

Abstract
Current genetic screening methods for inherited eye diseases are concentrated on the coding exons of known disease genes (gene panels, clinical exome). These tests have a variable and often limited diagnostic rate depending on the clinical presentation, size of the gene panel and our understanding of the inheritance of the disorder (with examples described in this issue). There are numerous possible explanations for the missing heritability of these cases including undetected variants within the relevant gene (intronic, up/down-stream and structural variants), variants harbored in genes outside the targeted panel, intergenic variants, variants undetectable by the applied technology, complex/non-Mendelian inheritance, and nongenetic phenocopies. In this article we further explore and review methods to investigate these sources of missing heritability.
Keywords: enhancer, regulatory, variant
Inherited ocular disease represent a wide spectrum of conditions, from malformations to degeneration. These represent a significant health burden among rare diseases, with ocular malformations occurring in 1:10,000 individuals and retinal degenerations in 1:2,000–3,000. Despite knowledge of hundreds of disease-associated genes, genetic testing for these conditions varies widely, from 20% for anophthalmia/microphthalmia (Chassaing et al., 2014), to nearly 70% for retinal degenerations (Carss et al., 2017; Ellingford et al., 2016). However, this largely relies on querying variants in coding sequences for previously mapped genes, which constitute 1.5%–2% of coding DNA. Here, we describe recent efforts in understanding the noncoding genomic regions, in particular the pathogenesis of splicing, transcriptional, and regulatory elements. Better comprehension of these noncoding regions will improve the yield of clinical molecular diagnostics by better matching clinical diagnoses and revealing additional patterns of disease mechanisms.
1 |. CRYPTIC SPLICE ALTERATION AND OPHTHALMIC DISEASES
Stargardt macular dystrophy (STGD1) is a well characterized autosomal recessive retinal dystrophy with the majority of disease caused by biallelic variants in the ABCA4 gene (Allikmets et al., 1997). However, up to 30% of cases remain unresolved or with a missing second allele following screening of the coding exons of the gene (Sangermano et al., 2019). Extra-exonic variants, in particular deep intronic cryptic splice variants, are now well characterized as a cause of STGD1, as demonstrated in the recent study by Khan and colleagues (Khan et al., 2020) showing that 25% of STGD1 cases carried an intronic or structural variant in the ABCA4 gene. This example highlights the importance of considering regions outside the coding exons in the pathogenesis of inherited diseases.
Historically, the introns of genes have been largely ignored in genetic testing due to their size, high frequency of variation and our poor knowledge of their function at the nucleotide level. This in combination with a paucity of population variant data meant that until recently, intronic variations were difficult to interpret. However, there are multiple examples of well characterized intronic variants in retinal diseases, identified through various strategies (den Hollander et al., 2006; Mayer & Aguilera, 1990; Mayer et al., 2016; van den Hurk et al., 2003). Now, with access to whole genome sequencing in research and clinical laboratories (Turnbull et al., 2018; Turro et al., 2020) and public availability of large population genome datasets such as gnomAD, researchers are beginning to apply similar variant rarity filtering strategies regularly performed in exome filtering pipelines to noncoding variants in order to identify candidate-disease variants in rare diseases (Carss et al., 2017; Cassini et al., 2019; Khan et al., 2017; Verdura et al., 2020). To date, the reports have broadly identified noncoding alleles in recessive retinal diseases (either homozygous noncoding alleles or the presence of second noncoding allele in an individual carrying a coding mutation) and noncoding variants that cause activation of a deep intronic splice site leading to pseudoexon incorporation in the transcript.
Effective, large-scale interpretation of noncoding variants remains challenging, due to the larger variant number and lower conservation found in intronic compared to exonic regions, and our limited understanding of the function of introns. Therefore, the key to unraveling pathogenic intronic mutations will be accurate tools to predict the effect of such variants.
Recent advances in the application of machine learning for splice prediction (Cheng et al., 2019; Jagadeesh et al., 2019; Jaganathan et al., 2019; Lee et al., 2017; Xiong et al., 2015) mean that more accurate characterization of large-scale variant data is possible (Ellingford et al., BioRXIV). Validation of high priority variants should still be performed with in vitro studies, such as transcript analysis from patient derived RNA or cells or in vitro gene splicing assays for genes with inaccessible tissue-specific expression.
2 |. COPY NUMBER AND STRUCTURAL VARIANT ANALYSIS
Many gene panel, exome and genome sequencing pipelines incorporate structural variant (SV) and/or copy number variant (CNV) surveillance tools including read depth analysis algorithms (examples: ExomeDepth for targeted panel and exome analysis, CANVAS for WGS analysis) and split read analysis algorithms (example: MANTA for WGS analysis, targeted panels and WES rarely capture the breakpoint/s of SV/CNVs).
Simple deletions spanning one or more exons can be effectively detected using read depth-based approaches and gene panel/exome analysis (Ellingford et al., 2017; Marchuk et al., 2018; Patel et al., 2019; Plagnol et al., 2012; Rajagopalan, Murrell, Luo, & Conlin, 2020). However, the ability to detect and characterize SV/CNVs is greatly enhanced with WGS due to the complete and even coverage of the genome (using PCR-free technology), thereby preserving the dosage of the genome for effective analysis of loss/gains. In addition, coverage of breakpoint regions allows effective characterization of deletions, tandem duplications, translocations, and inversions, to the single nucleotide. This includes any additional loss/gain at the breakpoint and complex rearrangements by incorporating an algorithm to analyze split read data (Arno et al., 2016; Ba-Abbad et al., 2016; Carss et al., 2017; Sanchis-Juan et al., 2018).
Standard paired-end read sequencing generates read pairs on the forward and reverse strand (approx. 70–200 bp) flanking an unsequenced insert region (approx. 400 bp). When mis-aligned to the reference genome due to the presence of an SV/CNV, this paired-end read structure will display a characteristic alteration in orientation, including altered insert size or read direction, specific for the SV/CNV type. This enables accurate characterization of rearrangements and easy visualization of the breakpoints using a genome viewer such as the Integrative Genomics Viewer [IGV, (Robinson et al., 2011; Thorvaldsdottir, Robinson, & Mesirov, 2013)].
It is estimated that SV/CNVs account for a significant proportion of the missing heritability in IRD (Carss et al., 2017; Ellingford et al., 2016) and these methods represent effective tools to characterize them. However, it is complicated to interpret SV/CNVs that do not directly impact a coding exon or known regulatory region of a gene; such entirely intronic or intergenic variants may nevertheless play an important role in gene regulation and Mendelian diseases (Cipriani et al., 2017).
