y Acad. Sci. 2012;1265:36–45. doi: 10.1111/j.1749-6632.2012.06669.x. [DOI] [PubMed] [Google Scholar]
- Eagle A.L., Manning C.E., Williams E.S., Bastle R.M., Gajewski P.A., Garrison A., Wirtz A.J., Akguen S., Brandel-Ankrapp K., Endege W. Circuit-specific hippocampal DeltaFosB underlies resilience to stress-induced social avoidance. Nat. Commun. 2020;11:4484. doi: 10.1038/s41467-020-17825-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow M.S., Dong H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink C.C., Bayer K.U., Myers J.W., Ferrell J.E., Jr., Schulman H., Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Golden S.A., Covington H.E., 3rd, Berton O., Russo S.J. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011;6:1183–1191. doi: 10.1038/nprot.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri A.R., Holmes A. Finding translation in stress research. Nat. Neurosci. 2015;18:1347–1352. doi: 10.1038/nn.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y., Shi S.H., Esteban J.A., Piccini A., Poncer J.C., Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Higuchi F., Uchida S., Yamagata H., Abe-Higuchi N., Hobara T., Hara K., Kobayashi A., Shintaku T., Itoh Y., Suzuki T., Watanabe Y. Hippocampal MicroRNA-124 enhances chronic stress resilience in mice. J. Neurosci. 2016;36:7253–7267. doi: 10.1523/JNEUROSCI.0319-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa T., Mitsushima D., Kaneko R., Hayashi Y. Stoichiometry and phosphoisotypes of hippocampal AMPA-type glutamate receptor phosphorylation. Neuron. 2015;85:60–67. doi: 10.1016/j.neuron.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I., Tennant R.S., Helton R., Marr R.A., Singer O., Redwine J.M., Ellison J.A., Schadt E.E., Verma I.M., Lockhart D.J., Barlow C. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005;438:662–666. doi: 10.1038/nature04250. [DOI] [PubMed] [Google Scholar]
- Jimenez J.C., Su K., Goldberg A.R., Luna V.M., Biane J.S., Ordek G., Zhou P., Ong S.K., Wright M.A., Zweifel L. Anxiety cells in a hippocampal-hypothalamic circuit. Neuron. 2018;97:670–683 e676. doi: 10.1016/j.neuron.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler K.S., Karkowski L.M., Prescott C.A. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- Kheirbek M.A., Drew L.J., Burghardt N.S., Costantini D.O., Tannenholz L., Ahmari S.E., Zeng H., Fenton A.A., Hen R. Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron. 2013;77:955–968. doi: 10.1016/j.neuron.2012.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirbek M.A., Klemenhagen K.C., Sahay A., Hen R. Neurogenesis and generalization: a new approach to stratify and treat anxiety disorders. Nat. Neurosci. 2012;15:1613–1620. doi: 10.1038/nn.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V., Nestler E.J. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonte B., Engmann O., Purushothaman I., Menard C., Wang J., Tan C., Scarpa J.R., Moy G., Loh Y.E., Cahill M. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017;23:1102–1111. doi: 10.1038/nm.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfumey L., Pardon M.C., Laaris N., Joubert C., Hanoun N., Hamon M., Cohen-Salmon C. 5-HT1A autoreceptor desensitization by chronic ultramild stress in mice. Neuroreport. 1999;10:3369–3374. doi: 10.1097/00001756-199911080-00021. [DOI] [PubMed] [Google Scholar]
