

Abstract
Peptidomimetics have gained great attention for their function as protein–protein interaction (PPI) inhibitors. Herein, we report the design and investigation of a series of right-handed helical heterogeneous 1:1 α/Sulfono-γ-AA peptides as unprecedented inhibitors for p53-MDM2 and p53-MDMX. The most potent helical heterogeneous 1:1 α/Sulfono-γ-AA peptides were shown to bind tightly to MDM2 and MDMX, with Kd of 19.3 and 66.8 nM, respectively. Circular dichroism spectra, 2D-NMR spectroscopy, and the computational simulations suggested that these helical sulfono-γ-AA peptides could mimic the critical side chains of p53 and disrupt p53/MDM2 PPI effectively. It was noted that these 1:1 α/Sulfono-γ-AA peptides were completely resistant to proteolytic degradation, boosting their potential for biomedical applications. Furthermore, effective cellular activity is achieved by the stapled 1:1 α/Sulfono-γ-AA peptides, evidenced by significantly enhanced p53 transcriptional activity and much more induced level of MDM2 and p21. The 1:1 α/Sulfono-γ-AA peptides could be an alternative strategy to antagonize a myriad of PPIs.
Graphical Abstract
INTRODUCTION
In the endeavor of searching and designing molecules that bind to important biological targets such as proteins with high affinity and specificity, peptides predominately serve as the leads.1,2 This is particularly true for the identification of molecules that modulate protein–protein interactions (PPIs) in which large, flat, and noncontiguous interfaces are involved. However, natural peptides are deemed not suitable for the therapeutic development due to their intrinsic drawbacks such as susceptibility to proteolytic degradation.3 To alleviate the problem, non-natural sequence-specific peptidomimetics have emerged to be a viable alternative strategy.4–14 Compared to their natural counterparts, the peptidomimetics could not only retain or mimic the folding domain of the peptides such as helices but also display distinct structures and functions. Additionally, the chemodiversity is enhanced due to the ease of introducing diverse and unnatural functional side chains.3,15 Of course, thanks to their unnatural backbones, they are generally resistant to enzymatic hydrolysis, which enhances bioavailability.16 Furthermore, peptidomimetic foldamers could mimic the primary, secondary, and even tertiary structures of peptides and proteins, and their potential application in biomedical science is enhanced through the structure-based design.17,18 In the past two decades, significant efforts have been made to develop new classes of peptidomimetics and successfully employ them into the study of biomolecular recognition and protein–protein interactions (PPIs).19–23 However, it remains an urgent task to exploit new peptidomimetic foldamers bearing new molecular scaffolds and frameworks due to current limited availability and application of peptidomimetics.
To advance the field of peptidomimetics, we have recently developed a new class of foldamer, γ-AA peptides (oligomers of N-acylated-N-aminoethyl amino acids), which were created based on the molecular scaffold γ-chiral PNA but with an enlarged chemodiversity to mimic peptides rather than nucleic acids.3,17 γ-AA peptides are highly resistant to enzymatic hydrolysis and have shown excellent cellular translocation capability.15,16,25–27 The modular synthesis of γ-AA peptides and their enormous chemodiversity make them excellent candidates for identification of valuable molecular probes or drug candidates through combinatorial library screening.15,28,29 More importantly, as a subclass of γ-AA peptides, sulfono-γ-AA peptides have been found to adopt a series of unprecedented helical secondary structures, stabilized by both intramolecular hydrogen bonding and the curved nature of sulfonamido moieties in the molecular framework,24,30–32 endowing them with superior folding propensity to the α-helix in solution. As such, sulfono-γ-AA peptides could be ideal candidates that could be rationally designed to mimic helical domains of proteins and disrupt medicinally related PPIs. Indeed, the homogeneous sulfono-γ-AA peptides, which form the left-handed 414 helical structure,31 have been shown to mimic the critical residues of the helical domain of BCL9 and disrupt the β-catenin/B cell lymphoma 9 protein–protein interaction (PPI)16 and to mimic GLP-1 to recognize GLP1-R and decrease blood glucose.33 Recently, we have also demonstrated that they could be designed to mimic those important residues of the p53 helix and antagonize p53/MDM2 PPI.27 However, even if the results are highly promising, the mimicry of a right-handed α-helix by a left-handed helix may not be ideal, particularly when residues on the multiface of the helix are involved in interactions at the protein–protein interface. Previous structural studies show that 1:1 heterogeneous α/Sulfono-γ-AA peptides adopt a robust right-handed 413 windmill-shaped helices with a helical pitch of 5.34 Å (Figure 1A–E),30,34 implying their close similarity to the α-helix (pitch: 5.34 Å). Inspired by this desired structural information, we speculated that 1:1 heterogeneous α/Sulfono-γ-AA peptides could be a new class of helix mimetics as potential PPI inhibitors. In this paper, as a proof of concept, for the first time, we explored the heterogeneous 1:1 α/Sulfono-γ-AA peptides for the mimicry of the α-peptide helix. It is known that p53 is a tumor suppressor that plays a prominent role in the oncogenic transformation process and protects higher organisms from cancer. MDM2 and MDMX share considerable structural homology, binding to the N-terminus of p53 and leading to its transcriptional activity inhibition and degradation.35 Recent findings suggest that overexpression of MDM2 and MDMX is a key factor that results in multiple human cancers, making them the promising target for antitumor drug development.22,36–40 Hence, p53–MDM2 and p53–MDMX protein–protein interactions were selected as the model applications to evaluate the feasibility and practicality of our new developed foldamer.
Figure 1.
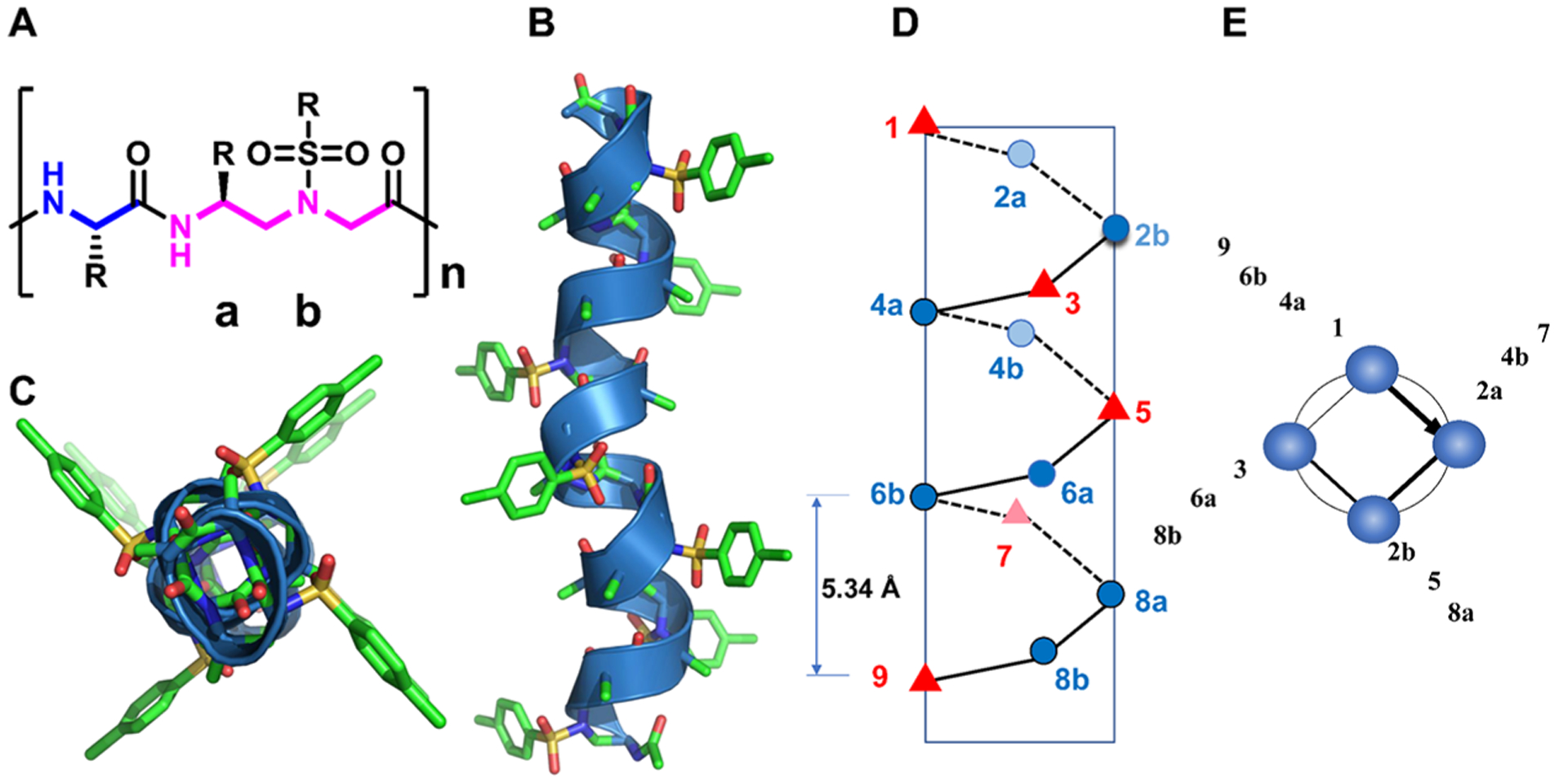
(A) Chemical structure of 1:1 α/Sulfono-γ-AA peptides. a and b denote the chiral side chain and the sulfonamido side chain from the building block, respectively. (B) The crystal structure of a 1:1 α/Sulfono-γ-AA peptide.24 (C) Top view of panel (D,E). The schematic representation of the distribution of side chains from 1:1 α/Sulfono-γ-AA peptides. (D) Side view. (E) Top view.
