

Abstract
Acrodysostosis refers to a rare heterogeneous group of bone dysplasias that share skeletal features, hormone resistance, and intellectual disability. Two genes have been associated with acrodysostosis with or without hormone resistance (PRKAR1A and PDE4D). Severe intellectual disability has been reported with acrodysostosis but brain malformations and ichthyosis have not been reported in these syndromes. Here we describe a female patient with acrodysostosis, intellectual disability, cerebellar hypoplasia, and lamellar ichthyosis. The patient has an evolving distinctive facial phenotype and childhood onset ataxia. X-rays showed generalized osteopenia, shortening of middle and distal phalanges, and abnormal distal epiphysis of the ulna and radius. Brain magnetic resonance imaging showed cerebellar atrophy without other brainstem abnormalities. Genetic workup included nondiagnostic chromosomal microarray and skeletal dysplasia molecular panels. These clinical findings are different from any recognized form of acrodysostosis syndrome. Whole exome sequencing did not identify rare or predicted pathogenic variants in genes associated with known acrodysostosis, lamellar ichthyosis, and other overlapping disorders. A broader search for rare alleles absent in healthy population databases and controls identified two heterozygous truncating alleles in FBNL7 and PPM1M genes, and one missense allele in the NPEPPS gene. Identification of additional patients is required to delineate the mechanism of this unique disorder.
Keywords: acrodysostosis, cerebellar ataxia, cerebellar atrophy, lamellar ichthyosis, skeletal dysplasia
1 |. INTRODUCTION
Acrodysostosis (OMIM #101800), a rare form of peripheral dysostosis, was first described by Maroteaux and Malamut in 1968 (Maroteaux & Malamut, 1968). Although most cases are sporadic, there is evidence of the effect of paternal age that suggests the phenotype is originated from de novo mutations (Jones, Smith, Harvey, Hall, & Quan, 1975), and it could explain sporadic occurrence (Steiner & Pagon, 1992). It is a pleiotropic disease, characterized by skeletal (brachycephaly, malar flattening, anteverted nares, brachydactyly, lumbar stenosis of the spine), endocrine (hypothyroidism, hypogonadism, menstrual disorders), and neurological (developmental delay, intellectual disability) manifestations (Robinow et al., 1971).
Pathogenic dominant mutations in only two genes PRKAR1A (protein kinase, cyclic adenosine monophosphate [cAMP]-dependent, regulatory type I, alpha) (OMIM *188830) and PDE4D (phosphodiesterase 4D, cAMP-specific) (OMIM *600129) have been associated with acrodysostosis (Lee et al., 2012; Michot et al., 2012). Patients with acrodysostosis can have variable degrees of intellectual disability with or without resistance to multiple hormones. Severe intellectual disability has been reported with acrodysostosis but brain malformations, ataxia, and skin abnormalities are not reported features (Sezer, Sutbeyaz, Koseoglu, Aras, & Akin, 2009). There is also a limited group of syndromic entities, such the Sjögren–Larsson syndrome (OMIM #270200), that encompass ichthyosis, intellectual disability, short stature, spastic paraparesis, and dysmorphic features including midface hypoplasia (Drozdowski, Latkowski, Zachara, & Wojcicki, 2017; Dutra, de Aquino, & Barsottini, 2009).
Midface hypoplasia, brachymetacarpia, and brachyphalangia can also be caused by pathogenic variants in GNAS (OMIM *139320), HOXD13 (OMIM *142989), and PDE3A (OMIM *122805) related disorders. Acrodysostosis has been reported in other skeletal dysplasias (Giedion, 2002) and isolated single case reports of individuals with Gaucher disease type IIIc (OMIM #231005) (Abrahamov, Elstein, & Zimran, 2000), and 5-alpha-reductase deficiency (OMIM *607306) (Gupte, Kher, Kanade, Bharucha, & Sagade, 1994). Several other case reports are found in the literature suggesting the possibility of a dual diagnosis rather than a true association. Here we describe a sporadic new syndrome in an adult female with progressive acrodysostotis-like skeletal dysplasia, childhood onset cerebellar ataxia, cerebellar atrophy, mild intellectual disability, and lamellar ichthyosis.
2 |. RESULTS
2.1 |. Clinical report
A 58-year-old female was referred to genetics for the initial evaluation of dysmorphic features, hand abnormalities, short stature, gait difficulties, and tremors. Prenatal history was not available as her parents are deceased. The proband was the second child of non-consanguineous parents of Colombian ethnicity. Family history was relevant for early death of her older sister at age 6 months from diphtheria. This sister also had congenital torticollis but no reported skeletal, hand, or facial anomalies. No family history of congenital anomalies, short stature, ataxia, or learning difficulties.
Proband has global developmental delay that, as a child, was characterized by delayed walking at 2 years and first words at 3 years. Proband struggled academically but learned to read and basic single digit addition. Handwriting was poor given hand anomalies and coordination difficulties. She executes basic activities of daily living, understands complex instructions, carries on a conversation, and has clear and structured speech. She does not manage her own finances or more complex tasks. Proband has not undergone psychometric testing.
The proband has a history of tremors, coordination difficulties, and an ataxic gait. Proband holds caregiver’s arms for distance walking. She does not use wheelchair, frame, or cane.