The addition of emerging technologies, such as long-read or single molecule sequencing, that allow sequencing of genomic DNA up to >100Kb in a single read, is an exciting prospect to analyze noncoding regions (reviewed in (Mantere, Kersten, & Hoischen, 2019)). These powerful technologies enable effective de novo assembly of an individual’s genome, read through of complex rearrangements and the potential to read through regions intractable to current short-read technologies (Sanchis-Juan et al., 2018; Vache et al., 2020).
3 |. GENE EXPRESSION
Gene expression information is important evidence for prioritizing candidate disease-associated genes and variation. Exome and genome sequencing detect hundreds of thousands of coding variants and millions of noncoding variants. Even after filtering for frequency in the general population or gene constraint to missense or truncating variation in such databases as gnomAD, multiple candidate variants exist. A complementary strategy to prioritizing filtered variant sets is expression or lack thereof in ocular tissues. Vertebrate expression data is extremely valuable as gene identity is well-conserved across multiple animal model systems, including nonhuman primate, mouse, and zebrafish. Mouse expression databases, made possible by collating decades of publications using gene expression arrays and in situ hybridization experiments, are available at Mouse Genome Informatics. Murine homologue expression data is available for gene-by-gene queries. Expression data are also available for zebrafish, frog, and fruit fly at different developmental and adult stages.
Human gene expression datasets have been made available more recently. RNA-seq is a massive parallel sequencing technique which can be used for quantifiable comparisons of gene expression levels between tissues. The Genotype-Tissue Expression (GTEx) project compiles RNA-seq data from 54 nondiseases human tissues from nearly 1,000 donors. Notably, ocular tissues were not included in this dataset. To address this, investigators at the National Eye Institute (National Institutes of Health) created eyeIntegration, a compilation of publicly deposited RNA-seq datasets from developing and adult human ocular tissues, and compared expression levels to nonocular tissues in GTEx. Subsequently, transcript-level data, de novo transcriptome data, and single cell data have been added to the website (Bryan et al., 2018).
Importantly, tissue-specific transcripts exist for several genes implicated in retinal degeneration. RPGR (OMIM 312610) ORF15 is an open-reading frame with expression specifically in retinal cell types and harbors the majority of disease-associated alleles with this form of X-linked retinitis pigmentosa (Neidhardt et al., 2007). Similarly, several retina-enriched transcripts were described for RPGRIP1 (OMIM 605446), associated with autosomal recessive Leber congenital amaurosis and cone-rod dystrophy (Lu & Ferreira, 2005), including causal noncoding variants that alter splicing. Notably, as discussed above, deep intronic alleles have been found in several retinal-expressed genes, which subsequently were shown to generate cryptic exons with functional implications for inherited retinal dystrophies (Bauwens et al., 2019; Braun et al., 2013; Sangermano et al., 2019; Weisschuh et al., 2020; Zernant et al., 2014). As such, ocular-specific transcripts and deep intronic alleles reveal a biological link between genetic variation and tissue-specific gene expression.
While expression of a gene during ocular development or postnatally is a priori evidence of involvement in these tissues, this does not infer that mutation of this gene is necessary or sufficient to cause a disease in these tissues. Expression data is also used to validate the impact of variants on gene expression, which correlates with partial or total loss-of-function. Genome sequencing coupled to RNA-seq can be used to evaluate deep intronic and splicing changes genome-wide for deleterious variants causing exon skipping or inclusion of cryptic exons, and, in some studies, RNA-seq can be used alone to infer DNA-level variants altering splicing (Gonorazky et al., 2016). In this manner, RNA sequencing can be integrated into clinical molecular diagnostics for rare diseases.
Genome-wide association studies using single nucleotide polymorphism genotyping to compare thousands of cases versus controls can detect risk alleles for common disorders, such as age-related macular degeneration (AMD). Following detection of the first risk locus in the CFH gene (OMIM 134370), now 52 rare and common variants associated with AMD have been discovered (Klein et al., 2005). To correlate these phenotype-related variants with alterations of gene expression, transcriptome data from cases and controls are directly compared to generate expression quantitative trait loci (eQTLs). In a recent study, over 4,000 eQTLs were detected in postmortem retinas from individuals with AMD compared to those from unaffected donors (Ratnapriya et al., 2019). These eQTLs correlated significantly with 6 of the previously reported AMD risk loci from GWAS studies, thereby refining the functional implications of more than 10% of previously reported risk alleles.
Thus, expression data can be of value to prioritize candidate genes, detect splicing changes, and infer relationships between genomic variation and functional implications on transcriptional and splicing regulation.
4 |. GENOMIC APPROACHES TO DISCOVER REGULATORY REGIONS OF GENES THAT CAUSE EYE DISEASES
While molecular genetic studies of the coding regions of genes are now commonplace to discover variants associated with diseases, discovery of such variants within noncoding regions that influence, or control, gene expression is still in its infancy. Axenfeld-Rieger Syndrome can serve as an example of this approach.
