

Abstract
Momelotinib is a potent inhibitor of JAK1 and JAK2 that demonstrated efficacy in patients with primary and secondary myelofibrosis. This phase 2, open-label, randomized study evaluated the efficacy and safety of oral once-daily momelotinib (100 mg and 200 mg) for the treatment of polycythemia vera (PV) and essential thrombocythemia (ET). The primary endpoint for PV was overall response rate (ORR), defined as the proportion of patients with hematocrit < 45%, white blood cell count < 10 × 109/L, platelet count ≤400 × 109/L, and resolution of palpable splenomegaly, each lasting ≥4 weeks. The definition of ORR for ET excluded the hematocrit component. A total of 39 patients (28 PV, 11 ET) were enrolled, with 28 patients receiving ≥12 weeks of treatment. The study was terminated due to limited efficacy. Two patients (ORR 5.1%) met the primary efficacy endpoint (both PV 200 mg). Predose plasma levels of momelotinib were stable over time. A total of 31 (79.5%) patients experienced momelotinib-related adverse events (AEs), the most frequent being headache (23.1%), dizziness (18.0%), somnolence (15.4%), nausea (15.4%), and fatigue (15.4%). Three patients experienced serious AEs (7.7%), with 1 considered related to momelotinib (dyspnea). Peripheral neuropathy occurred in 7 (17.9%) patients (4 PV, 3 ET).
Keywords: Momelotinib, Polycythemia vera, Essential thrombocythemia, Phase 2 study
1. Introduction
Polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (MF) belong to a group of BCR-ABL1-negative myeloproliferative neoplasms (MPNs) [1], characterized by excessive clonal myeloproliferation, resulting, characteristically, in erythrocytosis in PV and thrombocytosis in ET [1,2]. Other disease characteristics may include leukocytosis, splenomegaly, thrombosis, bleeding, microcirculatory symptoms, and several constitutional symptoms (headache, fatigue, dizziness, pruritus, and sweating) [2,3]. Standard therapies for PV and ET are ineffective or intolerable in some patients [4,5]. Therefore, there is a need for improved, biologically targeted therapies.
A single acquired substitution of valine for phenylalanine in codon 617 in the Janus kinase (JAK) 2 gene (JAK2V617F) is frequently present in patients with PV (about 97%) or ET (about 57%) [6–9]. This mutation renders JAK2 kinase constitutively active, disrupts Signal Transducer and Activator of Transcription (STAT) signaling, and leads to cell transformation [7,9,10]. Higher burden of JAK2V617F is associated with pruritus and an increased risk of fibrotic transformation in PV [11] and arterial and venous thrombosis in ET [12]. Less common MPN-associated mutations include JAK2 exon 12 mutation (seen in PV), and calreticulin or myeloproliferative leukemia virus oncogene mutations (seen in ET) [2,13]. All of these mutations activate the JAK-STAT pathway in hematopoietic cells. A hyperactive JAK-STAT pathway, therefore, is a unifying biological abnormality in MPNs and is a target for new drug development (ie, JAK inhibitors).
Several small-molecule compounds developed to inhibit activity of JAK2 have demonstrated efficacy in patients with MF, PV, and ET [14–20], but many had further development stalled due to their toxicity (eg, neurological side effects) and/or limited efficacy [21]. One of the newer compounds, momelotinib (formerly CYT387), is an aminopyrimidine derivative that inhibits activity of JAK1 and JAK2 [22,23], and demonstrates activity in a mouse model of JAK2V617F-driven PV-like MPN [24] and in patients with primary and secondary MF [25,26].
This paper presents results of an open-label phase 2 study of momelotinib in patients with PV and ET. The study was terminated early due to limited efficacy. This paper discusses possible explanations for why momelotinib is efficacious in MF, but did not benefit patients with PV or ET.
2. Methods
2.1. Patients
Male or nonpregnant, nonlactating females, ≥18 years old diagnosed with either PV or ET, as defined by the 2008 WHO Diagnostic Criteria, requiring treatment, were eligible for this study [1]. Patients were required to have direct bilirubin ≤2x upper limit of normal (ULN); liver transaminases ≤3x ULN; calculated creatinine clearance of ≥45 mL/min; an Eastern Cooperative Oncology Group performance status of 0, 1, or 2; and a life expectancy > 24 weeks. Key exclusion criteria included prior splenectomy, major surgery within 28 days of first dose of study drug, myeloproliferative neoplasm-directed therapy (other than aspirin, hydroxyurea, anagrelide, and/or phlebotomy) within 21 days of first dose of study drug, use of strong cytochrome P450 3A4 inhibitors or inducers within 1 week of the first dose of study drug, prior use of a JAK1 or JAK2 inhibitor, hepatitis B or C infection, human immunodeficiency virus-positive, unresolved (grade > 1) non-hematologic toxicities from prior therapies, or the presence of peripheral neuropathy grade ≥2. All patients provided signed informed consent.
2.2. Study design
This was an open-label, randomized phase 2 study (ClinicalTrials.gov # NCT01998828). Patients with PV or ET were randomized via interactive voice/web response system 1:1 to receive either 100 or 200 mg of oral momelotinib once daily. Momelotinib 200 mg tablets were chosen for their equivalence to 300-mg capsules (data not shown), which demonstrated effectiveness in MF patients [25,26]. The 100-mg tablet of momelotinib achieved approximately 50% exposure compared with the 200-mg capsule, and was expected to be efficacious based on preliminary dose-response analyses (data not shown). The 100-mg dose was administered in this study to determine whether a lower dose would be sufficient to treat PV or ET compared with the dose needed to treat MF.
