

Abstract
Background/Aims:
Structural and functional alterations in mitochondria, particularly, the inner mitochondrial membrane (IMM) plays a critical role in mitochondria-mediated cell death in response to cardiac ischemia-reperfusion (IR) injury. The integrity of IMM can be affected by two potential intra-mitochondrial factors: i) mitochondrial matrix swelling, and ii) proteolytic cleavage of the long optic atrophy type 1 (L-OPA1), an IMM-localized dynamin-like GTPase engaged in the regulation of structural organization and integrity of the mitochondrial cristae. However, the relationship between these two factors in response to oxidative stress remains unclear. Here, we elucidated the effects of cardiac IR injury on L-OPA1 cleavage and OMA1 activity.
Methods:
Langendorff-mode perfused isolated rat hearts were subjected to 25-min of global ischemia followed by 90-min reperfusion in the presence or absence of XJB-5-131 (XJB, a mitochondria-targeting ROS scavenger) and sanglifehrin A (SfA, a permeability transition pore inhibitor).
Results:
XJB in combination with SfA increased post-ischemic recovery of cardiac function and reduced mitochondrial ROS production at 30- and 60-min reperfusion and affected mitochondrial swelling. L-OPA1 levels were reduced in IR hearts; however, neither XJB, SfA, and their combination prevented IR-induced reduction of L-OPA1 cleavage. Likewise, IR increased the OMA1 enzymatic activity, which remained unchanged in the presence of XJB and/or SfA.
Conclusion:
IR-induced cardiac and mitochondrial dysfunctions are associated with OMA1 activation and L-OPA1 cleavage. However, XJB, SfA, and their combination do not prevent these changes despite improved heart and mitochondria function, thus, suggesting that different mechanisms can be implicated in L-OPA1 processing in response to cardiac IR injury.
Keywords: Cardiac ischemia-reperfusion, Mitochondria, Optic atrophy type 1 protein, Reactive oxygen species, Mitochondrial swelling, Permeability transition pores
Introduction
Mitochondria play a crucial role in cellular energy metabolism, particularly, in high energy demanding tissues such as the heart, where it occupies nearly 30% of cell volume. Coronary heart diseases, including myocardial infarction, is the leading cause of mortality and morbidity worldwide [1]. The clinical standard of care in patients with myocardial infarction includes a set of reperfusion strategies that indeed induces additional damage to the heart known as “reperfusion injury” [2]. Mitochondrial dysfunction plays a central role in the pathogenesis of cardiac ischemia-reperfusion (IR) injury when severe oxidative stress accompanied by Ca2+ overload induces the formation of the mitochondrial permeability transition pores (mPTP) in the inner mitochondrial membrane (IMM). mPTP opening causes depolarization of the IMM that, in turn, impairs oxidative phosphorylation and further enhances mitochondrial ROS (mtROS) production [3, 4]. Hence, the structural and functional integrity of the IMM changes in response to mPTP opening, which promotes mitochondrial matrix swelling as a result of increased colloid-osmotic pressure. Therefore, regulation of mitochondrial volume is essential for the maintenance of the structural and functional integrity of the IMM [5].
Mitochondria are considered highly dynamic organelles that are constantly changing their shape to mediate the formation of continuous networks in response to cellular demands for ATP [6]. They undergo structural modifications to repair or eliminate dysfunctional components through fission-fusion and mitophagy and, together with mitochondrial biogenesis, to maintain the mitochondrial quality control [7]. Mitochondrial fission involves the translocation of dynamin-related protein 1 from the cytoplasm to mitochondria, where it interacts with mitochondrial fission 1 protein at the fission site, which oligomerizes and tightens around the mitochondria. This leads to constriction of the outer mitochondrial membrane and fission of the mitochondrion into two independent organelles, followed by selective elimination of the damaged mitochondria through mitophagy [8-10]. Mitochondrial fusion is responsible for the merging of two mitochondria to produce one healthy mitochondrion through the elimination of dysfunctional components including lipids, proteins, and DNA [11, 12]. The optic atrophy 1 (OPA1) is localized in the IMM, and, in addition to mitochondrial fusion, it also participates in cristae morphogenesis, oxidative phosphorylation, and mitochondrial DNA stability [13]. OPA1 mutations cause type 1 optic atrophy, a dominantly inherited optic neuropathy that is associated with the advance loss in visual perception and blindness [14]. OPA1 deficiency leads to alterations of cristae structure and fusion impairment in cells [15, 16]. Moreover, a global loss of OPA1 decreases cell proliferation, causes fragmentation of mitochondria, and reduces mitochondrial membrane potential (ΔΨm) and respiratory capacity [17, 18]. Studies on mutant mice with a 50% reduction of OPA1 showed alterations in mitochondrial morphology as well as cristae indicating the importance of OPA1 for the regulation of mitochondrial structure and function [15]. However, the mechanisms underlying the OPA1-mediated regulation of mitochondrial structural organization and function remain to be elucidated.
OPA1 exists in five different isoforms in the heart mitochondria. Two long-OPA1 (L-OPA1) isoforms are further cleaved into three soluble short-OPA1 (S-OPA1); the mixture of both long and short forms facilitates mitochondrial fusion. In humans, L-OPA1 contains two sites of cleavage, the S1 site, which is cleaved by the protease OMA1, and the S2 site, which is cleaved by YME1L [19]. Under physiological conditions, equimolar stability between L-OPA1 and S-OPA1 is achieved, given the activity of the proteases. However, the balance is affected in response to pathological stimuli leading to dysfunction of mitochondria [20, 21]. OMA1 has been shown to undergo autocatalysis to produce an unstable short-OMA1 (S-OMA1) in response to stress stimuli. The S-OMA1 is further stabilized by membrane depolarization and stimulates proteolytic cleavage of L-OPA1. In vitro studies revealed that an increase in mtROS production can act as a signal for the OMA1 activation upon YME1L depletion [19]. The mechanism by which OMA1 is activated remains to be clarified; recent identification of a stress-sensing domain might serve as a clue to unravel the modulation of mitochondrial dynamics through OPA1 processing [13].