Short stature was noted during childhood but no diagnostic evaluations were performed. Menarche at the age of 12 years, regular menstrual cycles, and menopause at age 50 years. No history of pregnancies. The proband underwent hip replacement surgery after a hip fracture at age 58. At age 62, she developed glaucoma with loss of visual acuity. She also had scleroatrophic gallbladder detected through abdominal ultrasonography.
She was first noted to have atopic dermatitis and hyperthyroidism at age 33. She was treated with methimazole for 6 years and then received treatment with radioactive iodine. She was placed on levothyroxine replacement therapy. Skin abnormalities persisted after the treatment of thyroid disease. She was evaluated by dermatology and diagnosed with lamellar ichthyosis.
On physical examination at last evaluation, her height was 145 cm (<3 centile, −3 SD), weight was 37 kg (<3 centile, −3 SD), head circumference was 53 cm (10th centile, −1.2 SD), arm span was 124.5 cm, superior segment was 76 cm, and the inferior segment was 69 cm. Arm span/height ratio was 0.85 and upper segment/lower segment ratio was 1.1. The cranium was brachycephalic, long face, malar flattening, wide and depressed nasal bridge, thickened alae nasi, anteverted nares, smooth, and long philtrum (Figure 1a,b). Palate was high, no uvula abnormalities, and normal hairline. Skin was dry, scaly, and flaky consistent with lamellar ichthyosis primarily affecting the lower extremities and back (Figure 1c). Extremities have acromelic shortening, tapered fingers, trident hand configuration, deep creases, short palms, and normal nails (Figure 1d,e). Feet were broad with short toes, hammertoes, clinodactyly, bilateral tibial deviation of hallux, and flat feet. Neurological examination was remarkable for rapid frequency intentional tremor and reduced amplitude. She also had dysmetria, and dysdiadochokinesia. She exhibited increased gait width base, reduced walking speed, and used frequent arm extension to steady herself on walls and furniture. Normal tone and muscle strength, no cranial nerve abnormalities, symmetric deep tendon reflexes and normal sensation.
FIGURE 1.
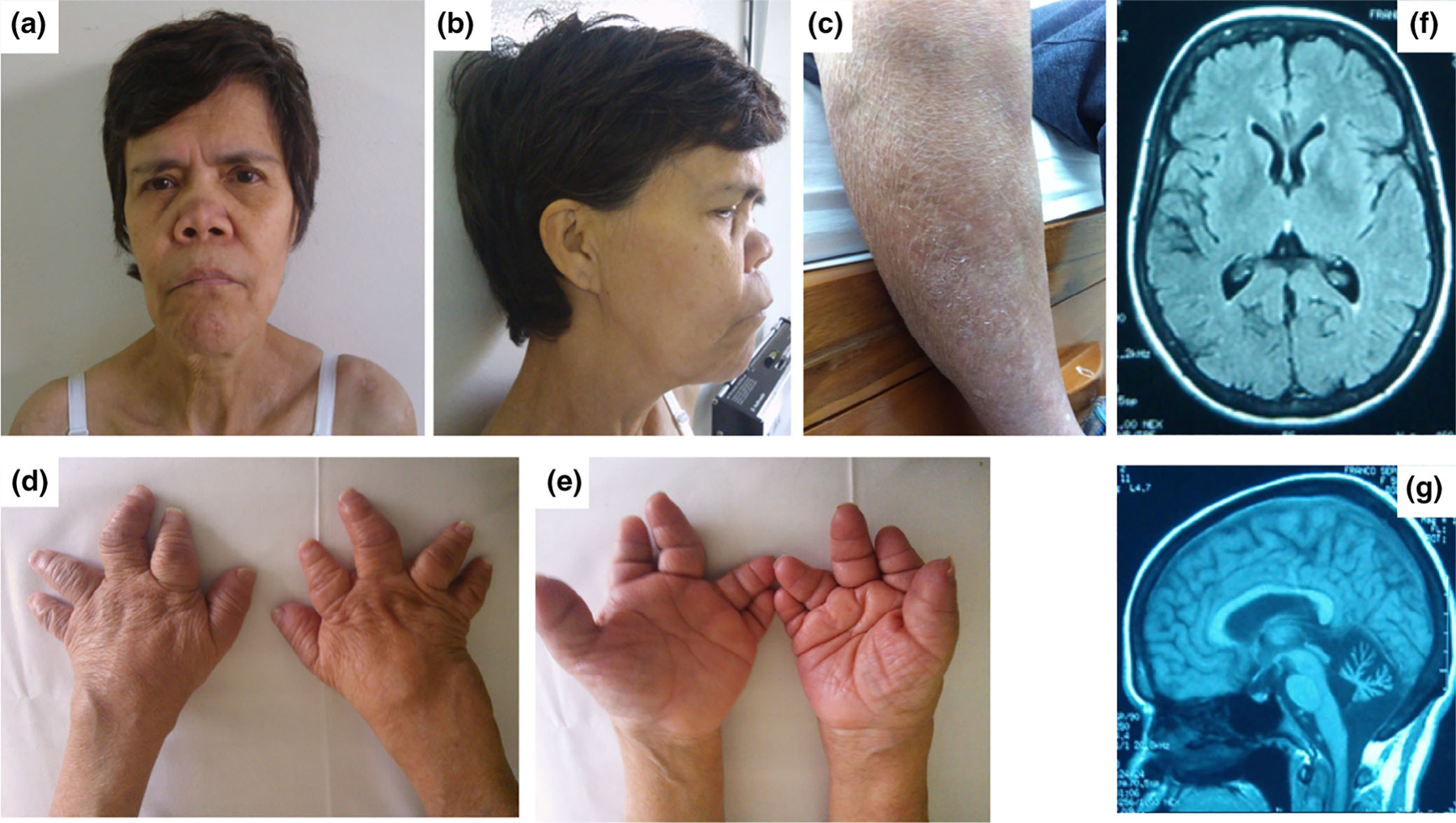
Acrodysostosis-like skeletal dysplasia with cerebellar atrophy and ichthyosis phenotype. (a) Frontal and (b) lateral views of face describing dysmorphic features including long face, malar flattening, wide and depressed nasal bridge with thickened alae nasi, anteverted nares, and smooth and long philtrum. (c) Lamellar ichthyosis located predominantly in the legs. (d, e) Hand anomalies demonstrate short, tapered fingers, clinodactyly, and ulnar deviation conferring a trident configuration. (f) Brain MRI showing cortical atrophy and wide sulci, and (g) cerebellar atrophy without brainstem involvement [Color figure can be viewed at wileyonlinelibrary.com]
Physical exam and facial features are more pronounced over the years (Figure 2a–f) and her parents did not show any dysmorphic abnormalities (Figure 2g,h). No pectus deformity, abdominal exam showed no organomegaly, and genitalia examination was Tanner V.