All patients were observed for 4 h after the first dose of momelotinib due to possible first-dose hypotension, which was reported within hours after first dose and returned to baseline within 24 h in a previous study on momelotinib in MF patients [26]. In the event of a grade 3 or 4 toxicity considered related to momelotinib, the treatment was interrupted for a maximum of 28 days and was restarted following resolution with dose reduced by 50 mg. If a recurrent grade 3 or 4 toxicity related to momelotinib occurred in patients already taking the 100-mg dose, the treatment was discontinued permanently. The treatment was administered for 24 weeks or until study discontinuation. Following completion of 24 weeks of treatment or study discontinuation, patients were followed for safety and disease status for 30 days. Patients had the option of continuing with maintenance treatment beyond 24 weeks at the clinically beneficial and/or tolerated dose, at investigators’ discretion (separate rollover study). This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice.
2.3. Study endpoints and assessments
The primary efficacy endpoint was the overall response rate (ORR), defined for the PV cohort as the proportion of patients with hematocrit < 45% in the absence of phlebotomy, white blood cell (WBC) count < 10 × 109/L, platelet count ≤400 × 109/L, and resolution of palpable splenomegaly; each lasting for at least 4 weeks. For the ET cohort, ORR was defined as WBC count < 10 × 109/L, platelet count ≤400 × 109/L, and resolution of palpable splenomegaly; each criterion met for at least 4 weeks. For patients to be considered responders, all criteria had to be met at some point during the treatment period (not necessarily for the same 4 weeks).
A key secondary endpoint was confirmed ORR for PV or ET, defined as the proportion of responders who maintained the overall response for 12 weeks (all criteria not necessarily met for the same 12 weeks). Other secondary endpoints included proportion of patients with hematocrit < 45% in the absence of phlebotomy, WBC < 10 × 109/L, platelet count ≤400 × 109/L, and resolution of palpable splenomegaly, all lasting for at least 4 weeks; and proportion of patients with ≥ 10-point decrease from baseline in the modified Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS) lasting for at least 12 weeks.
Following screening, patients received an electronic diary to complete the modified MPN-SAF TSS daily through the week 24 or study drug discontinuation visit. Clinical laboratory and disease assessments were performed at the scheduled visits every other week during the first 8 weeks of treatment, and every 4 weeks thereafter.
To monitor the pharmacokinetics of momelotinib and its metabolites, plasma samples were collected prior to dosing at 2, 4, 8, 12, 16, 20, and 24 weeks. Safety was evaluated by characterization of laboratory abnormalities and adverse events (AEs), which were graded using the Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03.
JAK2V617F allele burden was determined in whole blood by allele-specific quantitative real-time polymerase chain reaction (Cancer Genetics Inc., Rutherford, NJ). Relevant plasma markers were analyzed by immunoassay (Myriad MAPv2.0, Austin, TX).
2.4. Statistical analyses
Efficacy was assessed in the intent-to-treat analysis set, which included all randomized patients. The pharmacokinetic analysis set included all patients who received ≥ 1 dose of momelotinib and had ≥ 1 momelotinib plasma concentration measurement. Safety analyses included all patients who received ≥ 1 dose of momelotinib, with study treatment assignment designated according to the actual treatment received.
For each treatment group, ORR and corresponding 90% exact confidence intervals, confirmed ORR, and proportions of patients meeting the criteria of secondary endpoints were calculated using the binomial distribution. Resolution of splenomegaly was defined as 100% reduction in spleen size postbaseline, with baseline spleen size defined as the last spleen size measurement by palpation from the baseline period prior to randomization. Patients who met hematocrit (PV only), WBC, and platelet criteria could be counted as responders if their postbaseline spleen size (0– < 5) remained unchanged.
Baseline and postbaseline MPN-SAF TSS was defined as the average of daily total symptom score (TSS) from the 7-day period. Daily TSS was defined as the sum of 7 individual symptom scores (each on a 0–10 point scale) collected on the same day. The steady state trough plasma concentrations (at 22–26 h postdose) of momelotinib and its metabolites were summarized by dose and nominal sampling time using descriptive statistics.
3. Results
3.1. Patient disposition
From March to October 2014, 39 patients (28 PV, 11 ET) were enrolled. The study was terminated in February 2015 due to limited efficacy. By then, all enrolled patients either completed at least 12 weeks of treatment or withdrew early from the study. All data collected up to week 24 were analyzed.
All patients received at least 1 dose of momelotinib and 15/39 (38.5%) completed the study (10 PV, 5 ET; Fig. 1). The most common reasons for discontinuations were AEs, study termination, investigator’s decision, and withdrawal of consent (Fig. 1). In total, 28/39 (71.8%) patients received at least 12 weeks of treatment with momelotinib and 12/39 (30.8%) received 24 weeks of treatment.
Fig. 1.
