

Abstract
Alcohol use disorder (AUD) is highly comorbid with depression. Withdrawal from chronic alcohol drinking results in depression and understanding brain molecular mechanisms that drive withdrawal-related depression is important for finding new drug targets to treat these comorbid conditions. Here, we performed RNA sequencing of the rat hippocampus during withdrawal from chronic alcohol drinking to discover key signaling pathways involved in alcohol withdrawal-related depressive-like behavior. Data were analyzed by weighted gene co-expression network analysis to identify several modules of co-expressed genes that could have a common underlying regulatory mechanism. One of the hub, or highly interconnected, genes in module 1 that increased during alcohol withdrawal was the transcription factor, signal transducer and activator of transcription 3 (Stat3), a known regulator of immune gene expression. Total and phosphorylated (p)STAT3 protein levels were also increased in the hippocampus during withdrawal after chronic alcohol exposure. Further, pSTAT3 binding was enriched at the module 1 genes Gfap, Tnfrsf1a, and Socs3 during alcohol withdrawal. Notably, pSTAT3 and its target genes were elevated in the postmortem hippocampus of human subjects with AUD when compared with control subjects. To determine the behavioral relevance of STAT3 activation during alcohol withdrawal, we treated rats with the STAT3 inhibitor stattic and tested for sucrose preference as a measure of anhedonia. STAT3 inhibition alleviated alcohol withdrawal-induced anhedonia. These results demonstrate activation of STAT3 signaling in the hippocampus during alcohol withdrawal in rats and in human AUD subjects, and suggest that STAT3 could be a therapeutic target for reducing comorbid AUD and depression.
Subject terms: Molecular neuroscience, Addiction
Introduction
Withdrawal from chronic alcohol drinking elicits negative emotional states such as anxiety, anhedonia, and depression1,2. These psychological states are detrimental to achieving abstinence from alcohol use and are associated with increased risk for relapse to alcohol drinking3,4. The hippocampus is an alcohol-sensitive brain region involved in mood regulation and cognition5–8. Brain imaging of human subjects with alcohol use disorder (AUD) has found reductions in hippocampal volume and altered hippocampal connectivity7, which likely contribute to cognitive and emotional problems observed in those with AUD. In rodent models of AUD, chronic alcohol exposure and withdrawal alters the hippocampus at the cellular and molecular level9–15. Chronic alcohol drinking increases the expression of inflammatory mediators, such as cytokines and chemokines, increases oxidative stress, and induces excitotoxicity and reactive gliosis16,17. This constellation of changes is associated with altered synaptic function, reduced neurogenesis, and ultimately, neurodegeneration16, as well as increased depressive-like behaviors and learning and memory deficits during alcohol withdrawal10,18,19.
Activation of the innate immune system during withdrawal from chronic alcohol use is particularly interesting because an overactive immune response has also been observed in human subjects with major depressive disorder20,21, and in rodent models of stress-induced depression21, suggesting that a brain pro-inflammatory response might be a common mechanism linking AUD and depression during alcohol withdrawal. Cytokines and chemokines bind to cell surface receptors, resulting in the activation and nuclear translocation of transcription factors such as the nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) complex and signal transducer and activator of transcription 3 (STAT3)22,23. These transcription factors bind to specific DNA sequences to increase the expression of genes involved in the immune response, such as chemokines, cytokines, and their receptors, thus amplifying the immune response. Activation of the NF-κB complex has been observed in the hippocampus of human subjects with AUD and in rodent models of AUD24,25. Interestingly, STAT3 plays a role in depressive-like behaviors in mice26,27, and polymorphisms in the human STAT3 gene are associated with the response to antidepressants28. Acute and chronic alcohol exposure activates STAT3 in the mouse hippocampus29,30. However, it is unknown whether STAT3 in the hippocampus regulates expression of immune response genes and development of depression-like behaviors during ethanol withdrawal after chronic exposure.
We therefore used an unbiased transcriptomic approach to identify gene expression changes in the rat hippocampus during chronic ethanol exposure and withdrawal, with the aim of identifying genes and signaling pathways that could contribute to alcohol withdrawal-induced depressive-like behavior. We identified STAT3 as a “hub” gene that is increased in the hippocampus during alcohol withdrawal. STAT3 was activated in hippocampal astrocytes during alcohol withdrawal and was more highly associated with the promoter of glial fibrillary acidic protein (Gfap), which is a hallmark of activated astrocytes, and the promoters of several immune-related genes whose expression also increased during alcohol withdrawal. We used a pharmacological inhibitor of STAT3 to demonstrate that activation of STAT3 during alcohol withdrawal is behaviorally relevant to anhedonia, a key symptom of depression31. Finally, we examined the activation of STAT3 and gene expression in the postmortem hippocampus of human AUD subjects.
Materials and methods
Animals
Adult male Sprague Dawley rats (75–85 days old) were purchased from Harlan (Indianapolis, IN, USA) and individually housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle (lights on a 6 am), with food and water provided ad libitum prior to beginning the chronic ethanol treatment protocol. Animal care was conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and all experimental procedures were approved by the University of Illinois at Chicago Animal Care Committee.
Chronic ethanol treatment
When rats reached ~250–300 g body weight, they were fed with the nutritionally complete Lieber-DeCarli control or ethanol liquid diet (Bio-Serv; Frenchtown, NJ, USA) as their only source of food and fluid as described by us previously32,33. Rats were randomly assigned to treatment groups. Rats were fed 80 ml/day control diet for 3 days and then were randomly assigned to different treatment groups. The control group continued with the control diet for 16 days, while the ethanol groups were gradually introduced to ethanol over 7 days (1.8–8.1%). Rats were then maintained on a 9% ethanol diet for 15 days. Ethanol-withdrawn rats were switched to control liquid diet for 24 h after removal of the ethanol liquid diet whereas ethanol diet-fed (0 h withdrawal) continued on an ethanol diet for one additional day. Rats were pair fed and their liquid diet intake and body weights were closely monitored. We have previously reported blood alcohol levels (BALs) in the range of 172–198 mg% for ethanol-diet fed (no withdrawal) rats, whereas the BALs after 24 h of ethanol withdrawal were undetectable32,33. Investigators were not blinded to treatment groups.
Tissue collection
Rats were anesthetized with isoflurane and rapidly decapitated. Brains were removed and sectioned into a 1-mm-thick section using a rat brain matrix (Zivic Instruments, Pittsburgh, PA, USA). The dorsal hippocampus (located 2–4 mm posterior to bregma) was dissected, rapidly frozen on dry ice and stored at −80 oC.