RESULTS AND DISCUSSION
Peptide Design and Activity Test.
p53/MDM2 PPI has been a testing base for validation of helical peptidomimetics for the mimicry of the α-helix. As shown in Figure 2, three crucial residues of p53, Phe19, Trp23, and Leu26, on one face of the p53 helix, make multiple hydrophobic contacts at the deep pocket of the MDM2 cleft. It is demonstrated that molecules that can reproduce the binding modes of these three residues are expected to bind MDM2 tightly.41–45 As shown in Figure 1D, the pitch of 1:1 right-handed α/Sulfono-γ-AA peptides is 5.34 Å, essentially the same as that for the α-helix. Due to the characteristic of four side chains per turn (Figure 1E), we could select any face of the helix to mimic p53 (Figure 2).
Figure 2.
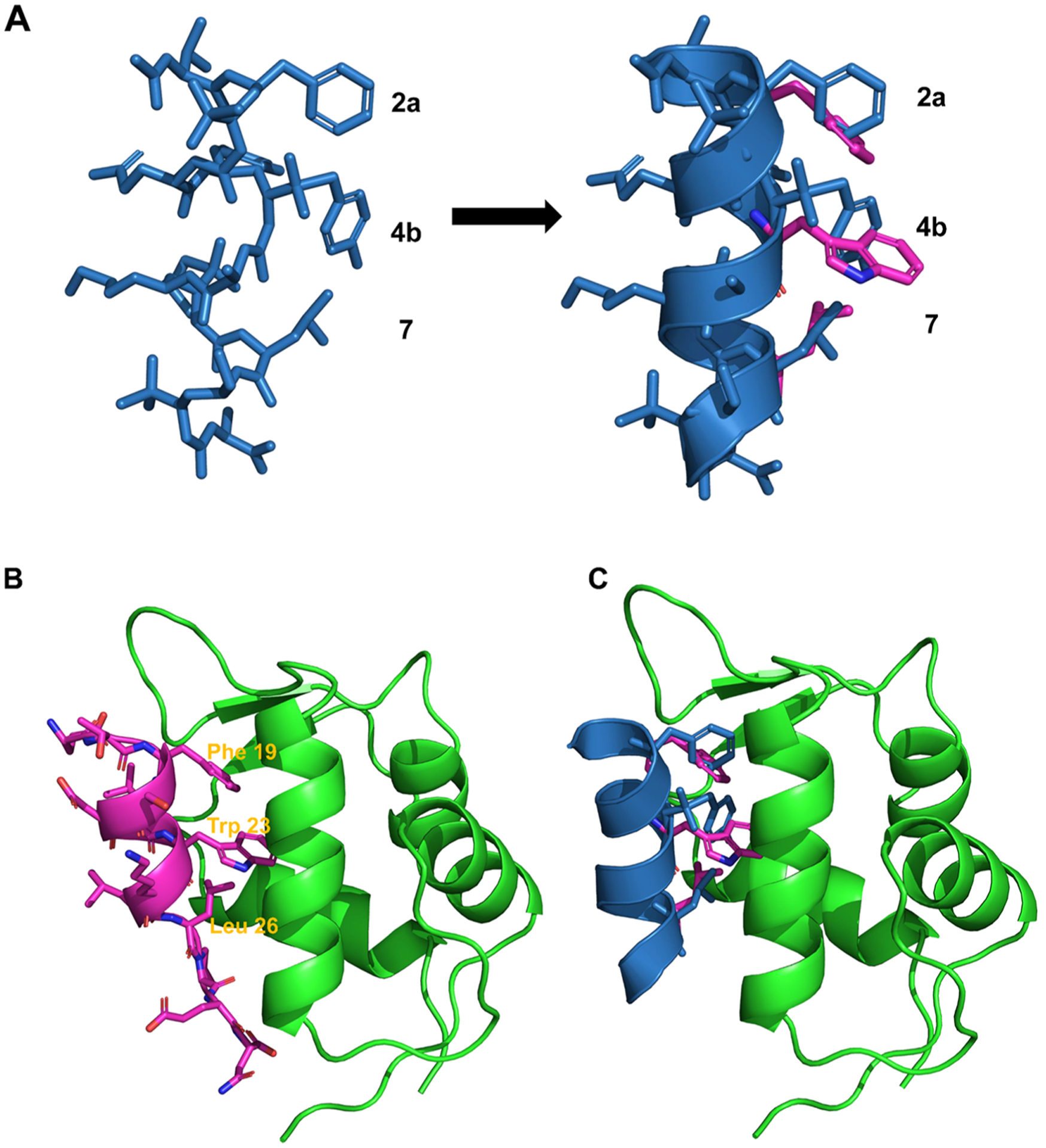
(A) Superimposition of the p53 α-helix (purple) with a designed 1:1 α/Sulfono-γ-AA peptide (blue) (a PDB file is shown in the Supporting Information). (B) The structure overlay of p53 (purple) binding and MDM2 (green) (PDB: 1YCR). (C) Overlay of the p53 helix (purple) with a 1:1 α/Sulfono-γ-AA peptide (blue) on the binding of MDM2 (green) (a PDB file is shown in the Supporting Information).
As an initial attempt, we randomly selected 2a, 4b, and 7 of 1:1 α/sulfono-γ-AA peptides to mimic F19, W23, and L26 in p53. 2a comes from the chiral side chain of the sulfono-γ-AA building block (Scheme 1, compounds 1–3) to mimic F19, 7 is the leucine amino acid residue to mimic L26, and 4b possesses sulfonyl side chains such as sulfonyl-2-naphthalene to mimic the W23 (Scheme 1, compounds 1–3). To our delight, the first peptide 1, which contains the sulfonyl-2-naphthalene group, shows a Kd of 19.3 nM, revealing an almost 18-fold higher binding affinity toward MDM2 than the p53 (16–29) peptide (Kd, 360 nM, Scheme 1; Figure S2). It strongly suggested that the approach of 1:1 α/Sulfono-γ-AA peptides for the mimicry of the α-helix is viable. We next probed the effect of the side chains on positions 2a and 7. When the Trp group is used to mimic Phe on the 2a position (compound 2, Scheme 1), Kd = 82.7 nM, suggesting that although both indole and phenyl groups are hydrophobic, a phenyl group (compound 1) is more preferable to mimic F19 in p53. Interestingly, replacing the Leu 7 in compound 1 with the cyclobutylmethyl group residue (Scheme 1, compound 3), the activity is still slightly weaker than that of 1 (Scheme 1, Kd = 72.6 nM), implying that Leu is the optimized residue. Subsequently, we explored the importance of the side chains at the position 4b. It is noted that compound 4 bearing the sulfonyl-1-naphthalene side chain induces a dramatic drop in the binding affinity (Scheme 1, Kd = 482 nM), possibly due to the short group that could not make hydrophobic contact with the p53 binding domain of MDM2. The hypothesis is supported by the result that when changing this position to sulfonyl-TPE groups, the binding affinity of compound 5 becomes 6-fold potent as p53 (Scheme 1, Kd = 62 nM). Instead of the naphthalene moiety, the effect of the phenyl group, as well as the substituent effect on its aromatic ring, was also investigated at the 4b position. Interestingly, although their binding affinities are all less than that of 1, they bind more tightly to MDM2 than the p53 peptide.
Scheme 1.
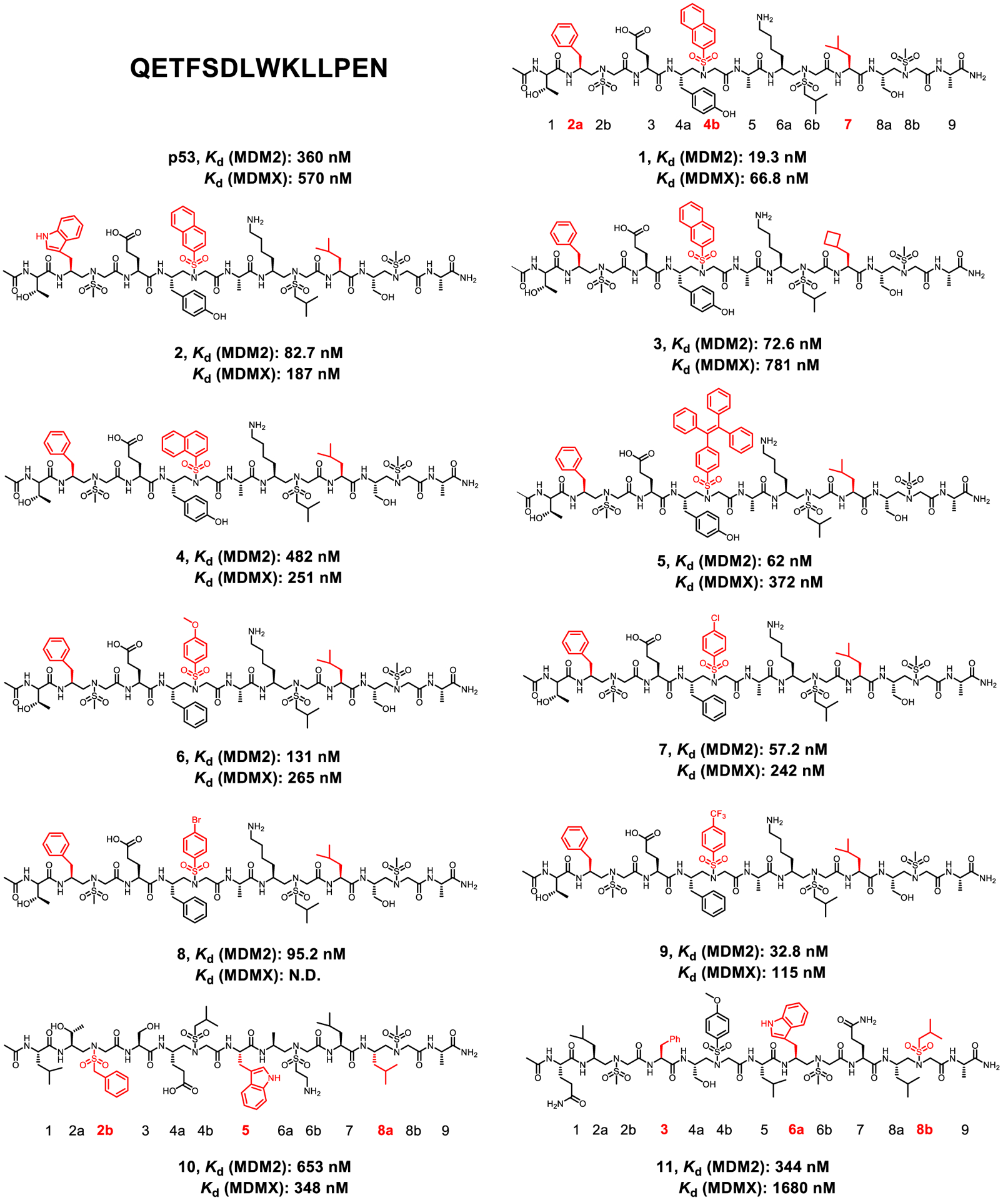
Structures of the p53 and 1:1 α/Sulfono-γ-AA peptide 1–11
It is intriguing that the binding affinity reveals the relationship of 4-CF3 > 4-Cl > 4-Br > 4-OCH3 (compounds 9, 7, 8, and 6; Scheme 1). We speculated that the stronger electron-withdrawing group may facilitate the interaction with the MDM2 binding pocket. As shown in Figure 1D, since the 1:1 α/Sulfono-γ-AA peptide helical scaffold has four faces, we postulated that residues on the other faces, such as 2b-5–8a and 3–6a-8b, could also be used for the mimicry of three hot-spot side chains F19, W23, and L26 in p53. As such, we synthesized compound 10 (Scheme 1) in which the side chains on the positions of 2b, 5, and 8a were designed to mimic F19, W23, and L26 in p53. To our disappointment, the binding affinity decreased sharply compared to that of 1 (Scheme 1, Kd = 653 nM). Also, if the side chains at the positions of 3, 6a, and 8b are used to mimic those three crucial residues in p53, although the activity is slightly improved (compound 11, Scheme 1, Kd = 344 nm) and comparable to that of the p53 peptide, it is still much less potent than compound 1. These findings suggest that the precise arrangement of side chains on each helical face of 1:1 α/Sulfono-γ-AA peptides is not identical. In the meantime, the binding affinity is also highly affected by the neighboring side chains that are not directly involved in the binding to the target protein. The IC50 values of these compounds are shown in Figure S4.