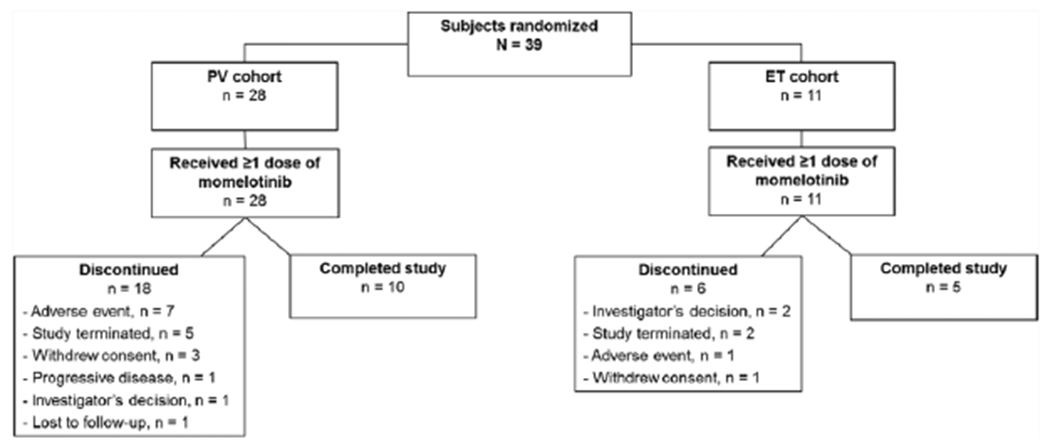
Patient disposition.
ET, essential thrombocythemia; PV, polycythemia vera.
The majority of enrolled patients were male (59.0%) and white (79.5%), with a median age (range) of 62.1 (34.4–81.0) years in the PV cohort and 51.1 (37.8–73.8) years in the ET cohort (Table 1). The median time from diagnosis was 5.2 years for PV patients and 6.6 years for ET patients. Prior PV- or ET-directed therapies (hydroxyurea, interferon, interferon-alpha 2A or 2B, pipobroman, or aspirin) were used by 12 (42.9%) PV patients and 5 (45.5%) ET patients, with a median of 1 prior therapy per cohort. In the PV cohort, 17 (60.7%) had a hematocrit ≥45%, 12 (42.9%) had platelet counts ≥400 × 109/L, 16 (57.1%) had WBC counts ≥10 × 109/L, and 7 (25.0%) had spleen size ≥5 cm below the costal margin. In the ET cohort, 11 (100%) had platelet counts ≥400 × 109/L, 1 (9.1%) had WBC counts ≥10 × 109/L, and none had spleen size ≥ 5 cm below the costal margin. The JAK2V617F mutation was evident in 45.5% of ET patients (n = 11) and 82.1% of PV patients (n = 28). The baseline mean ± standard deviation JAK2V617F allele burden was 18.9 ± 22.3% in PV patients and 3.6 ± 4.1% in ET patients. Spleen size was more variable in PV patients (Table 1).
Table 1.
Demographic and baseline clinical characteristics.
| PV Cohort |
ET Cohort |
|||||
|---|---|---|---|---|---|---|
| 100 mg (N = 14) | 200 mg (N = 14) | Total (N = 28) | 100 mg (N = 5) | 200 mg (N = 6) | Total (N = 11) | |
| Age [years] | ||||||
| Median (Range) | 61.1 (34.4–81.0) | 62.1 (51.7–75.2) | 62.1 (34.4–81.0) | 69.0 (37.8–73.8) | 47.8 (39.6–70.7) | 51.1 (37.8–73.8) |
| Hematocrit [%] | ||||||
| Median (Range) | 44.0 (36.0–54.0) | 47.5 (39.0–55.0) | 45.5 (36.0–55.0) | 42.0 (30.0–45.0) | 42.5 (38.0–45.0) | 42.0 (30.0–45.0) |
| Platelets [x 109/L] | ||||||
| Median (Range) | 446.5 (107.0–955.0) | 282.5 (116.0–877.0) | 357.5 (107.0–955.0) | 698.0 (592.0–1055.0) | 747.0 (516.0–765.0) | 739.0 (516.0–1055.0) |
| WBC count [x 109/L] | ||||||
| Median (Range) | 12.0 (5.2–71.7) | 9.4 (3.6–31.9) | 10.1 (3.6–71.7) | 6.2 (3.0–10.9) | 7.4 (5.6–8.9) | 7.1 (3.0–10.9) |
| Spleen Size [LCM], n (%) | ||||||
| 0 cm | 8 (57.1) | 11 (78.6) | 19 (67.9) | 4 (80.0) | 6 (100.0) | 10 (90.9) |
| > 0– < 5 cm | 1 (7.1) | 0 | 1 (3.6) | 1 (20.0) | 0 | 1 (9.1) |
| ≥5 – < 10 cm | 3 (21.4) | 2 (14.3) | 5 (17.9) | 0 | 0 | 0 |
| ≥ 10 cm | 1 (7.1) | 1 (7.1) | 2 (7.1) | 0 | 0 | 0 |
| Not Done | 1 (7.1) | 0 | 1 (3.6) | 0 | 0 | 0 |
| JAK2V617F Mutation Status, n (%) | ||||||
| Positive | 9 (64.3) | 14 (100.0) | 23 (82.1) | 2 (40.0) | 3 (50.0) | 5 (45.5) |
| Negative | 2 (14.3) | 0 | 2 (7.1) | 3 (60.0) | 3 (50.0) | 6 (54.5) |
| Unknown | 2 (14.3) | 0 | 2 (7.1) | 0 | 0 | 0 |
| Missing | 1 (7.1) | 0 | 1 (3.6) | 0 | 0 | 0 |
ET, essential thrombocythemia; LCM, left costal margin; PV, polycythemia vera; WBC, white blood cell.