4.1 |. Identification of the genetic basis of Axenfeld–Rieger syndrome
Axenfeld–Rieger Syndrome (ARS) is a rare autosomal dominant eye disease that affects 1/10,000–1/20,000 people, regardless of ethnicity (Seifi & Walter, 2018). Patients with ARS present with ocular features that can include iris hypoplasia, misplaced pupils, full thickness tears in the iris (polycoria), adhesions between the iris and the cornea, and a displaced Schwalbe line. Patients may also present with nonocular malformations of the teeth, jaw and umbilicus, as well as cerebellar, hearing and heart defects (Chrystal & Walter, 2019). More than 50% of ARS patients present with glaucoma that is often recalcitrant to normally prescribed glaucoma medications (Strungaru, Dinu, & Walter, 2007). Linkage analyses of large families in which ARS was segregating was used to map genes responsible for the disease in these families (Gould, Mears, Pearce, & Walter, 1997; Mears, Mirzayans, Gould, Pearce, & Walter, 1996; Semina et al., 1996a; Walter, Mirzayans, Mears, Hickey, & Pearce, 1996). Subsequently, mutations of PITX2 (pituitary homeobox protein 2; (Semina et al., 1996b) and FOXC1 (forkhead box C1; (Mears et al., 1998; Mirzayans et al., 2000; Nishimura et al., 1998) were shown to cause ARS. Molecular characterizations have shown that mutations within the coding regions of either gene typically result in loss of protein functions which include impaired nuclear localization, DNA binding, protein–protein interactions, and transactivation capacity (Footz, Idrees, Acharya, Kozlowski, & Walter, 2009; Kozlowski & Walter, 2000; Lines et al., 2004; Murphy et al., 2004; Saleem, Banerjee-Basu, Berry, Baxevanis, & Walter, 2001; Saleem, Banerjee-Basu, Berry, Baxevanis, & Walter, 2003a; Saleem, Banerjee-Basu, Murphy, Baxevanis, & Walter, 2004; Saleem, Murphy, Liebmann, & Walter, 2003b). However, there are reports of PITX2 mutations resulting in a gain of function effect (Priston et al., 2001; Saadi et al., 2006). Gene copy number changes, and insertions and deletions within the coding regions of PITX2 (Flomen et al., 1997, 1998; Lines et al., 2004; Semina, Datson, et al., 1996a) and FOXC1 gene (Chanda et al., 2008; D’Haene et al., 2011; Lehmann et al., 2000) have also been found in ARS patients, consistent with the concept that too much or too little PITX2 or FOXC1 can result in ARS (Walter, 2003). However, only 40% of ARS patients have mutations involving the coding regions of PITX2 or FOXC1. To investigate the missing heredity, other candidate genes have been examined for additional ARS-associated disease-causing mutations. Mutational screening of three candidate genes (FOXC2, P32, and PDP2) that encode proteins that interact with FOXC1 or PITX2 (Acharya, Huang, Fleisch, Allison, & Walter, 2011; Huang et al., 2008; Strungaru et al., 2011) did not detect mutations in ARS patients, suggesting that these genes do not contribute to the missing heredity of ARS. PAX6 deletions were initially reported to be associated with ARS (Riise, Storhaug, & Brondum-Nielsen, 2001), but this observation was not reproduced upon further investigations by the same investigators using improved reagents (Riise, D’Haene, De Baere, Gronskov, & Brondum-Nielsen, 2009). Recently, mutations within the coding regions of two additional genes, PRDM5 and COL4A1 (Micheal et al., 2016; Sibon et al., 2007), have been suggested to result in a small fraction of ARS patients (less than 1%). Thus, despite expanded insertion/deletion investigations of the FOXC1 and PITX2 coding regions and mutation screening of additional candidate genes, the molecular defect in over half of ARS patients remains unknown.
In an effort to discover additional sources of the missing ARS disease-associated heritability, researchers turned to investigations of the cis-regions that regulate the expression of PITX2 and FOXC1. However, like most human genes, the elements that regulate the expression of PITX2 and FOXC1 are largely unknown or are experimentally unverified. Therefore, researchers have turned to model organisms to identify and characterize the function of such gene regulatory elements. Volkmann and colleagues identified 13 regions potentially controlling PITX2 expression, through comparison of the genomes of human and zebrafish (Volkmann et al., 2011). Investigation of these putative regulatory regions identified a group of patients with structural variants of subsets of these regions in ARS patients known to not have coding region changes of PITX2 or FOXC1 (Protas et al., 2017). Subsequent deletion of some of these PITX2 regions in zebrafish, using CRISPR-Cas9 gene editing, yielded animals with phenotypes overlapping with those of ARS patients. These data are thus consistent with the hypothesis that deletion of upstream regulatory elements can cause ARS in patients with normal PITX2 coding regions (Protas et al., 2017; Volkmann et al., 2011). Importantly, these results also indicate that mutations of noncoding regions of known genes, rather than mutations of unknown genes, could explain a substantial proportion of ARS patients with unknown etiology.
4.2 |. Tools and resources to discover structural variations associated with human ocular disease
While the example of discovery that structural variation of regulatory elements can explain some of the missing heritability for ARS, detection and validation of such elements remains challenging. For PITX2, Volkmann’s approach was to inspect a 1.6 Mb interval containing the PITX2 gene for conserved noncoding sequences with 80–90% identity between the human and zebrafish species. Further comparisons indicated that 12/13 elements detected in this manner also had high levels of sequence conservation in the chicken and mouse genomes, and that the elements were unlikely to be parts of transcripts since their sequences were absent from zebrafish or human expression databases. The ability of all 13 elements to regulate expression was then tested by cloning each element upstream of a GFP promoter plasmid containing 1.9 kb of the basic PITX2 promoter. Transient transfection of these reports in zebrafish embryos demonstrated GFP expression patterns that overlapped with that of endogenous PITX2. Importantly deletion of some of these elements using CRISPR-Cas9 produced animals with ARS-like features, providing reciprocal evidence of the key role of these elements in regulating PITX2 expression. This information was then used to support investigation of the role of these regulatory elements in ARS. The usefulness of the results of these laborious experiments to provide explanations for the missing heritability of ARS was then confirmed with the detection of noncoding structural variations involving these elements in ARS patients (Protas et al., 2017; Volkmann et al., 2011).
Fortunately, resources have considerably advanced since the research of Volkmann and colleagues to discover and evaluate the regulatory regions of genes such as PITX2. As an example, we conducted an analysis to discover potential regulatory regions upstream of FOXC1. Analysis of such conserved elements, as was done for PITX2, could identify ARS-associated variation near FOXC1 that would be missed by regular DNA sequencing of coding regions.
We used the NCBI Basic Local Alignment Search Tool (BLAST) for nucleotides to identify regions of similarity between DNA sequences of human and mouse. Our query was 1 megabase upstream of the FOXC1 gene within GRCh38 chromosome 6 at NC_000006.11:609,915–1,609,915. The database for this search was “Nucleotide collection (nr/nt)” which we used to compare human sequences against the mouse DNA sequence database. BLAST default parameters were used. Regions of low compositional complexity were masked as these regions may cause spurious or misleading results. Results were manually filtered to eliminate hits corresponding to gene coding regions, and sequences that did not map to mouse chromosome 13 (syntenic to human chromosome 6p25). Using these criteria, 6 out of the total of 55 BLAST hits of homology between human and mouse databases were selected for further analyses (Figure 1).
FIGURE 1.