FIGURE 2.
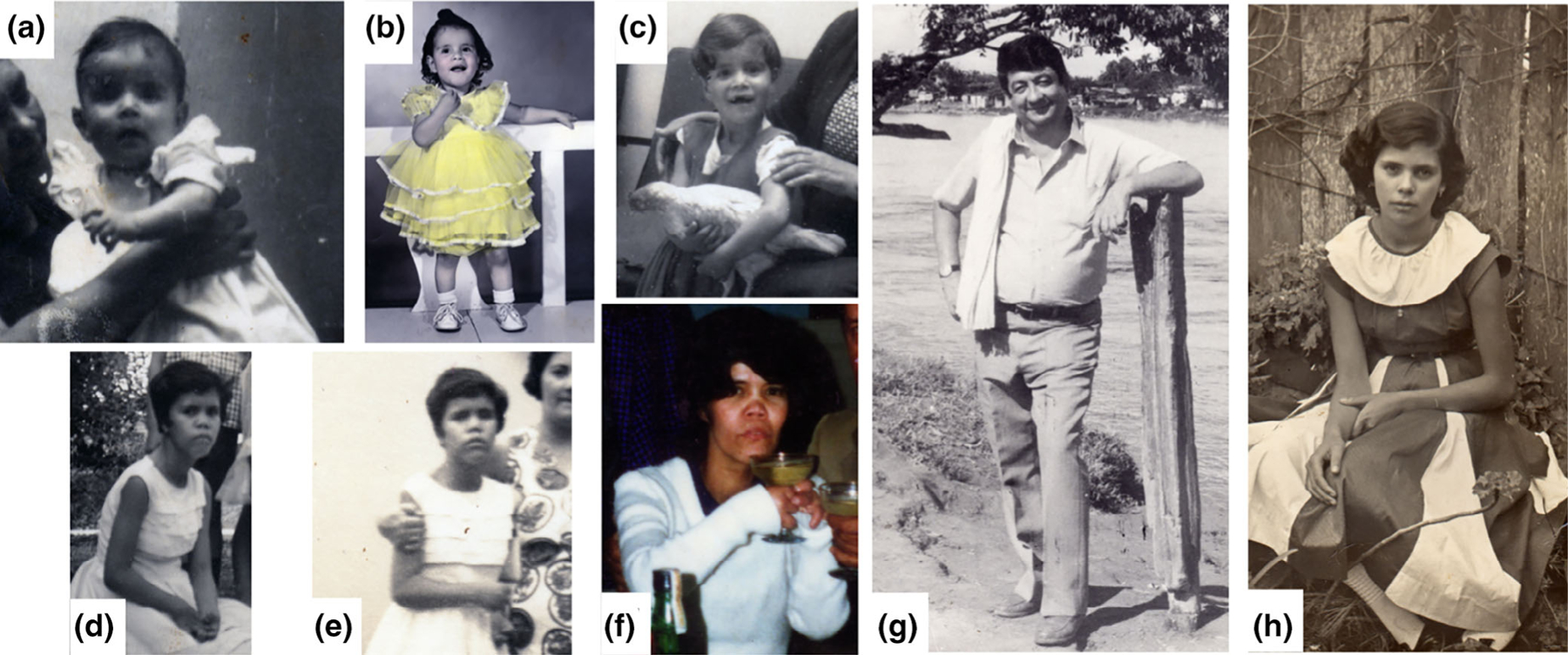
Age progression of acrodysotosis-like phenotype. Proband at different ages, showing evolving phenotype over the years. (a) 9 months, (b) 3 years, (c) 5 years, (d, e) 16 years, and (f) 35 years. No facial or acrodysotosis of proband’s parents, (g) father and (h) mother [Color figure can be viewed at wileyonlinelibrary.com]
Brain magnetic resonance imaging (MRI) showed cerebellar atrophy without brainstem involvement. Cerebral hemispheres had cortical atrophy and wide sulci (Figure 1f,g).