3.2. Efficacy
Two PV patients treated with momelotinib (both taking the 200-mg daily dose) and none of the ET patients met the criteria for overall response. The total ORR for the study was 2/39 (5.1%) (Table 2). Among patients with abnormal values at baseline, there was 1 response each for hematocrit, WBC, and platelet count in PV patients and 1 WBC response in ET patients (Table 2). Two PV patients and none of the ET patients achieved spleen response (12 cm reduced to 0 cm and 5 cm reduced to 0 cm, respectively).
Table 2.
Efficacy findings.
| PV Cohort |
ET Cohort |
|||||
|---|---|---|---|---|---|---|
| 100 mg (N = 14) | 200 mg (N = 14) | Total (N = 28) | 100 mg (N = 5) | 200 mg (N = 6) | Total (N = 11) | |
| Overall response | ||||||
| n | 14 | 14 | 28 | 5 | 6 | 11 |
| Yes, n (%) | 0 | 2 (14.3) | 2 (7.1) | 0 | 0 | 0 |
| 90% CI | 0–19.3 | 2.6–38.5 | 1.3–20.8 | 0–45.1 | 0–39.3 | 0–23.8 |
| Hematocrit response, ≥ 45% at baseline | ||||||
| n | 7 | 10 | 17 | N/A | N/A | N/A |
| Yes, n (%) | 0 | 1 (10.0) | 1 (5.9) | N/A | N/A | N/A |
| 90% CI | 0–34.8 | 0.5–39.4 | 0.3–25.0 | N/A | N/A | N/A |
| Platelet response, ≥ 400 x 109/L at baseline | ||||||
| n | 7 | 5 | 12 | 5 | 6 | 11 |
| Yes, n (%) | 0 | 1 (20.0) | 1 (8.3) | 0 | 0 | 0 |
| 90% CI | 0–34.8 | 1–65.7 | 0.4–33.9 | 0–45.1 | 0–39.3 | 0–23.8 |
| WBC count response, ≥ 10 x 109/L at baseline | ||||||
| n | 10 | 6 | 16 | 1 | 0 | 1 |
| Yes, n (%) | 0 | 1 (16.7) | 1 (6.3) | 1 (100.0) | 0 | 1 (100.0) |
| 90% CI | 0–25.9 | 0.9–58.2 | 0.3–26.4 | 5–100 | N/A | 5–100 |
| Spleen responsea | ||||||
| n | 4 | 3 | 7 | 0 | 0 | 0 |
| Yes, n (%) | 1 (25.0) | 1 (33.3) | 2 (28.6) | 0 | 0 | 0 |
| 90% CI | 1.3–75.1 | 1.7–86.5 | 5.3–65.9 | N/A | N/A | N/A |
| Response in modified MPN-SAF TSS, n (%) | ||||||
| Yes | 0 | 1 (7.1) | 1 (3.6) | 0 | 1 (16.7) | 1 (9.1) |
| No | 14 (100.0) | 13 (92.9) | 27 (96.4) | 5 (100.0) | 5 (83.3) | 10 (90.9) |
| JAK2V617F allele burden response in patients with JAK2V617F mutation at baseline | ||||||
| Median baseline allele burden | ||||||
| n | 22 | 5 | ||||
| % (range) | 21.8 (1.4, 73.2) | 5.3 (0.3, 10.3) | ||||
| Median change in allele burden at week 24, | ||||||
| n | 13 | 3 | ||||
| % (range) | 99 (49, 172)b | 155 (62, 179)b | ||||
CI, confidence interval; ET, essential thrombocythemia; JAK, Janus kinase; MPN-SAF TSS, Modified Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score; N/A, not available; PV, polycythemia vera; WBC, white blood cell.
Overall Response: Patient must meet hematocrit, WBC, platelet, and spleen response criteria in PV cohort; must meet WBC, platelet, and spleen response in ET cohort at some point during the treatment period. These criteria do not have to be met for the same 4 weeks.
Hematocrit response: Hematocrit < 45% in the absence of phlebotomy that lasts ≥ 4 weeks (applicable to PV cohort only).
Platelet response: Platelet count ≤ 400 × 109/L that lasts ≥ 4 weeks.
WBC response: WBC < 10 × 109/L that lasts ≥ 4 weeks.
Spleen response: Resolution or 50% reduction of palpable splenomegaly postbaseline.
MPN-SAF TSS response: Proportion of patients with ≥ 10-point decrease from baseline in modified MPN-SAF TSS that lasted ≥ 12 weeks.
Patients with baseline spleen size ≥ 5 cm are evaluated for spleen response and serve as denominator for spleen response rate calculation.
Not significant by Wilcoxon test.
There was 1 confirmed overall response (2.6%) in the 200-mg PV cohort. Overall, 2/39 (5.1%) patients had a ≥10-point decrease in the modified MPN-SAF TSS that lasted ≥12 weeks (Table 2). Momelotinib treatment did not significantly change JAK2V617F allele burden for patients that were JAK2V617F-positive at baseline (Table 2). At 4 weeks, plasma C-reactive protein was 2-fold decreased in both cohorts (Supplemental Table 1). Changes for other inflammatory biomarkers at 4 weeks are included in Supplemental Table 1.