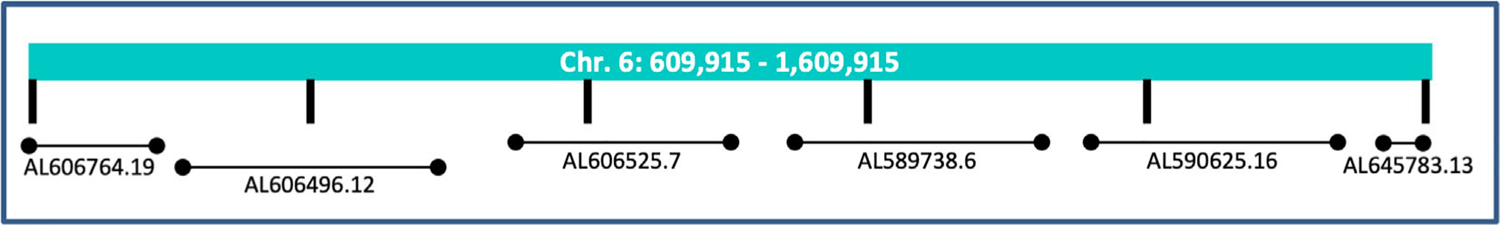
The location and names of six mouse BAC clones containing noncoding genomic DNA sequences homologous to the human genome upstream of FOXC1. Vertical lines indicate 200 kb segments, which black horizontal lines indicate position of mouse BACs containing regions of similarly to human GRCh38 chromosome 6 at NC_000006.11:609,915–1,609,915. BAC clone names are identified below the horizontal lines
These six hits were genomic BAC clones mapped to mouse chromosome 13. Each BAC contained smaller regions of homology larger than 100 bp and varying in length between 158 and 1,662 bp (Supplementary Table 1), for a total of 45 conserved regions. Sequences identified in our analysis have 78.46%–85.47% homology between mouse and human.
In preliminary investigations, we next determined if these 45 conserved elements were associated with known structural variation of the human genome. Since ARS is rare, with a frequency of less than 1/100,000 in the population, we expect that any ARS-associated structural variations would also have low frequency. We therefore searched 1 megabase upstream of the FOXC1 gene within the NCBI dbVar database to identify human genomic structural variations larger than 50 bp from published studies (Figure 2). A total of 10 copy number variants (CNVs Table 1), reported with 1, 2, or 3 variant calls in dbVar, were found within the 1 megabase region upstream of FOXC1 that overlapped with any of the 45 conserved elements. Several other CNVs are known in the 1 megabase upstream region, however, these did not overlap with any of the conserved elements. CNVs that involve the FOXC1 coding region were excluded since these would be automatically considered pathogenic for an autosomal dominant disease such as ARS.
FIGURE 2.
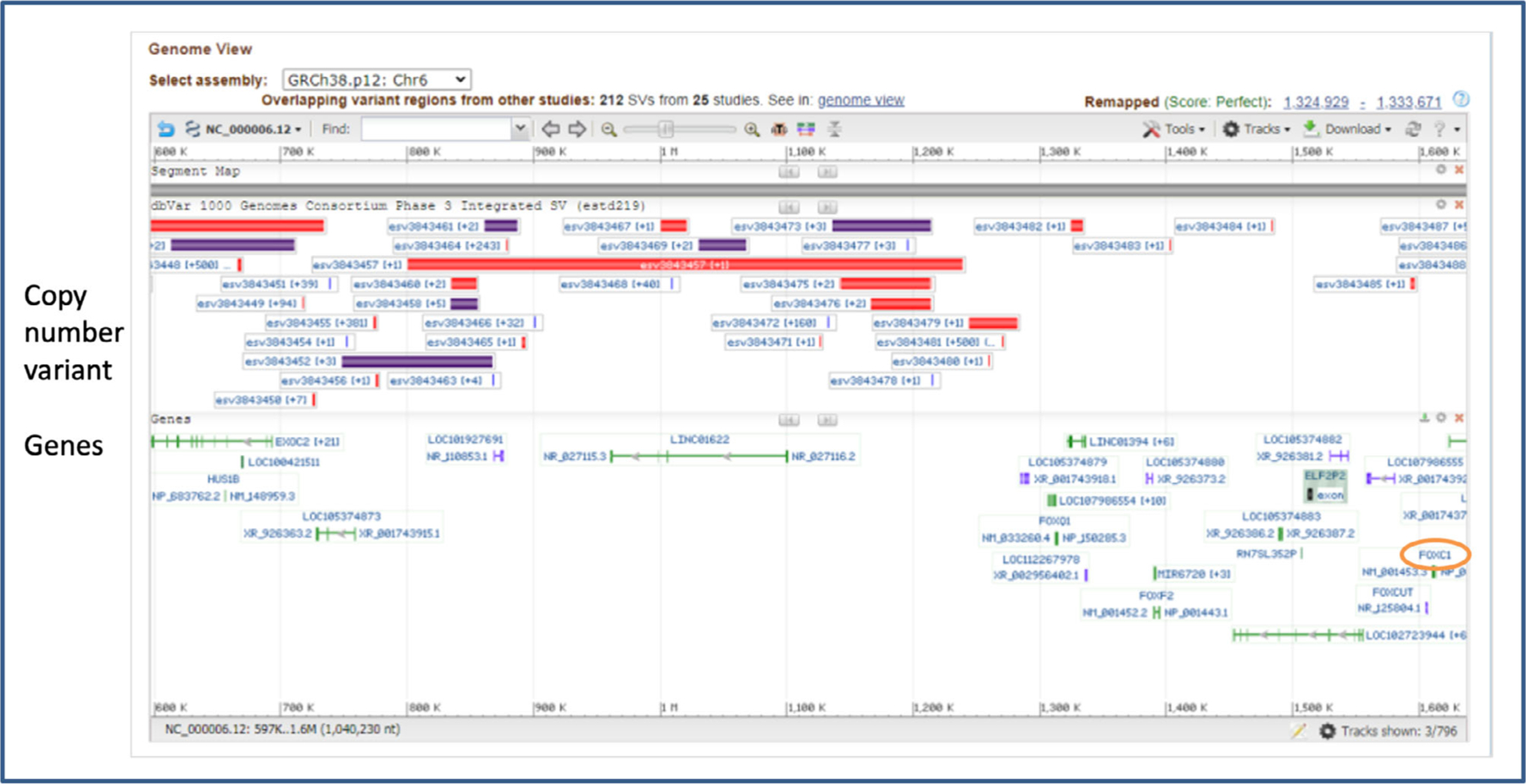
Identification of known structural CNVs in the 1 megabase region upstream of the FOXC1 gene. Figure is a screen capture of Sequence Viewer displaying CNVs reported in the NCBI dbVar database and the location of genes. The FOXC1 gene is circled in orange for orientation
TABLE 1.