- Lee H.K., Takamiya K., He K., Song L., Huganir R.L. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGates T.A., Kvarta M.D., Tooley J.R., Francis T.C., Lobo M.K., Creed M.C., Thompson S.M. Reward behaviour is regulated by the strength of hippocampus-nucleus accumbens synapses. Nature. 2018;564:258–262. doi: 10.1038/s41586-018-0740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Zhou T., Liao L., Yang Z., Wong C., Henn F., Malinow R., Yates J.R., 3rd, Hu H. betaCaMKII in lateral habenula mediates core symptoms of depression. Science. 2013;341:1016–1020. doi: 10.1126/science.1240729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Lee B., Liu R.J., Banasr M., Dwyer J.M., Iwata M., Li X.Y., Aghajanian G., Duman R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.Y., Sann S.B., Zhou K., Nabavi S., Proulx C.D., Malinow R., Jin Y., Tsien R.Y. Optogenetic inhibition of synaptic release with chromophore-assisted light inactivation (CALI) Neuron. 2013;79:241–253. doi: 10.1016/j.neuron.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J., Yasuda R., Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher M.P., Matta J.A., Gu S., Seierstad M., Bredt D.S. Getting a handle on neuropharmacology by targeting receptor-associated proteins. Neuron. 2017;96:989–1001. doi: 10.1016/j.neuron.2017.10.001. [DOI] [PubMed] [Google Scholar]
- Maher M.P., Wu N., Ravula S., Ameriks M.K., Savall B.M., Liu C., Lord B., Wyatt R.M., Matta J.A., Dugovic C. Discovery and characterization of AMPA receptor modulators selective for TARP-gamma8. J. Pharmacol. Exp. Ther. 2016;357:394–414. doi: 10.1124/jpet.115.231712. [DOI] [PubMed] [Google Scholar]
- Makino Y., Johnson R.C., Yu Y., Takamiya K., Huganir R.L. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc. Natl. Acad. Sci. U S A. 2011;108:8450–8455. doi: 10.1073/pnas.1105261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen B.S. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann. N. Y Acad. Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- McEwen B.S., Bowles N.P., Gray J.D., Hill M.N., Hunter R.G., Karatsoreos I.N., Nasca C. Mechanisms of stress in the brain. Nat. Neurosci. 2015;18:1353–1363. doi: 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh A.M., Sullivan P.F., Lewis C.M. Uncovering the genetic architecture of major depression. Neuron. 2019;102:91–103. doi: 10.1016/j.neuron.2019.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood A.J., Gladding C.M., Pouladi M.A., Kaufman A.M., Hines R.M., Boyd J.D., Ko R.W., Vasuta O.C., Graham R.K., Hayden M.R. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008S0896-6273(10)00013-9. [DOI] [PubMed] [Google Scholar]
- Mozhui K., Karlsson R.M., Kash T.L., Ihne J., Norcross M., Patel S., Farrell M.R., Hill E.E., Graybeal C., Martin K.P. Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. J. Neurosci. 2010;30:5357–5367. doi: 10.1523/JNEUROSCI.5017-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy C., Maitra M., Tanti A., Suderman M., Theroux J.F., Davoli M.A., Perlman K., Yerko V., Wang Y.C., Tripathy S.J. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 2020;23:771–781. doi: 10.1038/s41593-020-0621-y. [DOI] [PubMed] [Google Scholar]
- Nestler E.J., Barrot M., DiLeone R.J., Eisch A.J., Gold S.J., Monteggia L.M. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Nestler E.J., Hyman S.E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama T., Kitamura T., Roy D.S., Itohara S., Tonegawa S. Ventral CA1 neurons store social memory. Science. 2016;353:1536–1541. doi: 10.1126/science.aaf7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacchioni A.M., Vallone J., Worley P.F., Kalivas P.W. Neuronal pentraxins modulate cocaine-induced neuroadaptations. J. Pharmacol. Exp. Ther. 2009;328:183–192. doi: 10.1124/jpet.108.143115jpet.108.143115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Coreano N., Bolkan S.S., Pierce G.M., Blackman D.R., Hardin W.D., Garcia-Garcia A.L., Spellman T.J., Gordon J.A. Direct ventral hippocampal-prefrontal input is required for anxiety-related neural activity and behavior. Neuron. 