As MDMX is also involved in p53 binding and affecting the p53 signaling pathway, the binding affinities of these helical mimetics to MDMX are obtained. As shown in Scheme 1 and Figure S3, most of the 1:1 α/Sulfono-γ-AA peptides have shown good Kd values to MDMX compared to p53, demonstrating that these peptides are potential dual-inhibitors to both MDM2 and MDMX.
CD (Circular Dichroism) Spectra.
In order to evaluate the helical propensity of these peptides in solution, the circular dichroism (CD) spectra are next recorded in PBS (100 μM) in the range of 195–260 nm. As shown in Figure 3, most of our compounds revealed a pronounced maximum at 198–202 nm and a minimum at 212 nm, suggesting that these peptides (1–11) adopt similar right-handed helical conformations.34 Interestingly, a minimum of less than 200 nm was observed for p53 (16–29), signifying a random coil with almost no α-helix population. The data may explain why 1:1 α/Sulfono-γ-AA peptides are better MDM2 binders than p53.
Figure 3.
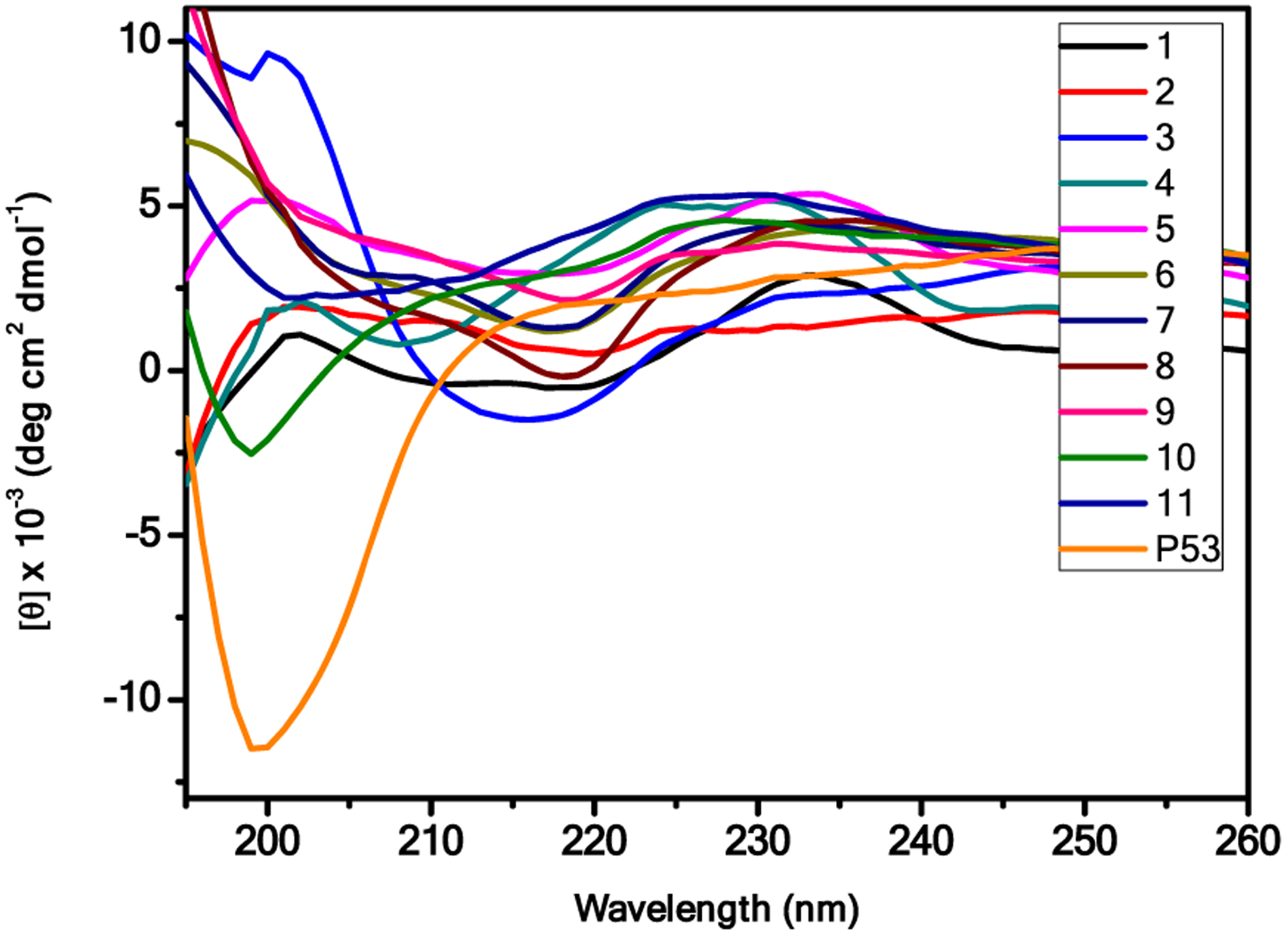
CD spectra of the p53 and compounds 1–11.
HSQC NMR of Lead Peptide 9 in Complex with MDM2.
As both 1 and 9 have comparable binding activities whereas 9 has slightly better solubility than 1, we chose 9 for NMR study to further elucidate its binding to MDM2. Nuclear magnetic resonance (NMR) spectroscopy was used to determine if the compound 9 binding site on MDM217–125 was similar to the binding site for the p53 transactivation domain (TAD). Figure 4A demonstrates the overlay of 1H–15N HSQC spectra before (blue) and after (red) the addition of increasing concentrations of 9. We measured the amide proton and nitrogen chemical shift changes of a uniformly 15N-labeled sample of MDM217–125 after a stoichiometric excess of 9 was added. Figure 4B shows the average chemical shift changes in ppm for the amide proton and nitrogen resonances in MDM217–125. The average chemical shift change for all the detectable MDM217–125 resonances when bound to 9 was 0.048 ppm. Figure 4C,D shows the structure of MDM217–125 in orange bound to p53TAD15–29 in green.46 Figure 4C shows MDM217–125 residues with chemical shift changes greater than 0.048 ppm when bound to 9 colored red; 33 residues had chemical shift changes above the average. Figure 4D shows MDM217–125 residues with chemical shift changes greater than 0.048 ppm when bound to p53TAD1–73 colored red; 52 residues had chemical shift changes above the average.
Figure 4.