List of the 10 copy number variants in the 1 megabase region upstream of FOXC1 that overlapped with any of the 45 conserved elements
| Human dbVar CNV accession | Variant calls in 1000 genome |
|---|---|
| esv3843457 | 1 |
| esv3843465 | 1 |
| esv3843467 | 1 |
| esv3843482 | 1 |
| esv3843461 | 2 |
| esv3843469 | 2 |
| esv3843475 | 2 |
| esv3843476 | 2 |
| esv3843452 | 3 |
| esv3843473 | 3 |
Note: Indicated to the right are the numbers of variant calls in dbVar as reported from the 1,000 Genome project
For illustration, a 200 kb region is shown as an example in Figure 3. Five of the conserved elements (numbers 22–26 of Supplementary Table 1) are located in this region upstream of FOXC1. These five conserved elements are known to reside within several previously reported CNVs. Rare CNVs, such as esv3843471, reported once in the dbVar database (Table 1) might be associated with ARS. In contrast, esv3843472 (which does not overlap with any conserved element), is much less likely to be associated with a rare disease such as ARS since it was reported with more than 160 variant calls in dbVar. This information is useful for evaluation of the possible pathogenicity of CNVs found in an ARS panel of patients involving these 45 conserved elements. For example, discovery of a CNV similar to esv3843472 in this patient panel would likely be excluded from further investigation. In contrast, conserved elements discovered to be involved in CNVs within the patient panel, but which are unknown or with few variant calls in dbVar, could be prioritized for further investigations.
FIGURE 3.
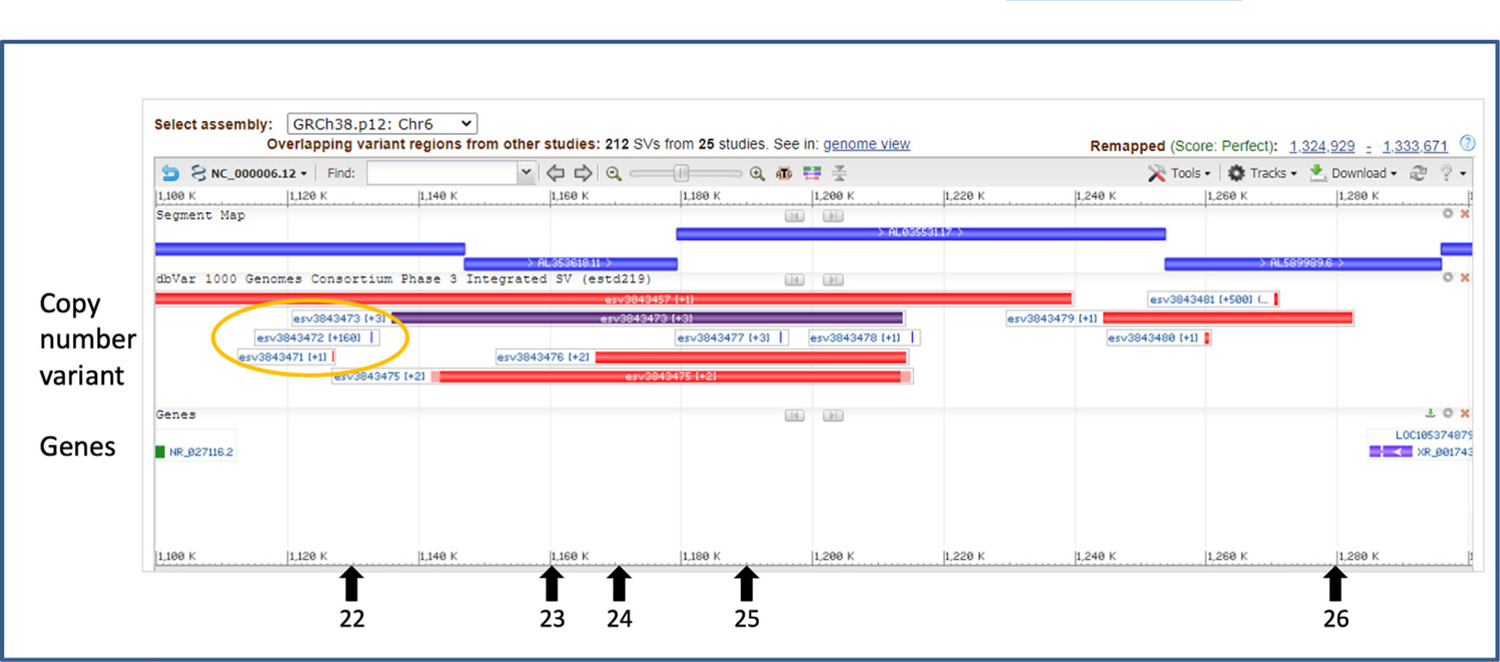
Known CNVs within a 200 kb region upstream of FOXC1 involving DNA sequences conserved between human and mouse. Figure is a screen capture of the output of Sequence Viewer showing CNVs reported in the dbVar database and location of genes. The locations of the five conserved elements are indicated below with black arrows. Three known CNVs that neighbor several of these conserved elements are circled in orange as examples. CNVs such as esv3843471, reported once in the dbVar database, might be associated with ARS. In contrast, esv3843472, reported more than 160 times, is much less likely to be associated with ARS
4.3 |. Cautionary note regarding the general applicability of these approaches
Identification and validation of regulatory elements, nevertheless, remains a challenge. While structural variants are more disruptive than single nucleotide variation, common sequencing approaches (e.g., short-read sequencing) fail to detect most larger deletions and insertions and nearly all inversions (Turner & Eichler, 2019). Understanding the impact of miRNA binding of such regulatory elements in eye development and disease is also important (Liu, Huang, Britton, & Chen, 2020). As well, not all gene regions are easily analyzed using the in silico methods described above, due to the presence of large amounts of repetitive DNA sequences, low complexity DNA sequences, and neighboring gene rich regions. For example, analysis of FOXC1 in the manner described by Volkmann and colleagues (Volkmann et al., 2011) did not result in the identification of noncoding, nontranscribed DNA sequences with high homology between humans and zebrafish (Rezaie and Walter, unpublished data). Thus, for some genes, brute force methods that analyze the consequence of expression of upstream regions, or the observation of deletions/duplications of regions not including the coding regions of genes, are still required at least currently.
