

Abstract
Huntington’s disease is a progressive neurodegenerative disease caused by expansion of the polyglutamine domain in the first exon of huntingtin (HttEx1). The extent of expansion correlates with disease progression and formation of amyloid-like protein deposits within the brain. The latter display polymorphism at the microscopic level, both in cerebral tissue and in vitro. Such polymorphism can dramatically influence cytotoxicity, leading to much interest in the conditions and mechanisms that dictate the formation of polymorphs. We examine conditions that govern HttEx1 polymorphism in vitro, including concentration and the role of the non-polyglutamine flanking domains. Using electron microscopy, we observe polymorphs that differ in width and tendency for higher-order bundling. Strikingly, aggregation yields different polymorphs at low and high concentrations. Narrow filaments dominate at low concentrations that may be more relevant in vivo. We dissect the role of N- and C-terminal flanking domains using protein with the former (httNT or N17) largely removed. The truncated protein is generated by trypsin cleavage of soluble HttEx1 fusion protein, which we analyze in some detail. Dye binding and solid-state NMR studies reveal changes in fibril surface characteristics and flanking domain mobility. Higher order interactions appear facilitated by the C-terminal tail, while the polyglutamine forms an amyloid core resembling those of other polyglutamine deposits. Fibril-surface-mediated branching, previously attributed to secondary nucleation, is reduced in absence of httNT. A new model for the architecture of the HttEx1 filaments is presented and discussed in context of the assembly mechanism and biological activity.
Keywords: Huntington’s disease, amyloid, supramolecular assembly, TEM, MAS ssNMR
Introduction
Huntington’s disease (HD) is one of several heritable diseases that are characterized by the abnormal expansion of a CAG trinucleotide repeat that codes for a polyglutamine (polyQ) stretch or domain in a mutant protein [1]. In HD, polyQ expansion occurs within the first exon (HttEx1) of the huntingtin protein (htt) [Fig. 1(a ,b)], which results in the deposition of htt N-terminal fragments (including HttEx1) as neuronal inclusion bodies. The misfolding and deposition of the mutant protein is generally associated with a toxic gain-of-function that contributes to neuronal degradation in HD [2, 3]. The mature protein deposits formed in HD reflect an amyloid type molecular architecture, which they have in common with protein aggregates found in Parkinson’s and Alzheimer’s disease patients [1, 4–6]. Similar to the protein misfolding and amyloid formation processes in those disorders, their HD counterparts are considered disease relevant due to their ability to contribute to disease toxicity, disease propagation and disease onset [7–10]. Simultaneously, there is significant evidence that some protein deposits may reduce cellular toxicity, and even contribute to cellular rescue mechanisms [11–13]. As a consequence of these findings there is much interest in understanding the underlying molecular processes in hopes of finding means to control and modulate various aspects of these as-yet incurable disorders.
Fig. 1. Huntingtin exon 1 and MBP-based fusion protein.