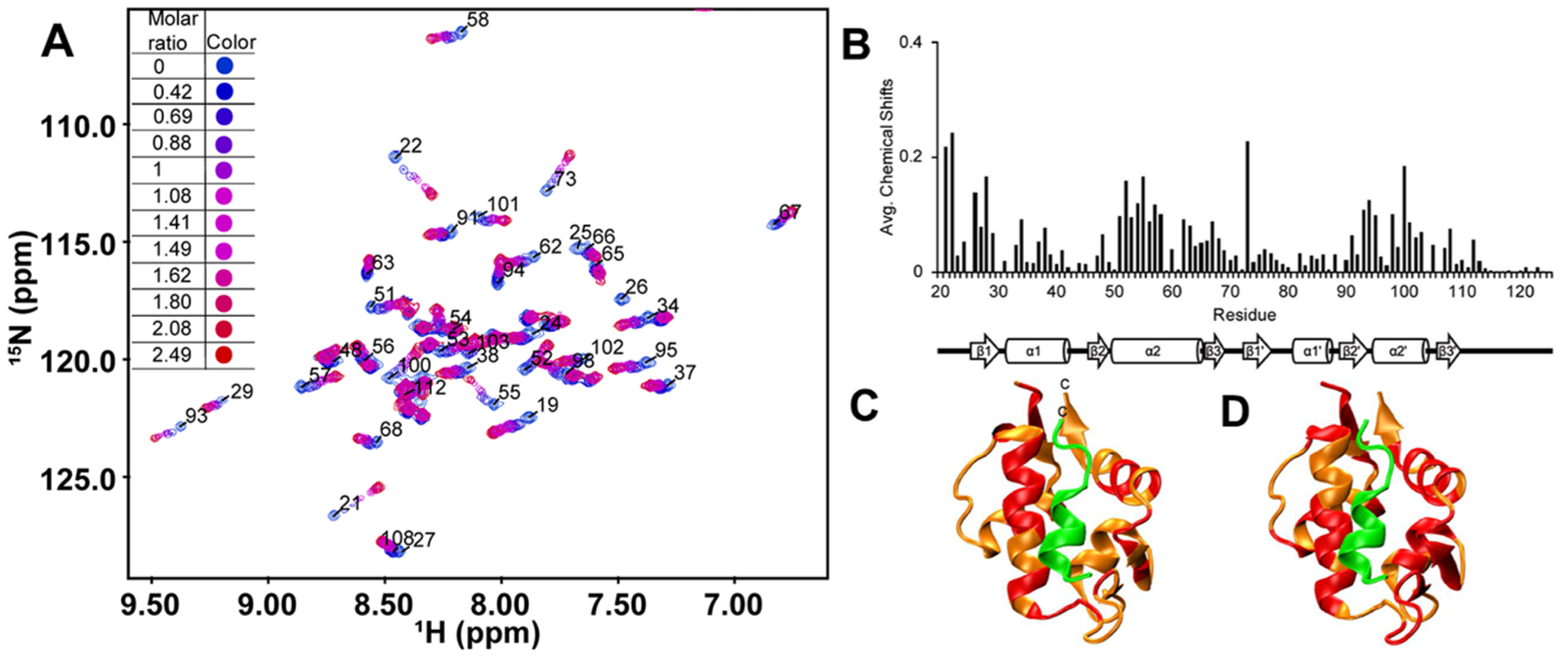
(A) Overlay of 1H–15N HSQC spectra for before (blue) and after (red) the addition of increasing concentrations of 9. (B) Average chemical shift changes in ppm (parts per million) of the MDM2 p53 binding domain (residues 17–125) when bound to 9. The 2° structure of MDM217–125 is shown below the graph. (C) Ribbon structure of MDM217–125 (orange) and p53TAD (green, residues 15–29) (PDB: 1YCR). Residues colored red have chemical shifts above the average of 0.048 ppm when bound to 9. (D) Ribbon structure of MDM217–125 (orange) and p53TAD15–29 (green) (PDB: 1YCR). Residues colored red have chemical shifts above the average of 0.048 ppm when bound to p53TAD1–73.
MDM217–125 β1 and β2 had shifts in the presence of 9 and p53TAD1–73, as well as β3’. α1, β1’, β2’, and α2’ had more residues with shifts above the 0.048 ppm threshold in the presence of p53TAD1–73 than 9 and β3 had shifts only in the presence of 9 while α1’ had shifts only in the presence of p53TAD1–73. α2 had shifts in the presence of both 9 and p53TAD1–73. For 9, most of the large chemical shift changes were localized to the N-terminal half of α2. For p53TAD1–73, most of the large chemical shift changes were localized to the C-terminal half of α2. Based on these results, it appears that the binding site for 9 is similar to p53TAD1–73 but not identical. We are currently using this data to optimize peptide design.
Stapled 1:1 α/Sulfono-γ-AA Peptide Induces Activation of p53 in Cells.
To investigate the abilities of the compounds in activating p53 in cells, stapled 1:1 lactam-bridged α/Sulfono-γ-AA peptides 12–15 were synthesized via a side chain cross-linking strategy based on the most potent sequences 1 and 2 (Figure 5A–C) in the hope of enhancing cell permeability.47 The IC50s of compounds 12–15 are 11.7, 9.4, 4.9, and 9.0 μM, respectively (Figure S4), which was better than those of corresponding linear compounds 1 and 2. The luciferase reporter assay was employed to check the efficiency of wild-type p53 in activating the BP-100 MDM2 promoter, which constructed into the promoting sequence of luciferase (Figure S9). Unlike the linear compounds 1 and 2, enhanced luciferase activities (1.5–2-fold) were observed in the cells treated with stapled sequences. U2OS cells expressing endogenous wild-type p53 under negative regulation by both MDM2 and MDMX were treated with the stapled peptides. Treatment with all four stapled compounds led to increased p53 levels to various degrees. Modest induction of p53 target genes p21 and MDM2 were also detected, suggesting increased activities of p53 due to these compounds (Figure 5D). The modest effect of the compounds in activating p53 in cell culture suggests that further optimization is needed to improve cell permeability and fully exploit their MDM2 binding ability. Overall, the results demonstrated the inhibitory function of these compounds for MDM2 in vitro, as well as their potential to be further developed into cell-permeable activators of p53 for cancer treatment.
Figure 5.
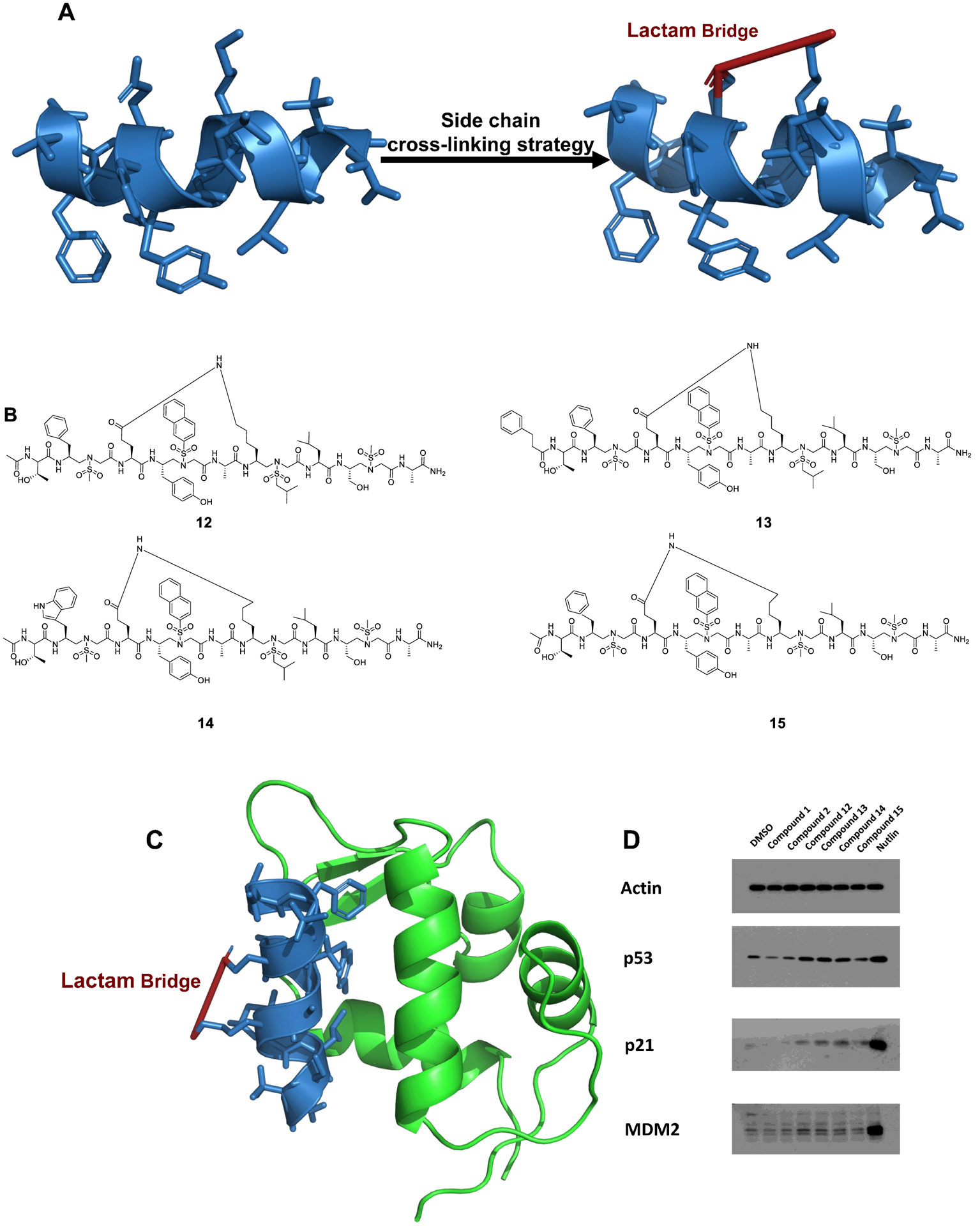
(A) Design and synthesis of the stapled sequences (a PDB file is shown in the Supporting Information). (B) The structures of the stapled 1:1 α/Sulfono-γ-AA peptides 12–15. (C) Overlay of the stapled 1:1 α/Sulfono-γ-AA peptide (blue) on the binding of MDM2 (a PDB file is shown in the Supporting Information). (D) Activation of cellular p53 by compounds 1 and 2 and stapled p53 helical mimetics. U2OS osteosarcoma cells expressing endogenous wild-type p53 were treated with indicated compounds at 30 μM for 16 h. The cells were analyzed by Western blot for the level of p53 pathway markers.
Enzymatic Stability Study.
One distinctive characteristic of peptidomimetics is the remarkable resistance to enzymatic degradation, which has been proven in our previous study.16 Herein, the stability of the 1:1 α/Sulfono-γ-AA peptide was evaluated by using p53 (16–29) as the control peptide. 0.1 mg/mL of compound 1 and p53 were incubated with 0.1 mg/mL pronase in 100 mM ammonium bicarbonate buffer (pH 7.8) at 37 °C for 24 h. After being monitored by HPLC-MS (Figures S6–S7), the peptide p53 was found to be completely degraded by pronase as multiple unidentified peaks with no intact peptide remaining were detected. Meanwhile, the peptide 1 showed negligible degradation, indicating that these 1:1 α/Sulfono-γ-AA peptides were of great stability against proteolysis.
