

Abstract
Although cell lineage information is fundamental to understanding organismal development, very little direct information is available about humans. We performed high-depth (250X) whole-genome sequencing of multiple tissues from three individuals to identify hundreds of somatic single nucleotide variants (sSNVs). Using these variants as “endogenous barcodes” in single cells, we reconstructed early embryonic cell divisions. Targeted sequencing of clonal sSNVs in different organs (~25,000X) and in >1,000 cortical single cells, as well as snRNA-seq and snATAC-seq of ~100,000 cortical single cells, demonstrated asymmetric contributions of early progenitors to extraembryonic tissues, distinct germ layers, and organs. Our data suggest onset of gastrulation at an effective progenitor pool of ~170 cells and ~50–100 founders for the forebrain. Thus, mosaic mutations provide a permanent record of human embryonic development at remarkably high-resolution.
One Sentence Summary:
Bulk and single-cell detection of mosaic variants in multiple organs resolve post-zygotic lineages to reveal embryonic development.
Although recent strategies using DNA editing have used molecular barcodes as clonal markers to map the developmental processes of proliferation, migration, and tissue formation (1), such methods are not applicable to understanding human development. Although single-cell RNA-seq methods have been used to analyze transcriptional changes and cell differentiation during human development (2), they are inadequate for lineage tracing, leaving global lineage patterns in humans still largely unexplored. Here, to examine developmental ancestries and clonal composition across the body, we characterized somatic single nucleotide variants (sSNVs), which are suitable as lineage markers because they accumulate with each cell division (3) and most mutations are predicted to be functionally silent (4, 5).
High-depth whole-genome sequencing (WGS, >250X per sample) was performed for five bulk DNA samples from a 17-year old male (ID: UMB1465) who died with no medical diagnosis—prefrontal cortex (PFC, Section 2) grey-matter (GM) and white-matter (WM), heart, spleen, and liver (>1250X total; Fig. 1A, Table S1). Similarly, >250X WGS was also performed for PFC and two visual cortex samples (Brodmann area 17 and 18, BA17, BA18) from two additional individuals, a 15-year old female (UMB4638) and a 42-year old female (UMB4643). Applying MosaicForecast, a machine-learning algorithm (4), to bulk data and integrating with previously published single-cell WGS (6, 7), we identified 516 total sSNVs (8) (Table S2). Among the 297 sSNVs detected in individual 1465, 65 (22%) were found across all tissues and 181 (61%) in at least two (Fig. 1B, Table S2). All 65 widely-shared sSNVs showed alternate allele frequency (AAF) >1%, with 38 (58%) showing >3% (Fig. 1B, Table S2). Sensitivity estimates suggest that our approach achieved nearly 100% sensitivity for detecting sSNVs of 3–30% AAF (8) (Fig. 1C, Fig. S1A–C). Most sSNVs were predicted to be functionally neutral (only 2/297 sSNVs in 1465 were exonic, Table S3), and thus represent unbiased lineage markers.