4.4 |. Future directions
As our whole genome sequence databases become deeper, it will be possible to use additional new methods to detect regulatory elements. Comparisons of the distribution of mutations in noncoding regions between large numbers of people in the general population could allow identification of noncoding regions under evolutionary constraints, some of which could be key cis-acting regulatory regions. Improvements to the ability to predict transcription factor binding in the context of chromatin will also improve the detection of regulatory elements. Deeper eQTL and chromatin state data, from a substantially wider array of tissues and organisms, will also likely yield multiple new regulatory elements when combined with the data from the above methods. Nevertheless, validation of the functional role of these putative regulatory elements will continue to require in vitro and in vivo wet laboratory testing, at least for the foreseeable future. Even more importantly, we currently lack methods to combine the knowledge of rare coding and noncoding regulatory variants with environmental risk factors that together underlie complex polygenic traits. This ability will be essential to understand the basis of common disease.
Supplementary Material
ACKNOWLEDGMENT
Portions of this manuscript were supported by a grant to M.A.W. from the Canadian Glaucoma Research Society.
Funding information
Canadian Glaucoma Research Society
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interests.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Acharya M, Huang L, Fleisch VC, Allison WT, & Walter MA (2011). A complex regulatory network of transcription factors critical for ocular development and disease. Human Molecular Genetics, 20(8), 1610–1624. [DOI] [PubMed] [Google Scholar]
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, … Leppert M (1997). Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science, 277(5333), 1805–1807. [DOI] [PubMed] [Google Scholar]
- Arno G, Agrawal SA, Eblimit A, Bellingham J, Xu M, Wang F, … Chen R (2016). Mutations in REEP6 cause autosomal-recessive retinitis Pigmentosa. American Journal of Human Genetics, 99(6), 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba-Abbad R, Arno G, Carss K, Stirrups K, Penkett CJ, Moore AT, … Holder GE (2016). Mutations in CACNA2D4 cause distinctive retinal dysfunction in humans. Ophthalmology, 123(3), 668–671. e662. [DOI] [PubMed] [Google Scholar]
- Bauwens M, Garanto A, Sangermano R, Naessens S, Weisschuh N, De Zaeytijd J, … De Baere E (2019). ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genetics in Medicine, 21(8), 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, … Stone EM (2013). Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Human Molecular Genetics, 22(25), 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan JM, Fufa TD, Bharti K, Brooks BP, Hufnagel RB, & McGaughey DM (2018). Identifying core biological processes distinguishing human eye tissues with precise systems-level gene expression analyses and weighted correlation networks. Human Molecular Genetics, 27(19), 3325–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, … Raymond FL (2017). Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. American Journal of Human Genetics, 100(1), 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassini TA, Duncan L, Rives LC, Newman JH, Phillips JA, Koziura ME, … Undiagnosed Diseases Network. (2019). Whole genome sequencing reveals novel IGHMBP2 variant leading to unique cryptic splice-site and Charcot-Marie-tooth phenotype with early onset symptoms. Molecular Genetics & Genomic Medicine, 7(6), e00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, Asai-Coakwell M, Ye M, Mungall AJ, Barrow M, Dobyns WB, … Lehmann OJ (2008). A novel mechanistic spectrum underlies glaucoma-associated chromosome 6p25 copy number variation. Human Molecular Genetics, 17(22), 3446–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri JL, Boespflug-Tanguy O, … Calvas P (2014). Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clinical Genetics, 86(4), 326–334. [DOI] [PubMed] [Google Scholar]
- Cheng J, Nguyen TYD, Cygan KJ, Celik MH, Fairbrother WG, Avsec Z, & Gagneur J (2019). MMSplice: Modular modeling improves the predictions of genetic variant effects on splicing. Genome Biology, 20(1), 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrystal PW, & Walter MA (2019). Aniridia and Axenfeld-Rieger syndrome: Clinical presentations, molecular genetics and current/emerging therapies. Experimental Eye Research, 189, 107815. [DOI] [PubMed] [Google Scholar]
- Cipriani V, Silva RS, Arno G, Pontikos N, Kalhoro A, Valeina S, … Moore AT (2017). Duplication events downstream of IRX1 cause North Carolina macular dystrophy at the MCDR3 locus. Scientific Reports, 7(1), 7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Haene B, Meire F, Claerhout I, Kroes HY, Plomp A, Arens YH, … De Baere E (2011). Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Investigative Ophthalmology & Visual Science, 52(1), 324–333. [DOI] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, … Cremers FP (2006). Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. American Journal of Human Genetics, 79(3), 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, … Black GC (2016). Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology, 123(5), 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford JM, Campbell C, Barton S, Bhaskar S, Gupta S, Taylor RL, … Black GC (2017). Validation of copy number variation analysis for next-generation sequencing diagnostics. European Journal of Human Genetics, 25(6), 719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flomen RH, Gorman PA, Vatcheva R, Groet J, Barisic I, Ligutic I, … Nizetic D (1997). Rieger syndrome locus: A new reciprocal translocation t(4;12)(q25;q15) and a deletion del(4)(q25q27) both break between markers D4S2945 and D4S193. Journal of Medical Genetics, 34(3), 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flomen RH, Vatcheva R, Gorman PA, Baptista PR, Groet J, Barisic I, … Nizetic D (1998). Construction and analysis of a sequence-ready map in 4q25: Rieger syndrome can be caused by haploinsufficiency of RIEG, but also by chromosome breaks approximately 90 kb upstream of this gene. Genomics, 47(3), 409–413. [DOI] [PubMed] [Google Scholar]
- Footz T, Idrees F, Acharya M, Kozlowski K, & Walter MA (2009). Analysis of mutations of the PITX2 transcription factor found in patients with Axenfeld-Rieger syndrome. Investigative Ophthalmology & Visual Science, 50(6), 2599–2606. [DOI] [PubMed] [Google Scholar]