In this study, we elucidated an associative link between mitochondrial swelling (mPTP opening) and proteolytic cleavage of L-OPA1 in response to cardiac IR injury. Since IR-induced oxidative stress can provoke both mPTP induction (mitochondrial swelling) and OMA1 activation (L-OPA1 cleavage), we applied XJB-5-131 (XJB, a mitochondria-targeting ROS scavenger) and sanglifehrin A (SfA, a PTP inhibitor) to elucidate the contribution of mtROS and mPTP to the proteolytic cleavage of L-OPA1. We have previously shown that these compounds demonstrate protective effects against cardiac IR injury [22, 23]. We suggested that XJB and SfA could exert a synergistic effect via simultaneous inhibition of mtROS accumulation and mPTP-induced mitochondrial swelling that would attenuate OMA1 activation and L-OPA1 cleavage during cardiac IR injury. Our results demonstrated that cardiac and mitochondrial dysfunctions induced by IR injury correlate with the degradation of L-OPA1 and activation of OMA1. However, the beneficial effects of XJB and SfA are not associated with inhibition of OMA1 activation and L-OPA1 cleavage. Likely, in addition to mitochondrial swelling and mtROS, other mechanisms are involved in L-OPA1 cleavage during cardiac IR injury.
Materials and Methods
Animals
Adult Sprague Dawley male rats (275–325 g) were purchased from Taconic (Hillside, NJ, USA). Animals were housed in individual cages in a temperature-controlled room under a regular light-dark cycle. Water and food were provided ad libitum. All experiments were performed according to protocols approved by the UPR Medical Sciences Campus and Institutional Animal Care and Use Committee (IACUC) and conformed to the National Research Council Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (2011, eighth edition). All chemicals were handled with the appropriate personal protective equipment in accordance with the regulations established by the Biosafety and Biosecurity Committee of the UPR Medical Sciences Campus.
Langendorff-mode heart perfusion
Hearts were isolated and perfused according to the Langendorff-mode technique as described previously [24]. Briefly, rats were anesthetized with the anesthetic cocktail (in mg/kg body weight: 4.2 xylazine, 87.5 ketamine, and 87.5 acepromazine) administered intraperitoneally. The heart was rapidly excised, connected to the Langendorff-perfusion setup, and perfused at a constant flow (10-12 mL/min per gram heart weight) with Krebs-Henseleit solution (KHS) containing (in mM): 1.2 KH2PO4, 1.2 MgSO4, 2.0 CaCl2, 4.8 KCl, 118 NaCl, 25 NaHCO3, and 11 glucose. The buffer was equilibrated at 95% O2 and 5% CO2, pH 7.4, at 37°C. All hearts were perfused for 30-min (equilibration period), followed by 25-min of ischemia and 90-min of reperfusion. In certain groups, the hearts were perfused with KHS containing the mitochondria-targeting electron and ROS scavenger, XJB, and the mPTP inhibitor, SfA, that were administered 10-min before ischemia and kept during the entire reperfusion period. The studies were performed in the following groups (see Fig. 1): i) control group (no IR), hearts were perfused with KHS supplemented with DMSO; ii) IR group, hearts were subjected to IR and perfused with KHS supplemented with DMSO; iii) IRX group, hearts were subjected to IR in the presence of XJB (0.2 μM); iv) IRS group, hearts were subjected to IR in the presence of SfA (0.5 μM); and v) IRXS group, hearts were subjected to IR in the presence of XJB (0.2 μM) and SfA (0.5 μM). XJB and SfA were dissolved in DMSO. SfA was generously provided by Novartis Pharma AG (Basel, Switzerland). XJB was received from our collaborator Dr. Peter Wipf (University of Pittsburgh, PA).
Fig. 1.
Design of experiments. Groups: i) control, non-ischemic hearts perfused continuously for 145-min (n=7); ii) IR, hearts subjected to 25-min ischemia followed by 90-min reperfusion (n=6); iii) IRX, hearts subjected to 25-min ischemia followed by 90-min reperfusion in the presence of 0.2 μM XJB (n=6); iv) IRS, hearts subjected to 25-min ischemia followed by 90-min reperfusion in the presence of 0.5 μM SfA (n=7); and v) IRXS, hearts subjected to 25-min ischemia followed by 90-min reperfusion in the presence of XJB and SfA (n=7). Details are given in Materials and Methods.
To examine cardiac function, a water-filled balloon inserted to the left ventricle (LV) was connected to a pressure transducer. Functional cardiac parameters, including LV systolic pressure (LVSP), end-diastolic pressure (LVEDP), heart rate (HR), the rate of contraction and relaxation (± dP/dt), and aortic pressure (AP) were continuously monitored using the Labscribe2 Data Acquisition Software (iWorx 308T, Dover, NH, USA). LV developed pressure (LVDP) was calculated as the difference between LVSP and LVEDP (LVDP = LVSP - LVEDP). Post-ischemic recovery of LVDP was calculated as a percentage of pre-ischemic values. The rate pressure product (RPP) was calculated as the product of LVDP and HR (RPP = LVDP × HR) to estimate cardiac work.