Lower bone density is noted in x-rays of cervical and thoracic spine, hips, and long bones showed osteopenia (Figure 3a–h). Dual-energy X-ray absorptiometry (DXA) was performed. The total hip densitometry was 0.555 g/cm2 with a T-score of −3.7 (Z-score −1.7) consistent with osteoporosis for her age. Bone metabolic test were normal including calcium 10.2 mg/dL (reference range 8.4–10.6), phosphorus 4.2 mg/dL (reference range 2.5–4.6), and PTH 63.7 pg/ml (reference range 14–65). Mild right convexity lumbar scoliosis, L3–L4 intervertebral space narrowing and syndesmophytes (Figure 3c). Hand X-rays showed irregular carpal bones, bilateral shortening of middle and distal phalanges (F2–F5), and radiolucent inclusion bodies in the epiphysis of metacarpal bones and proximal phalanges (Figure 3g). Upper extremity X-rays showed slightly short and bowed forearms (Figure 3h).
FIGURE 3.
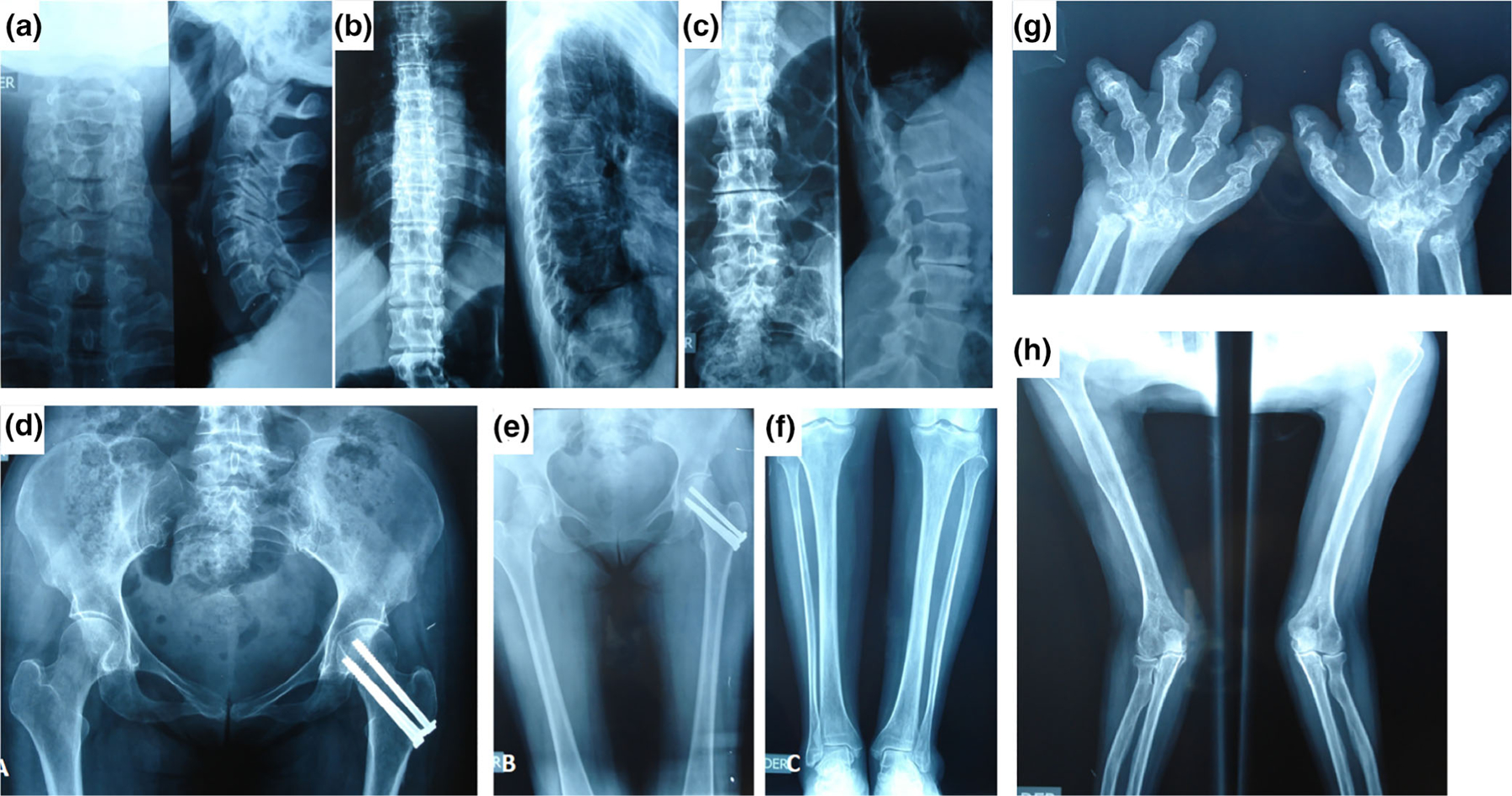
Roentgenograms characterizing skeletal findings. X-rays of (a) cervical, (b) thoracic, and (c) lumbar spine of the patient, showing minor lumbar scoliosis with right convexity, L3-L4 intervertebral space narrowing, syndesmophytes, and absent platyspondyly. (d) X-ray of hips showing fracture s/p surgery and (e) osteopenia of long bones. (g) Hand X-rays show shortness of all phalanges except the proximal phalanx III. The metacarpals are not disproportionately short, the shafts of metacarpals V are wide. The articular surfaces of all tubular bones are irregular and tented in the phalanges with radial deviation of the middle phalanx of the third and ulnar deviation of the fourth fingers. The carpal bones are misshaped. The proximal row of carpal bones is not visualized. (h) Upper extremity x-rays show slightly short and bowed forearms [Color figure can be viewed at wileyonlinelibrary.com]
Additional abnormal studies include echocardiogram with diastolic dysfunction, aortic valve sclerosis with minimal regurgitation, mitral valve sclerosis, and elevated pulmonary artery pressure (29 mmHg). EKG also showed abnormal progression of R wave in V1–V4 leads. EMG and nerve conduction studies were normal.