RNA-Seq and bioinformatics
RNA-Seq was performed on dorsal hippocampi from 6 rats per group (control, ethanol, and withdrawal). Total RNA was isolated using the miRNeasy Mini Kit (Qiagen) and RNA integrity numbers were determined using a TapeStation instrument (Agilent, Santa Clara, CA, USA). cDNA libraries were prepared using the TruSeq RNA Library Preparation Kit (Illumina, San Diego, CA, USA) and were paired-end sequenced on the HiSeq 4000 System (Illumina) Approximately 100,000,000 raw reads were obtained per sample. The libraries were prepared and sequenced in the DNA Services laboratory of the Roy J. Carver Biotechnology Center at the University of Illinois at Urbana-Champaign. Sequences were aligned to the NCBI Rnor 6.0 genome (annotation 106) using STAR (v. 2.5.2a). A total of 18,118 genes had edgeR’s34 TMM-normalized values >0.5 and were kept for differential expression (DE) analysis. After adjusting for surrogate variables35, we used limma-voom36 to calculate a one-way ANOVA across all three treatments. Weighted gene co-expression network analysis (WGCNA) was performed in order to group genes into modules showing similar expression patterns across samples. Principle components analysis of genes in modules was used to calculate the “eigengene” value. Clusters or modules with similar expression patterns were merged, resulting in a total of 53 modules. STRING37 and Enrichr38 were used for pathway analysis. RNA-Seq data are available in Gene Expression Omnibus (GEO) database, accession number GSE171051.
Quantitative real-time PCR (qPCR)
qPCR was performed on RNA from dorsal hippocampi from 10 rats per group (control, ethanol, and withdrawal; six were samples from the RNA-Seq experiment plus an additional four samples). Total RNA was isolated from frozen tissue using TRIzol (Thermo Fisher Scientific), further purified using the miRNeasy Mini kit (Qiagen) and converted to cDNA using iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA, USA). For postmortem hippocampus tissue, qPCR was performed in 13 controls, 13 AUD subjects with no blood alcohol levels at the time of death, and 7 AUD subjects with positive blood alcohol level (demographic details are in Table 1). Total RNA was isolated from frozen tissue using the miRNeasy Mini kit (Qiagen) then converted to cDNA using M-MLV Reverse Transcriptase (Invitrogen). SYBR Green Supermix (Bio-Rad) was used for qPCR with the primers listed in Supplemental Methods.
Table 1.
Demographic characteristics of control and alcohol use disorder (AUD) subjects.
| Samples used for pSTAT3 IHC | Samples used for qPCR | ||||
|---|---|---|---|---|---|
| Subjects | Control | AUD | Control | AUD | AUD-BAL |
| n | 14 | 14 | 13 | 13 | 7 |
| Age | 61.6 ± 2.2 | 61.1 ± 2.4 | 57 ± 2 | 60 ± 1.8 | 49 ± 2.8# |
| Sex (n) | F (4), M (10) | F (4), M (10) | F (2), M (11) | F (4), M (9) | F (1), M (6) |
| PMI (h) | 34.4 ± 3.7 | 36.7 ± 3.8 | 35 ± 4.2 | 39 ± 5 | 40 ± 4.4 |
| pH | 6.6 ± 0.08 | 6.6 ± 0.06 | 6.6 ± 0.08 | 6.6 ± 0.08 | 6.7 ± 0.16 |
| BMI | 31.3 ± 1.7 | 25.8 ± 1.4* | 31 ± 1.5 | 25 ± 1.9 | 29 ± 2.0 |
| Total drinking years | 36.9 ± 3.3 | 35.9 ± 2.7 | 35 ± 3.1 | 38 ± 1.8 | 28 ± 3.2 |
| Ethanol daily use (g) | 17.5 ± 5.6 | 159.8 ± 32.6*** | 14 ± 4.9 | 179 ± 35** | 258 ± 73*** |
| Drinks per week | 10.9 ± 4.7 | 78.7 ± 18.2** | 8.5 ± 3.6 | 87 ± 19* | 147 ± 53** |
| Cigarette pack years | 16.9 ± 6.6 | 40.0 ± 7.3* | 25 ± 9.7 | 39 ± 5.1 | 30 ± 4.7 |
| Cause of death (n) | Cardiac (10), cancer (1), cardiovascular (1), respiratory (1), vascular (1) | Cardiac (8), hepatic (1), infection (2), respiratory (2), toxicity (1) | Cardiac (10), vascular (1), cardiovascular (1), toxicity (1) | Cardiac (6), respiratory (3), hepatic (1), toxicity (1), infection (1), stroke (1) | Cardiac (3), toxicity (3), unknown (1) |
Data are shown as mean ± SEM.
BMI body mass index, F female, M male, PMI postmortem interval (hours).
*p < 0.05, **p < 0.01, ***p < 0.001 by unpaired two-tailed Student’s t-test when compared to control. #p < 0.01 when compared to AUD.
Immunohistochemistry (IHC)
Rats (3 per group) were euthanized using a commercial euthanasia solution containing pentobarbital and transcardially perfused with 4% paraformaldehyde. Brains were processed for IHC with antibodies to phosphorylated (p)STAT3 (phosphorylated at tyrosine 705, Cell Signaling Technology, RRID: AB_2198588) and GFAP (Thermo Fisher Scientific, RRID: AB_2532994) as detailed in Supplemental Methods. Human brain slices (10 male and 4 female from each group) were deparaffinized and heated for antigen retrieval. The same pSTAT3 antibody and method used for rat sections were performed on human slices.
Western blot
Western blots were performed on hippocampal lysates from 6 rats per group using standard procedures with primary antibodies to STAT3 and pSTAT3 (Cell Signaling Technology, RRID: AB_331757 and RRID: AB_2491009), and β-actin (Sigma-Aldrich RRID: AB_476744) as detailed in Supplemental Methods. Blots were imaged on the Odyssey Fc Dual-Mode Imaging system (LI-COR, Lincoln, NE, USA).
Chromatin immunoprecipitation (ChIP)
ChIP was performed on dorsal hippocampi from 14 rats per group. Frozen tissue was homogenized in phosphate-buffered saline (PBS) and fixed in 1% methanol-free formaldehyde for 5 min at 37 oC. Homogenate was lysed with SDS buffer (1% SDS, 50 mM Tris-HCl, pH 8, 10 mM EDTA) for 10 min at 4 oC and sonicated (Covaris, Woburn, MA, USA) to shear chromatin to 200 bp fragments. Chromatin (100 μg) was diluted in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8, 167 mM NaCl) and an aliquot was removed as input. Samples were pre-cleared with protein A/G PLUS-agarose beads (Santa Cruz Biotechnology, Dallas, TX, USA) prior to immunoprecipitation (IP). IP was performed overnight at 4 oC with an antibody to pSTAT3 (5 μl, #9145; Cell Signaling Technology; RRID: AB_2491009) and protein A/G PLUS-agarose. IPs were washed with ChIP dilution buffer and chromatin isolated using Chelex 100 resin (Bio-Rad). qPCR was performed as described above with the primers indicated in Supplementary methods. Relative enrichment of pSTAT3 binding was calculated by normalizing to input using 2−ΔΔCT method.