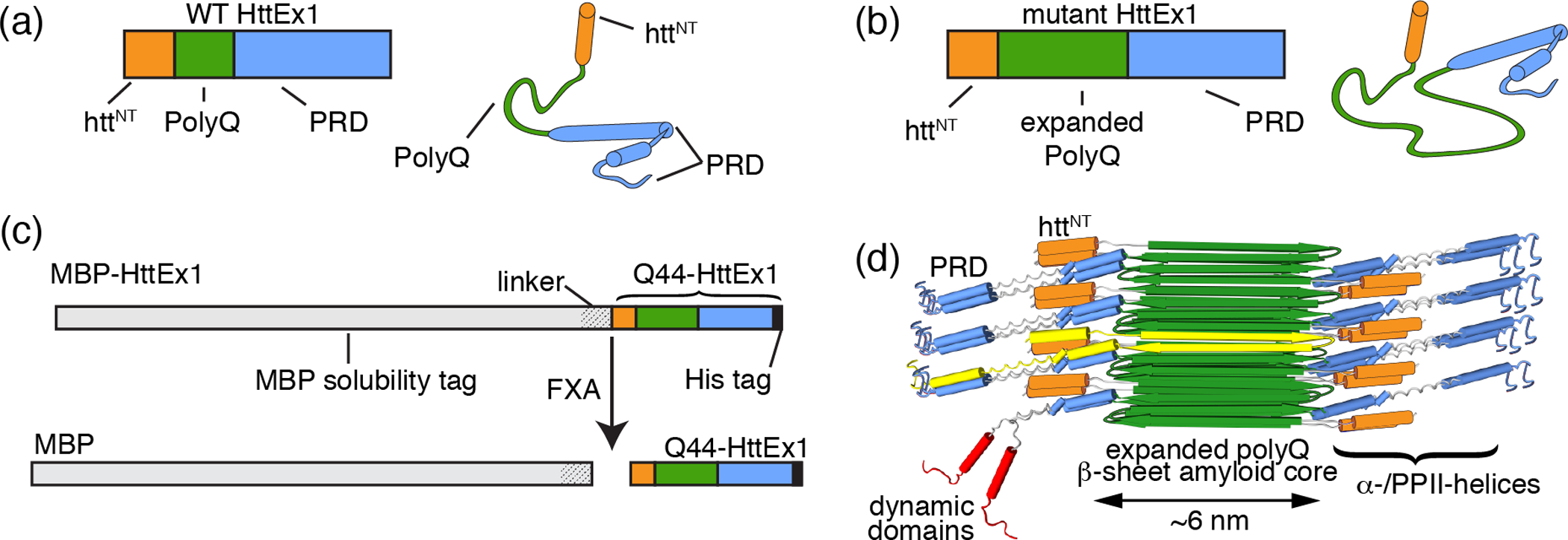
Primary and secondary structure schematics of wild type HttEx1 (a) and mutant HttEx1 (b) showing the httNT domain (orange; α-helix), polyQ domain (green; intrinsically disordered), and the proline rich domain (PRD, blue; intrinsically disordered with PPII helices). Mutant Q44-HttEx1 contains an expanded polyQ domain. (c) Top: Primary structure schematic of the employed MBP-HttEx1 fusion protein, with C-terminal His tag marked (black). Bottom: HttEx1 monomer and free MBP are released from the MBP-HttEx1 fusion protein by proteolytic cleavage using Factor Xa (FXa) protease. (d) Model of the previously determined fibril architecture of single-filament narrow (~ 6 nm wide) HttEx1 fibrils formed at 37 °C, with a single monomer highlighted in yellow. An example of local dynamic domains is depicted in red. The His tag is not shown. Panel (d) is adapted from Lin, H. K., Boatz, J. C., Krabbendam, I. E., Kodali, R., Hou, Z., Wetzel, R., Dolga, A. M., Poirier, M. A., van der Wel, P. C. A. Fibril polymorphism affects immobilized non-amyloid flanking domains of huntingtin exon1 rather than its polyglutamine core, Nature Communications 85462. Copyright (2017) Lin, H. K., et. al. [36]
Reports of varying levels of disease toxicity observed in cellular and organismal studies of HD may be explained by the phenomenon of amyloid polymorphism seen in vitro for these and other amyloidogenic proteins [1]. Polymorphism is a key feature of amyloid-forming proteins, where the same protein or peptide is capable of forming a range of distinct oligomeric, fibrillar or non-fibrillar deposits in absence of changes in the primary or chemical structure [14]. The obtained structure is dependent on the environmental conditions, with important factors being pH, temperature and the presence or absence of agitation during aggregation. Identical proteins can aggregate into differently structured amyloid states, or even into either amyloid or amorphous deposits, depending on e.g. the pH [15, 16]. Importantly, such polymorphic deposits can have different levels of neurotoxicity, although the molecular rationale for these toxicity differences remains as yet opaque [17–19]. Their formation is expected to result from distinct aggregation pathways populated by transient species, which in turn display differing levels of neurotoxicity and variable lifetimes. Some of these transient species may be important contributors to disease toxicity. It has been proposed that the overall toxicity in amyloid diseases may be reduced by modulating key aggregation pathways to limit the formation of oligomer precursors [20, 21].