CONCLUSIONS
In this paper, we have designed and synthesized a series of unprecedented right-handed helical 1:1 α/Sulfono-γ-AA peptide inhibitors based on the single crystal structure. The feasibility and practicality of the new helical peptidomimetic scaffolds were evaluated by mimicking the p53 helix for targeting MDM2 and MDMX. The activities of these peptides were evaluated by Kd. Besides, through the 2D-NMR experiment, the lead compound 9 was proven to interact with the p53-binding pocket of MDM2. The induction of p53 accumulation and its transcriptional targets p21 and MDM2 were also detected among the stapled 1:1 α/Sulfono-γ-AA peptide, suggesting that these peptides have the potential to be further developed into in vivo p53 activators. Furthermore, these peptidomimetics have shown great resistance toward the enzymatic degradation, which also make them to be promising therapeutic agents. This work provided a streamlined approach to discover potent peptidomimetic inhibitors of a myriad of protein–protein interactions.
EXPERIMENTAL SECTION
General Information.
Fmoc-protected amino acids were purchased from Chem-impex (Wood Dale, IL). Rink-Amide-MBHA resin (0.64 mmol/g) was purchased from GL Biochem (Shanghai, China). 1-Hydroxybenzotriazole wetted with no less than 20 wt % water (HOBt), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and N,N-diisopropylethylamine (DIPEA) were purchased from Oakwood Chemical (Estill, SC). Tetraphenylethylene was purchased from Alfa Aesar. Chlorosulfonic acid was purchased from Sigma Aldrich. Solid phase synthesis was conducted in peptide synthesis vessels on a Burrell Wrist-Action shaker. γ-AA peptides were analyzed and purified on a Waters Breeze 2 HPLC system installed with both the analytic module (1 mL/min) and preparative module (16 mL/min), by employing a method using a 5–100% linear gradient of solvent B (0.1% TFA in acetonitrile) in solvent A (0.1% TFA in water) over 40 min followed by 100% solvent B over 10 min. Then the pure peak was collected and lyophilized on a Labcono lyophilizer. The purity of the compounds was determined to be >95% by analytical HPLC. High-resolution mass spectra were obtained on an Agilent 6220 using electrospray ionization time-of-flight (ESI-TOF). 1H NMR spectra were recorded at 400 MHz using TMS as the internal standard. 13C NMR spectra were recorded at 100 MHz using TMS as the internal standard. The multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), multiplet (m). Coupling constants are reported in hertz (Hz).
Preparation of Sulfono-γ-AA Building Blocks.
The sulfono-γ-AA building blocks were synthesized based on a previous report.16 As shown in Scheme S1, the initial starting materials were Fmoc-protected amino acids. Sulfono-γ-AA building blocks 4–10 and 15 were synthesized based on route 1, while sulfono-γ-AA building blocks 1–3 and 11–14 were synthesized based on route 2.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(tert-butoxy)phenyl)propyl)-n-(naphthalen-2-ylsulfonyl)glycine (BB1).
1H NMR (400 MHz, DMSO-d6): δ 8.38 (d, J = 12 Hz, 1H), 8.05–8.13 (m, 4H), 7.82 (s, 2H), 7.63–7.65 (m, 3H), 7.23–7.35 (m, 6H), 7.04 (d, J = 8 Hz, 1H), 6.94 (d, J = 8 Hz, 1H), 6.71 (d, J = 8 Hz, 1H), 6.69 (d, J = 8 Hz, 1H), 4.12–4.18 (m, 2H), 3.8 (s, 2H), 3.52–3.57 (m, 2H), 3.36–3.43 (m, 1H), 3.14–3.23 (m, 1H), 2.71–2.84 (m, 1H), 1.11 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 170.8, 155.9, 153.6, 144.2, 141.1, 132.1, 130.4, 130.0, 129.8, 129.7, 129.2, 128.3, 128.1, 127.9, 127.8, 127.4, 125.6, 123.7, 123.0, 120.5, 115.3, 77.9, 65.6, 52.1, 51.9, 49.4, 47.0, 31.7, 28.9, 16.6. HRMS (ESI) ([M + H]+) Calcd. for C40H41N2O7S: 693.2634, found: 693.2650.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(tert-butoxy)phenyl)propyl)-N-(naphthalen-1-ylsulfonyl)glycine (BB2).
1H NMR (400 MHz, DMSO-d6): δ 8.52 (d, J = 8 Hz, 1H), 8.18 (d, J = 4 Hz, 1H), 8.02–8.09 (m, 2H), 7.82 (s, 2H), 7.56–7.66 (m, 5H), 7.27–7.36 (m, 5H), 6.97 (d, J = 8 Hz, 1H), 6.85 (d, J = 8 Hz, 1H), 6.71 (d, J = 8 Hz, 2H), 6.58 (d, J = 4 Hz, 1H), 4.08–4.26 (m, 2H), 4.02–4.06 (m, 1H), 3.72 (s, 2H), 3.33–3.49 (m, 2H), 2.58–2.72 (m, 1H), 2.37–2.46 (m, 2H), 1.11 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 170.6, 155.9, 153.6, 144.2, 141.1, 135.4, 134.5, 130.3, 129.9, 129.4, 129.1, 128.5, 128.4, 128.0, 127.4, 127.2, 125.6, 124.9, 123.7, 120.5, 115.3, 77.9, 67.4, 65.7, 51.78, 49.2, 47.1, 37.4, 37.1, 31.7, 28.9, 27.7 HRMS (ESI) ([M + H]+) Calcd. for C40H41N2O7S: 693.2634, found: 693.2650.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(tert-butoxy)phenyl)propyl)-N-((4-(1,2,2-triphenylvinyl)phenyl)-sulfonyl)glycine (BB3).
1H NMR (400 MHz, DMSO-d6): δ 7.85 (d, J = 8 Hz, 2H), 7.60 (d, J = 4 Hz, 2H), 7.47 (d, J = 4 Hz, 2H), 7.37 (s, 2H), 7.27–7.31 (m, 4H), 7.05–7.08 (m, 12H), 6.88–6.96 (m, 5H), 6.75 (d, J = 8 Hz, 2H), 6.62 (d, J = 8 Hz, 1H), 4.14–4.18 (m, 1H), 4.08–4.11 (m, 2H), 4.01–4.05 (m, 2H), 3.85–3.92 (m, 2H), 3.28–3.32 (m, 1H), 3.03–3.09 (m, 1H), 2.67–2.75 (m, 1H), 1.14 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 170.6, 155.9, 153.6, 148.1, 144.2, 143.0, 142.8, 141.1, 139.4, 137.4, 131.7, 131.1, 130.9, 130.1, 128.4, 128.3, 128.0, 127.4, 127.3, 126.9, 125.6, 123.8, 120.5, 77.9, 65.8, 51.7, 49.5, 47.1, 37.5, 31.7, 28.9 HRMS (ESI) ([M + H]+) Calcd. for C56H53N2O7S: 897.3573, found: 897.3582.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenyl-propyl)-N-((4-chlorophenyl)sulfonyl)glycine (BB4).
1H NMR (400 MHz, DMSO-d6): δ 12.82 (s, 1H), 7.83 (d, J = 8 Hz, 2H), 7.73 (d, J = 8 Hz, 2H), 7.47 (t, J = 4 Hz, 4H), 7.33–7.38 (m, 2H), 7.19–7.24 (m, 7H), 7.13 (d, J = 4 Hz, 1H), 4.13 (s, 1H), 4.07–4.09 (m, 2H), 3.97–4.02 (m, 1H), 3.84 (s, 1H), 3.35 (q, J = 12 Hz, 2H), 3.17 (q, J = 12 Hz, 1H), 2.83–2.87 (m, 1H), 2.55–2.61 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.5, 155.9, 144.2, 141.1, 139.0, 138.8, 129.7, 129.5, 129.3, 128.5, 128.0, 127.4, 126.5, 125.6, 120.5, 65.7, 52.2, 51.7, 49.3, 47.1, 37.9 HRMS (ESI) ([M + H]+) Calcd. for C32H30ClN2O6S: 605.1513, found: 605.1520.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenyl-propyl)-N-((4-bromophenyl)sulfonyl)glycine (BB5).
1H NMR (400 MHz, DMSO-d6): δ 12.83 (s, 1H), 7.83 (d, J = 8 Hz, 2H), 7.72 (d, J = 8 Hz, 2H), 7.65 (d, J = 4 Hz, 2H), 7.54–7.56 (m, 2H), 7.36 (q, J = 12 Hz, 2H), 7.19–7.29 (m, 8H), 7.13 (d, J = 4 Hz, 1H), 4.13(s, 1H), 4.07–4.08 (m, 2H), 3.97–4.01 (m, 1H), 3.84 (s, 1H), 3.34 (q, J = 12 Hz, 1H), 3.18 (q, J = 12 Hz, 1H), 2.84 (q, J = 12 Hz, 1H), 2.55–2.61 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.5, 155.9, 144.2, 141.1, 139.2, 139.0, 129.5, 129.4, 128.5, 128.0, 127.4, 126.5, 125.6, 120.5, 65.7, 52.2, 51.7, 49.3, 47.1, 37.9. HRMS (ESI) ([M + H]+) Calcd. for C32H30BrN2O6S: 649.1008, found: 649.1001.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenyl-propyl)-N-((4-(trifluoromethyl)phenyl)sulfonyl)glycine (BB6).