Sucrose preference test
Sucrose preference testing was performed on 14–16 rats per group (control-vehicle n = 16, withdrawal-vehicle n = 14, control-stattic n = 16, and withdrawal-stattic n = 16). Bottles containing control liquid diet were removed from the home cages at 24 h of withdrawal (of both ethanol-withdrawn and control rats) and replaced with two standard water bottles containing either a 0.5% sucrose (w/v) solution or water for 1 h. Stattic (Selleck Chemicals, Houston, TX, USA) was administered intraperitoneally at a dose of 50 mg/kg in vehicle (2% DMSO, 30% PEG300), 2 h prior to testing sucrose preference. Dorsal hippocampi were dissected after the completion of the sucrose preference test and subjected to either western blots or ChIP with pSTAT3 antibody to demonstrate stattic efficacy.
Human subjects
Postmortem hippocampal sections and frozen hippocampus from subjects diagnosed with AUD and control subjects (n = 10 males and 4 females per group for IHC; n = 13 control, 13 AUD, and 7 AUD with positive blood alcohol levels for qPCR) were obtained from the New South Wales Brain Tissue Resource Centre (University of Sydney, Australia). Demographic characteristics of subjects are shown in Table 1. Individuals were diagnosed with AUD according to DSM-IV criteria. Control cases were matched with AUD for sex, age, race, and postmortem interval. IHC was used for pSTAT3 staining in sections as described above. One AUD subject was removed from the pSTAT3 analysis because it was identified as a statistical outlier using the ROUT method (Q = 1%). RNA was isolated from frozen tissue and analyzed by qPCR as described above.
Statistical analysis
The sample size was determined by a power analysis based on effect sizes from our previous experience with molecular and behavioral measures. Statistical testing was performed using Prism 8 (GraphPad, San Diego, CA, USA). Data were analyzed by one-way ANOVA, followed by post hoc Tukey’s or Kruskal–Wallis tests, or two-way ANOVA, followed by post hoc Sidak’s test. p < 0.05 was considered statistically significant. Statistical outliers were determined using the ROUT method and removed prior to performing ANOVAs. For correlations, two-tailed Pearson’s correlations were performed using PASW v.18 software (SPSS). Rat experiments (except for RNA-Seq) were repeated 2–3 times and the data are presented as the mean ± SEM from combined experiments.
Results
Increased expression of Stat3 and its transcriptional target genes in the hippocampus of ethanol withdrawn rats after chronic exposure
We performed RNA-Seq to discover gene expression changes in the hippocampus of rats in withdrawal from chronic alcohol exposure. RNA-Seq was performed on the hippocampus from control, ethanol (no withdrawal), and ethanol plus withdrawal (24 h) groups. The largest number of differentially expressed (DE) genes were in the control vs. withdrawal comparison (925 out of 18,118 detected; q < 0.1), while only 65 genes were DE between control and chronic ethanol, and 474 genes were DE between chronic ethanol and withdrawal (Fig. 1A–C). These results indicate that more genes are altered in the hippocampus during withdrawal from chronic ethanol than during chronic ethanol exposure.
Fig. 1. Increased expression of Stat3 and its transcriptional target genes in the hippocampus of ethanol withdrawn rats after chronic exposure.

Rats were fed control liquid diet (C), ethanol liquid diet (E), or ethanol liquid diet followed by 24 h of withdrawal from ethanol diet (W). The dorsal hippocampus was dissected and RNA isolated and subjected to RNA sequencing (RNA-Seq, n = 6) or qPCR (n = 10). A–C Volcano plots of genes differentially expressed between (A) C and E, (B) E and W, and (C) C and W by RNA-Seq. Red and blue dots indicate genes that were increased and decreased, respectively, in each comparison with q ≤ 0.1. D Heat map of 53 modules identified by weighted gene coexpression network analysis (WGCNA) with eigengene q-values (−log10) in C vs. E, C vs. W, and W vs. E comparisons. E Heat map showing expression of 1794 genes in module 1 in C, E, and W conditions. Each column is from an individual rat. Blue and red indicate decreased and increased expression levels, respectively. F Cytoscape plot of STRING protein–protein interaction analysis showing a module 1 cluster with the first shell of interactors with STAT3. G–L Relative expression by qPCR of (G) Stat3, (H) Gfap, (I) Socs3, (J) Tnfrsf1a, (K) Timp1 and (L) Osmr in C, E, and W conditions. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, and ****p < 0.0001 by Tukey’s test after one-way ANOVA.
We next performed WGCNA39 to identify modules of co-expressed genes, with the rationale that members of a module could be regulated by a common molecular mechanism. We identified 53 modules (Fig. S1 and Table S1), with many of these modules exhibiting a significant association with at least one of the treatment conditions (Fig. 1D). The module 1 (1794 genes) eigengene value was significantly higher during withdrawal when compared with the control and ethanol conditions (Fig. 1E and Fig. S2, p < 0.05), and 186 genes in module 1 were elevated during withdrawal with a q value cutoff of 0.1 when compared with the control condition (Table S2). The top 5 enriched KEGG pathways for these 186 genes were TNF signaling, spliceosome, Kaposi’s sarcoma-associated herpesvirus infection, PI3K-Akt signaling, and human papillomavirus infection (Table S3), indicating alterations in the immune response and RNA splicing. We next performed a STRING analysis37 on these 186 genes to determine if there were any known protein–protein interactions and observed two clusters of highly interconnected genes. Genes in one cluster encoded RNA splicing factors, consistent with a top enriched pathway by KEGG. The second cluster contained the transcription factor STAT3 as a central node that was highly interconnected with other genes in the cluster (Fig. 1F). Many of these genes are known transcriptional targets of STAT3, including Socs3, Tnfrsf1a, Timp1, Icam, Ctgf, and Osmr40–45. Closer examination of the 186 DE genes in module 1 also revealed increased expression of Gfap during withdrawal. GFAP is a marker for reactive astrocytes and another transcriptional target of STAT346. These results indicate altered RNA splicing, immune response, and STAT3 signaling in the hippocampus during withdrawal from chronic alcohol exposure.
To validate the RNA-Seq results, we performed qPCR of Stat3, Gfap, Tnfrsf1a, Timp1, and Osmr in hippocampal RNA from the three treatment groups and found that the expression of these genes was significantly elevated in the withdrawal compared to the control group (Fig. 1G–L). Tnfrsfa1 expression was also higher in the chronic ethanol compared to the control group (Fig. 1J). Thus, the expression of Stat3 and some of its known transcriptional target genes are elevated in the hippocampus during ethanol withdrawal after chronic exposure.