The aggregation behavior of polyQ peptides, mutant HttEx1 and mutant proteins from other polyQ diseases share features common to amyloidogenic proteins. They aggregate via β-sheet rich nuclei, which form after a lag phase that may be populated with oligomeric species [22]. Fibril elongation rapidly follows the formation of nuclei [18, 23]. The lag phase can be bypassed or shortened in the presence of “seeds”, which are preformed fibrils and fibril fragments that act as nuclei for monomers and multimers that have not yet aggregated [4]. Recent studies implicate seeding processes in vivo in disease propagation in various amyloid-associated neurodegenerative disorders [8, 9, 24, 25]. HD-associated protein deposits may be transmitted between neurons in HD patients as the disease progresses [8, 9]. In this context it is important to note that different types of fibril polymorphs are expected (and observed) to have different levels of seeding ability.
The aggregates formed by mutant HttEx1 and other polyQ proteins display polymorphism, as detected by a variety of experimental techniques. Two such techniques, which also proved essential in detailing the polymorphs of Aβ1–40 and other amyloid forming proteins, are transmission electron microscopy (TEM) and magic angle spinning solid state NMR (MAS ssNMR) [6, 26–29]. TEM studies reveal a significant level of heterogeneity in mutant HttEx1 fibrils formed in vitro or in cells, which limits the resolution achievable by structural analyses [30–32]. Different types of HttEx1 fibrils are formed, based on their overall morphology and fiber width, and depending on local environmental conditions [14, 19, 33]. A notable feature of mutant HttEx1 is that its polymorphic behavior is highly influenced by dynamic domains that flank the aggregates’ amyloid core [Fig. 1 (d)]. Interestingly, the domains that decorate the surface of the deposits are considered crucial for the biological properties of the polymorphs [12]. In mutant HttEx1, the expanded polyQ domain makes up the amyloid core of the fibrils, featuring an architecture that is antiparallel β-sheet, highly rigid, and dehydrated. MAS ssNMR experiments revealed dynamic flanking domains exposed on the fiber surface that lack β-sheet structure: the 17 residue N-terminal domain (httNT or N17) and a proline-rich C-terminal domain (PRD) containing PPII helices and flexible random coil (RC) residues [6, 34–39]. These domains are also of interest in mechanistic studies of HttEx1 aggregation as they have opposing influences on aggregation kinetics [22, 36, 40, 41]. Outside of the disease-associated deposition process, the httNT domain is very important for htt function, acting as a binding partner for membranes, chaperones, and other proteins with roles in membrane trafficking, cell signaling, and the regulation of gene expression and transcription [42–45]. Additionally, the httNT domain is the target of several post-translational modifications that directly influence htt function [46, 47]. We previously reported two temperature-dependent polymorphs of HttEx1 fibrils containing 44 residues in the polyQ domain (Q44-HttEx1) that can be distinguished by TEM and MAS ssNMR [36]. Intriguingly, the polyQ fingerprint seen by MAS ssNMR is the same for both temperature-dependent polymorphs, indicating that they share an analogous polyQ core [36]. The httNT domain and PPII helices in the PRD are immobilized with restricted motion proximal to the polyQ domain and the PRD as a whole presents variable dynamics and solvent accessibility. The main differences between the fiber polymorphs were found in the dynamics and accessibility of the C-terminal PRD, which appeared to mediate a type of supramolecular polymorphism [48] in the fiber architecture. We presented a model of the HttEx1 fibril architecture, reproduced in Fig. 1 (d), that integrates information obtained from TEM, MAS ssNMR and antibody binding assays. This model reflects both similarities and specific differences with fibril models reported by others [39, 49]. However, all current models of HttEx1 fibril architecture are limited in their molecular detail or lack a three-dimensional perspective. A better understanding of the structure of these protein inclusions is important for modeling their mechanism of formation and for explaining their propensity to form polymorphs, their biological activity, and their ability to recruit other proteins.