Isolation of mitochondria
Mitochondria were isolated according to the protocol described previously [23]. Briefly, at the end of reperfusion, the hearts were rapidly removed, and ventricles were homogenized with a Polytron homogenizer at 1,500 rpm for 5-sec in ice-cold sucrose buffer containing (in mM): 300 sucrose, 10 Tris-HCl, and 2 EGTA, at pH 7.4. Mitochondria were isolated from the homogenate by centrifugation at 2,000 × g for 3-min in sucrose buffer supplemented with 0.5% BSA, using a benchtop centrifuge to remove cell debris. Then, the supernatant was centrifuged at 10,000 × g for 6-min to sediment the mitochondrial suspension and washed again under the same conditions in sucrose buffer (BSA-free). The final pellet containing mitochondria was resuspended in the sucrose buffer. Inhibitors cocktail were added accordingly to each mitochondrial sample right after finishing isolation.
mPTP opening
The swelling of mitochondria as an indicator of mPTP opening in the presence or absence of Ca2+ was determined freshly isolated mitochondria (50 μg) by monitoring the decrease in light scattering at 525 nm as previously described, with minor modifications [23]. The swelling buffer contained (in mM): 125 KCl, 20 Tris-base, 2 KH2PO4, 1 MgCl2, 1 EGTA, 5 α-ketoglutarate, 5 L-malate, pH 7.1. Mitochondrial swelling was measured at 37°C using the SpectraMax Microplate Reader (Molecular Devices) by adding Ca2+ to a total (accumulative) concentration of 100, 200, 300, and 800 μM with 5-min intervals. The rates of swelling were presented as an absorbance value (A525) per minute (ΔA525·min−1·mg−1).
Mitochondrial ROS production
To estimate mtROS production, H2O2 levels were measured in freshly isolated mitochondria (50 μg) at 37°C by the SpectraMax Microplate Reader (Molecular Devices). The mitochondria were incubated at 37°C in incubation buffer (in mM): 125 KCl, 20 Tris-base, 2 KH2PO4, 1 MgCl2, 1 EGTA, 5 α-ketoglutarate, 5 L-malate, pH 7.1. Production of H2O2 was measured in the medium containing 0.1 mM Ampliflu™ Red (Sigma), 50 mM sodium phosphate pH 7.4, 0.2 U/mL HRP. External Ca2+ was added with 5-min intervals to increase a total (accumulative) concentration of the ion in the incubation medium up to 100, 200, 300, and 800 μM. The fluorescence intensity of Ampliflu™ Red was determined at the excitation/emission of 560 nm/590 nm. Results were expressed as pmol of H2O2 per min in 1 mg of protein.
Mitochondrial respiration rates
Measurement of mitochondrial respiration was performed at 37°C using a YSI Oxygraph (Yellow Springs OH, USA) model 5300 equipped with a Clark-type oxygen electrode. Oxygen consumption rates were recorded and analyzed using Chart5 (Powerlab, Colorado Springs, CO, USA). Mitochondria were suspended in a buffer containing (in mM): 125 KCl, 20 MOPS, 10 Tris-HCl, 0.5 EGTA, and 2 KH2PO4, pH 7.2, supplemented with the following substrates to measure complex I: 2.5 mM α-ketoglutarate and 1 mM L-malate. Respiration rates were measured in the absence (state 2) and presence (state 3) of 1 mM ADP. The O2 consumption was calculated in the absence (state 2) or presence of ADP (state 3), and the respiratory control index (RCI) by Lardy was calculated as the ratio of state 3 to state 2.
SDS-PAGE and Western blotting
Equal amounts of mitochondrial proteins were resolved by SDS-PAGE and transferred overnight to nitrocellulose membranes (GE Healthcare Bio-Sciences). The membranes were immunoblotted with OPA1 (BD Biosciences, #612607) and OMA1 (Santa Cruz, # sc-515788). Images were acquired using the Odyssey CLx Infrared Imaging System (LI-COR Biosciences). Image analysis was performed using the Image Studios Lite (Version 5.2.5) from LI-COR. L-OPA1 and S-OPA1 levels were quantified as a percentage of total OPA1, whereas OMA1 levels were normalized to VDAC or porin, a mitochondrial housekeeping protein (Cell Signaling Technology, #46615).
OMA1 activity assay
OMA1 activity assay was performed by the technique developed recently [25]. The assay utilizes an OPA1 fluorogenic reporter substrate that contains a fluorogenic MCA moiety on the N-terminus and a DNP quencher moiety on the C-terminus (LifeTein LLC). Briefly, 5 μg of each mitochondrial sample was resolved in the OMA1 activity assay buffer, containing (in mM): 50 Tris-HCl and 40 KCl, pH 7.5. Reaction medium contained 200 μM N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (the zinc chelator) and 5 μM OPA1 fluorogenic reporter substrate. The assay was run with a final volume of 100 μL using black opaque 96 well plates (Costar). Relative fluorescence was recorded at 37°C in 5-min intervals for 30-min, using a fluorescent plate reader (SpectraMax M2e, Molecular Devices equipped with SoftMax Pro v5 software) with excitation/emission of 320/405 nm. OMA1 activation was proportional to the fluorescence released as a result of cleavage of the OPA1 fluorogenic reporter substrate by OMA1. Results are presented in relative fluorescence units.
Statistical analysis
Data were analyzed using one-way, two-way, and repeated measures ANOVA, as well as a t-student test for comparison of independent treatments using Graphpad prism. Data are presented as mean ± SE. P<0.05 was considered statistically significant. The number of biological samples but not technical replicates was used as a sample size.