Increased total and phosphorylated STAT3 protein in the hippocampus of ethanol withdrawn rats after chronic exposure
To determine if increased Stat3 mRNA during ethanol withdrawal is functionally relevant, we measured protein levels of STAT3 and pSTAT342 in the hippocampus of control, ethanol, and ethanol-withdrawn rats by western blot. Total STAT3 and pSTAT3 were significantly higher in the ethanol withdrawal compared to the control group (Fig. 2A–C). We examined the hippocampal structures that contained pSTAT3 using IHC and observed pSTAT3 in the stratum moleculare and stratum radiatum (Fig. 2D–I and Fig. S3), areas that are enriched in astrocytes47. It is interesting to note that pSTAT3 was not strictly limited to the hippocampus and was observed in other regions such as the hypothalamus in the same sections (data not shown). We next examined whether pSTAT3 was located in astrocytes, microglia, or neurons in the hippocampus by performing immunofluorescent staining with antibodies to pSTAT3 and either the astrocytic maker, GFAP, the microglial marker, IBA1, or the neuronal marker, NeuN. Nuclear pSTAT3 was observed in cells immunoreactive for GFAP (Fig. 2J), but not NeuN (Fig. S4). pSTAT3 was also observed in IBA1-positive cells, although only 9% of pSTAT3-positive cells were also labeled with IBA1 (Fig. S4), compared with 86% of pSTAT3-positive cells labeled with GFAP. These results indicate that elevations in pSTAT3 in the hippocampus during withdrawal from chronic alcohol may be abundant in astrocytes.
Fig. 2. Increased total and phosphorylated STAT3 protein in the hippocampus of ethanol withdrawn rats after chronic exposure.

Rats were fed control liquid diet (C), ethanol liquid diet (E), or ethanol liquid diet followed by 24 h of withdrawal from ethanol diet (W). The dorsal hippocampus was either (A–C) dissected and protein lysates analyzed by western blot (n = 6 per group) or (D–J) processed for immunohistochemistry (n = 3 per group). A Representative western blot showing (top to bottom) merged image of phosphorylated (p)STAT3 and STAT3, STAT3 (red), pSTAT3 (green), and β-actin (red). Molecular weight markers are indicated in kDa. B Levels of STAT3 relative to β-actin. C Levels of pSTAT3 relative to β-actin. Data are presented as the mean ± SEM. *p < 0.05 and **p < 0.01 when compared with control by Kruskal–Wallis test after one-way ANOVA. Representative images of hippocampus showing pSTAT3-positive cells in brown in (D–F) control and (G–I) ethanol withdrawn rats. Panels (D) and (G) are images at ×5 magnification with a scale bar representing 100 μm. Boxes show areas that are magnified in (E–F) and (H–I), with scale bar representing 50 μm. sg stratum granulosum, SM stratum moleculare, SLM stratum lacunosum moleculare, SR stratum radiatum, H hilus. J Representative immunofluorescent image of pSTAT3 in green, colocalized with the astrocytic marker glial fibrillary acidic protein (GFAP) in red. White arrows show localization of pSTAT3 in a GFAP-expressing cell. Scale bar, 50 μm.
Increased pSTAT3 enrichment at target genes in the hippocampus of ethanol withdrawn rats after chronic exposure
We hypothesized that the increase in pSTAT3 in the hippocampus of rats in ethanol withdrawal would result in enrichment of pSTAT3 at those genes whose expression was increased during withdrawal. To test this, we performed ChIP with an antibody to pSTAT3, followed by qPCR with primers flanking known or predicted STAT3 consensus sequences in Tnfrsf1a, Gfap, and Socs3. pSTAT3 was significantly enriched at a predicted STAT3 motif in the first intron of Tnfrsf1a, 1192 bp downstream of the transcriptional start site, in both the ethanol and withdrawal groups (Fig. 3A), consistent with the elevated mRNA levels of Tnfrsf1a under both conditions (Fig. 1J). Enrichment of pSTAT3 at this location was specific, as pSTAT3 was not enriched at two other predicted STAT3 binding motifs located further downstream. pSTAT3 was also more highly enriched during withdrawal at a STAT3 motif in the Gfap promoter located 1214 bp upstream of the transcriptional start site (Fig. 3B), and at a STAT3 motif located in the first intron of the Socs3 gene (Fig. 3C). These results demonstrate that pSTAT3 associates with specific target genes during alcohol withdrawal and implicates pSTAT3 in transcriptional activation of these genes during withdrawal.
Fig. 3. Increased pSTAT3 enrichment at target genes in the hippocampus of ethanol withdrawn rats after chronic exposure.
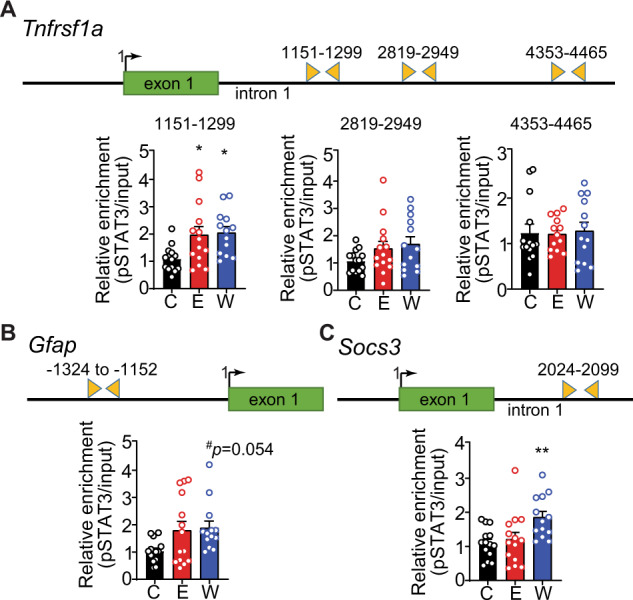
Rats were fed control liquid diet (C, n = 13), ethanol liquid diet (E, n = 14), or ethanol liquid diet (n = 13) followed by 24 h of withdrawal from ethanol diet (W). The dorsal hippocampus was dissected and chromatin immunoprecipitation (ChIP) was performed using an antibody to phosphorylated STAT3 (pSTAT3), followed by qPCR with primers surrounding predicted STAT3 binding sites. Shown above each graph is a schematic of each gene with primer locations indicated by triangles. A Bar diagram shows relative pSTAT3 enrichment in the first intron of Tnfrsf1a between 1151 and 1299 bp downstream of the transcription start site but not at sites further downstream (2819–2949 and 4353–4465). Bar diagrams show relative pSTAT3 enrichment at the B Gfap promoter and C first intron of Socs3. Data are presented as the mean ± SEM. *p < 0.05 and **p < 0.01 compared with control by Tukey’s test after one-way ANOVA.