3.3. Pharmacokinetics
Steady state trough plasma concentrations of momelotinib were stable, with moderate variations, throughout the study. The 200-mg dose generally resulted in higher trough plasma concentrations than the 100-mg dose (Supplemental Table 2). Between weeks 2 and 24, the range of median plasma concentration was 12.1 to 26.8 ng/mL in the 100-mg PV and ET cohorts, and 22.3 to 51.3 ng/mL in the 200-mg PV and ET cohorts (Fig. 2). A similar trend was noted for momelotinib metabolites (not shown). The 2 patients who achieved overall response were not outliers in plasma momelotinib levels.
Fig. 2.
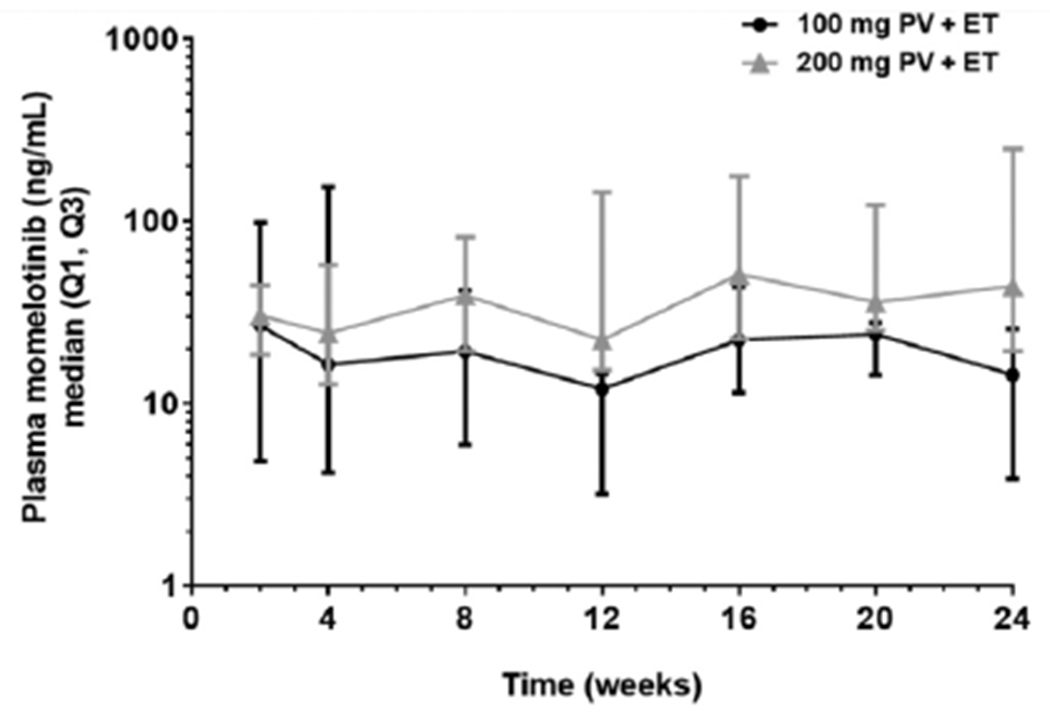
Momelotinib plasma concentration over time by dose.
ET, essential thrombocythemia; PV, polycythemia vera; Q1, first quartile; Q3, third quartile.
3.4. Safety
The median (min, max) duration of exposure to momelotinib was 143.5 (3.0, 175.0) days for PV patients and 167.0 (54.0, 169.0) days for ET patients. Of 39 patients enrolled in the study, 19 received momelotinib 100 mg/day (PV, n = 14; ET, n = 5) and 20 patients received momelotinib 200 mg/day (PV, n = 14; ET, n = 6). A total of 12/39 (30.8%) patients received momelotinib for ≥24 weeks. One PV patient treated with momelotinib 200 mg had a dose interruption due to AEs (asthma, hemoptysis, and noncardiac chest pain; none considered related to momelotinib). Six patients (PV, n = 4; ET, n = 2) had a dose reduction at the investigator’s discretion (2 due to an AE [both in the PV 200 mg cohort] and 4 due to other reasons, including withdrawal necessitating dose taper). AEs leading to dose reduction were fatigue, nausea, impaired concentration, dizziness, dyspnea, and peripheral sensory neuropathy in 1 patient (considered related to momelotinib); and fatigue, pain, and dizziness in another patient (all related to momelotinib).
In total, 36/39 (92.3%) patients reported at least 1 AE (Supplemental Table 3). The most frequent AEs were headache (5/14, [35.7%]), somnolence (4/14, [28.6%]), and hypertension (3/14, [21.4%]) in the PV 100 mg cohort; and dizziness (6/14, [42.9%]), fatigue (4/14, [28.6%]), headache (4/14, [28.6%]), hypertension (4/14, [28.6%]), and nausea (4/14, [28.6%]) in the PV 200 mg cohort. In the ET 200 mg cohort, the most common AEs were dizziness (3/6, [50.0%]), headache (3/6, [50.0%]), and thrombocytosis (3/6, [50.0%]). Headache, diarrhea, thrombocytosis, and pain in extremity each occurred in 1/5 (20.0%) patients (all in the ET 100 mg cohort). Thrombocytosis was likely related to lack of treatment efficacy.