1H NMR (400 MHz, DMSO-d6): δ 12.86 (s, 1H), 7.95 (d, J = 8 Hz, 2H), 7.88 (d, J = 8 Hz, 2H), 7.83 (d, J = 8 Hz, 2H), 7.55 (d, J = 8 Hz, 2H), 7.36 (q, J = 12 Hz, 2H), 7.19–7.29 (m, 7H), 7.13 (d, J = 4 Hz, 1H), 4.13(s, 1H), 4.07–4.08 (m, 2H), 3.96–4.01 (m, 1H), 3.86 (s, 1H), 3.39 (q, J = 12 Hz, 2H), 3.23 (q, J = 12 Hz, 1H), 2.85 (q, J = 12 Hz, 1H), 2.57–2.62 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 170.4, 155.9, 144.2, 144.1, 143.8, 141.1, 138.9, 129.5, 128.5, 128.4, 127.9, 127.4, 126.7, 126.5, 125.6, 125.3, 122.7, 120.5, 65.7, 52.2, 51.7, 49.3, 47.1, 37.9. HRMS (ESI) ([M + H]+) Calcd. for C33H30F3N2O6S: 639.1777, found: 639.1780.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenyl-propyl)-N-(methylsulfonyl)glycine (BB7).
1H NMR (400 MHz, DMSO-d6): δ 7.80 (d, J = 8 Hz, 2H), 7.53 (d, J = 8 Hz, 2H), 7.33 (t, J = 8 Hz, 2H), 7.23 (q, J = 12 Hz, 3H), 7.16 (d, J = 4 Hz, 4H), 7.08–7.11 (m, 1H), 4.12 (d, J = 8 Hz, 2H), 4.05–4.06 (m, 1H), 4.00–4.03 (m, 2H), 3.87 (s, 1H), 3.31 (q, J = 12 Hz, 1H), 3.14(q, J = 16 Hz, 1H), 2.88 (s, 3H), 2.79–2.853 (m, 1H), 2.49–2.55 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 171.3, 156.1, 144.2, 144.1, 139.1, 129.5, 128.5, 127.9, 127.4, 126.4, 125.5, 120.5, 65.7, 51.9, 51.6, 49.0, 47.1, 37.9. HRMS (ESI) ([M + H]+) Calcd. for C27H29N2O6S: 509.1746, found: 509.1750.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-(methylsulfonyl)glycine (BB8).
1H NMR (400 MHz, DMSO-d6): δ 7.80 (d, J = 8 Hz, 2H), 7.60 (d, J = 4 Hz, 2H), 7.34 (t, J = 4 Hz, 2H), 7.24–7.27 (m, 2H), 7.06 (d, J = 12 Hz, 1H), 4.27 (d, J = 8 Hz, 2H), 4.13–4.15 (m, 1H), 3.89 (s, 2H), 3.64 (s, 2H), 3.16 (q, J = 16 Hz, 1H), 3.01 (q, J = 12 Hz, 1H), 2.85 (s, 3H), 1.46–1.47 (m, 1H), 1.13–1.20 (m, 1H), 0.76 (q, J = 12 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.3144.3, 141.1, 127.9, 127.4, 125.5, 120.5, 65.5, 51.9, 48.8, 48.1, 47.2, 41.2, 24.6, 23.6, 21.9. HRMS (ESI) ([M + H]+) Calcd. for C24H31N2O6S: 475.1903, found: 475.1910.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-(isobutylsulfonyl)glycine (BB9).
1H NMR (400 MHz, DMSO-d6): δ 7.81 (d, J = 8 Hz, 2H), 7.61 (q, J = 8 Hz, 2H), 7.34 (t, J = 8 Hz, 2H), 7.23–7.27 (m, 2H), 7.08 (d, J = 12 Hz, 1H), 4.28–4.31 (m, 1H), 4.19–4.24 (m, 1H), 4.12–4.15 (m, 1H), 3.90 (s, 2H), 3.62–3.65 (m, 1H), 3.19 (q, J = 12 Hz, 1H), 3.05 (q, J = 16 Hz, 1H), 2.89 (t, J = 8 Hz, 2H), 1.96–2.04 (m, 1H), 1.46–1.48 (m, 1H), 1.14–1.21 (m, 2H), 0.9 (q, J = 8 Hz, 6H), 0.77 (q, J = 8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.4, 144.3, 144.1, 141.1, 127.9, 127.4, 125.4, 120.5, 65.5, 59.3, 51.8, 48.6, 47.9, 47.2, 24.6, 23.6, 22.5, 21.9. HRMS (ESI) ([M + H]+) Calcd. for C27H37N2O6S: 517.2372, found: 517.2386.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-((tert-butoxycarbonyl)amino)hexyl)-N-(isobutylsulfonyl)glycine (10BB).
1H NMR (400 MHz, DMSO-d6): δ 7.81 (d, J = 8 Hz, 2H), 7.63 (q, J = 4 Hz, 2H), 7.34 (t, J = 8 Hz, 2H), 7.25–7.28 (m, 2H), 7.10 (d, J = 8 Hz, 1H), 6.66 (s, 1H), 4.19–4.26 (m, 2H), 4.14–4.16 (m, 1H), 3.92 (s, 2H), 3.55 (s, 2H), 3.20–3.24 (m, 1H), 3.05–3.11 (m, 1H), 2.90–2.95 (m, 2H), 2.83–2.88 (m, 2H), 1.98–2.05 (m, 1H), 1.29 (s, 9H), 1.18–1.24 (m, 5H), 0.9 (q, J = 8 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.4, 155.9, 144.2, 141.1, 128.0, 127.4, 125.6, 120.5, 77.8, 65.6, 59.4, 51.5, 49.9, 48.6, 48.5, 47.2, 31.9, 29.7, 28.6, 24.6, 23.1, 22.6. HRMS (ESI) ([M + H]+) Calcd. for C32H46N3O8S: 632.3006, found: 632.3009.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1-(tert-butoxycarbonyl)-1H-indol-3-yl)propyl)-N-(methylsulfonyl)-glycine (BB11).
δ 7.96 (d, J = 8 Hz, 1H), 7.78 (q, J = 4 Hz, 2H), 7.62 (d, J = 8 Hz, 1H), 7.49 (d, J = 4 Hz, 3H), 7.30–7.33 (m, 3H), 7.23–7.26 (m, 1H), 7.17 (q, J = 16 Hz, 3H), 4.12–4.15 (m, 2H), 4.08–4.09 (m, 1H), 3.90 (s, 2H), 3.42 (q, J = 12 Hz, 1H), 3.23 (q, J = 12 Hz, 1H), 2.91 (s, 3H), 2.87 (s, 1H), 2.66–2.72 (m, 1H), 1.5 (s, 1H), 1.47 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 171.3, 171.2, 158.9, 158.6, 156.3, 149.4, 144.1, 144.0, 141.1, 135.1, 130.9, 127.9, 127.8, 127.4, 125.5, 120.4, 117.8, 83.8, 65.9, 52.1, 50.1, 49.2, 47.1, 27.9. HRMS (ESI) ([M + H]+) Calcd. for C34H38N3O8S: 648.2380, found: 648.2371.
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butoxy)propyl)-N-((4-methoxyphenyl)sulfonyl)glycine (BB12).
1H NMR (400 MHz, DMSO-d6): δ 7.77 (d, J = 4 Hz, 6H), 7.33 (t, J = 4 Hz, 2H), 7.24–7.27 (m, 2H), 7.06 (d, J = 8 Hz, 1H), 6.68 (d, J = 8 Hz, 2H), 4.23–4.25 (m, 2H), 4.16 (d, J = 8 Hz 1H), 3.96–4.10 (m, 2H), 3.74 (s, 1H), 3.69 (s, 3H), 3.42 (d, J = 12 Hz 1H), 3.20–3.22 (m, 3H), 1.02 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 170.7, 162.8, 156.2, 144.2, 144.1, 131.5, 129.7, 128.0, 127.4, 125.6, 120.4, 114.6, 72.9, 65.9, 61.8, 55.9, 55.8, 51.1, 49.4, 47.1, 27.5. HRMS (ESI) ([M + H]+) Calcd. for C31H37N2O8S: 597.2271, found: 597.2268.
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butoxy)propyl)-N-(methylsulfonyl)glycine (BB13).
1H NMR (400 MHz, DMSO-d6): δ 7.82 (d, J = 8 Hz, 2H), 7.64 (d, J = 8 Hz, 2H), 7.36 (t, J = 4 Hz, 2H), 7.27 (t, J = 8 Hz, 2H), 7.11 (d, J = 8 Hz, 1H), 4.25–4.26 (m, 2H), 4.16–4.18 (m, 1H), 3.9–4.2 (m, 2H), 3.70 (s, 1H), 3.33–3.38 (m, 1H), 3.13–3.24 (m, 3H), 2.9 (s, 3H), 1.05 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.2, 144.2, 128.0, 127.4, 125.6, 120.5, 73.0, 65.8, 61.2, 50.9, 49.1, 48.9, 47.1, 27.6. HRMS (ESI) ([M + H]+) Calcd. for C25H33N2O7S: 505.2008, found: 505.2012.
N-((2R,3S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butoxy)butyl)-N-(benzylsulfonyl)glycine (BB14).
1H NMR (400 MHz, DMSO-d6): δ 7.81 (d, J = 8 Hz, 2H), 7.65 (d, J = 8 Hz, 2H), 7.34–7.35 (m, 5H), 7.28–7.29 (m, 3H), 7.17–7.24 (m, 2H), 4.27–4.38 (m, 4H), 4.17 (t, J = 8 Hz, 1H), 3.92–3.87 (m, 1H), 3.77 (s, 1H), 3.52–3.55 (m, 3H), 3.9 (t, J = 12 Hz, 1H), 1.07 (s, 9H), 0.88 (d, J = 8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.5, 144.3, 141.1, 131.3, 130.1, 128.7, 128.5, 128.0, 127.4, 125.6, 125.5, 120.5, 120.4, 73.8, 67.2, 65.8, 58.5, 54.8, 49.2, 47.5, 47.3, 28.5, 17.9. HRMS (ESI) ([M + H]+) Calcd. for C32H39N2O7S: 595.2478, found: 595.2468.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propyl)-N-((2-((tert-butoxycarbonyl)amino)ethyl)sulfonyl)glycine (BB15).