Treatment with a STAT3 inhibitor during withdrawal decreases anhedonia
To determine if blocking STAT3 activity reduces anhedonia during withdrawal from chronic alcohol, we treated rats with the STAT3 inhibitor stattic (50 mg/kg, i.p), 22 h into ethanol withdrawal, and tested them for anhedonia using the sucrose preference test 2 h later. Vehicle-treated rats in ethanol withdrawal had decreased sucrose preference compared to vehicle-treated control rats. Treatment with stattic normalized sucrose preference in rats in ethanol withdrawal (Fig. 4A), indicating that STAT3 inhibition decreased anhedonia during withdrawal. After the completion of the sucrose preference test, we verified the efficacy of stattic by measuring levels of pSTAT3 and enrichment of pSTAT3 at the Socs3 gene by western blot and ChIP, respectively. As expected, pSTAT3 in the hippocampus of vehicle-treated rats in ethanol withdrawal was higher than in vehicle-treated, control diet-fed rats, while pSTAT3 in the hippocampus of rats treated with stattic during ethanol withdrawal was comparable to control-diet fed rats (Fig. 4B). pSTAT3 was enriched at the Socs3 gene in vehicle-treated rats and decreased by stattic treatment (Fig. 4C). These results indicate that inhibition of STAT3 can reduce ethanol withdrawal-induced anhedonia.
Fig. 4. Treatment with a STAT3 inhibitor during withdrawal decreases anhedonia as measured by the sucrose preference test.

Rats were fed control or ethanol liquid diet for 15 days followed by withdrawal for 24 h. Stattic (50; mg/kg, i.p.) or vehicle was administered 2 h prior to testing anhedonia. Hippocampus was dissected immediately after behavioral testing and analyzed for phosphorylated STAT3 (pSTAT3) by western blot and enrichment of pSTAT3 at the Socs3 promoter using chromatin immunoprecipitation (ChIP)-qPCR. A Sucrose preference test, n = 14–16. *p < 0.05 by Sidak’s test after two-way ANOVA (stattic, F(1, 58) = 4.46, p = 0.039; withdrawal, F(1, 58) = 8.88, p = 0.0042; interaction, F(1, 58) = 1.88, p = 0.18). B pSTAT3 protein levels, n = 8. ***p < 0.001 by Sidak’s test after two-way ANOVA (stattic × withdrawal interaction, F(1, 28) = 17.5, p = 0.0003). C Enrichment of pSTAT3 at Socs3 gene after ChIP-PCR, n = 5–8. *p < 0.05, main effect of stattic by two-way ANOVA (F(1, 23) = 6.56, p = 0.018). Data are presented as the mean ± SEM.
Increased phosphorylated (p)STAT3 and expression of STAT3 target genes in the postmortem hippocampus of human subjects with AUD
We next investigated whether our findings in rats translate to humans by performing IHC with pSTAT3 antibody on postmortem hippocampus from 14 controls and 14 AUD subjects. AUD subjects had significantly lower body mass index, higher ethanol daily use at the time of death, drinks per week, and cigarette pack-years compared with controls (Table 1). Notably, the number of pSTAT3-positive nuclei in the hippocampus was significantly higher in individuals with AUD (Fig. 5). We reasoned that an increase in activated STAT3 would reflect elevated expression of its target genes in the hippocampus of AUD subjects. To test this, we compared DE genes from previously published microarray data of hippocampus from human AUD and control subjects48 (GSE44456) with the DE genes in our rat withdrawal vs. control comparison. Gene homologs were matched based on gene symbols, and concordant gene pairs were determined based on an FDR < 0.2 and log2 fold changes in the same direction for both species. We found 39 concordant DE genes between the rat and human data (Fig. S5, Table S4). Several of these genes were present in rat WGCNA module 1, including TNFRSF1A, TAGLN2, PECAM1, OSMR, IFITM2, and SLC14A1. Specifically, the STAT3 target genes, TNFRSF1A43 and OSMR45, were significantly DE in both rat and human hippocampus. Other STAT3 targets, SOCS340 and TIMP141, were also concordantly DE in the rat and human datasets, although they did not meet the FDR < 0.2 cutoff in our analysis (q = 0.23 for both).
Fig. 5. Increased phosphorylated (p)STAT3 and expression of STAT3 target genes in the postmortem hippocampus of human subjects with AUD.

A–D Representative hippocampal sections showing immunohistochemistry with a pSTAT3 antibody (brown spots) from (A, C) control and (B, D) AUD subjects. Arrows point to examples of nuclei positive for pSTAT3. Scale bar, 50 μm. M male, F female. E Quantification of pSTAT3-positive cells in control and AUD subjects. Data are presented as the mean ± SEM, n = 10 males per group and 4 females per group. *p < 0.05 main effect of condition by two-way ANOVA (F(1, 23) = 6.7, p = 0.016). F–K Relative expression of F STAT3, G GFAP, H SOCS3, I TNFRSF1A, J TIMP1, and K OSMR by qPCR in human postmortem hippocampus of control (n = 13), AUD (n = 13), and AUD subjects with blood alcohol levels at the time of death (AUD-BAL, n = 7). *p < 0.05 when compared to control by post hoc Tukey’s test after one-way ANOVA.
We extended these studies in human postmortem hippocampus from AUD and control subjects by performing qPCR to measure expression of STAT3, GFAP, SOCS3, TNFRSF1A, TIMP1, and OSMR mRNA in human AUD (n = 20) and control (n = 13) hippocampus (Table 1, Fig. 5). The AUD samples were divided based on whether there were measurable blood alcohol levels (BALs) at the time of death (13 AUD and 7 AUD-BAL [mean BAL = 0.221 ± 0.062%; range = 0.03–0.43%]) because of differences in gene expression that we observed in the rat hippocampus in the ethanol and withdrawal conditions. STAT3, SOCS3, TIMP1, and OSMR levels were significantly increased in AUD compared to control hippocampus, but not in AUD-BAL compared to control hippocampus (Fig. 5). Next, we examined correlations between STAT3 and GFAP, SOCS3, TNFRSF1A, TIMP1, and OSMR transcript levels in all samples. STAT3 transcript was significantly correlated with TNRFSF1A (r = 0.777, p < 0.01), SOCS3 (r = 0.746, p < 0.01), OSMR (r = 0.922, p < 0.01), and TIMP1 (r = 0.772, p = 0.01) transcripts (Fig. S6), suggesting coregulation of these genes. The correlations remained significant in the control, AUD, and AUD-BAL groups even when the groups were analyzed separately (Table S5). STAT3 expression was not significantly correlated with alcohol drinking measures of total drinking years, ethanol daily use, or a number of drinks per week (Table S5). Overall, our results provide evidence for activation of the STAT3 signaling pathway in the hippocampus of individuals with AUD similar to our preclinical findings, and suggest that this signaling pathway could be a target for AUD treatment.