2016;89:857–866. doi: 10.1016/j.neuron.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt G.M., Nguyen R., Bang J.Y., Aqrabawi A.J., Tran M.M., Seo D.K., Richards B.A., Kim J.C. Bidirectional control of anxiety-related behaviors in mice: role of inputs arising from the ventral Hippocampus to the lateral septum and medial prefrontal cortex. Neuropsychopharmacology. 2017;42:1715–1728. doi: 10.1038/npp.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Chavez A.E., Mineur Y.S., Morimoto-Tomita M., Lutzu S., Kim K.S., Picciotto M.R., Castillo P.E., Tomita S. CaMKII phosphorylation of TARPgamma-8 is a mediator of LTP and learning and memory. Neuron. 2016;92:75–83. doi: 10.1016/j.neuron.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi G., Gao D., Wu D., Wang Y., Lei H., Zeng W., Gao Y., Yu H., Xiong R., Jiang T. Posterior basolateral amygdala to ventral hippocampal CA1 drives approach behaviour to exert an anxiolytic effect. Nat. Commun. 2020;11:183. doi: 10.1038/s41467-019-13919-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli M., Yan Z., McEwen B.S., Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangon C.M., Fortes S., Lelievre V., Leroux P., Plaisant F., Joubert C., Lanfumey L., Cohen-Salmon C., Gressens P. Chronic mild stress during gestation worsens neonatal brain lesions in mice. J. Neurosci. 2007;27:7532–7540. doi: 10.1523/JNEUROSCI.5330-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N., Byrd K., Petralia R.S., Elias G.M., Adesnik H., Tomita S., Karimzadegan S., Kealey C., Bredt D.S., Nicoll R.A. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat. Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- Rumpel S., LeDoux J., Zador A., Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Russo S.J., Murrough J.W., Han M.H., Charney D.S., Nestler E.J. Neurobiology of resilience. Nat. Neurosci. 2012;15:1475–1484. doi: 10.1038/nn.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seney M.L., Huo Z., Cahill K., French L., Puralewski R., Zhang J., Logan R.W., Tseng G., Lewis D.A., Sibille E. Opposite molecular signatures of depression in men and women. Biol. Psychiatry. 2018;84:18–27. doi: 10.1016/j.biopsych.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S., Lubeck E., Zhou W., Cai L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse Hippocampus. Neuron. 2016;92:342–357. doi: 10.1016/j.neuron.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Powers A., Bradley B., Ressler K.J. Gene x environment determinants of stress- and anxiety-related disorders. Annu. Rev. Psychol. 2016;67:239–261. doi: 10.1146/annurev-psych-122414-033408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline Y.I., Liston C., McEwen B.S. Parsing the Hippocampus in depression: chronic stress, hippocampal volume, and major depressive disorder. Biol. Psychiatry. 2019;85:436–438. doi: 10.1016/j.biopsych.2019.01.011. [DOI] [PubMed] [Google Scholar]
- Sheng M., McFadden G., Greenberg M.E. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Slattery D.A., Cryan J.F. Modelling depression in animals: at the interface of reward and stress pathways. Psychopharmacology (Berl) 2017;234:1451–1465. doi: 10.1007/s00213-017-4552-6. [DOI] [PubMed] [Google Scholar]
- Southwick S.M., Charney D.S. The science of resilience: implications for the prevention and treatment of depression. Science. 2012;338:79–82. doi: 10.1126/science.1222942. [DOI] [PubMed] [Google Scholar]
- Strange B.A., Witter M.P., Lein E.S., Moser E.I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 2014;15:655–669. doi: 10.1038/nrn3785. [DOI] [PubMed] [Google Scholar]