2.2 |. Genetic and variant analysis
Chromosomal examination from peripheral blood lymphocytes revealed normal female karyotype 46,XX. Array-CGH genotyping (GRCh37/hg19) showed a region of homozygosity in chromosome 2p25.3p25.2 spanning 6.7 Mb with boundaries from 50,813 to 6,727,777. Whole exome sequencing was undertaken as previously described (Hufnagel et al., 2015) and analysis was performed with Ingenuity Variant Analysis (Qiagen). Because parental and other familial samples were unavailable, analysis was performed as a singleton sample compared to public allele frequency databases (ExAC [Lek et al., 2016], 1,000 genomes [Genomes Project et al., 2015], NHLBI ESP [Fu et al., 2013]) and 217 exomes from unrelated healthy or differentially phenotypic individuals, including 66 Colombian individuals. An allele cutoff of 0.0001 (1:10,000) was used due to the unique aspects of the condition. No rare and predicted pathogenic variants were found in genes associated with acrodysostosis (PDE4D, PRKAR1A), lamellar icthyosis (ABCA12, NIPAL4, PNPLA1, LIPN, ST14, TGM1, CERS3, ALOX12B, ALOXE3, CYP4F22), Conradi–Hunermann syndrome (EBP), and Sjögren–Larsson syndrome (ALDH3A1, ALDH3A2). A broader search for rare genetic variants associated with osteochondrodysplasias, osteolysis, cerebellar atrophy, or cerebellar ataxia (see Section 3 and Table 1 for selected rare candidate rare variants) did not reveal pathogenic mutations in known disease genes associated with these phenotypes. In addition, no apparently homozygous variants were detected in these genes to suggest an autosomal recessive disorder in the region of homozygosity of chromosome 2p25.3p25.2 (Table 2). Of the rare alleles present in the patient, 27 variants were completely absent from healthy population databases and controls including 25 missense, one intronic, and two truncating. Of these, heterozygous truncating alleles were detected in FBLN7 (c.243C>A;p.Cys81Ter) and PPM1M (c.597+1_597+4delGTGA). No matches were available for these candidate genes.
TABLE 1.
Rare candidate variants from whole exome analysis from proband
| Gene | HGVSc | Protein change | gnomAD AF | Zygosity | Pathogenic prediction | pLI | LOEUF | Missense Z-score |
|---|---|---|---|---|---|---|---|---|
| FBLN7 | NM_001128165.1:c.243C>A | p.Cys81Ter | Absent | Het | Damaging (4 pathogenic predictions from DANN, GERP, LRT and MutationTaster) | 0 | 0.96 | 1.15 |
| PPM1M | NM_144641.3:c.597 + 4_597 + 7delAGTG | NA | Absent | Het | NA | 0 | 0.95 | 0.37 |
| NPEPPS | NM_006310.4: c.757T>C | p.Tyr253His | Absent | Het | Damaging (DANN, EIGEN, FATHMM, M-CAP, MutationAssessor, MutationTaster, PrimateAI and SIFT) | 1 | 0.06 | 3.98 |
| P2RY2 | NM_002564.3:c.646G>T | p.Val216Phe | 0.000008192 | Het | Uncertain (6 pathogenic predictions from DANN, GERP, LRT, MutationTaster, PROVEAN and SIFT, conflicting with four benign predictions from dbNSFP. FATHMM, MetaLR, MetaSVM and MutationAssessor] | 0.08 | 0.91 | 0.2 |
| LPO | NM_006151.2:c.2T>C | p.Met1Thr | 0.00001061 | Het | Uncertain (3 pathogenic predictions from GERP, MutationTaster and SIFT, conflicting with 5 benign predictions from DANN, dbNSFP. FATHMM, LRT, MetaLR and MetaSVM) | 0 | 1.15 | 0.24 |
| SLC19A1 | NM_194255.4: c.1594_1595insGCCCCG | p.Gln532delinsArgProGlu | Absent | Het | NA | 0 | 1.16 | 0.61 |
| CLEC4M | NM_001144909.1:c.421C>G | p.Arg141Gly | Absent | Het | Benign (5 benign predictions from dbNSFP. FATHMM, MetaLR, MetaSVM, MutationTaster and PROVEAN [vs 2 pathogenic predictions from MutationAssessor and SIFT]) | 0 | 1.12 | 0.92 |
| ACSM4 | NM_001080454.1:c.1456G>A | p.Glu486Lys | 0.00001219 | Het | Pathogenic (8 pathogenic predictions from DANN, GERP, MetaLR, MetaSVM, MutationAssessor, MutationTaster, PROVEAN and SIFT [vs 1 benign prediction from dbNSFP.FATHMM]) | 0 | 1.24 | 0.91 |
| TRAPPC8 | NM_014939.3:c.2077G>T | p.Ala693Ser | 0.000009489 | Het | Uncertain (5 pathogenic predictions from DANN, GERP, LRT, MutationAssessor and MutationTaster, conflicting with 5 benign predictions from dbNSFP.FATHMM, MetaLR, MetaSVM, PROVEAN and SIF) | 1 | 0.14 | 1.21 |
A follow-up with less stringent allele cutoff of 0.001 (1:1,000) was used for exome re-analysis. We observed a total of 167 non-synonymous variants including seven truncating variants, three splice affecting variants, one in-frame duplication, and 156 missense variants. Among missense variants, 35 were prioritized which were predicted to be damaging by SIFT and PolyPhen2.0 and had a CADD score >15. We did not observe any plausible variants in genes known to cause disorders which overlap our patient’s phenotypic features, whether inherited in a dominant or recessive manner. We have included 47 prioritized variants as a supplemental table with pLI, LOEUF, and gnomAD missense Z-scores (Table 1). Of these, the following heterozygous missense allele in the NPEPPS gene (c.757T>C;p. Tyr253His) had a low LOEUF (0.06) and high missense Z-score (3.98).