Discussion
Using a genome-wide transcriptomic approach, we identified STAT3 as a key transcriptional regulator in the rat hippocampus that is involved in anhedonia during withdrawal from chronic alcohol drinking. Activation of STAT3 during alcohol withdrawal in the hippocampus primarily occurred in astrocytes, as demonstrated by the majority of pSTAT3-positive cells colocalizing with the reactive astrocyte marker GFAP. Of note, the results we obtained using a preclinical model of dependence translate to humans, as an increased number of pSTAT3-labeled cells was observed in the hippocampus of human subjects with AUD compared with control subjects and known STAT3 target genes (SOCS3, TIMP1, and OSMR) were expressed at higher levels in the hippocampus of AUD subjects. These results indicate that STAT3 signaling is activated not only in the rat hippocampus after chronic ethanol exposure, but also in the hippocampus of individuals with AUD.
The RNA-Seq data pointed to the enrichment of genes in several biological pathways in the rat hippocampus during alcohol withdrawal, including those involved in RNA splicing and the response to viral infection (i.e., immune response pathways). These results are consistent with transcriptomic profiling of the hippocampus of mice in withdrawal from chronic intermittent ethanol vapor exposure49,50. Notably, the transcriptome of postmortem hippocampus of human subjects with AUD has been analyzed and, similar to our results, changes in genes related to RNA processing and immune response pathways have been found48,51. The behavioral implications and molecular details of altered expression of RNA splicing factors have not been investigated in the context of chronic alcohol exposure and withdrawal, although Wolfe et al.52 demonstrated that the synaptic transcriptome is changed in the hippocampus of mice treated acutely with ethanol, resulting in differential exon usage. Chronic alcohol exposure and withdrawal, by changing the expression of splicing factors, could conceivably alter exon usage, leading to alterations in synaptic proteins and ultimately impacting depression-like behavior. This requires further investigation.
With regard to the enrichment of immune response pathways in our rat RNA-Seq data, activation of the neuroimmune system after chronic alcohol exposure is a common theme across species and has been observed in rodent models of AUD and in postmortem brain from humans with AUD16,17,48,53. Here, we determined that STAT3 protein and mRNA levels increase, that STAT3 is activated, and that Tnfrsf1a (encoding tumor necrosis factor receptor 1A), Osmr (encoding oncostatin M receptor), and Socs3 (encoding suppressor of cytokine signaling 3) are STAT3-regulated immune-response genes that are induced in the rat hippocampus upon chronic alcohol exposure and withdrawal. A previous study of the human hippocampal AUD transcriptome reported increased expression of the STAT3 target genes TNFRSF1A, SOCS3, TIMP1, and OSMR48 in AUD subjects vs. controls, consistent with our findings showing increased expression of most of these genes in the hippocampus of AUD vs. control subjects. Given that we observed increased pSTAT3 in the hippocampus of AUD subjects, it seems likely that the increased expression of these genes in the human AUD hippocampus is mediated by activated STAT3. Thus, STAT3 is a transcriptional mediator of neuroimmune activation in the hippocampus in response to chronic alcohol exposure. Of note, the increase in pSTAT3 was highly variable during withdrawal in rat hippocampus and in the hippocampus of human subjects with AUD. The source of this variability is not known but could reflect innate differences in the neuroimmune response to chronic alcohol use and withdrawal.
The majority of pSTAT3 in the hippocampus co-localized with GFAP, demonstrating that STAT3 activation primarily occurs in astrocytes in response to withdrawal from chronic alcohol. Further evidence for astrocytic activation of STAT3 during alcohol withdrawal is provided by our finding that pSTAT3 is enriched at the Gfap promoter. GFAP is a marker of reactive astrocytes and a known STAT3 target gene46. Astrocytes are key mediators of the brain immune response54, and STAT3 is considered an important regulator of astrocyte reactivity55. Reactive astrocytes exhibit transcriptional states that have been defined as neurotoxic, called “A1”, or neuroprotective, called “A2”56,57. A1 astrocytes are found in many neurodegenerative and autoimmune diseases, whereas A2 astrocytes have been observed after ischemic stroke57. STAT3 and GFAP have been described as “pan-injury” markers of reactive astrocytes57,58. STAT3 can play a protective or detrimental role after nervous system injury or neurodegeneration59–61. It remains to be determined whether the increase in pSTAT3 in hippocampal astrocytes after chronic alcohol drinking and withdrawal is pro- or anti-inflammatory, and whether it might contribute to alcohol-induced neurodegeneration and related behavioral sequelae.
It is important to note that although we observed increased pSTAT3 in astrocytes, there was also a small percentage (<10%) of pSTAT3 observed in microglia in the rat hippocampus during withdrawal from chronic alcohol. We cannot rule out the possibility that microglial-expressed STAT3 may be involved in the neuroimmune62 and anhedonic responses during withdrawal from chronic alcohol. Depression has been postulated to be a disease mediated by microglia63,64. Indeed, knockout of the Stat3 gene in microglia alleviates depression-like behavior in mice27. Further investigation will be necessary to disentangle whether activation of STAT3 signaling in microglia, astrocytes, or both cell types, promotes depression-like behavior during withdrawal from chronic alcohol exposure.
Socs3 was one STAT3 target gene whose expression increased during withdrawal from chronic alcohol exposure. Socs3 transcription increases after cytokine receptor engagement and STAT3 activation. SOCS3 acts in a negative feedback loop to inhibit STAT3 and provide a molecular brake on an overactive immune response65,66. The induction of Socs3 during withdrawal from chronic alcohol drinking might be a mechanism of limiting pro-inflammatory signaling. Consistent with its role as a negative regulator of STAT3, knockout of Socs3 in mouse astrocytes have the opposite effect of Stat3 knockout on inflammation in a spinal cord injury model67. Further investigation is necessary to understand the role of SOCS3 in alcohol withdrawal-induced neuroinflammation and the fine balance between STAT3 and SOCS3 signaling in pro- vs. anti-inflammatory signaling in the brain in response to alcohol. Interestingly, expression of Lif and Osmr, both encoding STAT3 activators68,69, were increased during alcohol withdrawal and in WGCNA module 1 (Fig. 1F and Table S2). They could be involved in activating STAT3 in the hippocampus during alcohol withdrawal.
The behavioral relevance of activated STAT3 in the hippocampus during alcohol withdrawal was established using STAT3 inhibitor (stattic) treatment. Rats in alcohol withdrawal exhibited an anhedonic phenotype, as reported by us earlier10. Interestingly, treatment with stattic alleviated alcohol withdrawal-induced anhedonia, indicating that activated STAT3 may promote anhedonia. Targeting STAT3 pharmacologically has the potential to dampen an overactive neuroimmune response in AUD and treat comorbid AUD and depression in humans. Although stattic is not suitable for use in humans, other STAT3 inhibitors are being developed for cancer therapy and other disease indications70 and could conceivably be repurposed for AUD.