In the current work, we used aggregation assays, TEM, MAS ssNMR and model building to extend our structural and kinetic studies of the HttEx1 and its aggregation propensity and polymorphism. Key aims were to understand in more detail the parameters that underpin the known polymorphism of HttEx1 fibrils [18, 33, 36] and to develop a better model of their structure. We describe new polymorphs that we had not reported previously and find additional parameters that correlate with fibril polymorphism. Notably, we report a significant role for protein concentration on the aggregation pathway, which has potential implications for the biological context and will be discussed in relation to prior studies of the multi-stage HttEx1 misfolding process [22, 50]. We also probe the role of the flanking domain httNT using an N-terminally truncated version of HttEx1. Despite a change in aggregation kinetics some features of the fibrils are preserved while others are significantly modified, pinpointing the distinct role of the N- and C-terminal flanking domains. Integrating our current and prior data, we present an improved and more detailed model of the HttEx1 filament structure and discuss it in context of the mutant protein’s self-assembly mechanisms in vitro and in vivo.
Results
Concentration of MBP-Q44-HttEx1 affects width of mature fibrils
The fibrillization temperature is a well-known source of polymorphism for mutant HttEx1 fibrils, with reported impacts on neurotoxicity [18, 36]. We previously reported that variation in the average fibril width is a key marker of polymorphism in mature Q44-HttEx1 fibrils formed at similar concentrations but different temperatures. Depending on conditions, ‘narrow’ fibrils (average 5 – 7 nm) [Fig. 1 (d)] were generated at 37 °C and ‘wide’ fibrils (average 15 – 16 nm) at 22 °C. Significant differences in the exposure and dynamics of the flanking domains pointed to a tight bundling of narrow protofilaments to form the wide fibrils [36]. Having noticed a surprising observation that the fibril width also seemed to depend on the initial protein concentration, we set out to probe in more detail some experimental parameters that influence HttEx1 polymorphism. To do so, we again employed our protease-cleavable HttEx1 fusion protein construct that enables the on-demand release of the aggregation-prone Q44-HttEx1 [34, 40]. We allowed released Q44-HttEx1 to aggregate at 37 °C at a range of concentrations (14.3 – 98.9 μM) and measured the average fibril widths by negative stain TEM after ≥ 3 days. Narrow fibrils with a relatively well-defined with of 4 – 6 nm are primarily observed at 14.3 μM [Fig. 2 (a), Fig. S1 (a–b)]. At 28.6 μM (Fig. 2(b) we still observe relatively narrow fibrils (< 10 nm width) as the dominant species, but they are more heterogeneous as reflected in a broader width distribution in the histogram. Moreover, we observe a much less populated co-existing population of fibrils (14 – 17 nm in width) with a morphology reminiscent of 15 – 16 nm ‘wide’ fibrils previously observed to form at 22 °C [36] [Fig. 2 (b)]. Upon increasing the concentration to 98.9 μM we observe two populations of fibrils [Fig. 2 (c), Fig. 1 (c)] with narrow fibrils with average widths of 5 – 7 nm and wide fibrils with average widths of 21 – 23 nm. The latter fibrils show a substantial degree of heterogeneity in the histogram of width measurements, while the narrower fibril class is relatively well defined in width. We note that measurements of fibril widths from negative stain TEM images can vary according to differences in stain accumulation at the exterior surface of the fibrils as well as any penetration into the structures. This limits the precision, especially in these heterogeneous samples featuring fiber-fiber interactions and bundling. The reported image analysis was done by two independent investigators, in an attempt to account for the analysis subjectivity, which resulted in variation of the measured width in the range of 1–2 nm. Importantly this variability in width for each fibril class is significantly smaller than differences in width between the fibril classes that we identified here (Fig. 2) and previously [36].
Fig. 2. Q44-HttEx1 fibril polymorphism is dependent on the monomer concentration and molar ratio of fusion protein to protease.