1H NMR (400 MHz, DMSO-d6): δ 7.81 (d, J = 8 Hz, 2H), 7.62 (s, 2H), 7.34 (t, J = 8 Hz, 2H), 7.26–7.28 (m, 2H), 7.16–7.18 (m, 1H), 6.83 (s, 1H), 4.23 (t, J = 8 Hz, 2H), 4.14 (s, 1H), 3.68 (s, 2H), 3.25 (s, 2H), 3.17 (s, 3H), 1.29 (s, 9H), 1.00 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.0, 155.7, 144.3, 141.1, 128.0, 127.4, 125.6, 120.5, 78.5, 65.7, 52.4, 51.5, 48.8, 47.2, 45.7, 35.2, 28.5, 18.6. HRMS (ESI) ([M + H]+) Calcd. for C27H36N3O8S: 562.2223, found: 562.2216.
Preparation of the α/Sulfono-γ-AA Peptide.
The synthesis of the α/sulfono-γ-AA peptides was carried out on 150 mg of Rink Amide-MBHA resin (0.64 mmol/g) at room temperature (Scheme S2). The resin was soaked in DMF for 5 min, and then the Fmoc protecting group was deprotected in 20% piperidine in DMF solution (2 mL, 10 min × 2). The resin was thoroughly washed with DCM (3 mL, 10 min × 3) and DMF (3 mL, 10 min × 3). After that, Fmoc-deprotected amino acid (2 equiv.), HOBt (4 equiv.), and DIC (4 equiv.) were added and shaken in 2 mL of DMF. After 3 h, the resin was washed with DCM and DMF and then treated with 20% piperidine/DMF solution (10 min × 2). Next, a solution of the α/Sulfono-γ-AA peptide building block (2 equiv.), HOBt (4 equiv.), and DIC (4 equiv.) in 2 mL of DMF was added and reacted for 4 h, and then the Fmoc protecting group was removed as stated above. The reaction cycles were repeated until the desired α/Sulfono-γ-AA peptides were synthesized. The N-terminus of the sequence was capped with acetic anhydride (1 mL) in pyridine (2 mL,15 min) followed by treatment with TFA/DCM (4 mL, 1:1, v/v) for 2.5 h. The cleavage solution was collected, and the resin was washed with DCM (3 mL × 2). The solution was combined and evaporated under air flow to give the crude product, which was analyzed and purified using a Waters HPLC system. The gradient eluting method of 5 to 100% of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over 40 min was performed. All the α/Sulfono-γ-AA peptides were obtained with moderate yields after prep-HPLC purification.
For the FITC-labeled α/sulfono-γ-AA peptide synthesis, after attaching the last α-amino acid, the Fmoc protecting group was then removed. FITC (1.2 equiv.) in 2 mL of DMF and DIPEA (6 equiv.) were added to the resin and shaken overnight to complete the reaction. After washing with DMF (×3) and DCM (×3), the resin was cleaved using TFA/DCM (6 mL, 1:1, v/v) for 3 h. Then, the pure FITC-labeled α/Sulfono-γ-AA peptides were obtained using the same abovementioned method.
Fluorescence Polarization Competition Assays.
The binding affinities (Kd) of the AA peptides were investigated by fluorescence polarization (FP). GST-MDM2-1-150 containing human MDM2 was expressed in E. coli as previously described by us. FP was carried out by incubating the 50 nM FITC-labeled AA peptide with MDM2/MDMX (0 to 1 μM) in 1× PBS with 0.1% Pluronic F-68. Dissociation constants (Kd) were determined by plotting the fluorescence anisotropy values as a function of protein concentration, and the plots were fitted to the following equation, where Lst is the concentration of the peptide, and x stands for the concentration of the protein. The experiments were performed in triplicate and repeated three times.
Circular Dichroism.
Circular dichroism (CD) spectra were measured on an Aviv 215 circular dichroism spectrometer using a 1 mm path length quartz cuvette, and compound solutions in PBS buffer (or in trifluoroethanol) were prepared using dry weight of the lyophilized solid followed by dilution to give the desired concentration (100 uM) and solvent combination. 10 scans were averaged for each sample, and three independent experiments were conducted, and the spectra were averaged. The final spectra were normalized by subtracting the average blank spectra. Molar ellipticity [θ] (deg·cm2·dmol−1) was calculated using the equation
where θobs is the measured ellipticity in millidegree, n is the number of side groups, l is the path length in centimeter (0.1 cm), and c is the concentration of the sulfono-γ-AA peptide in molar unit.
Luciferase Reporter Assay.
U2OS cells (p53 wild-type osteosarcoma) with stably integrated BP100-luc (p53-responsive luciferase reporter) and CMV-lacZ (internal control for cell mass and toxicity) were treated with compounds for 18 h. Luciferase and lacZ activities were determined, and the ratio of luc/lacZ indicates p53 transcriptional activity. Positive control Nutlin was used at 4 and 8 μM, respectively.
Western Blotting.
To detect proteins by Western blotting, cells were lysed in lysis buffer (50 mM Tris–HCl [pH 8.0], 150 mM NaCl, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× protease inhibitor cocktail) and centrifuged at 4 °C for 10 min at 14,000g. The supernatant was boiled in Laemmli sample buffer for 5 min and subjected to SDS-PAGE and Western blotting to detect the expression of proteins indicated in the figures using the corresponding antibodies.
Enzymatic Stability Study.
Lead compounds and peptide control p53 (0.1 mg/mL) were incubated with 0.1 mg/mL pronase in 100 mM ammonium bicarbonate buffer (pH 7.8) at 37 °C for 24 h. Then, the reaction mixtures were concentrated in a speed vacuum at medium temperature to remove water and ammonium bicarbonate. The resulting residues were redissolved in H2O/CH3CN and analyzed on a Waters analytical HPLC system with a 1 mL/min flow rate and 5 to 100% linear gradient of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over the duration of 50 min. The UV detector was set to 215 nm.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by NIH 9R01AI152416 and NIH 5R01AG056569.
ABBREVIATIONS USED
- DMF
dimethylformamide
- DCM
dichloromethane
- DIPEA
N, N-diisopropylethylamine
- EDC
ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FITC
fluorescein isothio-cyanate
- HOBt
hydroxybenzotriazole
- IC50
half maximal inhibitory concentration
- Kd
dissociation constant
- MDM2
mouse double minute 2
- MDMX
also known as MDM4
- TFA
trifluoroacetic acid
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01638.
Preparation of sulfono-γ-AA building blocks; preparation of α/Sulfono-γ-AA peptides; fluorescence polarization competition assays; circular dichroism; enzymatic stability study; 1H–15N HSQC NMR of lead peptide 9 in complex with MDM2; luciferase reporter assay; 1H and 13C NMR spectra of the sulfono-γ-AA peptide building blocks (PDF)
PDB file of p53 α-helix with a designed 1:1 α/Sulfono-γ-AA peptide and MDM2 (PDB)
PDB file of stapled sequence binding with MDM2 (PDB)
Molecular formula strings and some data (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01638
The authors declare no competing financial interest.
Contributor Information
Junhao Lu, Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612, United States.
Pirada Higbee, Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, Florida 33620, United States.
Timothy Odom, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Gary Daughdrill, Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, Florida 33620, United States.
Jiandong Chen, Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612, United States.