The most common AEs considered related to momelotinib treatment were headache, dizziness, somnolence, nausea, and fatigue (Table 3). Dizziness and somnolence were more common in PV patients (6 each) than in ET patients (1 with dizziness). Severe (grade ≥ 3) AEs included hypertension, dizziness, fatigue, and bone pain (Table 3). All AEs of interest were grade 1 and included peripheral neuropathy occurring in 7 (17.9%) patients (PV, n = 4; ET, n = 3) and 1 case of cataract (PV). In total, 9/39 (23.1%) patients discontinued the study due to at least 1 AE. In 7/39 (18.0%) patients, AEs leading to discontinuation were considered related to momelotinib (MF [likely progression of disease], palpitations, pain in extremity, fatigue, increased platelet count, thrombocytosis, and dyspnea). One patient died due to cardiopulmonary arrest, which was considered unrelated to study treatment after review by the medical safety monitor.
Table 3.
Frequency of severe TEAEs of grade ≥ 3 and momelotinib-related AEs in ≥ 2 patients.
| PV Cohort |
ET Cohort |
|||||
|---|---|---|---|---|---|---|
| 100 mg (N = 14) | 200 mg (N = 14) | Total (N = 28) | 100 mg (N = 5) | 200 mg (N = 6) | Total (N = 11) | |
| No. (%) of patients with any grade ≥ 3 TEAE | 4 (28.6) | 6 (42.9) | 10 (35.7) | 0 | 1 (16.7) | 1 (9.1) |
| Hypertension | 2 (14.3) | 2 (14.3) | 4 (14.3) | 0 | 0 | 0 |
| Dizziness | 0 | 1 (7.1) | 1 (3.6) | 0 | 1 (16.7) | 1 (9.1) |
| Fatigue | 0 | 1 (7.1) | 1 (3.6) | 0 | 1 (16.7) | 1 (9.1) |
| Bone pain | 1 (7.1) | 0 | 1 (3.6) | 0 | 0 | 0 |
| Cardiorespiratory arrest | 1 (7.1) | 0 | 1 (3.6) | 0 | 0 | 0 |
| Dyspnea | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (9.1) |
| Night sweats | 1 (7.1) | 0 | 1 (3.6) | 0 | 0 | 0 |
| Pain in extremity | 1 (7.1) | 0 | 1 (3.6) | 0 | 0 | 0 |
| Platelet count increased | 0 | 1 (7.1) | 1 (3.6) | 0 | 0 | 0 |
| Thrombocytosis | 0 | 1 (7.1) | 1 (3.6) | 0 | 0 | 0 |
| Urinary bladder polyp | 0 | 1 (7.1) | 1 (3.6) | 0 | 0 | 0 |
| No. (%) of patients with any TEAE related to MMB | 9 (64.3) | 13 (92.9) | 22 (78.6) | 4 (80.0) | 5 (83.3) | 9 (81.8) |
| Headache | 3 (21.4) | 4 (28.6) | 7 (25.0) | 1 (20.0) | 1 (16.7) | 2 (18.2) |
| Dizziness | 0 | 6 (42.9) | 6 (21.4) | 0 | 1 (16.7) | 1 (9.1) |
| Somnolence | 4 (28.6) | 2 (14.3) | 6 (21.4) | 0 | 0 | 0 |
| Nausea | 1 (7.1) | 3 (21.4) | 4 (14.3) | 0 | 2 (33.3) | 2 (18.2) |
| Fatigue | 0 | 4 (28.6) | 4 (14.3) | 0 | 2 (33.3) | 2 (18.2) |
| Diarrhea | 1 (7.1) | 1 (7.1) | 2 (7.1) | 1 (20.0) | 2 (33.3) | 3 (27.3) |
| Vomiting | 1 (7.1) | 2 (14.3) | 3 (10.7) | 0 | 2 (33.3) | 2 (18.2) |
| Pruritus | 2 (14.3) | 2 (14.3) | 4 (14.3) | 0 | 0 | 0 |
| Dyspnea | 2 (14.3) | 1 (7.1) | 3 (10.7) | 0 | 1 (16.7) | 1 (9.1) |
| Abdominal pain | 0 | 1 (7.1) | 1 (3.6) | 0 | 2 (33.3) | 2 (18.2) |
| Peripheral sensory neuropathy | 1 (7.1) | 1 (7.1) | 2 (7.1) | 1 (20.0) | 0 | 1 (9.1) |
| Pain in extremity | 1 (7.1) | 1 (7.1) | 2 (7.1) | 1 (20.0) | 0 | 1 (9.1) |
| Cough | 2 (14.3) | 0 | 2 (7.1) | 0 | 1 (16.7) | 1 (9.1) |
| Asthenia | 0 | 2 (14.3) | 2 (7.1) | 0 | 0 | 0 |
| Rhinorrhea | 2 (14.3) | 0 | 2 (7.1) | 0 | 0 | 0 |
| No. of patients with any grade ≥ 3 TEAE related to MMB | 1 (7.1) | 3 (21.4) | 4 (14.3) | 0 | 1 (16.7) | 1 (9.1) |
AE, adverse event; ET, essential thrombocythemia; MMB, momelotinib; PV, polycythemia vera; TEAE, treatment-emergent adverse event.