3 |. DISCUSSION
We are reporting a patient with a novel unique syndromic entity consisting of progressive facial and skeletal features reminiscent of acrodysostosis, intellectual disability, ichthyosis, cerebellar atrophy, ataxia, cardiac valvar calcification, elevated pulmonary artery resistance, and unusual hands. The constellation of multisystem findings in this patient have not been reported in other known Mendelian disorders.
Although our patient shows a phenotype related with acrodysostosis, she has ichthyosis and cerebellar atrophy that are not present in this disease. Exome testing did not identify mutations in the PDE4D or PRKAR1A genes known to cause acrodysostosis. These mutations can either reduce the amount of RIα protein, with a subsequent depletion of Protein Kinase A (Linglart et al., 2011), or alter the modulation of the levels of cAMP, generating resistance to several hormones (Lee et al., 2012). Individuals with PDE4D related acrodysostosis have a more pronounced midface hypoplasia and intellectual disability compared to PRKAR1A individuals (Michot et al., 2018).
Lamellar ichthyosis and hand deformities are reported in psoriatic arthritis but onset is typically later in life. The proband’s hand abnormalities and skin findings were present during early childhood and continue to progress during her life. Another possible diagnostic entity is Sjögren–Larsson syndrome. The overlapping features include short stature, ichthyosis and brain/cerebellar atrophy, however, additional symptoms belonging to Sjögren–Larsson syndrome like corneal opacity, thoracic kyphosis, spasticity, and seizures were absent in our patient (Fuijkschot et al., 2012). This syndrome is an inborn error of lipid metabolism caused by several types of mutations of the ALDH3A2 (aldehyde dehydrogenase three family, member A2) gene, that produces an enzymatic deficit of fatty aldehyde dehydrogenase (FALDH) which were not identified in the exome study. In normal conditions, this protein acts freely or as a part of the NAD oxidoreductase complex (FAO), or inactivating B4 Leukotriene (LTB4) by ω-oxidation, important in the oxidation of fatty aldehides to fatty acids (Lamari, Mochel, Sedel, & Saudubray, 2013).
Myhre syndrome (OMIM #139210) is characterized by muscle hypertrophy, midface hypoplasia, blepharophimosis, prognathism, intellectual disability, short stature, brachydactyly, and cardiovascular defects with a striking fibroproliferative response to surgical intervention (Lin et al., 2016). Cerebellar atrophy has been reported in a single individual with Myhre syndrome (Bachmann-Gagescu, Hisama, & Yuen, 2011). Dysmorphism was not reminiscent of Myhre and exome did not identify SMAD4 disease causative variants.
Conradi–Hünermann Syndrome (OMIM #302960) is the most well-known chondrodysplasia punctata type. This is an X-linked dominant disorder caused by pathogenic variants in the EBP gene (Braverman et al., 1999). Conradi–Hünermann Syndrome can present with midface hypoplasia, transient congenital ichthyosis, cataracts, alopecia, rizhomelic shortening of limbs, and intellectual disability. Radiographic stippling was not observed in the present case, and skin manifestations are not typical of this disease.
Consistent with the assertion that this is a new syndrome, we did not detect pathogenic alleles in genes associated with these known disorders or disorders associated with the individual phenotypes. While many rare variants were detected in exome analysis, one limitation was that parental and other familial samples were not available for segregation. However, in FBLN7 and PPM1M we detected truncating alleles, which may be informative in the absence of family history. FBLN7 haploinsufficiency has been implicated in the craniofacial and congenital heart defects in 2q13 deletion syndrome, which was phenocopied in zebrafish morpholino experiments (Russell et al., 2014). While this patient did not have congenital heart defects, these features have been noted to be incompletely penetrant in 2q13 deletion syndrome. This FBLN7 nonsense allele may account for the mild dysmorphic facial features and mild microcephaly. While patients with 2q13 deletion syndrome do not exhibit skeletal dysplasia, it is unclear whether this FBLN7 allele results in isolated gene haploinsufficiency or produces a stable truncated protein with additional effects. FBLN7 is expressed in developing cartilage and teeth, and so there may be a role for this gene. Fibulins form intermolecular bridges within the extracellular matrix and serve as mediators for cellular processes and tissue remodeling. Little is known about PPM1M, a magnesium-dependent protein phosphatase expressed in testis, heart, lung, kidney, brain, and liver (Komaki et al., 2003). Expression in the developing skeleton has not been observed.