Results
Cardiac function
First, we examined the effects of XJB and SfA, either alone or in combination, on the post-ischemic recovery of cardiac function (Fig. 2). Untreated hearts subjected to IR injury (IR group) showed a 48% (P<0.05) less LVDP at the end of 90-min reperfusion. Comparative analysis of the post-ischemic recovery of cardiac function from the present and previous studies [23] demonstrates that LVDP values after a 60-min and 90-min reperfusion were significantly different (~25% for 60-min vs. ~45% for 90-min reperfusion) in untreated IR hearts. These data indicate that post-ischemic recovery of cardiac function increases with the extension of the reperfusion. Notably, sustained reperfusion decreased cardiac function over time (after 60-min of reperfusion) in the presence of XJB and/or SfA. Therefore, no significant differences were observed in cardiac recovery (LVDP and RPP) between the treatment and non-treatment groups at the end of reperfusion. However, the combination of XJB and SfA (IRXS group) increased post-ischemic recovery of LVDP by 26% and 22% (P<0.05 for both) more than IR at 30- and 60-min reperfusion (Fig. 2A). Results from XJB-treated hearts (IRX group) showed a 17% (P<0.05) and 18% post-ischemic recovery of LVDP more than IR at 20- and 60-min reperfusion, respectively. However, no differences were seen between IR+SfA and IR groups. SfA decreased HR during 20- to 50-min of reperfusion (P<0.05) (Fig. 2B) that could contribute to the post-ischemic recovery of cardiac function in this group. Analysis of RPP (Fig. 2C), which represents cardiac work, revealed a 60% reduction in the IR group compared to the control hearts (P<0.05). Also, XJB alone (IRX group), and in combination with SfA (IRXS group), had a similar trend, being increased at the beginning of reperfusion at 20-min, and 20- to 50-min, respectively. It should be noted the extent of post-ischemic recovery of the hearts in the presence of SfA or XJB at the end of 90-min reperfusion was similar to that shown previously for 60- min reperfusion [23]. Thus, these data show that the combination of XJB and SfA exerted the best improvement in post-ischemic recovery of cardiac function.
Fig. 2.

Cardiac function. A, Left ventricular development pressure (LVDP). B, Heart rate (HR). C, Rate pressure product (RPP). LVDP is the difference between left ventricular systolic pressure and left ventricular diastolic pressure, and it is presented as a percentage of pre-ischemic values. RPP was calculated as the product of LVDP and HR (RPP=LVDP*HR), and it is presented as mmHg*bpm/1000. Groups: Con, control (black circle); IR, ischemia-reperfusion (red square); IRX, IR+XJB (orange triangle); IRS, IR+SfA (blue diamond); IRXS, IR+XJB+SfA (green hexagon). *P<0.05, **P<0.01 vs. Con; &P<0.05 vs. IRX; +P<0.05, ++P<0.01 vs. IRS; #P<0.05, ##P<0.01 vs. IRXS. n= 6-7 per each group.
Mitochondrial respiration, ROS production, and mPTP opening
Mitochondrial respiration rates for complex I were determined in mitochondria isolated from untreated and treated hearts (Fig. 3A-C). Cardiac IR had no significant effect on state 2 (Fig. 3A) whereas it reduced state 3 (Fig. 3B) respiration rate by 49% (P<0.01) compared to control hearts. Hearts treated with either XJB or SfA showed no differences in state 3, whereas hearts treated with XJB and SfA simultaneously demonstrated a 50% (P<0.05) more state 3 compared to untreated IR hearts. Additionally, the RCI of mitochondria (Fig. 3C) was reduced by 37% (P<0.001) by the end of reperfusion (IR group) compared to control, with no differences between treatment and non-treatment groups.
Fig. 3.

Mitochondrial respiration and mtROS production. State 2 (A) and State 3 (B) respiration rates, and respiratory control index (RCI, C) for complex I, and basal (no Ca2+, D) and Ca2+-induced (E, F) mtROS production rates. Ca2+-induced mtROS production rates are shown for all time points (E) and for the maximum (25 min) measurement time (F) Mitochondrial respiration rates were measured in isolated mitochondria using substrates for complexes I (α-ketoglutarate and L-malate) in the absence (state 2) or presence of ADP (state 3). Oxygen consumption is defined as nmol of O2 per minute in 1 mg of protein. RCI was measured as the ratio of state 3 to state 2. Basal and Ca2+-induced mtROS production was measured with Ampliflu Red for 25 min with 5-min intervals by monitoring the increase in fluorescence at excitation/emission of 560/590 nm. Groups: Con, control (black circle); IR, ischemia-reperfusion (red square); IRX, IR+XJB (orange triangle); IRS, IR+SfA (blue diamond); IRXS, IR+XJB+SfA (green hexagon). *P<0.05, **P<0.01 all groups vs. Con; &P<0.05 IRX vs. IR; #P<0.05, ##P<0.01 IRXS vs. IR. n= 6-7 per each group.
Next, we evaluated the effects of XJB and/or SfA on mtROS levels. Analysis by Ampliflu Red fluorescence revealed that basal (no Ca2+ stimulated) H2O2 production rates were significantly higher in IR compared to control and reached a nearly 4-fold increase (P<0.01) by the end of the measurement period (25 min; Fig. 3D). Basal H2O2 production rates in mitochondria isolated from XJB- or SfA-treated hearts remained at the levels similar to untreated IR hearts. However, treatment of the heart with XJB and SfA in combination reduced H2O2 production by 37% (P<0.05 vs. IR) at the end of the measurement period. Ca2+ added directly to mitochondrial suspension in vitro further stimulated mtROS production. Mitochondria isolated from IR hearts showed a 3.7-fold increase in Ca2+-induced mtROS production (P<0.01 vs. control) (Fig. 3E). When comparing all treatment groups to IR at the end of the measurement period, XJB reduced mtROS production by 28% (P<0.05), while a combination of XJB with SfA reduced it by 42% (P<0.01) (Fig. 3F). This data demonstrates that the combination of XJB and SfA was more effective in decreasing basal and Ca2+-stimulated mtROS production during IR injury. Like cardiac function, we observed the differences in H2O2 levels between the current and previous studies [23] due to variabilities in the reperfusion time (60-min vs. 90 min) used in these studies that affect functional and metabolic parameters of the heart and mitochondria. Furthermore, our studies demonstrate that the therapeutic efficiency of SfA and XJB on the cardiac IR can be affected depending on the reperfusion time. SfA and XJB can be protective in early reperfusion, but their protective effects may be weakened with the extension of the reperfusion time.