The majority of recorded laboratory abnormalities were grade 1 or 2. Grade ≥ 3 events included 2 cases of hyperuricemia and 1 case each of elevated triacylglycerol lipase and hyperkalemia (both in PV patients). Decreased lymphocytes occurred in 1 ET patient. None of these events were considered clinically significant.
4. Discussion
The results presented herein indicate that momelotinib treatment has very limited activity as therapy for PV or ET. Out of 39 patients enrolled in this study, 2 patients (5.1%) met the primary composite endpoint (ORR), with only 1 confirmed response. Both responders were PV patients receiving momelotinib 200 mg/day. Single component hematocrit, WBC, or platelet count responses were noted in 3 patients in the PV cohort and 1 patient in the ET cohort (WBC response). Two PV patients and none of the ET patients achieved a spleen response. In total, 1 PV patient and 1 ET patient had a ≥ 10-point decrease from baseline in modified MPN-SAF TSS that lasted ≥12 weeks.
This finding contrasts previously published nonclinical and clinical data. Momelotinib inhibited JAK1 and JAK2 at nanomolar concentrations in in vitro assays, and reduced ex vivo formation of erythroid colonies from JAK2V617F-positive PV patients [22]. In a mouse model of JAK2V617F-driven PV-like MPN, momelotinib reduced JAK2V617F allele burden and disease symptoms; and restored normal spleen size, hematocrit, white blood cell counts, and cytokine levels [24].
Two open-label phase 1/2 clinical trials support the efficacy of momelotinib in treatment of MF in patients with high- or intermediaterisk primary or post-PV/ET MF [25,26]. Spleen responses were achieved in 45.8% to 48% patients and anemia responses in 45% to 59% of patients [25,26]. Moreover, among patients with baseline transfusion-dependence, 51.7% achieved 8-week transfusion-independence [26] and 21.1% to 67% achieved 12-week transfusion-independence.
The explanation for this apparent discrepancy in the activity of momelotinib in different MPNs is unclear. The data reported for other JAK2 inhibitors vary. Lestaurtinib (CEP-701) achieved only a modest response rate (14%) in patients with PV and ET [19]. Trials evaluating TG101348 (SAR302503) in PV and ET patients (ClinicalTrials.gov#NCT01420783) [27] were halted due to toxicity [28]. Gandotinib (LY-2784544) was well tolerated and showed some clinical improvement in PV and ET patients in a phase 1 study [29]. Ruxolitinib has demonstrated efficacy in patients with PV in phase 2 and 3 studies [14,17,30] and is approved for PV patients who have resistance or intolerance to hydroxyurea treatment [31].
There are notable differences in patients and study design between this study and the previous phase 2 and phase 3 ruxolitinib trials. First, the patients in ruxolitinib trials were resistant to or intolerant of hydroxyurea treatment [14,17,30], whereas this study did not require this for enrollment. Second, our primary endpoint had a different definition of response for PV patients (hematocrit, WBC, platelet, and spleen responses) compared with the ruxolitinib trials (hematocrit response without phlebotomy or hematocrit response with reduction in spleen size).
The pharmacokinetic results of momelotinib 200 mg in PV and ET patients are generally consistent with findings in MF patients taking the equivalent 300-mg once-daily capsules, which showed efficacy in MF through JAK1/2 inhibition [25]. An additional dose (100 mg) of momelotinib was tested to identify the optimal dosage, which was discovered to be 200 mg. It is possible that treatment with a 200-mg momelotinib starting dose for longer than 24 weeks in patients with more advanced PV would have shown a greater overall response rate. In conclusion, momelotinib 100 mg or 200 mg was well tolerated in PV/ET patients for up to 24 weeks, but had limited efficacy.
Supplementary Material
Acknowledgements
This study was supported by Gilead Sciences, Inc., Foster City, CA. Medical writing and editorial was provided by Claire Daniele, PhD, and Ewa Wandzioch, PhD, of AlphaBioCom, LLC, King of Prussia, PA, and funded by Gilead Sciences, Inc., Foster City, CA.
Author disclosures
SV and RM received research support from Gilead Sciences, Inc. RM received consulting fees from Novartis, Ariad, and Galena Biopharma; and research support from Incyte Corporation, CTI BioPharma, Promedior, and Celgene. MG served on an advisory board or received a speaker fee from AOP Orphan Pharmaceuticals, Novartis, Shire, Sanofi, Roche, and Amgen. CBB, JK, JM, LS, and YX are full-time employees of Gilead Sciences, Inc. AB, SC, and DH have no conflicts of interest to declare.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.Org/10.1016/j.leukres.2017.05.002.