- Gonorazky H, Liang M, Cummings B, Lek M, Micallef J, Hawkins C, … Dowling JJ (2016). RNAseq analysis for the diagnosis of muscular dystrophy. Annals of Clinical Translational Neurology, 3(1), 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould DB, Mears AJ, Pearce WG, & Walter MA (1997). Autosomal dominant Axenfeld-Rieger anomaly maps to 6p25. American Journal of Human Genetics, 61(3), 765–768. [PMC free article] [PubMed] [Google Scholar]
- Huang L, Chi J, Berry FB, Footz TK, Sharp MW, & Walter MA (2008). Human p32 is a novel FOXC1-interacting protein that regulates FOXC1 transcriptional activity in ocular cells. Investigative Ophthalmology & Visual Science, 49(12), 5243–5249. [DOI] [PubMed] [Google Scholar]
- Jagadeesh KA, Paggi JM, Ye JS, Stenson PD, Cooper DN, Bernstein JA, & Bejerano G (2019). S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. Nature Genetics, 51(4), 755–763. [DOI] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, … Farh KK (2019). Predicting splicing from primary sequence with deep learning. Cell, 176(3), 535–548 e524. [DOI] [PubMed] [Google Scholar]
- Khan AO, Becirovic E, Betz C, Neuhaus C, Altmuller J, Maria Riedmayr L, … Bolz HJ (2017). A deep intronic CLRN1 (USH3A) founder mutation generates an aberrant exon and underlies severe usher syndrome on the Arabian peninsula. Scientific Reports, 7(1), 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Cornelis SS, Pozo-Valero MD, Whelan L, Runhart EH, Mishra K, … Cremers FPM (2020). Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genetics in Medicine, 22(7), 1235–1246. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, … Hoh J (2005). Complement factor H polymorphism in age-related macular degeneration. Science, 308(5720), 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski K, & Walter MA (2000). Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Human Molecular Genetics, 9(14), 2131–2139. [DOI] [PubMed] [Google Scholar]
- Lee M, Roos P, Sharma N, Atalar M, Evans TA, Pellicore MJ, … Cutting GR (2017). Systematic computational identification of variants that activate Exonic and Intronic cryptic splice sites. American Journal of Human Genetics, 100(5), 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann OJ, Ebenezer ND, Jordan T, Fox M, Ocaka L, Payne A, … Bhattacharya SS (2000). Chromosomal duplication involving the forkhead transcription factor gene FOXC1 causes iris hypoplasia and glaucoma. American Journal of Human Genetics, 67(5), 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines MA, Kozlowski K, Kulak SC, Allingham RR, Heon E, Ritch R, … Walter MA (2004). Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Investigative Ophthalmology & Visual Science, 45(3), 828–833. [DOI] [PubMed] [Google Scholar]
- Liu CH, Huang S, Britton WR, & Chen J (2020). MicroRNAs in vascular eye Diseases. International Journal of Molecular Sciences, 21(2), 649. 10.3390/ijms21020649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, & Ferreira PA (2005). Identification of novel murine- and human-specific RPGRIP1 splice variants with distinct expression profiles and subcellular localization. Investigative Ophthalmology & Visual Science, 46(6), 1882–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantere T, Kersten S, & Hoischen A (2019). Long-read sequencing emerging in medical genetics. Frontiers in Genetics, 10, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchuk DS, Crooks K, Strande N, Kaiser-Rogers K, Milko LV, Brandt A, … Berg JS (2018). Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One, 13(12), e0209185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer AK, Rohrschneider K, Strom TM, Glockle N, Kohl S, Wissinger B, & Weisschuh N (2016). Homozygosity mapping and whole-genome sequencing reveals a deep intronic PROM1 mutation causing cone-rod dystrophy by pseudoexon activation. European Journal of Human Genetics, 24(3), 459–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer VW, & Aguilera A (1990). High levels of chromosome instability in polyploids of Saccharomyces cerevisiae. Mutation Research, 231(2), 177–186. [DOI] [PubMed] [Google Scholar]
- Mears AJ, Mirzayans F, Gould DB, Pearce WG, & Walter MA (1996). Autosomal dominant iridogoniodysgenesis anomaly maps to 6p25. American Journal of Human Genetics, 59(6), 1321–1327. [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, … Walter MA (1998). Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. American Journal of Human Genetics, 63(5), 1316–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheal S, Khan MI, Islam F, Akhtar F, Qamar R, Tassignon MJ, … den Hollander AI (2016). Identification of mutations in the PRDM5 gene in brittle cornea syndrome. Cornea, 35(6), 853–859. [DOI] [PubMed] [Google Scholar]
- Mirzayans F, Gould DB, Heon E, Billingsley GD, Cheung JC, Mears AJ, & Walter MA (2000). Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. European Journal of Human Genetics, 8(1), 71–74. [DOI] [PubMed] [Google Scholar]
- Murphy TC, Saleem RA, Footz T, Ritch R, McGillivray B, & Walter MA (2004). The wing 2 region of the FOXC1 forkhead domain is necessary for normal DNA-binding and transactivation functions. Investigative Ophthalmology & Visual Science, 45(8), 2531–2538. [DOI] [PubMed] [Google Scholar]
- Neidhardt J, Glaus E, Barthelmes D, Zeitz C, Fleischhauer J, & Berger W (2007). Identification and characterization of a novel RPGR isoform in human retina. Human Mutation, 28(8), 797–807. [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, … Sheffield VC (1998). The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nature Genetics, 19(2), 140–147. [DOI] [PubMed] [Google Scholar]
- Patel A, Hayward JD, Tailor V, Nyanhete R, Ahlfors H, Gabriel C, … Sowden JC (2019). The Oculome panel test: Next-generation sequencing to diagnose a diverse range of genetic developmental eye disorders. Ophthalmology, 126(6), 888–907. [DOI] [PubMed] [Google Scholar]
- Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, … Nejentsev S (2012). A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics, 28(21), 2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priston M, Kozlowski K, Gill D, Letwin K, Buys Y, Levin AV, … Heon E (2001). Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Human Molecular Genetics, 10(16), 1631–1638. [DOI] [PubMed] [Google Scholar]