Analysis of mitochondrial Ca2+-induced swelling after IR-injury was assessed through the measurement of mPTP opening as a marker of swelling. The decrease in absorbance was monitored after consecutive Ca2+ additions with 5-min intervals to reach an accumulative Ca2+ concentration of 100, 200, 300, and 800 μM (Fig. 4A). Interestingly, mitochondria isolated from the IR hearts treated with SfA, XJB, or their combination showed similar to untreated IR hearts the swelling in the presence of 200 μM Ca2+ (Fig. 4B). The lack of differences between the treated and untreated hearts can be explained, at least in part, by increased Ca2+ overload during the reperfusion time (90 min) that makes the mitochondria less sensitive to external Ca2+. Analysis of mitochondrial swelling rates induced by 300 μM of Ca2+ (Fig. 4C) demonstrated that mitochondria of hearts treated with SfA alone or in combination with XJB were less sensitive to swelling compared to untreated hearts (IR group). The less sensitivity to 300 μM of Ca2+ can also be explained with the pre-swelling induced by 200 μM Ca2+.
Fig. 4.

Mitochondrial Ca2+- induced swelling. Representative curves (A) and quantitative data (B, C) of mitochondrial swelling measured by monitoring the decrease in light scattering at 525 nm. To induce swelling of mitochondria, Ca2+ (arrows on the top, A) was added to achieve a final concentration of 100, 200, 300, and 800 μM in the swelling buffer. Mitochondrial swelling rates at 200 μM (B) and 300 μM (C) are expressed as ΔAbs per min in 1 mg of protein. Groups: Con, control (black line); IR, ischemia-reperfusion (red line); IRX, IR+XJB (orange line); IRS, IR+SfA (blue line); IRXS, IR+XJB+SfA (green line). +P<0.05 IRS vs.IR; #P<0.05 IRXS vs. IR. n=6 per each group.
Overall, these results demonstrate thatmitochondrial dysfunction, as evidenced by diminished mitochondrial respiration, high ROS, and swelling can be prevented by XJB and SfA during cardiac IR injury.
OPA1 and OMA1 protein expression,and OMA1 activity
Protein levels of OPA1 and OMA1 were analyzed in mitochondrial samples isolated from control and untreated and treated IR hearts. The results of L-OPA1 immunoblotting revealed both L-OPA1 and S-OPA1 in all samples (Fig. 5A, B). Protein levels of L-OPA1 presented as a percentage of total OPA1 showed a 10% (P<0.01) decrease in untreated IR hearts (IR group) compared to control (no IR) hearts (Fig. 5C). L-OPA1 expression in hearts treated with XJB, SfA, or both was also decreased compared to control (P<0.01), meaning that none of the compounds were able to prevent L-OPA1 proteolytic cleavage. As a result, protein levels of S-OPA1 normalized to total OPA1 had a 10% increase (P<0.01) in IR hearts compared to control hearts. However, no differences in S-OPA1 levels were observed among untreated (IR group) and treated groups (Fig. 5D). Quantitative data of OMA1, normalized to voltage-dependent anion channel or porin, demonstrated that IR-induced changes in the mature OMA1 (~40KDa) expression had a positive correlation with OPA1; OMA1 protein levels were reduced by 19% (P<0.01) in IR hearts compared to control (Fig. 6A, B). The reductions of matured OMA1 protein levels were associated with a 42% (P<0.05) increase of the OMA1 enzymatic activity in untreated IR hearts (Fig. 6C). However, like L-OPA1, no changes in protein levels and activity of OMA1 were observed in IR hearts treated with XJB, SfA, or their combination. These data demonstrate that although IR induced L-OPA1 cleavage was associated with OMA1 activation, XJB or SfA alone or in combination were not able to prevent or restore L-OPA1 levels to normal values after global ischemia.
Fig. 5.

OPA1 protein levels. A, Representative immunoblots of L-OPA1 and S-OPA1 in mitochondria isolated from control and IR hearts. Mitochondrial samples were run on 3 different gels in the following order: top blot: control and IR (n=4 per group); middle blot: IR, IRX, and IRS (n=4 for IR, n=5 for IRX and IRS); bottom blot: IR and IRXS (n=4 for IR, n=5 for IRX and IRXS). Mitochondrial samples from the same individual IR hearts were run in all 3 gels. OPA1 samples were normalized to the total expression of OPA1. B, Representative immunoblot image showing all five OPA1 isoforms expressed in the hearts. Lines 1 and 2 represent two L-OPA1 isoforms whereas lines 3, 4, and 5 show three S-OPA1 isoforms. C, Quantitative data of L-OPA1. D, Quantitative data of S-OPA1. L-OPA1 and S-OPA1 levels were calculated based on densitometry analysis using LI-COR Image Studio Lite. OPA1 levels were calculated as the proportion of L-OPA1 (C) and S-OPA1 (D) to the total OPA1 (L-OPA1+S-OPA1). Groups: con, control; IR, ischemia-reperfusion; IRX, IR+XJB; IRS, IR+SfA; IRXS, IR+XJB+SfA. *P<0.05, **P<0.01 all groups vs. Con. n=4-5 per each group.
Fig. 6.