Fig. 1. Mosaic events of human development.
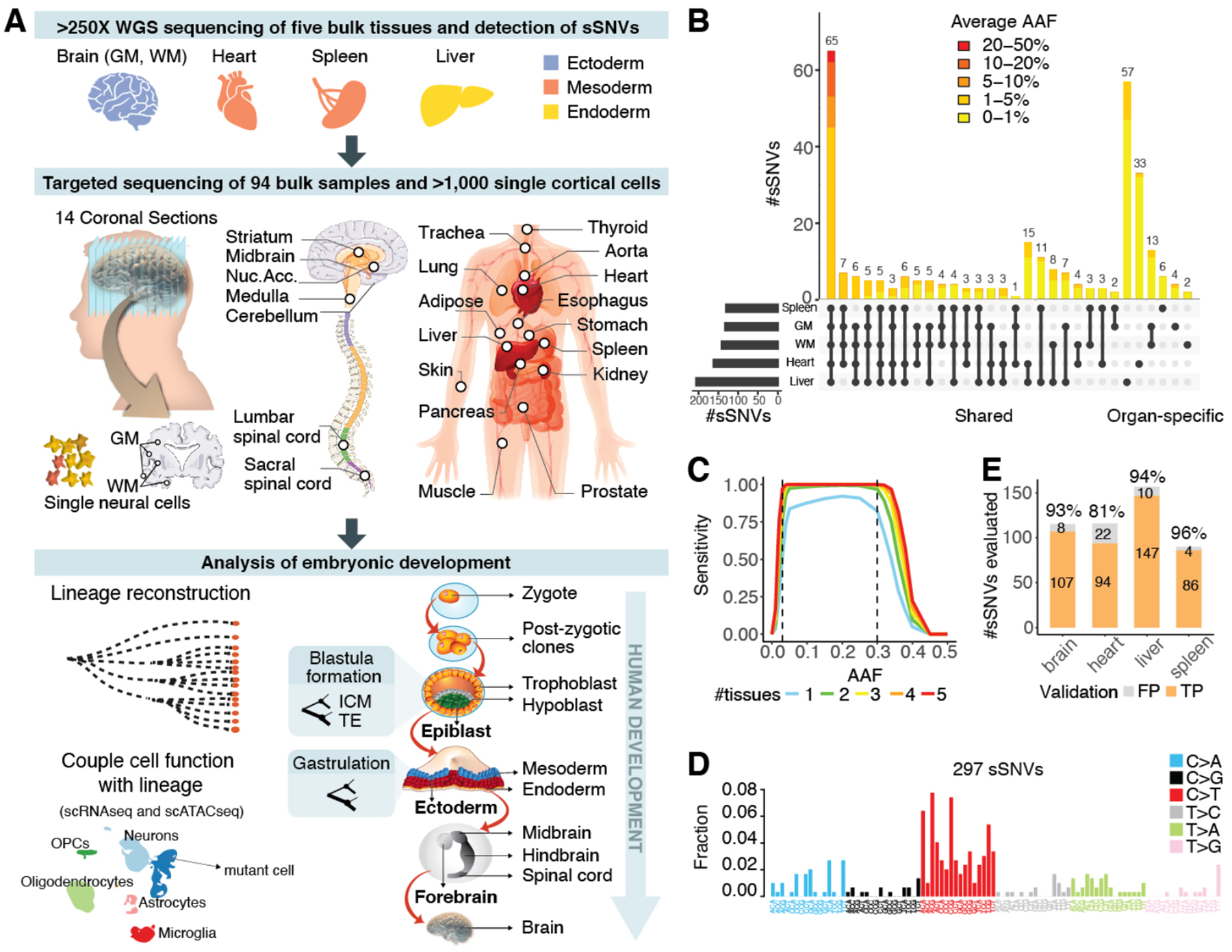
(A) Schematic of the workflow for individual 1465. (B) Number and AAF of sSNVs detected across samples from case 1465. (C) Sensitivity of MosaicForecast in detecting sSNVs from five 250X WGS data. (D) Trinucleotide context profile of the identified sSNVs. (E) Numbers of true positive (TP) and false positive (FP) sSNVs present in the WGS data validated by deep amplicon-sequencing.
Clonal sSNVs in all organs showed similar base substitution patterns, with 55% being C>T substitutions (Fig. 1D, Fig. S1D–E). The trinucleotide context resembled that of sSNVs seen in proliferating tissues and cancer, e.g., clock-like Signature 1 in the COSMIC catalog (9), which likely reflects faulty repair of cytosine deamination in cycling cells (5, 7). Liver-specific variants were more common than heart- or brain-specific variants (57, 33 and 19, respectively), consistent with known patterns of clonal amplification and replacement of hepatic units from resident stem cells (10), whereas spleen-specific variants were the fewest (Fig. 1B, Table S2). Amplicon-based targeted sequencing (~25,000X on average) of 94 samples from 17 organs (Fig. 1A, Table S1) reidentified most sSNVs (>93%) when the same biopsy used for WGS was profiled (Table S1), or slightly less when distinct tissue biopsies were profiled (81%); overall, 196/229 (86%) of targeted variants were validated (Fig. 1E, Fig. S1F, Table S4).
Single-cell WGS data of 20 single neurons (6, 7) from 1465 resolved 82/297 sSNVs into branching clades or clones, producing a lineage tree that spans early post-zygotic cell generations and traces the origin of each mutation back to the embryo (Fig. 2A, Fig. S2A, Tables S2, S5). As expected, earlier sSNVs showed higher mosaic fractions (MF, fractions of cells carrying the variant, defined as 2×bulk AAF for autosomal SNV), with the MFs from daughter clades summing to that of the mother clone. Similar patterns of early lineage were also identified in the two additional individuals based on bulk WGS and single cell (7, 11) analysis (Fig. 2B, C, Fig. S2B, C, Table S5). In 1465, we identified the first eight post-zygotic progenitors corresponding to the third cell generation (c1-c8; with c5-c6 not fully resolved and annotated as a second-generation clone)—with the MFs of c1-c8 summing to ≈100%, suggesting that all major early lineages were captured—and traced their relative contributions to each organ (Fig. 2D, Fig. S2D) (8). Contributions of c1-c8 were highly unequal across organs, with c4 undetected in heart and spleen while c3 and c8 together contributed >50% of the cellular content (Fig. 2D).