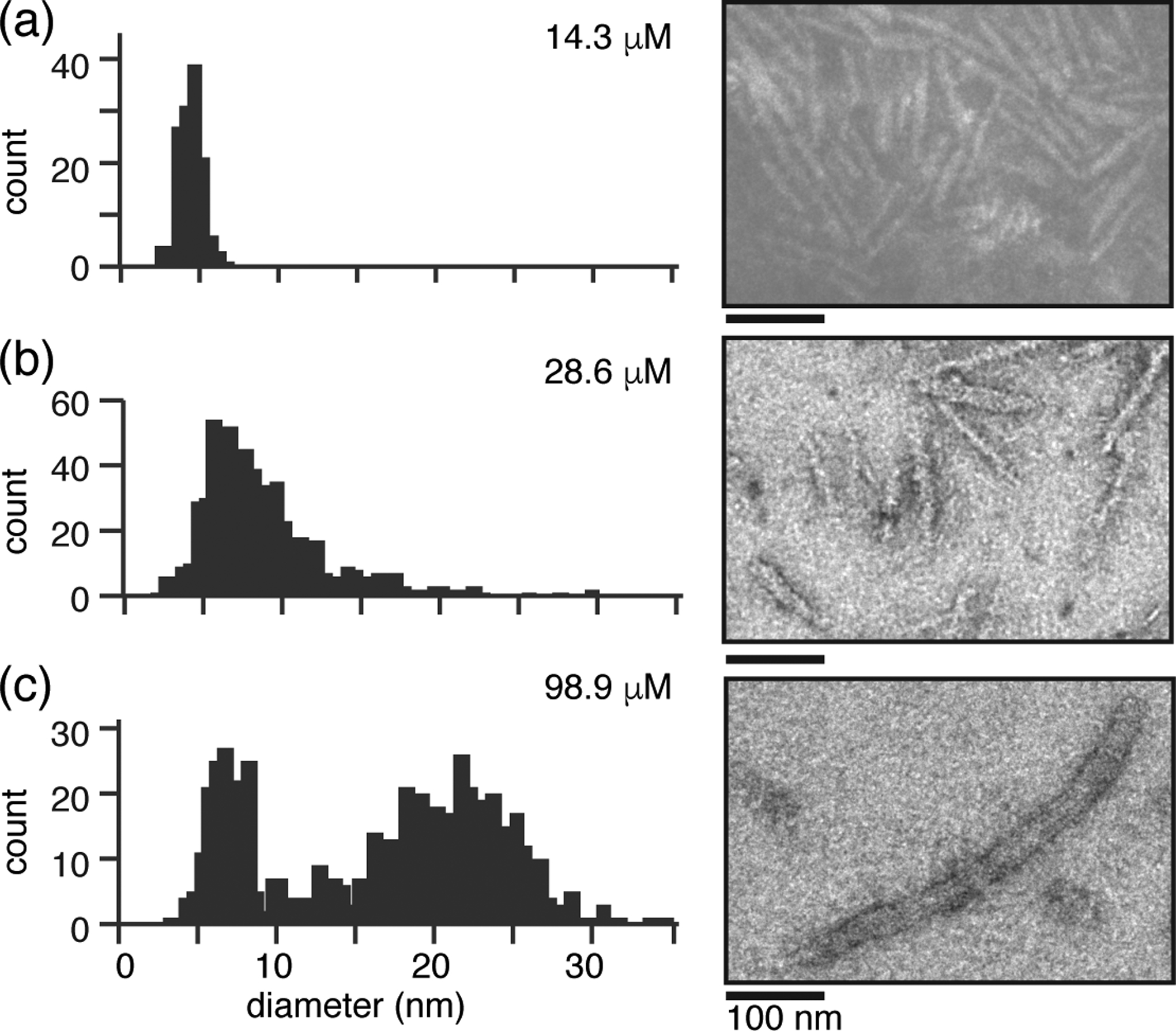
(a-c) Histograms depicting a range of widths observed by negative stain TEM of Q44-HttEx1 fibrils, with one representative image shown per sample. Fibrils were prepared at 37 °C following cleavage of (a) 14.3 μM (62.5:1 FP:P), (b) 28.6 μM (62.5:1 FP:P), and (c) 98.9 μM (42.5:1 FP:P) MBP-Q44-HttEx1 by FXa. FP:P indicates the molar ratio of MBP-Q44-HttEx1 to FXa. See Fig. S1 for additional TEM images.