Supplementary information
Acknowledgements
This work was funded by grants from the National Institute on Alcohol Abuse and Alcoholism (P50 AA022538 to A.W.L., S.C.P., and D.R.G.; U01 AA020912 to A.W.L.; and T32 AA026577 to K.H.) and the National Center for Advancing Translational Science (UL1 TR002003 to M.M.C.). S.C.P. is also supported by the senior research career scientist award from Department of Veterans Affairs. We would like to thank Alvaro J. Hernandez and Christopher J. Fields at the Roy J. Carver Biotechnology Center at the University of Illinois at Urbana-Champaign for RNA sequencing and help with bioinformatics, respectively. We would also like to thank the Fluorescence Imaging Core at the University of Illinois at Chicago and Luana Martins de Carvalho for assistance with qPCR. The Center for Alcohol Research in Epigenetics received human postmortem tissues from the New South Wales Brain Tissue Resource Centre at the University of Sydney, which is funded by the National Institute on Alcohol Abuse and Alcoholism (R28 AA012725). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-021-01421-8.
References
- 1.Petit G, Deschietere G, Loas G, Luminet O, de Timary P. Link between anhedonia and depression during early alcohol abstinence: gender matters. Alcohol Alcohol. 2020;55:71–77. doi: 10.1093/alcalc/agz090. [DOI] [PubMed] [Google Scholar]
- 2.Petit G, et al. Gender differences in affects and craving in alcohol-dependence: a study during alcohol detoxification. Alcohol Clin. Exp. Res. 2017;41:421–431. doi: 10.1111/acer.13292. [DOI] [PubMed] [Google Scholar]
- 3.Greenfield SF, et al. The effect of depression on return to drinking: a prospective study. Arch. Gen. Psychiatry. 1998;55:259–265. doi: 10.1001/archpsyc.55.3.259. [DOI] [PubMed] [Google Scholar]
- 4.Oliva F, et al. Gender differences in anxiety and depression before and after alcohol detoxification: anxiety and depression as gender-related predictors of relapse. Eur. Addict. Res. 2018;24:163–172. doi: 10.1159/000490046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akil H, et al. Treatment resistant depression: a multi-scale, systems biology approach. Neurosci. Biobehav Rev. 2018;84:272–288. doi: 10.1016/j.neubiorev.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mira RG, et al. Alcohol impairs hippocampal function: from NMDA receptor synaptic transmission to mitochondrial function. Drug Alcohol Depend. 2019;205:107628. doi: 10.1016/j.drugalcdep.2019.107628. [DOI] [PubMed] [Google Scholar]
- 7.Zahr NM, Pfefferbaum A, Sullivan EV. Perspectives on fronto-fugal circuitry from human imaging of alcohol use disorders. Neuropharmacology. 2017;122:189–200. doi: 10.1016/j.neuropharm.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfonso-Loeches S, Guerri C. Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Crit. Rev. Clin. Lab Sci. 2011;48:19–47. doi: 10.3109/10408363.2011.580567. [DOI] [PubMed] [Google Scholar]
- 9.Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010;30:8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen WY, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol. 2019;78:79–87. doi: 10.1016/j.alcohol.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewin SE, et al. Chronic intermittent ethanol exposure selectively increases synaptic excitability in the ventral domain of the rat hippocampus. Neuroscience. 2019;398:144–157. doi: 10.1016/j.neuroscience.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ezquer F, et al. Intranasal delivery of mesenchymal stem cell-derived exosomes reduces oxidative stress and markedly inhibits ethanol consumption and post-deprivation relapse drinking. Addict. Biol. 2019;24:994–1007. doi: 10.1111/adb.12675. [DOI] [PubMed] [Google Scholar]
- 13.Kane CJ, et al. Effects of ethanol on immune response in the brain: region-specific changes in aged mice. J. Neuroinflammation. 2013;10:66. doi: 10.1186/1742-2094-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kane CJ, et al. Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcohol Clin. Exp. Res. 2014;38:384–391. doi: 10.1111/acer.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberto M, Nelson TE, Ur CL, Gruol DL. Long-term potentiation in the rat hippocampus is reversibly depressed by chronic intermittent ethanol exposure. J. Neurophysiol. 2002;87:2385–2397. doi: 10.1152/jn.2002.87.5.2385. [DOI] [PubMed] [Google Scholar]
- 16.Crews FT, Vetreno RP. Neuroimmune basis of alcoholic brain damage. Int Rev. Neurobiol. 2014;118:315–357. doi: 10.1016/B978-0-12-801284-0.00010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erickson EK, Grantham EK, Warden AS, Harris RA. Neuroimmune signaling in alcohol use disorder. Pharm. Biochem. Behav. 2019;177:34–60. doi: 10.1016/j.pbb.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belmer A, Patkar OL, Lanoue V, Bartlett SE. 5-HT1A receptor-dependent modulation of emotional and neurogenic deficits elicited by prolonged consumption of alcohol. Sci. Rep. 2018;8:2099. doi: 10.1038/s41598-018-20504-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vetreno RP, Hall JM, Savage LM. Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiol. Learn Mem. 2011;96:596–608. doi: 10.1016/j.nlm.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dowlati Y, et al. A meta-analysis of cytokines in major depression. Biol. Psychiatry. 2010;67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Wohleb ES, Franklin T, Iwata M, Duman RS. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016;17:497–511. doi: 10.1038/nrn.2016.69. [DOI] [PubMed] [Google Scholar]
- 22.Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017;18:374–384. doi: 10.1038/ni.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mulero MC, Huxford T, Ghosh G. NF-kappaB, IkappaB, and IKK: integral components of immune system signaling. Adv. Exp. Med. Biol. 2019;1172:207–226. doi: 10.1007/978-981-13-9367-9_10. [DOI] [PubMed] [Google Scholar]
- 24.Yakovleva T, Bazov I, Watanabe H, Hauser KF, Bakalkin G. Transcriptional control of maladaptive and protective responses in alcoholics: a role of the NF-kappaB system. Brain Behav. Immun. 2011;25:S29–38. doi: 10.1016/j.bbi.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, W., Vetreno, R. P. & Crews, F. T. Hippocampal TNF-death receptors, caspase cell death cascades, and IL-8 in alcohol use disorder. Mol. Psychiatry10.1038/s41380-020-0698-4 (2020). [DOI] [PMC free article] [PubMed]