Fig. 2. Asymmetric contribution of early embryonic clones to the human body.
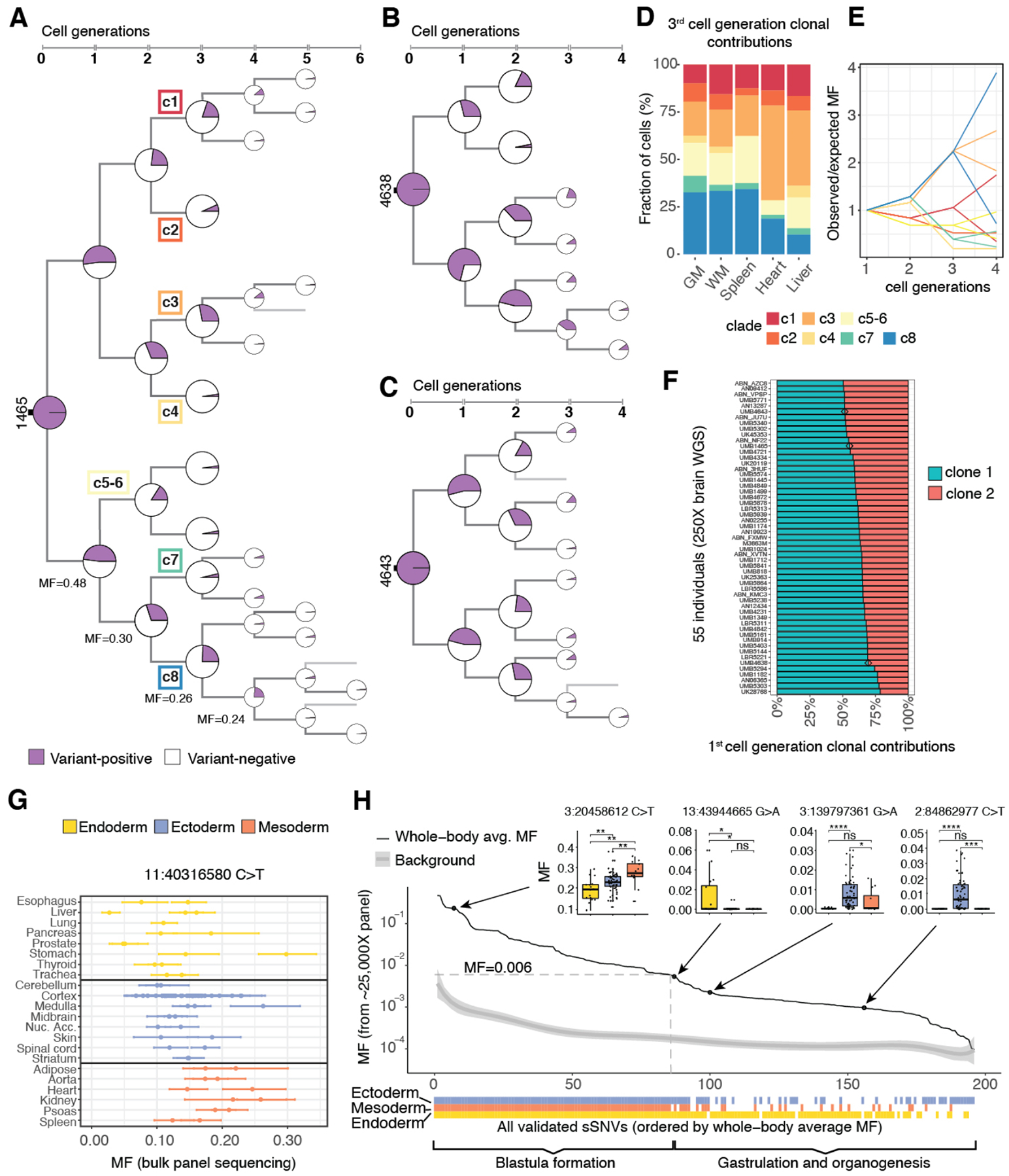
(A-C) Phylogenetic trees of individuals 1465, 4638, and 4643. The cell-generation numbers for later sSNVs (5th/6th) are likely to be underestimates due to the limited number of cells used for lineage reconstruction and the reduced power of detecting very low MF sSNVs. (D) 3rd cell generation clones (c1-c8) of 1465 show unequal contributions to specific organs (p-value <10−15, chi-square test), with the fraction of cells in each tissue contributed by clones c1-c8 normalized by summing to 100% (see Fig. S2D for non-normalized values). (E) Observed whole-body MFs for sSNVs from clades c1-c8 across 2–4 cell generations strongly deviate from expected values based on a symmetrical model of development. 95% CIs calculated with binomial sampling are reported in Table S2. (F) First cell generation clonal contributions are asymmetric and variable across 55 individuals (p<10−13, K-S test for the null hypothesis of symmetry). Individuals 1465, 4638 and 4643 are marked with a diamond. (G) High intra-organ fluctuation of MFs for early-embryonic mosaic variants, illustrated for chr11:40316580 C>T. (H) sSNVs restricted to one or two germ layers mark the beginning of gastrulation. 196 validated sSNVs are ordered “chronologically” by their whole-body MFs (8). MFs in different germ layers are compared in four examples (two-tailed Wilcoxon rank sum test; ns = non-significant; *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001).