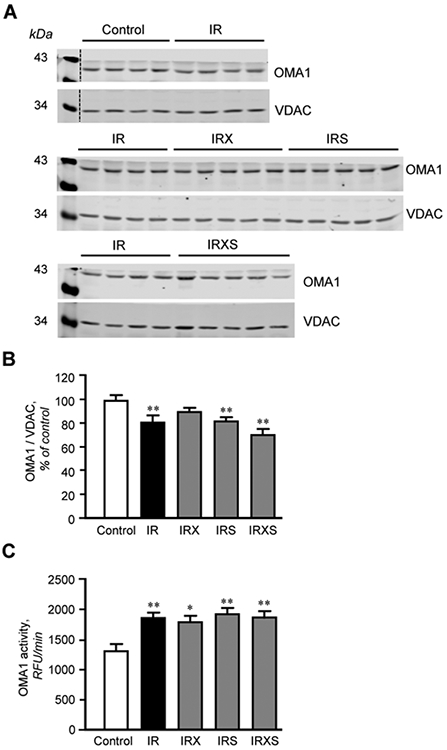
Protein levels and activity of OMA1. A, Representative immunoblots of OMA1 in mitochondria isolated from control and IR hearts. Mitochondrial samples were run on 3 different gels in the following order: top blot: control and IR (n=4 per group); middle blot: IR, IRX, and IRS (n=4 for IR, n=5 for IRX and IRS); bottom blot: IR and IRXS (n=4 for IR, n=5 for IRX and IRXS). Mitochondrial samples from the same individual IR hearts were run in all 3 gels. OMA1 samples were normalized to VDAC expression for each band, and mean values were presented as a percent of control. B, Quantitative data of OMA1 protein levels. C, The enzymatic activity of OMA1. OMA1 protein levels were calculated based on densitometry analysis using LI-COR Image Studio Lite and normalized to VDAC, a mitochondrial housekeeping protein. OMA1 activity was measured for 30 min at 37°C by spectrofluorometer at excitation/emission of 320/405 nm. RFU, relative fluorescence units. Groups: Con, control; IR, ischemia-reperfusion; IRX, IR+XJB; IRS, IR+SfA; IRXS, IR+XJB+SfA. *P<0.05, **P<0.01 vs. Con. n=4-5 for OMA1 protein expression studies (B) and n=4-6 for OMA1 assay (C), per each group.
Discussion
Although the role of L-OPA1 in mitochondrial cristae reshaping is well established, the relationship between mitochondrial volume changes and L-OPA1 cleavage remains to be elucidated. This study demonstrates that IR-induced heart and mitochondrial dysfunctions in the heart are associated with L-OPA1 cleavage and OMA1 activation. Also, we showed that cardioprotective effects of XJB and SfA on cardiac function in response to IR injury are not due to the prevention of OMA1 activation and OPA1 proteolytic cleavage. The treatment with XJB and SfA improved the state 3 respiration rate with no differences in the RCI after prolonged (90-min) reperfusion. Additionally, both basal and Ca2+-induced mtROS was significantly reduced in the presence of XJB and SfA, and mitochondria isolated from hearts treated with SfA alone or in combination with XJB demonstrated low vulnerability to swelling.
Several mitochondrial-mediated mechanisms are involved in the pathogenesis of cardiac IR-injury, including, but not limited to, increased mtROS production and calcium overload [26]. Mitochondrial swelling induced by mPTP opening, which involves depolarization of the IMM, cristae remodeling, ATP depletion, and ROS accumulation, could stimulate L-OPA1 cleavage observed in the present study. OMA1 is a stress-activated metalloprotease [27] that can be provoked by IR-induced mitochondrial dysfunction. We have recently shown that mPTP-dependent mitochondrial swelling stimulates L-OPA1 proteolytic cleavage in isolated mitochondria in vitro [28]. Moreover, OPA1 downregulation reduced mPTP opening with no effect on ROS production. These data suggest a new regulatory mechanism for L-OPA1 metabolism that mediates through the changes in matrix volume. Apparently, the crosstalk between mitochondrial swelling and L-OPA1 cleavage is more complex in cardiac IR in vivo and requires further studies. OPA1 deficiency in OPA1+/− mice have been shown to increase the sensitivity of the heart to IR injury [29].
Mitochondrial swelling is accompanied by increased ROS production during cardiac IR-injury; therefore, the use of mitochondria-targeted antioxidants has shown to elicit greater protection than non-targeted antioxidants [30]. A recently developed mitochondria-targeted ROS scavenger, XJB showed anti-apoptotic effects in mouse embryonic cells [31], improved skeletal muscle contractility [32], decreased the vulnerability of the heart to IR injury [22] in old rats, and protected against global cerebral IR [33]. XJB improved cardiac function, reduced mtROS, and prevented the disintegration of respiratory supercomplexes in rat hearts that were subjected to global ischemia followed by 60-min reperfusion [24]. The present study shows that XJB and SfA alone provide cardioprotection after global ischemia followed by 90-min reperfusion, however, when combined, they are more effective in cardiac recovery. The synergistic effects of the compounds can be explained by simultaneous reduction of mitochondrial ROS production and PTP-induced swelling in IR hearts. Notably, no differences in the RCI after prolonged (90-min) reperfusion indicate that mitochondrial oxygen consumption increased overall, however, the mitochondrial respiratory efficiency was not improved by reperfusion. This was due to state 2 respiration rate which remained high in untreated and treated IR groups due to Ca2+ overload induced by long reperfusion.
In response to oxidative stress, mitochondria undergo fragmentation associated with high mtROS production and apoptosis. Mitochondrial quality control mechanisms maintain mitochondrial health through mitochondrial biogenesis, degradation, and mitophagy that eliminate damaged mitochondria and increase the number of healthy mitochondria. However, the number of dysfunctional mitochondria increases due to impaired balance between fusion and fission of mitochondria in a variety of diseases, including coronary heart diseases [34]. Mitochondrial fusion and cristae morphogenesis regulated by OPA1 play a crucial role in the maintenance of the IMM topography and cristae structure [35]. The balance between L-OPA1 and S-OPA1 may regulate the assembly of respiratory supercomplexes, the multiprotein complexes that comprise of individual ETC complexes. In favor of this, OPA1 ablation in mouse embryonic fibroblasts has been shown to disintegrate the supercomplexes, and conversely, OPA1 overexpression in mice increased supercomplexes assembly [36]. Likewise, OPA1 deficiency impaired the supercomplex assembly and diminished ETC and oxidative phosphorylation in HEK-293 cells [28]. Consequently, a low state 3 respiration rate observed in mitochondria isolated from the IR hearts could be due to the proteolytic cleavage of L-OPA1 (Fig. 3A and Fig. 5).