- 26.Kong E, et al. STAT3 controls IL6-dependent regulation of serotonin transporter function and depression-like behavior. Sci. Rep. 2015;5:9009. doi: 10.1038/srep09009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon SH, et al. Dysfunction of microglial STAT3 alleviates depressive behavior via neuron-microglia interactions. Neuropsychopharmacology. 2017;42:2072–2086. doi: 10.1038/npp.2017.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong ML, Dong C, Maestre-Mesa J, Licinio J. Polymorphisms in inflammation-related genes are associated with susceptibility to major depression and antidepressant response. Mol. Psychiatry. 2008;13:800–812. doi: 10.1038/mp.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts AJ, et al. Increased IL-6 expression in astrocytes is associated with emotionality, alterations in central amygdala GABAergic transmission, and excitability during alcohol withdrawal. Brain Behav. Immun. 2019;82:188–202. doi: 10.1016/j.bbi.2019.08.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruol DL, Huitron-Resendiz S, Roberts AJ. Altered brain activity during withdrawal from chronic alcohol is associated with changes in IL-6 signal transduction and GABAergic mechanisms in transgenic mice with increased astrocyte expression of IL-6. Neuropharmacology. 2018;138:32–46. doi: 10.1016/j.neuropharm.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Der-Avakian A, Markou A. The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci. 2012;35:68–77. doi: 10.1016/j.tins.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J. Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC. Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int J. Neuropsychopharmacol. 2014;17:313–322. doi: 10.1017/S1461145713001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Lun AT, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline [version 2; peer review: 5 approved] F1000Res. 2016;5:1438. doi: 10.12688/f1000research.8987.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Law CW, Chen Y, Shi W, Smyth G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc. Natl Acad. Sci. USA. 1999;96:6964–6969. doi: 10.1073/pnas.96.12.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bugno M, et al. Identification of the interleukin-6/oncostatin M response element in the rat tissue inhibitor of metalloproteinases-1 (TIMP-1) promoter. Nucleic Acids Res. 1995;23:5041–5047. doi: 10.1093/nar/23.24.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caldenhoven E, et al. Activation of the STAT3/acute phase response factor transcription factor by interleukin-5. J. Biol. Chem. 1995;270:25778–25784. doi: 10.1074/jbc.270.43.25778. [DOI] [PubMed] [Google Scholar]
- 43.Egusquiaguirre SP, Yeh JE, Walker SR, Liu S, Frank DA. The STAT3 target gene TNFRSF1A modulates the NF-kappaB pathway in breast cancer cells. Neoplasia. 2018;20:489–498. doi: 10.1016/j.neo.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gressner OA, Peredniene I, Gressner AM. Connective tissue growth factor reacts as an IL-6/STAT3-regulated hepatic negative acute phase protein. World J. Gastroenterol. 2011;17:151–163. doi: 10.3748/wjg.v17.i2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ito K, et al. Gfap and Osmr regulation by BRG1 and STAT3 via interchromosomal gene clustering in astrocytes. Mol. Biol. Cell. 2018;29:209–219. doi: 10.1091/mbc.E17-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yanagisawa M, et al. Astrocyte differentiation of fetal neuroepithelial cells by interleukin-11 via activation of a common cytokine signal transducer, gp130, and a transcription factor, STAT3. J. Neurochem. 2000;74:1498–1504. doi: 10.1046/j.1471-4159.2000.0741498.x. [DOI] [PubMed] [Google Scholar]
- 47.Ogata K, Kosaka T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience. 2002;113:221–233. doi: 10.1016/S0306-4522(02)00041-6. [DOI] [PubMed] [Google Scholar]
- 48.McClintick JN, et al. Stress-response pathways are altered in the hippocampus of chronic alcoholics. Alcohol. 2013;47:505–515. doi: 10.1016/j.alcohol.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melendez RI, McGinty JF, Kalivas PW, Becker HC. Brain region-specific gene expression changes after chronic intermittent ethanol exposure and early withdrawal in C57BL/6J mice. Addict. Biol. 2012;17:351–364. doi: 10.1111/j.1369-1600.2011.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith ML, et al. Time-course analysis of brain regional expression network responses to chronic intermittent ethanol and withdrawal: implications for mechanisms underlying excessive ethanol consumption. PLoS ONE. 2016;11:e0146257. doi: 10.1371/journal.pone.0146257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Z, Yuan Q, Mash DC, Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl Acad. Sci. USA. 2011;108:6626–6631. doi: 10.1073/pnas.1018514108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolfe SA, et al. Ethanol and a rapid-acting antidepressant produce overlapping changes in exon expression in the synaptic transcriptome. Neuropharmacology. 2019;146:289–299. doi: 10.1016/j.neuropharm.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brenner E, et al. Single cell transcriptome profiling of the human alcohol-dependent brain. Hum. Mol. Genet. 2020;29:1144–1153. doi: 10.1093/hmg/ddaa038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in CNS inflammation. Neuron. 2020;108:608–622. doi: 10.1016/j.neuron.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ceyzeriat K, Abjean L, Carrillo-de Sauvage MA, Ben Haim L, Escartin C. The complex STATes of astrocyte reactivity: how are they controlled by the JAK-STAT3 pathway? Neuroscience. 2016;330:205–218. doi: 10.1016/j.neuroscience.2016.05.043. [DOI] [PubMed] [Google Scholar]
- 56.Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zamanian JL, et al. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Das S, Li Z, Noori A, Hyman BT, Serrano-Pozo A. Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflammation. 2020;17:227. doi: 10.1186/s12974-020-01898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson MA, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ceyzeriat K, et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018;6:104. doi: 10.1186/s40478-018-0606-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reichenbach N, et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019;11:e9665. doi: 10.15252/emmm.201809665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu Y, et al. Differential pro-inflammatory responses of astrocytes and microglia involve STAT3 activation in response to 1800 MHz radiofrequency fields. PLoS ONE. 2014;9:e108318. doi: 10.1371/journal.pone.0108318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng SL, Chen JG, Wang F. Microglia: a central player in depression. Curr. Med. Sci. 2020;40:391–400. doi: 10.1007/s11596-020-2193-1. [DOI] [PubMed] [Google Scholar]
- 64.Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci. 2015;38:637–658. doi: 10.1016/j.tins.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 65.Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front. Immunol. 2014;5:58. doi: 10.3389/fimmu.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J. Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okada S, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 68.Hsu MP, Frausto R, Rose-John S, Campbell IL. Analysis of IL-6/gp130 family receptor expression reveals that in contrast to astroglia, microglia lack the oncostatin M receptor and functional responses to oncostatin M. Glia. 2015;63:132–141. doi: 10.1002/glia.22739. [DOI] [PubMed] [Google Scholar]
- 69.Lapp DW, Zhang SS, Barnstable CJ. Stat3 mediates LIF-induced protection of astrocytes against toxic ROS by upregulating the UPC2 mRNA pool. Glia. 2014;62:159–170. doi: 10.1002/glia.22594. [DOI] [PubMed] [Google Scholar]
- 70.Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting Janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharm. Rev. 2020;72:486–526. doi: 10.1124/pr.119.018440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.