Changes in MFs across cell generations suggest highly asymmetrical segregation of the earliest progenitors between embryonic and extraembryonic tissues and to the several germ layers within the embryo. Instead of the expected two-fold reduction of MFs with cell division, observed MFs for one branch (c8) barely decreased (30%, 26% and 24%; p < 10−6, <10−22, <10−56, respectively; two-tailed binomial test); deviations from two-fold reduction were also observed in other branches (Fig. 2A, E, Fig. S2A) and in the two additional individuals (Fig. 2B, C, Fig. S2B, C). This pattern suggests unequal clonal partitioning during blastula formation, when extraembryonic tissues separate from embryonic tissue lineages (Fig. 1A). The observed MF asymmetries indicate that lineage segregation in human embryo might happen as early as the 2–4 cell stage, as suggested in the mouse (12–14). To further test this hypothesis, we analyzed published (11) bulk WGS data (250X) from 74 individuals. Our maximum likelihood estimates (8) indicate overall asymmetric contributions of the first cell generation clones to the human body with strong inter-individual variability, from a 50:50 symmetry in few individuals to a 20:80 asymmetry and potentially higher (Fig. 2F, Table S6). MFs of 196 sSNVs across 94 biopsies from 17 different organs (Table S1) from 1465 also revealed asymmetric contributions of early lineages to embryonic germ-layers during gastrulation (Fig. 1A, Fig. S3A–C, Table S4) (8). Relative contributions of several clades to organs of endoderm, ectoderm and mesoderm varied up to several-fold (Fig. S3B, C). Furthermore, multiple biopsies from the same organ showed striking intra-organ MF differences (Fig. 2G, Fig. S3D). For example, MFs for sSNV chr11:40316580 (C>T) ranged from 5% to 26% across cerebral cortex samples, suggesting highly variable local clonal amplification in all tissues (Fig. 2G).
The tissue distribution of sSNVs identifies the effective progenitor pool size at the onset of gastrulation. sSNVs with higher MFs were found in all organs and germ-layers (8) (Fig. 2H, Fig. S3E, Tables S4, S7), but as MFs decreased past ~0.6%, many sSNVs became undetectable in one or two germ-layers (Fig. 2H, Fig. S3E, Table S7), reflecting lineage divergence during gastrulation. The effective cell number at the time of mutation occurrence can be inferred as ~1/MF—thus 0.6% MF corresponds to ~170 epiblast cells. Despite the asymmetries of clonal contributions to various tissues, multiple germ layer-restricted variants gave similar estimates (Fig. 2H), and our in vivo estimates are consistent with counts from cultured human embryos (15).
The earliest brain-specific sSNVs provide similar estimates for the number of brain founder cells. Fourteen sSNVs were present in at least one of 64 central nervous system (CNS) samples but not in 30 non-CNS samples (Fig. 3A, Tables S1, S8), with ten showing significantly higher MFs in forebrain than other CNS regions (Fig. 3A, Table S8). The earliest-occurring sSNVs were confirmed from analysis of 1228 single cortical cells (88% are from PFC Section 2, thus forebrain MFs estimated from single cells may be biased) (8) (Table S9), of which 791 were successfully placed in a lineage tree (Fig. 3B, Figs. S4, S5, Table S9) with the neuronal and non-neuronal cells differentially distributed across the clades. The two earliest sSNVs showed wider presence in single cells (Fig. 3C, Fig. S5) and a higher overall bulk MF (~2.2%) than other CNS-specific mutations from the same c8 branch (Fig. 3A). We also examined CNS-specific sSNVs with the highest bulk MF (~1%) in clade c1 (Fig. 3A, D, Fig. S5). These early variants showed wide distribution across the forebrain (Fig. S6A–B) at relatively high MFs (Table S8) but were undetectable in most other samples. These variants therefore serve as markers of the first forebrain progenitors and, based on their average bulk MFs, the number of forebrain founder cells is estimated to be ~50–100, out of an estimated 600–1,300 epiblasts (Fig. 3E, Fig. S6C).