References
- [1].Tefferi A, Vardiman JW, Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 22 (2008) 14–22. [DOI] [PubMed] [Google Scholar]
- [2].Tefferi A, Barbui T, Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management, Am. J. Hematol. 90 (2015) 162–173. [DOI] [PubMed] [Google Scholar]
- [3].Mascarenhas J, Mughal TI, Verstovsek S, Biology and clinical management of myeloproliferative neoplasms and development of the JAK inhibitor ruxolitinib, Curr. Med. Chem. 19 (2012) 4399–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alvarez-Larran A, Martinez-Aviles L, Hemandez-Boluda JC, Ferrer-Marin F, Antelo ML, Burgaleta C, et al. , Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea, Ann. Hematol. 93 (2014) 2037–2043. [DOI] [PubMed] [Google Scholar]
- [5].Alvarez-Larran A, Pereira A, Cervantes F, Arellano-Rodrigo E, Hernandez-Boluda JC, Ferrer-Marin F, et al. , Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera, Blood 119 (2012) 1363–1369. [DOI] [PubMed] [Google Scholar]
- [6].Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. , Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders, Lancet 365 (2005) 1054–1061. [DOI] [PubMed] [Google Scholar]
- [7].James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. , A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434 (2005) 1144–1148. [DOI] [PubMed] [Google Scholar]
- [8].Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. , A gain-of-function mutation of JAK2 in myeloproliferative disorders, N. Engl. J. Med. 352 (2005) 1779–1790. [DOI] [PubMed] [Google Scholar]
- [9].Levine RL, Wadleigh M, Cools J, Ebert BL, Wemig G, Huntly BJ, et al. , Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis, Cancer Cell 7 (2005) 387–397. [DOI] [PubMed] [Google Scholar]
- [10].Sonbol MB, Firwana B, Zarzour A, Morad M, Rana V, Tiu RV, Comprehensive review of JAK inhibitors in myeloproliferative neoplasms, Ther. Adv. Hematol. 4 (2013) 15–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L, et al. , A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications, Leukemia 24 (2010) 1574–1579. [DOI] [PubMed] [Google Scholar]
- [12].Qin Y, Wang X, Zhao C, Wang C, Yang Y, The impact of JAK2V617F mutation on different types of thrombosis risk in patients with essential thrombocythemia: a meta-analysis, Int. J. Hematol. 102 (2015) 170–180. [DOI] [PubMed] [Google Scholar]
- [13].Kim SY, Im K, Park SN, Kwon J, Kim JA, Lee DS, CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable, Am. J. Clin. Pathol. 143 (2015) 635–644. [DOI] [PubMed] [Google Scholar]
- [14].Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. , Ruxolitinib versus standard therapy for the treatment of polycythemia vera, N. Engl. J. Med. 372 (2015) 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Verstovsek S, Kantaijian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. , Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis, N. Engl. J. Med. 363 (2010) 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. , A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis, N. Engl. J. Med. 366 (2012) 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Verstovsek S, Passamonti F, Rambaldi A, Barosi G, Rosen PJ, Rumi E, et al. , A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea, Cancer 120 (2014) 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. , JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis, N. Engl. J. Med. 366 (2012) 787–798. [DOI] [PubMed] [Google Scholar]
- [19].Hexner E, Roboz G, Hoffman R, Luger S, Mascarenhas J, Carroll M, et al. , Open-label study of oral CEP-701 (lestaurtinib) in patients with polycythaemia vera or essential thrombocythaemia with JAK2-V617F mutation, Br. J. Haematol. 164 (2014) 83–93. [DOI] [PubMed] [Google Scholar]
- [20].Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, et al. , Safety and efficacy of TGI01348, a selective JAK2 inhibitor, in myelofibrosis, J. Clin. Oncol. 29 (2011) 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Geyer HL, Mesa RA. Therapy for myeloproliferative neoplasms: when, which agent, and how, Hematol. Am. Soc. Hematol. Educ. Program 2014 (2014) 277–286. [DOI] [PubMed] [Google Scholar]
- [22].Pardanani A, Lasho T, Smith G, Bums CJ, Fantino E, Tefferi A, CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and pre-clinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia 23 (2009) 1441–1445. [DOI] [PubMed] [Google Scholar]
- [23].Monaghan KA, Khong T, Bums CJ, Spencer A, The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells, Leukemia 25 (2011) 1891–1899. [DOI] [PubMed] [Google Scholar]
- [24].Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, et al. , CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms, Blood 115 (2010) 5232–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pardanani A, Laborde RR, Lasho TL, Finke C, Begna K, Al-Kali A, et al. , Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis, Leukemia 27 (2013) 1322–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gupta V, Mesa RA, Deininger MW, Rivera CE, Sirhan S, Brachmann CB, et al. , A phase 1/2, open-label study evaluating twice-daily administration of momelotinib in myelofibrosis, Haematologica 102 (2017) 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tefferi A, Cortes JE, Hochhaus A, Kiladjian J-JM, Ruben A, Pardanani AD, Lebedinsky C, et al. , A randomized phase II, open label trial of orally administered SAR302503 in patients with polycythemia vera (PV) or essential thrombocythemia (ET) who are resistant or intolerant to hydroxyurea, J. Clin. Oncol. 30 (2012) TPS6641. abstract. [Google Scholar]
- [28].Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial, JAMA Oncol. 1 (2015) 643–651. [DOI] [PubMed] [Google Scholar]
- [29].Verstovsek S, Mesa RA, Salama ME, Giles JLK, Pitou C, Zimmermann AH, et al. , Phase I study of LY2784544, a JAK2 selective inhibitor, in patients with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET), Blood 122 (2013) 665 abstract. [Google Scholar]
- [30].Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T, et al. , Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study, Lancet Oncol. 18 (2017) 88–99. [DOI] [PubMed] [Google Scholar]
- [31].Jakafi® (ruxolitinib) Tablets, for Oral Use. Prescribing Information, Incyte, Wilmington, DE, USA, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.