Jianfeng Cai, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
REFERENCES
- (1).Wu Y-D; Gellman S Peptidomimetics. Acc. Chem. Res 2008, 41, 1231–1232. [DOI] [PubMed] [Google Scholar]
- (2).Jo H; Meinhardt N; Wu Y; Kulkarni S; Hu X; Low KE; Davies PL; DeGrado WF; Greenbaum DC Development of α-Helical Calpain Probes by Mimicking a Natural Protein–Protein Interaction. J. Am. Chem. Soc 2012, 134, 17704–17713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shi Y; Teng P; Sang P; She F; Wei L; Cai J γ-AApeptides: Design, Structure, and Applications. Acc. Chem. Res 2016, 49, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ko E; Liu J; Perez LM; Lu G; Schaefer A; Burgess K Universal Peptidomimetics. J. Am. Chem. Soc 2011, 133, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Huang ML; Benson MA; Shin SBY; Torres VJ; Kirshenbaum K Amphiphilic Cyclic Peptoids That Exhibit Antimicrobial Activity by Disrupting Staphylococcus aureus Membranes. Eur. J. Org. Chem 2013, 2013, 3560–3566. [Google Scholar]
- (6).Laursen JS; Engel-Andreasen J; Olsen CA β-Peptoid Foldamers at Last. Acc. Chem. Res 2015, 48, 2696–2704. [DOI] [PubMed] [Google Scholar]
- (7).Horne WS; Johnson LM; Ketas TJ; Klasse PJ; Lu M; Moore JP; Gellman SH Structural and Biological Mimicry of Protein Surface Recognition by α/β-peptide Foldamers. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14751–14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Berlicki Ł; Pilsl L; Wéber E; Mándity IM; Cabrele C; Martinek TA; Fülöp F; Reiser O Unique α,β- and α,α,β,β-Peptide Foldamers Based on cis-β-Aminocyclopentanecarboxylic Acid. Angew. Chem., Int. Ed 2012, 51, 2208–2212. [DOI] [PubMed] [Google Scholar]
- (9).Li X; Wu Y-D; Yang D α-Aminoxy Acids: New Possibilities from Foldamers to Anion Receptors and Channels. Acc. Chem. Res 2008, 41, 1428–1438. [DOI] [PubMed] [Google Scholar]
- (10).Karlsson AJ; Pomerantz WC; Weisblum B; Gellman SH; Palecek SP Antifungal Activity from 14-Helical β-Peptides. J. Am. Chem. Soc 2006, 128, 12630–12631. [DOI] [PubMed] [Google Scholar]
- (11).Kritzer JA; Lear JD; Hodsdon ME; Schepartz A Helical β-Peptide Inhibitors of the p53-hDM2 Interaction. J. Am. Chem. Soc 2004, 126, 9468–9469. [DOI] [PubMed] [Google Scholar]
- (12).Cheng RP; Gellman SH; DeGrado WF β-Peptides: From Structure to Function. Chem. Rev 2001, 101, 3219–3232. [DOI] [PubMed] [Google Scholar]
- (13).Seebach D; Gardiner J β-Peptidic Peptidomimetics. Acc. Chem. Res 2008, 41, 1366–1375. [DOI] [PubMed] [Google Scholar]
- (14).Bach AC; Espina JR; Jackson SA; Stouten PFW; Duke JL; Mousa SA; DeGrado WF Type II‘ to Type I β-Turn Swap Changes Specificity for Integrins. J. Am. Chem. Soc 1996, 118, 293–294. [Google Scholar]
- (15).Shi Y; Challa S; Sang P; She F; Li C; Gray GM; Nimmagadda A; Teng P; Odom T; Wang Y; van der Vaart A; Li Q; Cai J One-Bead–Two-Compound Thioether Bridged Macro-cyclic γ-AApeptide Screening Library against EphA2. J. Med. Chem 2017, 60, 9290–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sang P; Zhang M; Shi Y; Li C; Abdulkadir S; Li Q; Ji H; Cai J Inhibition of β-catenin/B Cell Lymphoma 9 Protein–protein Interaction using α-helix–mimicking Sulfono-γ-AApeptide Inhibitors. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 10757–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Niu Y; Hu Y; Li X; Chen J; Cai J γ-AApeptides: Design, Synthesis and Evaluation. New J. Chem 2011, 35, 542–545. [Google Scholar]
- (18).Li Y; Wu H; Teng P; Bai G; Lin X; Zuo X; Cao C; Cai J Helical Antimicrobial Sulfono-γ-AApeptides. J. Med. Chem 2015, 58, 4802–4811. [DOI] [PubMed] [Google Scholar]
- (19).Cromm PM; Spiegel J; Grossmann TN Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol 2015, 10, 1362–1375. [DOI] [PubMed] [Google Scholar]
- (20).Rezaei Araghi R; Ryan JA; Letai A; Keating AE Rapid Optimization of Mcl-1 Inhibitors using Stapled Peptide Libraries Including Non-Natural Side Chains. ACS Chem. Biol 2016, 11, 1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To K-H; Olson KA; Kesavan K; Gangurde P; Mukherjee A; Baker T; Darlak K; Elkin C; Filipovic Z; Qureshi FZ; Cai H; Berry P; Feyfant E; Shi XE; Horstick J; Annis DA; Manning AM; Fotouhi N; Nash H; Vassilev LT; Sawyer TK Stapled α–helical Peptide Drug Development: A Potent Dual Inhibitor of MDM2 and MDMX for p53-dependent Cancer Therapy. Proc. Natl. Acad. Sci. U. S. A 2013, 110, E3445–E3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhan C; Zhao L; Wei X; Wu X; Chen X; Yuan W; Lu W-Y; Pazgier M; Lu W An Ultrahigh Affinity d-Peptide Antagonist Of MDM2. J. Med. Chem 2012, 55, 6237–6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Grison CM; Miles JA; Robin S; Wilson AJ; Aitken DJ An α-Helix-Mimicking 12,13-Helix: Designed α/β/γ-Foldamers as Selective Inhibitors of Protein–Protein Interactions. Angew. Chem., Int. Ed 2016, 55, 11096–11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shi Y; Yin G; Yan Z; Sang P; Wang M; Brzozowski R; Eswara P; Wojtas L; Zheng Y; Li X; Cai J Helical Sulfono-γ-AApeptides with Aggregation-Induced Emission and Circularly Polarized Luminescence. J. Am. Chem. Soc 2019, 141, 12697–12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Niu Y; Bai G; Wu H; Wang RE; Qiao Q; Padhee S; Buzzeo R; Cao C; Cai J Cellular Translocation of a γ-AApeptide Mimetic of Tat Peptide. Mol. Pharmaceutics 2012, 9, 1529–1534. [DOI] [PubMed] [Google Scholar]
- (26).Shi Y; Parag S; Patel R; Lui A; Murr M; Cai J; Patel NA Stabilization of lncRNA GAS5 by a Small Molecule and Its Implications in Diabetic Adipocytes. Cell Chem. Biol 2019, 26, 319–330.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sang P; Shi Y; Lu J; Chen L; Yang L; Borcherds W; Abdulkadir S; Li Q; Daughdrill G; Chen J; Cai J α-Helix-Mimicking Sulfono-γ-AApeptide Inhibitors for p53–MDM2/MDMX Protein–Protein Interactions. J. Med. Chem 2020, 63, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu H; Li Y; Bai G; Niu Y; Qiao Q; Tipton JD; Cao C; Cai J gamma-AApeptide-based small-molecule ligands that inhibit Abeta aggregation. Chem. Commun 2014, 50, 5206–5208. [DOI] [PubMed] [Google Scholar]
- (29).Teng P; Zhang X; Wu H; Qiao Q; Sebti SM; Cai J Identification of novel inhibitors that disrupt STAT3-DNA interaction from a γ-AApeptide OBOC combinatorial library. Chem. Commun 2014, 50, 8739–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Teng P; Niu Z; She F; Zhou M; Sang P; Gray GM; Verma G; Wojtas L; van der Vaart A; Ma S; Cai J Hydrogen-Bonding-Driven 3D Supramolecular Assembly of Peptidomimetic Zipper. J. Am. Chem. Soc 2018, 140, 5661–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).She F; Teng P; Peguero-Tejada A; Wang M; Ma N; Odom T; Zhou M; Gjonaj E; Wojtas L; van der Vaart A; Cai J De Novo Left-Handed Synthetic Peptidomimetic Foldamers. Angew. Chem., Int. Ed 2018, 57, 9916–9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Teng P; Gray GM; Zheng M; Singh S; Li X; Wojtas L; van der Vaart A; Cai J Orthogonal Halogen-Bonding-Driven 3D Supramolecular Assembly of Right-Handed Synthetic Helical Peptides. Angew. Chem., Int. Ed 2019, 58, 7778–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sang P; Zhou Z; Shi Y; Lee C; Amso Z; Huang D; Odom T; Nguyen-Tran V; Shen W; Cai J The Activity of Sulfono-γ- AApeptide Helical Foldamers That Mimic GLP-1. Sci. Adv 2020, 6, No. eaaz4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Teng P; Ma N; Cerrato DC; She F; Odom T; Wang X; Ming LJ; van der Vaart A; Wojtas L; Xu H; Cai J Right-Handed Helical Foldamers Consisting of De Novo d-AApeptides. J. Am. Chem. Soc 2017, 139, 7363–7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Brown CJ; Lain S; Verma CS; Fersht AR; Lane DP Awakening Guardian Angels: Drugging the p53 Pathway. Nat. Rev. Cancer 2009, 9, 862–873. [DOI] [PubMed] [Google Scholar]
- (36).Li C; Pazgier M; Liu M; Lu WY; Lu W Apamin as a Template for Structure-Based Rational Design of Potent Peptide Activators of p53. Angew. Chem., Int. Ed 2009, 48, 8712–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liu M; Li C; Pazgier M; Li C; Mao Y; Lv Y; Gu B; Wei G; Yuan W; Zhan C; Lu W-Y; Lu W D-peptide Inhibitors of the p53–MDM2 Interaction for Ttargeted Molecular Therapy of Malignant Neoplasms. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 14321–14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liu M; Pazgier M; Li C; Yuan W; Li C; Lu W A Left-Handed Solution to Peptide Inhibition of the p53–MDM2 Interaction. Angew. Chem., Int. Ed 2010, 49, 3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chen S; Li X; Yuan W; Zou Y; Guo Z; Chai Y; Lu W Rapid Identification of Dual p53-MDM2/MDMX Interaction Inhibitors through Virtual Screening and Hit-based Substructure Search. RSC Adv. 2017, 7, 9989–9997. [Google Scholar]
- (40).Pazgier M; Liu M; Zou G; Yuan W; Li C; Li C; Li J; Monbo J; Zella D; Tarasov SG; Lu W Structural Basis for High-affinity Peptide Inhibition of p53 Interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4665–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen L; Borcherds W; Wu S; Becker A; Schonbrunn E; Daughdrill GW; Chen J Autoinhibition of MDMX by Intramolecular p53 Mimicry. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 4624–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wang PSP; Nguyen JB; Schepartz A Design and High-Resolution Structure of a β3-Peptide Bundle Catalyst. J. Am. Chem. Soc 2014, 136, 6810–6813. [DOI] [PubMed] [Google Scholar]
- (43).Guichard G; Cussol L; Mauran L; Buratto J; Belorusova AY; Neuville M; Osz J; Fribourg S; Fremaux J; Dolain C; Goudreau S; Rochel N Structural basis for alpha-helix mimicry and inhibition of protein-protein interactions with oligourea foldamers. Angew Chem Int Ed Engl 2020, DOI: 10.1002/anie.202008992. [DOI] [PubMed] [Google Scholar]
- (44).Murray JK; Gellman SH Targeting Protein–protein Interactions: Lessons from p53/MDM2. Biopol.-Pept. Sci 2007, 88, 657–686. [DOI] [PubMed] [Google Scholar]
- (45).Lao BB; Drew K; Guarracino DA; Brewer TF; Heindel DW; Bonneau R; Arora PS Rational Design of Topographical Helix Mimics as Potent Inhibitors of Protein-protein Interactions. J. Am. Chem. Soc 2014, 136, 7877–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kussie PH; Gorina S; Marechal V; Elenbaas B; Moreau J; Levine AJ; Pavletich NP Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [DOI] [PubMed] [Google Scholar]
- (47).Murage EN; Gao G; Bisello A; Ahn J-M Development of Potent Glucagon-like Peptide-1 Agonists with High Enzyme Stability via Introduction of Multiple Lactam Bridges. J. Med. Chem 2010, 53, 6412–6420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.