Fig. 3. Brain-specific sSNVs estimate the number of forebrain founder cells.
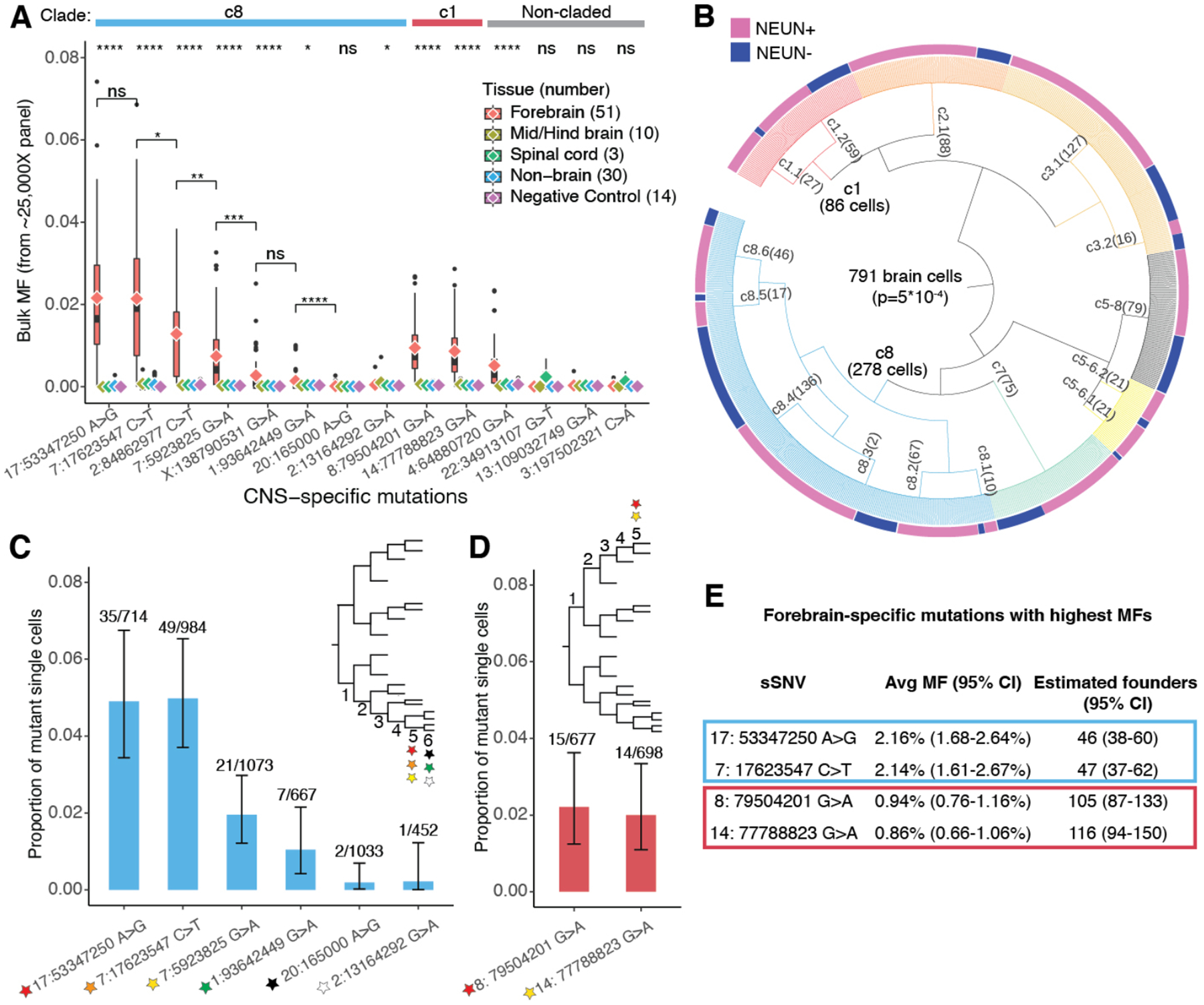
(A) MFs of 14 CNS-restricted sSNVs show significant enrichment of some variants in forebrain-derived samples (two-tailed Wilcoxon rank sum test, significance levels on the top). c8, c1 (Fig. 2A) and non-claded variants are indicated. chr17:53347250 A>G and chr7:17623547 C>T are the earliest brain-specific sSNVs in c8, based on average forebrain MFs (diamond symbols). The forebrain MFs between sSNVs are compared to estimate the likelihood that they arose at the same generation (two-tailed Wilcoxon rank sum test). (B) 791 single cells (out of 1228) are successfully assigned to lineage clades upon targeted sequencing of 37 sSNVs (8). NEUN+ and NEUN- cells are differentially distributed across clades (two-tailed Fisher’s exact test). (C) chr17:53347250 A>G and chr7:17623547 C>T are confirmed as the earliest lineage markers within c8 by single cell genotyping (shown are the number of mutant cells over the number of cells with >10X coverage at the position). (D) Same as (C) for c1. (E) Estimates of forebrain-founder cells based on average MFs (25,000X sequencing).