- Sumioka A., Brown T.E., Kato A.S., Bredt D.S., Kauer J.A., Tomita S. PDZ binding of TARPgamma-8 controls synaptic transmission but not synaptic plasticity. Nat. Neurosci. 2011;14:1410–1412. doi: 10.1038/nn.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto-Kimura S., Suzuki K., Horigane S.I., Kamijo S., Inoue M., Sakamoto M., Fujii H., Bito H. Calmodulin kinases: essential regulators in health and disease. J. Neurochem. 2017;141:808–818. doi: 10.1111/jnc.14020. [DOI] [PubMed] [Google Scholar]
- Thompson S.M., Kallarackal A.J., Kvarta M.D., Van Dyke A.M., LeGates T.A., Cai X. An excitatory synapse hypothesis of depression. Trends Neurosci. 2015;38:279–294. doi: 10.1016/j.tins.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tost H., Champagne F.A., Meyer-Lindenberg A. Environmental influence in the brain, human welfare and mental health. Nat. Neurosci. 2015;18:1421–1431. doi: 10.1038/nn.4108. [DOI] [PubMed] [Google Scholar]
- Tsankova N.M., Berton O., Renthal W., Kumar A., Neve R.L., Nestler E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Uchida S., Hara K., Kobayashi A., Fujimoto M., Otsuki K., Yamagata H., Hobara T., Abe N., Higuchi F., Shibata T. Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc. Natl. Acad. Sci. U S A. 2011;108:1681–1686. doi: 10.1073/pnas.10164241081016424108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S., Hara K., Kobayashi A., Otsuki K., Yamagata H., Hobara T., Suzuki T., Miyata N., Watanabe Y. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron. 2011;69:359–372. doi: 10.1016/j.neuron.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Uchida S., Martel G., Pavlowsky A., Takizawa S., Hevi C., Watanabe Y., Kandel E.R., Alarcon J.M., Shumyatsky G.P. Learning-induced and stathmin-dependent changes in microtubule stability are critical for memory and disrupted in ageing. Nat. Commun. 2014;5:4389. doi: 10.1038/ncomms5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S., Shumyatsky G.P. Synaptically localized transcriptional regulators in memory formation. Neuroscience. 2018;370:4–13. doi: 10.1016/j.neuroscience.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S., Teubner B.J.W., Hevi C., Hara K., Kobayashi A., Dave R.M., Shintaku T., Jaikhan P., Yamagata H., Suzuki T. CRTC1 nuclear translocation following learning modulates memory strength via exchange of chromatin remodeling complexes on the Fgf1 gene. Cell Rep. 2017;18:352–366. doi: 10.1016/j.celrep.2016.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S., Yamagata H., Seki T., Watanabe Y. Epigenetic mechanisms of major depression: targeting neuronal plasticity. Psychiatry Clin. Neurosci. 2018;72:212–227. doi: 10.1111/pcn.12621. [DOI] [PubMed] [Google Scholar]
- Wang J., Lv X., Wu Y., Xu T., Jiao M., Yang R., Li X., Chen M., Yan Y., Chen C. Postsynaptic RIM1 modulates synaptic function by facilitating membrane delivery of recycling NMDARs in hippocampal neurons. Nat. Commun. 2018;9:2267. doi: 10.1038/s41467-018-04672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner P. The validity of animal models of depression. Psychopharmacology (Berl) 1984;83:1–16. doi: 10.1007/bf00427414. [DOI] [PubMed] [Google Scholar]
- Yoshida K., Drew M.R., Mimura M., Tanaka K.F. Serotonin-mediated inhibition of ventral hippocampus is required for sustained goal-directed behavior. Nat. Neurosci. 2019;22:770–777. doi: 10.1038/s41593-019-0376-5. [DOI] [PubMed] [Google Scholar]
- Zhang K., Xu T., Yuan Z., Wei Z., Yamaki V.N., Huang M., Huganir R.L., Cai X. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci. Signal. 2016;9:ra123. doi: 10.1126/scisignal.aai7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data for the Western blots in the paper are available Mendeley Data https://doi.org/10.17632/m8zyb8mmtg.1.