- Protas ME, Weh E, Footz T, Kasberger J, Baraban SC, Levin AV, … Gould DB (2017). Mutations of conserved non-coding elements of PITX2 in patients with ocular dysgenesis and developmental glaucoma. Human Molecular Genetics, 26(18), 3630–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Murrell JR, Luo M, & Conlin LK (2020). A highly sensitive and specific workflow for detecting rare copy-number variants from exome sequencing data. Genome Medicine, 12(1), 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnapriya R, Sosina OA, Starostik MR, Kwicklis M, Kapphahn RJ, Fritsche LG, … Swaroop A (2019). Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nature Genetics, 51(4), 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riise R, D’Haene B, De Baere E, Gronskov K, & Brondum-Nielsen K (2009). Rieger syndrome is not associated with PAX6 deletion: A correction to Acta Ophthalmol Scand 2001: 79: 201–203. Acta Ophthalmologica, 87, 923. [DOI] [PubMed] [Google Scholar]
- Riise R, Storhaug K, & Brondum-Nielsen K (2001). Rieger syndrome is associated with PAX6 deletion. Acta Ophthalmologica Scandinavica, 79(2), 201–203. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, & Mesirov JP (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi I, Toro R, Kuburas A, Semina E, Murray JC, & Russo AF (2006). An unusual class of PITX2 mutations in Axenfeld-Rieger syndrome. Birth Defects Research. Part A, Clinical and Molecular Teratology, 76(3), 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, & Walter MA (2001). Analyses of the effects that disease-causing missense mutations have on the structure and function of the winged-helix protein FOXC1. American Journal of Human Genetics, 68(3), 627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, & Walter MA (2003a). Structural and functional analyses of disease-causing missense mutations in the forkhead domain of FOXC1. Human Molecular Genetics, 12(22), 2993–3005. [DOI] [PubMed] [Google Scholar]
- Saleem RA, Banerjee-Basu S, Murphy TC, Baxevanis A, & Walter MA (2004). Essential structural and functional determinants within the forkhead domain of FOXC1. Nucleic Acids Research, 32(14), 4182–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem RA, Murphy TC, Liebmann JM, & Walter MA (2003b). Identification and analysis of a novel mutation in the FOXC1 forkhead domain. Investigative Ophthalmology & Visual Science, 44(11), 4608–4612. [DOI] [PubMed] [Google Scholar]
- Sanchis-Juan A, Stephens J, French CE, Gleadall N, Megy K, Penkett C, … Carss KJ (2018). Complex structural variants in Mendelian disorders: Identification and breakpoint resolution using short- and long-read genome sequencing. Genome Medicine, 10(1), 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, … Cremers FPM (2019). Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genetics in Medicine, 21(8), 1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifi M, & Walter MA (2018). Axenfeld-Rieger syndrome. Clinical Genetics, 93(6), 1123–1130. [DOI] [PubMed] [Google Scholar]
- Semina EV, Datson NA, Leysens NJ, Zabel BU, Carey JC, Bell GI, … Murray JC (1996a). Exclusion of epidermal growth factor and high-resolution physical mapping across the Rieger syndrome locus. American Journal of Human Genetics, 59(6), 1288–1296. [PMC free article] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, … Murray JC (1996b). Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nature Genetics, 14(4), 392–399. [DOI] [PubMed] [Google Scholar]
- Sibon I, Coupry I, Menegon P, Bouchet JP, Gorry P, Burgelin I, … Goizet C (2007). COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Annals of Neurology, 62(2), 177–184. [DOI] [PubMed] [Google Scholar]
- Strungaru MH, Dinu I, & Walter MA (2007). Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Investigative Ophthalmology & Visual Science, 48(1), 228–237. [DOI] [PubMed] [Google Scholar]
- Strungaru MH, Footz T, Liu Y, Berry FB, Belleau P, Semina EV, … Walter MA (2011). PITX2 is involved in stress response in cultured human trabecular meshwork cells through regulation of SLC13A3. Investigative Ophthalmology & Visual Science, 52(10), 7625–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, & Mesirov JP (2013). Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull C, Scott RH, Thomas E, Jones L, Murugaesu N, Pretty FB, … Genomes P (2018). The 100 000 Genomes project: Bringing whole genome sequencing to the NHS. BMJ, 361, k1687. [DOI] [PubMed] [Google Scholar]
- Turner TN, & Eichler EE (2019). The role of De novo noncoding regulatory mutations in neurodevelopmental disorders. Trends in Neurosciences, 42(2), 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro E, Astle WJ, Megy K, Graf S, Greene D, Shamardina O, … Ouwehand WH (2020). Whole-genome sequencing of patients with rare diseases in a national health system. Nature, 583(7814), 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vache C, Puechberty J, Faugere V, Darmaisin F, Liquori A, Baux D, … Roux AF (2020). A 4.6 Mb inversion leading to PCDH15-LINC00844 and BICC1-PCDH15 fusion transcripts as a new pathogenic mechanism implicated in usher syndrome type 1. Front Genet, 11, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk JA, van de Pol DJ, Wissinger B, van Driel MA, Hoefsloot LH, de Wijs IJ, … Cremers FP (2003). Novel types of mutation in the choroideremia ( CHM) gene: A full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics, 113(3), 268–275. [DOI] [PubMed] [Google Scholar]
- Verdura E, Schluter A, Fernandez-Eulate G, Ramos-Martin R, Zulaica M, Planas-Serra L, … Pujol A (2020). A deep intronic splice variant advises reexamination of presumably dominant SPG7 cases. Annals of Clinical Translational Neurology, 7(1), 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann BA, Zinkevich NS, Mustonen A, Schilter KF, Bosenko DV, Reis LM, … Semina EV (2011). Potential novel mechanism for Axenfeld-Rieger syndrome: Deletion of a distant region containing regulatory elements of PITX2. Investigative Ophthalmology & Visual Science, 52(3), 1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MA (2003). PITs and FOXes in ocular genetics: The Cogan lecture. Investigative Ophthalmology & Visual Science, 44(4), 1402–1405. [DOI] [PubMed] [Google Scholar]
- Walter MA, Mirzayans F, Mears AJ, Hickey K, & Pearce WG (1996). Autosomal-dominant iridogoniodysgenesis and Axenfeld-Rieger syndrome are genetically distinct. Ophthalmology, 103(11), 1907–1915. [DOI] [PubMed] [Google Scholar]
- Weisschuh N, Sturm M, Baumann B, Audo I, Ayuso C, Bocquet B, … Kohl S (2020). Deep-intronic variants in CNGB3 cause achromatopsia by pseudoexon activation. Human Mutation, 41(1), 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, … Frey BJ (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science, 347 (6218), 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Xie YA, Ayuso C, Riveiro-Alvarez R, Lopez-Martinez MA, Simonelli F, … Allikmets R (2014). Analysis of the ABCA4 genomic locus in Stargardt disease. Human Molecular Genetics, 23(25), 6797–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.