Despite improved cardiac and mitochondrial function in the presence of XJB and SfA, OMA1 activation and L-OPA1 cleavage induced by IR injury were not prevented. The inability of either XJB or SfA to attenuate IR-induced L-OPA1 cleavage or to improve state 3 respiration suggests that other stimuli are responsible for the activation of OMA1 during IR injury. OMA1 is activated by a range of stimuli, including loss of ΔΨm, heat, and activation of unfolded protein response, among others [20, 21, 37]. Our data suggest that neither ROS nor PTP opening plays a causative role in OMA1 activation. OMA1 activation seems to be a complex process, which includes IR-induced loss of ΔΨm in combination with other stress factors. Increased S-OPA1 levels in response to IR injury might stimulate the fission machinery [38] leading to fragmentation of mitochondria thus, affecting the functionality [39] and morphology of mitochondria. However, other studies found that S-OPA1 can maintain mitochondrial energetics and structure [40] and could improve cell survival in oxidative stress conditions [41], indicating that S-OPA1 is required for the appropriate functioning of mitochondria. This, at least in part, might explain our results showing that significantly improved mitochondrial respiration and less sensitivity to swelling are not associated with changes in the L-OPA1 to S-OPA1 ratio. These findings suggest a certain role of S-OPA1 in maintaining mitochondrial functions in response to oxidative stress.
OMA1 activation is strongly enhanced upon mitochondrial dysfunction and respiratory deficiencies that, eventually, drive the loss of mitochondrial membrane potential. We examined the activation of OMA1 and found that indeed it is enhanced after IR injury. Activation of OMA1 was associated with a reduction of its protein levels (nearly 20%) that can be explained by autocatalytic cleavage of OMA1 upon activation. OMA1 activation induced by reduced ΔΨm, increased ROS production and decreased ATP levels in the IR heart mediates the conversion of OMA1 from the non-mature form (60 kDa pre-pro-OMA1) to the active matured form (~40 kDa pro-OMA1). The latter is thought to be a stress-sensitive protein that further undergoes autocatalytic cleavage to generate the active final form of OMA1, which is the one that processes L-OPA1 cleavage [20, 42]. The autocatalytic cleavage may reduce OMA1 levels while being activated. Interestingly, IR-induced alterations in both OMA1 activity and protein levels were not prevented in the presence of XJB, SfA or their combination suggesting that 1) these compounds are not able to normalize changes of OMA1 induced by prolonged reperfusion and/or 2) in addition to ROS and mPTP-induced mitochondrial swelling, other unknown factors are involved in OMA1 activation in response to oxidative stress in the heart. Studies performing genetic ablation of YME1L and other IMM-localized proteins have shown to activate OMA1, and that outer mitochondrial membrane permeabilization, via BAK and BAX oligomerization, also activates OMA1 [43]. Therefore, pro-apoptotic signaling serves as another regulatory pathway that affects cristae remodeling, and it could be through OMA1 activation and L-OPA1 processing. Furthermore, OMA1 downregulation prevents neuronal loss [44] and exerts protection against heart failure [45], suggesting a possible therapeutic potential of targeting OMA1 directly in mitochondrial health and disease. Epigallocatechin gallate, a phytochemical and non-specific inhibitor of OMA1, has demonstrated positive outcomes protecting cardiomyocytes against hypoxia-reperfusion injury by preventing OPA1 degradation [46]. Targeting OMA1 for therapeutic use would require a better understanding of all the stress-inducing factors that regulate its activation specifically in heart diseases.
Conclusion
This study provides strong evidence that IR injury activates OMA1 and induces proteolytic cleavage of L-OPA1 in Langendorff-perfused rat hearts. XJB in combination with SfA protects the heart against IR injury, but it is not associated with the prevention of OMA1 activation and OPA1 proteolytic cleavage. Likely, other alternative mechanisms are involved in the cardioprotective effects of the mitochondria-targeting compounds that have no crosstalk with OPA1 cleavage and OMA1 activation.
Acknowledgements
This study was supported by the National Institute of General Medical Sciences (Grants SC1GM128210 and R25GM061838) of the National Institutes of Health.
Abbreviations
- ETC
electron transport chain
- HR
heart rate
- IMM
inner mitochondrial membrane
- IR
ischemia-reperfusion
- L-OPA1
long-OPA1
- LVDP
left ventricular developed pressure
- ΔΨm
mitochondrial membrane potential
- mPTP
mitochondrial permeability transition pore
- mtROS
mitochondrial ROS
- RCI
respiratory control index
- ROS
reactive oxygen species
- RPP
rate pressure product
- SfA
sanglifehrin A
- S-OPA1
short-OPA1
- XJB
XJB-5-131
Footnotes
Disclosure Statement
Lee Ann MacMillan-Crow and the University of Arkansas for Medical Sciences (UAMS) have a financial interest in the OMA1 activity assay, which has been reviewed and approved in accordance with the UAMS Disclosure Statement policies.