NPEPPS encodes the puromycin-sensitive aminopeptidase (PSA), a zinc metallopeptidase which hydrolyzes amino acids from the N-terminus of its substrate. The protein has been localized to both the cytoplasm and to cellular membranes. PSA is the only cytosolic enzyme able to digest polyglutamine sequences (Bhutani, Venkatraman, & Goldberg, 2007). PSA inhibition increases levels of polyglutamine expanded ataxin-3, mutant alpha-synuclein, and superoxide dismutase-1 (Menzies et al., 2010). PSA has an important role in preventing the accumulation of aggregation-prone proteins and toxicity leading to neurodegeneration.
NPEPPS is ubiquitous but highly expressed in brain, cerebellum, and many other tissues including skeleton so there may be a role for this gene in human disease. PSA deficient mice showed dwarfism, increased anxiety, and impaired pain response (Osada et al., 1999). The body weight of the PSA deficient mice were <70% of the control littermates at 3 weeks after birth. No difference was identified in plasma levels of growth hormone, insulin growth factor I, thyroid hormone, and calcium metabolism (Osada et al., 1999). A previous study by Constam et al. 1995 reported that puromycin arrest cell cycle of COS cells and induces the cells to go into apoptosis (Constam et al., 1995). No studies have been performed to analyze the impact in skeletal development. Additional functional studies and patients with overlapping phenotypes are needed to determine if NPEPPS variants can cause human disease.
Causal variant detection in this case may be limited by technical factors. Whole exome sequencing remains variably successful in identifying causative mutation for genetic disorders in 25–40% of the patients (Sawyer et al., 2016). More recently, diagnostic utility of WES was found to be 41% for duo-, quad-, or trio-WES cases as compared to 28% singleton cases (Ji et al., 2019), which clearly indicates that the former approach has a higher diagnostic yield. However, another study focused on the evaluation of diagnostic efficiency of trio versus singleton WES did not show any major difference where diagnosis remained 36.7% and 40.0% for singleton and trio cases, respectively (Tan et al., 2019).
In conclusion, we have delineated a new syndrome of unknown genetic etiology, which may or may not be modified by FBLN7 or PPM1M truncating alleles or NPEPPS missense allele. Identification of additional patients and comparative genomics are required to delineate the mechanism of this unique disorder.
Supplementary Material
ACKNOWLEDGMENTS
The Authors thank the patient and her family for their efforts and participation. Written consent was obtained for the use of pictures and clinical findings. Intramural funding was received from National Eye Institute (R.B.H. and E.U.).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Abrahamov A, Elstein D, & Zimran A (2000). Type IIIc Gaucher disease and acrodysostosis. The Israel Medical Association Journal, 2(2), 182. [PubMed] [Google Scholar]
- Bachmann-Gagescu R, Hisama FM, & Yuen AL (2011). Myhre syndrome with ataxia and cerebellar atrophy. Clinical Dysmorphology, 20(3), 156–159. 10.1097/MCD.0b013e3283468043 [DOI] [PubMed] [Google Scholar]
- Bhutani N, Venkatraman P, & Goldberg AL (2007). Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. The EMBO Journal, 26(5), 1385–1396. 10.1038/sj.emboj.7601592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, … Valle D (1999). Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi–Hunermann syndrome. Nature Genetics, 22(3), 291–294. 10.1038/10357 [DOI] [PubMed] [Google Scholar]
- Constam DB, Tobler AR, Rensing-Ehl A, Kemler I, Hersh LB, & Fontana A (1995). Puromycin-sensitive aminopeptidase. Sequence analysis, expression, and functional characterization. The Journal of Biological Chemistry, 270(45), 26931–26939. 10.1074/jbc.270.45.26931 [DOI] [PubMed] [Google Scholar]
- Drozdowski PH, Latkowski I, Zachara MG, & Wojcicki P (2017). Binder syndrome: Clinical findings and surgical treatment of 18 patients at the Department of Plastic Surgery in Polanica Zdroj. Advances in Clinical and Experimental Medicine, 26(3), 427–437. 10.17219/acem/62123 [DOI] [PubMed] [Google Scholar]
- Dutra LA, de Aquino CC, & Barsottini OG (2009). Sjogren–larsson syndrome: Case report and review of neurologic abnormalities and ichthyosis. The Neurologist, 15(6), 332–334. 10.1097/NRL.0b013e3181906ff9 [DOI] [PubMed] [Google Scholar]
- Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, … Akey JM (2013). Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature, 493(7431), 216–220. 10.1038/nature11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuijkschot J, Theelen T, Seyger MM, van der Graaf M, de Groot IJ, Wevers RA, … Willemsen MA (2012). Sjogren–Larsson syndrome in clinical practice. Journal of Inherited Metabolic Disease, 35(6), 955–962. 10.1007/s10545-012-9518-6 [DOI] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, … Abecasis GR (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedion A (2002). Acrodysostosis and craniofacial conodysplasia are two separate bone dysplasias. Pediatric Radiology, 32(3), 214. 10.1007/s00247-001-0635-x [DOI] [PubMed] [Google Scholar]
- Gupte GL, Kher AS, Kanade SP, Bharucha BA, & Sagade SN (1994). Acrodysostosis with 5 alpha reductase deficiency: An unusual association. Indian Journal of Pediatrics, 61(3), 287–290. 10.1007/bf02752226 [DOI] [PubMed] [Google Scholar]