Analysis of sSNVs in 47 DNA samples spanning the rostro-caudal extent of the cerebral cortex (Fig. 1A, Table S1) confirmed previous descriptions of widespread clonal distribution at low MFs (6, 16), as well as suggesting broadly definable topographic variation between frontal (sections 1–7) and posterior cortex (sections 8–14) (8) (Fig. 4A, Table S8). sSNVs from early cell generations (1st-4th) were found in all rostro-caudal sections (8) (Fig. 4B, Fig. S6A–B), although their widely varying mosaic fractions highlighted unexpectedly large local nonuniformities in clonal amplification (Fig. 4B, Fig. S6A–B). Later (5th+/6th+ cell generation) sSNVs showed progressive restriction to frontal cortex (Fig. 4C, Fig. S6A–B) and finally the PFC, where they were discovered. Thus, while founder clones of the cortex show little topographic restriction for MFs of ~1% or higher, lower MF clones show evidence of broad differences in distribution from frontal to posterior regions, separated approximately by the Sylvian fissure and the central sulcus (Fig. 4D).
Fig. 4. Topographic patterns and function of embryonic clones in the rostro-caudal cerebral cortex.
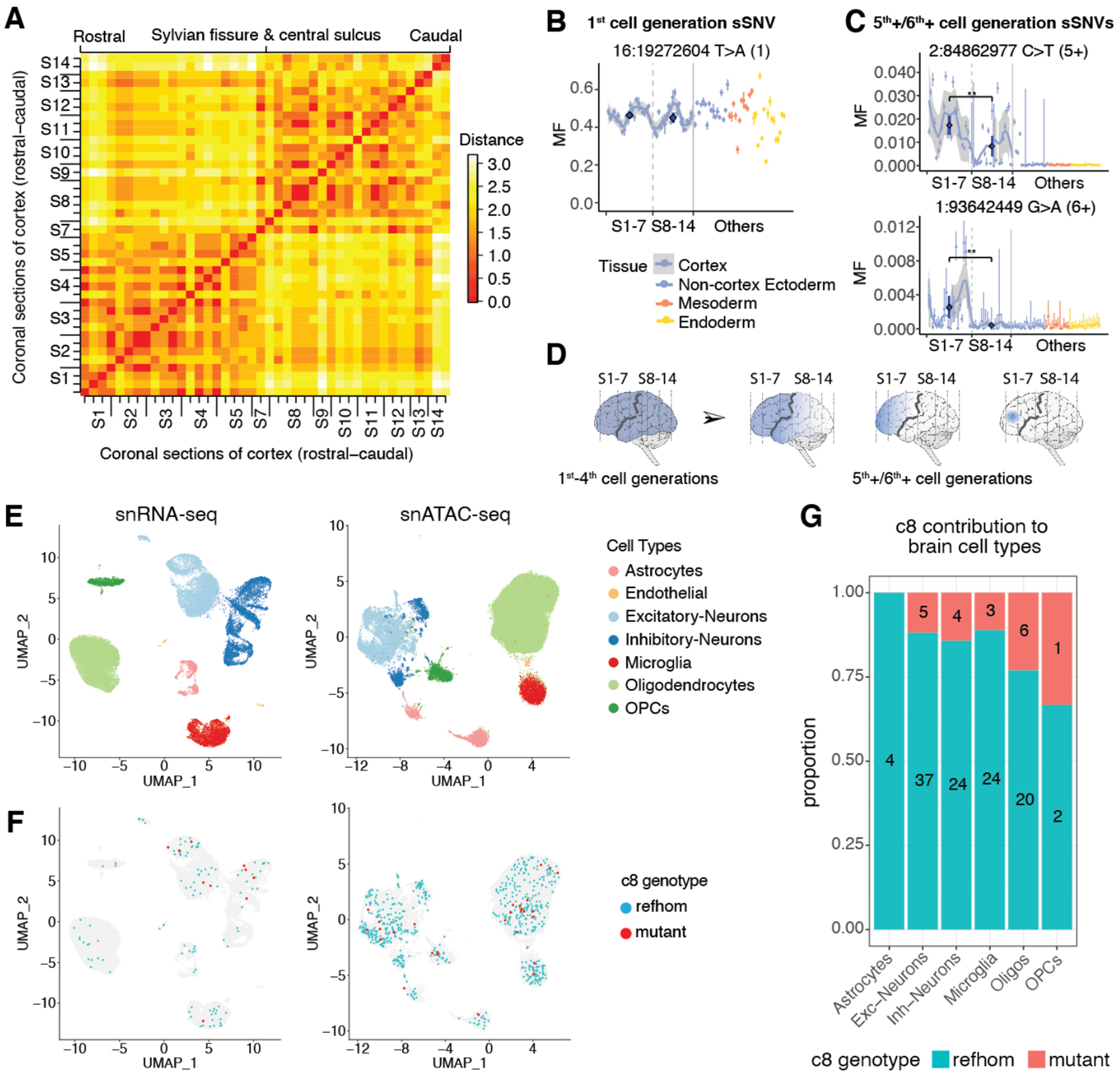
(A) Frontal regions (sections 1–7) and posterior regions (sections 8–14) form two broadly definable lineage clusters. Euclidian distances are computed based on the presence (score=1) or absence (score=0) of sSNVs. (8). (B) Earlier clones from the 1st to the 4th cell generations contribute to all rostro-caudal sections, as illustrated by an sSNV from 1st cell generation (Fig. S6A). The AAFs across sequential sections of cortex are shown with a confidence band. The average MFs (dark blue) in the two regions are compared using Wilcoxon rank sum test. (C) 5th+/6th+ cell-generation clones from the lineage tree show restriction in frontal cortex regions (Fig. S6B). (D) Successive subclones from 1st to 6th+ cell generations show progressive restriction to frontal cortical areas separated by Sylvian fissure and central sulcus (black line). (E) Clusters of major brain cell types identified by PFC snRNA-seq and snATAC-seq. (F) Distribution of reference homozygous (refhom) and mutant cells for clade c8 markers with >0 coverage across cell types. (G) Proportions of refhom cells and mutant cells for 4th cell generation clade c8 markers across brain cell types (p=0.58, Fisher’s exact test, see also Table S10).