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. : Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Yellon DM, Hausenloy DJ: Myocardial reperfusion injury. N Engl J Med 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 3.Halestrap AP, Clarke SJ, Javadov SA: Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res 2004;61:372–385. [DOI] [PubMed] [Google Scholar]
- 4.Bernardi P, Di Lisa F: The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 2015;78:100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Javadov S, Chapa-Dubocq X, Makarov V: Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion 2018;38:58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao YP, Zheng M: Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch Biochem Biophys 2019;663:214–219. [DOI] [PubMed] [Google Scholar]
- 7.Javadov S, Rajapurohitam V, Kilic A, Hunter JC, Zeidan A, Said Faruq N, et al. : Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol 2011;106:99–109. [DOI] [PubMed] [Google Scholar]
- 8.Mao K, Klionsky DJ: Mitochondrial fission facilitates mitophagy in Saccharomyces cerevisiae. Autophagy 2013;9:1900–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ji WK, Hatch AL, Merrill RA, Strack S, Higgs HN: Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. Elife 2015;4:e11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Low HH, Sachse C, Amos LA, Lowe J: Structure of a bacterial dynamin-like protein lipid tube provides a mechanism for assembly and membrane curving. Cell 2009;139:1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. : Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010;141:280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Weaver D, Shirihai O, Hajnoczky G: Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J 2009;28:3074–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacVicar T, Langer T: OPA1 processing in cell death and disease - the long and short of it. J Cell Sci 2016;129:2297–2306. [DOI] [PubMed] [Google Scholar]
- 14.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. : Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 2000;26:207–210. [DOI] [PubMed] [Google Scholar]
- 15.Song Z, Chen H, Fiket M, Alexander C, Chan DC: OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 2007;178:749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Chomyn A, Chan DC: Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 2005;280:26185–26192. [DOI] [PubMed] [Google Scholar]
- 17.Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, et al. : The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 2002;523:171–176. [DOI] [PubMed] [Google Scholar]
- 18.Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, et al. : Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res 2012;94:408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang K, Li H, Song Z: Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep 2014;15:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, et al. : Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J 2014;33:578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qi Y, Liu H, Daniels MP, Zhang G, Xu H: Loss of Drosophila i-AAA protease, dYME1L, causes abnormal mitochondria and apoptotic degeneration. Cell Death Differ 2016;23:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Escobales N, Nunez RE, Jang S, Parodi-Rullan R, Ayala-Pena S, Sacher JR, et al. : Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol 2014;77:136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang S, Javadov S: Association between ROS production, swelling and the respirasome integrity in cardiac mitochondria. Arch Biochem Biophys 2017;630:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang S, Lewis TS, Powers C, Khuchua Z, Baines CP, Wipf P, et al. : Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid Redox Signal 2017;27:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tobacyk J, Parajuli N, Shrum S, Crow JP, MacMillan-Crow LA: The first direct activity assay for the mitochondrial protease OMA1. Mitochondrion 2019;46:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binder A, Ali A, Chawla R, Aziz HA, Abbate A, Jovin IS: Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev Cardiovasc Ther 2015;13:1045–1057. [DOI] [PubMed] [Google Scholar]
- 27.Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. : Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 2009;187:1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jang S, Javadov S: OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020;51:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Page S, Niro M, Fauconnier J, Cellier L, Tamareille S, Gharib A, et al. : Increase in Cardiac Ischemia-Reperfusion Injuries in Opa1+/− Mouse Model. PLoS One 2016;11:e0164066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oyewole AO, Birch-Machin MA: Mitochondria-targeted antioxidants. FASEB J 2015;29:4766–4771. [DOI] [PubMed] [Google Scholar]
- 31.Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, et al. : Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J Am Chem Soc 2005;127:12460–12461. [DOI] [PubMed] [Google Scholar]
- 32.Javadov S, Jang S, Rodriguez-Reyes N, Rodriguez-Zayas AE, Soto Hernandez J, Krainz T, et al. : Mitochondria-targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 2015;6:39469–39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji J, Baart S, Vikulina AS, Clark RS, Anthonymuthu TS, Tyurin VA, et al. : Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 2015;35:319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, et al. : Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015;350:aad0116. [DOI] [PubMed] [Google Scholar]
- 35.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T et al. : OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006;126:177–189. [DOI] [PubMed] [Google Scholar]
- 36.Cogliati S, Frezza C, Soriano ME, Varanita T Quintana-Cabrera R, Corrado M, et al. : Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013;155:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rainbolt TK, Lebeau J, Puchades C, Wiseman RL: Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep 2016;14:2041–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olichon A, Guillou E, Delettre C, Landes T Arnaune-Pelloquin L, Emorine LJ, et al. : Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta 2006;1763:500–509. [DOI] [PubMed] [Google Scholar]
- 39.Kushnareva Y, Seong Y, Andreyev AY, Kuwana T Kiosses WB, Votruba M, et al. : Mitochondrial dysfunction in an Opa1(Q285STOP) mouse model of dominant optic atrophy results from Opa1 haploinsufficiency. Cell Death Dis 2016;7:e2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee H, Smith SB, Yoon Y: The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J Biol Chem 2017;292:7115–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H, Smith SB, Sheu SS, Yoon Y: The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J Biol Chem 2020;295:6543–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consolato F, Maltecca F, Tulli S, Sambri I, Casari G: m-AAA and i-AAA complexes coordinate to regulate OMA1, the stress-activated supervisor of mitochondrial dynamics. J Cell Sci 2018;131:jcs213546. [DOI] [PubMed] [Google Scholar]
- 43.Jiang X, Jiang H, Shen Z, Wang X: Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc Natl Acad Sci U S A 2014;111:14782–14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korwitz A, Merkwirth C, Richter-Dennerlein R, Troder SE, Sprenger HG, Quiros PM, et al. : Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J Cell Biol 2016;212:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Acin-Perez R, Lechuga-Vieco AV, Del Mar Munoz M, Nieto-Arellano R, Torroja C, Sanchez-Cabo F, et al. : Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med 2018;10:eaan4935. [DOI] [PubMed] [Google Scholar]
- 46.Nan J, Nan C, Ye J, Qian L, Geng Y, Xing D, et al. : EGCG protects cardiomyocytes against hypoxia-reperfusion injury through inhibition of OMA1 activation. J Cell Sci 2019;132:jcs220871. [DOI] [PubMed] [Google Scholar]