- Hufnagel RB, Arno G, Hein ND, Hersheson J, Prasad M, Anderson Y, … Ahmed ZM (2015). Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-moon syndromes. Journal of Medical Genetics, 52(2), 85–94. 10.1136/jmedgenet-2014-102856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Shen L, Bootwalla M, Quindipan C, Tatarinova T, Maglinte DT, … Gai X (2019). A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Cold Spring Harbor Molecular Case Studies, 5(2), a003756. 10.1101/mcs.a003756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Smith DW, Harvey MA, Hall BD, & Quan L (1975). Older paternal age and fresh gene mutation: Data on additional disorders. The Journal of Pediatrics, 86(1), 84–88. 10.1016/s0022-3476(75)80709-8 [DOI] [PubMed] [Google Scholar]
- Komaki K, Katsura K, Ohnishi M, Guang Li M, Sasaki M, Watanabe M, … Tamura S (2003). Molecular cloning of PP2Ceta, a novel member of the protein phosphatase 2C family. Biochimica et Biophysica Acta, 1630(2–3), 130–137. 10.1016/j.bbaexp.2003.09.004 [DOI] [PubMed] [Google Scholar]
- Lamari F, Mochel F, Sedel F, & Saudubray JM (2013). Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: Toward a new category of inherited metabolic diseases. Journal of Inherited Metabolic Disease, 36(3), 411–425. 10.1007/s10545-012-9509-7 [DOI] [PubMed] [Google Scholar]
- Lee H, Graham JM Jr., Rimoin DL, Lachman RS, Krejci P, Tompson SW, … Cohn DH (2012). Exome sequencing identifies PDE4D mutations in acrodysostosis. American Journal of Human Genetics, 90(4), 746–751. 10.1016/j.ajhg.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation C (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AE, Michot C, Cormier-Daire V, L’Ecuyer TJ, Matherne GP, Barnes BH, … Lindsay ME (2016). Gain-of-function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. American Journal of Medical Genetics. Part A, 170(10), 2617–2631. 10.1002/ajmg.a.37739 [DOI] [PubMed] [Google Scholar]
- Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, Cancel M, … Silve C (2011). Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. The New England Journal of Medicine, 364(23), 2218–2226. 10.1056/NEJMoa1012717 [DOI] [PubMed] [Google Scholar]
- Maroteaux P, & Malamut G (1968). Acrodysostosis. Presse Médicale, 76 (46), 2189–2192. [PubMed] [Google Scholar]
- Menzies FM, Hourez R, Imarisio S, Raspe M, Sadiq O, Chandraratna D, … Rubinsztein DC (2010). Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Human Molecular Genetics, 19(23), 4573–4586. 10.1093/hmg/ddq385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot C, le Goff C, Blair E, Blanchet P, Capri Y, Gilbert-Dussardier B, … Cormier-Daire V (2018). Expanding the phenotypic spectrum of variants in PDE4D/PRKAR1A: From acrodysostosis to acroscyphodysplasia. European Journal of Human Genetics, 26(11), 1611–1622. 10.1038/s41431-018-0135-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot C, le Goff C, Goldenberg A, Abhyankar A, Klein C, Kinning E, … Cormier-Daire V (2012). Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. American Journal of Human Genetics, 90(4), 740–745. 10.1016/j.ajhg.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada T, Ikegami S, Takiguchi-Hayashi K, Yamazaki Y, Katoh-Fukui Y, Higashinakagawa T, … Takeuchi T (1999). Increased anxiety and impaired pain response in puromycin-sensitive aminopeptidase gene-deficient mice obtained by a mouse gene-trap method. The Journal of Neuroscience, 19(14), 6068–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow M, Pfeiffer RA, Gorlin RJ, McKusick VA, Renuart AW, Johnson GF, & Summitt RL (1971). Acrodysostosis. A syndrome of peripheral dysostosis, nasal hypoplasia, and mental retardation. American Journal of Diseases of Children, 121(3), 195–203. [PubMed] [Google Scholar]
- Russell MW, Raeker MO, Geisler SB, Thomas PE, Simmons TA, Bernat JA, … Innis JW (2014). Functional analysis of candidate genes in 2q13 deletion syndrome implicates FBLN7 and TMEM87B deficiency in congenital heart defects and FBLN7 in craniofacial malformations. Human Molecular Genetics, 23(16), 4272–4284. 10.1093/hmg/ddu144 [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Hartley T, Dyment DA, Beaulieu CL, Schwartzentruber J, Smith A, … Boycott KM (2016). Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: Time to address gaps in care. Clinical Genetics, 89(3), 275–284. 10.1111/cge.12654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezer N, Sutbeyaz ST, Koseoglu F, Aras M, & Akin C (2009). Adult case of acrodysostosis with severe neurologic involvement. Journal of Back and Musculoskeletal Rehabilitation, 22(2), 125–129. 10.3233/BMR-2009-0223 [DOI] [PubMed] [Google Scholar]
- Steiner RD, & Pagon RA (1992). Autosomal dominant transmission of acrodysostosis. Clinical Dysmorphology, 1(4), 201–206. [PubMed] [Google Scholar]
- Tan TY, Lunke S, Chong B, Phelan D, Fanjul-Fernandez M, Marum JE, … White SM (2019). A head-to-head evaluation of the diagnostic efficacy and costs of trio versus singleton exome sequencing analysis. European Journal of Human Genetics, 27(12), 1791–1799. 10.1038/s41431-019-0471-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The data are not publicly available due to privacy or ethical restrictions.