Single nucleus (sn)RNA-seq and snATAC-seq data reveal cell-type classification, but the clusters can also be linked to genotypes. Although limited by the per-cell coverage sparsity, snATAC-seq reads were more uniformly distributed across the genome compared with snRNA-seq reads (Fig. S7A), suggesting that snATAC-seq may be better suited to detect sSNVs genome-wide (Fig. S7). At the 297 sSNV positions, 5.6% of snRNA-seq cells (1,933 of 34,325) and 12.8% of snATAC-seq cells (8,356 of 65,199) obtained coverage over at least one of the 297 sSNV loci (Table S10). To link cell-lineage information with cell types, we classified all ~100,000 cells into seven groups (Fig. 4E, Fig. S8 and S9) (8, 17) and checked cells with at least one lineage marker from Fig. S2A (Figs. 4F and S7B–F, Table S10). The sparse coverage of late-occurring variants generally prevents observations of lineage divergence with this approach, though a few trends of c8 contributions to distinct cell types were seen (Fig. 4E–G and Fig. S10). Our data point to the potential of newer methods for combining analysis of DNA and RNA (18, 19) at high-throughput to systematically analyze the formation of distinct cell types at scale in humans.
Our analysis shows that hundreds of sSNVs occurring over several post-zygotic cell divisions mark the landmarks of embryonic human development and inform the patterns of clonal distribution within and between organs and tissues. Although analysis of peripheral blood DNA had suggested asymmetries in the contribution of early post-zygotic clones to embryonic tissues (5), here we show sequential asymmetries and variabilities in clonal proliferation at later steps during gastrulation and organogenesis. The high intra-organ fluctuation of MFs (Fig. 2G, Fig. S3D) highlights a stochastic clonal pattern within and across all the tissues examined.
We found that clones generated by brain-specific progenitors have average MFs lower than 2.2% across the cortex, underscoring the need for single cell sequencing for their identification. Regional restrictions of sSNVs to the frontal lobe are seen at even lower MFs (≤0.6%). The observed dispersion of founder clones is consistent with previous estimates (19) that a given zone of the human cerebral cortex is formed from about 10 progenitors specified to form excitatory neurons that intermingle widely over a broad region of the cortex (6, 16, 19). Given the growing list of conditions associated with somatic mutations (20, 21), a deeper understanding of the patterns of cell lineage described here coupled with functional information will help elucidate the origin and consequence of mosaicism in these diseases.
Supplementary Material
Acknowledgments:
We thank R.S. Hill, J.E. Neil, D. Gonzalez, S. Yip, M. Joe, for assistance; S.R. Ehmsen for help with figure graphics, H. Gold, E. Maury, T. Shin for help on data analysis, A.Y. Huang and P. Li for sharing their snRNA-seq data, Walsh and Park lab members for discussion, especially R.E. Andersen, C.M. Dias, M.B. Miller and V.V. Viswanadham; the Boston Children’s Hospital Flow Cytometry Core and IDDRC Molecular Genetics Core, Biopolymers Facility and Research Computing at HMS. We thank the donors and their families for human tissues, obtained from the NIH NeuroBioBank at the University of Maryland.
Funding:
This work was supported by the NIMH through the Brain Somatic Mosaicism Network grant U01MH106883 (C.A.W., P.J.P.) the NINDS via R01NS032457 (C.A.W., P.J.P.), and the Allen Discovery Center program, a Paul G. Allen Frontiers Group advised program of the Paul G. Allen Family Foundation. Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center is funded by NIH grant U54HD090255. S.B. was supported by the Manton Center for Orphan Disease Research at Boston Children’s Hospital. J.G. was supported by a Basic Research Fellowship from the American Brain Tumor Association BRF1900016 and by the Brain SPORE grant P50CA165952. S.N.K. is a Stuart H.Q. & Victoria Quan Fellow at Harvard Medical School. C.A.W. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability: All genomic data is available from dbGaP under the accession number phs001485.v2.p1 and from National Institute of Mental Health Data Archive (DOI: 10.15154/1503337). Other materials are available through the authors upon reasonable request.
References and Notes:
- 1.Kalhor R et al. , Developmental barcoding of whole mouse via homing CRISPR. Science 361, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han X et al. , Construction of a human cell landscape at single-cell level. Nature 581, 303–309 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Rodin RE et al. , The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nature Neuroscience, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dou Y et al. , Accurate detection of mosaic variants in sequencing data without matched controls. Nat Biotechnol 38, 314–319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ju YS et al. , Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543, 714–718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lodato MA et al. , Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lodato MA et al. , Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Materials and methods are available as supplementary materials.
- 9.Tate JG et al. , COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang RR et al. , Hepatic stem cells with self-renewal and liver repopulation potential are harbored in CDCP1-positive subpopulations of human fetal liver cells. Stem Cell Res Ther 9, 29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodin RE et al. , The Landscape of Mutational Mosaicism in Autistic and Normal Human Cerebral Cortex. bioRxiv, 2020.2002.2011.944413 (2020). [Google Scholar]
- 12.Hupalowska A et al. , CARM1 and Paraspeckles Regulate Pre-implantation Mouse Embryo Development. Cell 175, 1902–1916 e1913 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White MD et al. , Long-Lived Binding of Sox2 to DNA Predicts Cell Fate in the Four-Cell Mouse Embryo. Cell 165, 75–87 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Piotrowska K, Zernicka-Goetz M, Role for sperm in spatial patterning of the early mouse embryo. Nature 409, 517–521 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Xiang L et al. , A developmental landscape of 3D-cultured human pre-gastrulation embryos. Nature 577, 537–542 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Evrony GD et al. , Cell lineage analysis in human brain using endogenous retroelements. Neuron 85, 49–59 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuart T et al. , Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nam AS et al. , Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 571, 355–360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang AY et al. , Parallel RNA and DNA analysis after deep sequencing (PRDD-seq) reveals cell type-specific lineage patterns in human brain. Proc Natl Acad Sci U S A, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh HY, Lee JH, Brain Somatic Mutations in Epileptic Disorders. Mol Cells 41, 881–888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baldassari S et al. , Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol 138, 885–900 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genovese G et al. , Mapping the human reference genome’s missing sequence by three-way admixture in Latino genomes. Am J Hum Genet 93, 411–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenna A et al. , The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cibulskis K et al. , Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karczewski KJ et al. , The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haeussler M et al. , The UCSC Genome Browser database: 2019 update. Nucleic Acids Res 47, D853–D858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karimzadeh M et al. , Umap and Bismap: quantifying genome and methylome mappability. Nucleic Acids Res 46, e120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larson DE et al. , SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saunders CT et al. , Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Koboldt DC et al. , VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poplin R et al. , A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 36, 983–987 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Zook JM et al. , Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data 3, 160025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M et al. , Comparison of multiple displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC) in single-cell sequencing. PLoS One 9, e114520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C et al. , Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356, 189–194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeileis A et al. , Regression Models for Count Data in R. 2008 27, 25 (2008). [Google Scholar]
- 36.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodge RD et al. , Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Icgc Tcga Pan-Cancer Analysis of Whole Genomes Consortium, Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenthal R et al. , DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L et al. , RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Inoue F et al. , A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res 27, 38–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.