We also performed a more extensive systematic analysis of fibril morphology as a function of the concentration and observed dramatic effects on the microscopic morphology of the resulting mature Q44-HttEx1 fibrils. Histograms depicting the distribution of fibril widths are shown in Fig. S2. At low monomer concentrations we see predominantly narrow fibrils (at 7.2– 28.6 μM MBP-Q44-HttEx1). A mix of ‘narrow’ and ‘wide’ fibrils (width > 16 nm) form at 78.8 – 98.9 μM MBP-Q44-HttEx1. However, narrow fibrils are also produced for the higher fusion protein concentration of 98.9 μM, when the cleavage rate is limited due to large fusion protein to protease (FP:P) molar ratios, resulting in low HttEx1 concentrations present during the initial stages of aggregation. Interestingly, at even lower concentrations of free HttEx1, ‘intermediate’ fibrils (between 9 and 16 nm) are observed (≤ 14.3 μM MBP-Q44-HttEx1 with FP:P of 510:1 or 1500:1). Thus, in summary, we see that polymorphic assembly of Q44-HttEx1 yields fibrils that can be classified by their averaged width and that this polymorphic behavior is sensitive to the HttEx1 concentration, with wider fibrils being dominant at high HttEx1 concentration.
Kinetics of Q44-HttEx1 monomer release by Factor Xa protease
Fibrillization is initiated in the above experiments by an approach employed in numerous HttEx1 in vitro studies where the Q44-HttEx1 monomers are released from a soluble fusion protein by proteolytic cleavage [Fig. 1 (c)] [18, 32, 36, 40, 51–56]. As such, the concentration of the Q44-HttEx1 monomer is changing during initial stages of the assay, leading to additional complexities in the kinetic analysis [51] and potential implications for the interpretation of concentration-dependent polymorphism. We analyzed the kinetics of monomer release using SDS-PAGE to monitor the disappearance of fusion protein and production of free MBP over time [Fig. S3]. At 14 – 15 μM MBP-HttEx1 and low FP:P molar ratios, the data show complete conversion to the free monomer state within 15 min to an hour, leading to detectable fibril formation within 24 h [Fig. S3 (a–d)]. A progressive delay in fibril formation is observed as the monomer release is slowed down. At 15,000:1 FP:P, no fusion protein cleavage or monomer release is detectable even after 41 hours [Fig. S3 (a)], and fibril formation is observed after 10 days. These findings reinforce and support prior reports indicating that the uncleaved fusion protein is not incorporated into the Q44-HttEx1 fibrils [36], in line with our own current and prior in-depth ssNMR studies of the fibrils (where no MBP signals are detected). Moreover, the effective monomer concentrations at highly substoichiometric P:FP ratios are lower than might be expected, at least during the crucial initial stages of the aggregation process.
Morphological analysis
Branching is apparent in some of the observed fibrils [Fig. 3 (a), left; Fig. S4] as also observed by others [51, 57]. This is significant because most amyloid fibrils do not branch, and indeed lack of branching is seen as a defining feature of amyloids [51]. Here, we see that branching mostly affects the wider Q44-httEx1 fibrils. Another, likely related, feature of the wider fibrils is that many of them show striations along their long axis. These striations are more easily visualized when we sum the gray values along the (aligned) fibril axis, as illustrated in Fig. 3 (b). We ascribe the striations to the presence of multiple bundled protofilaments [58]. Unlike the wide fibrils, the surface of both the narrow and intermediate fibrils frequently appears fuzzy, likely due to their exposed disordered flanking domains. It is possible that the narrow and intermediate fibrils also feature striations that are obscured by this fuzziness. Even within a single sample the characteristics of the Q44-HttEx1 fibrils can be heterogeneous. Two examples of narrow fibrils with respective average widths of 8.3 and 5.9 nm, and an intermediate fibril with a width of 16.8 nm, are shown in Fig. 3 (d). Fig. 3 (e) shows a fibril with varying width across the fibril axis; the center of the fibril is narrower than either end, which was also seen in fibrils from most other sample conditions. The increase in width at either fibril end in Fig. 3 (e) appears in quantified steps of a few nm, as opposed to a gradual change. These variations in widths between fibrils and along the length of individual fibrils provide further evidence of the presence of co-aligned protofilaments in the individual fibrils.
Fig. 3. Morphological analysis of Q44-HttEx1 fibrils.
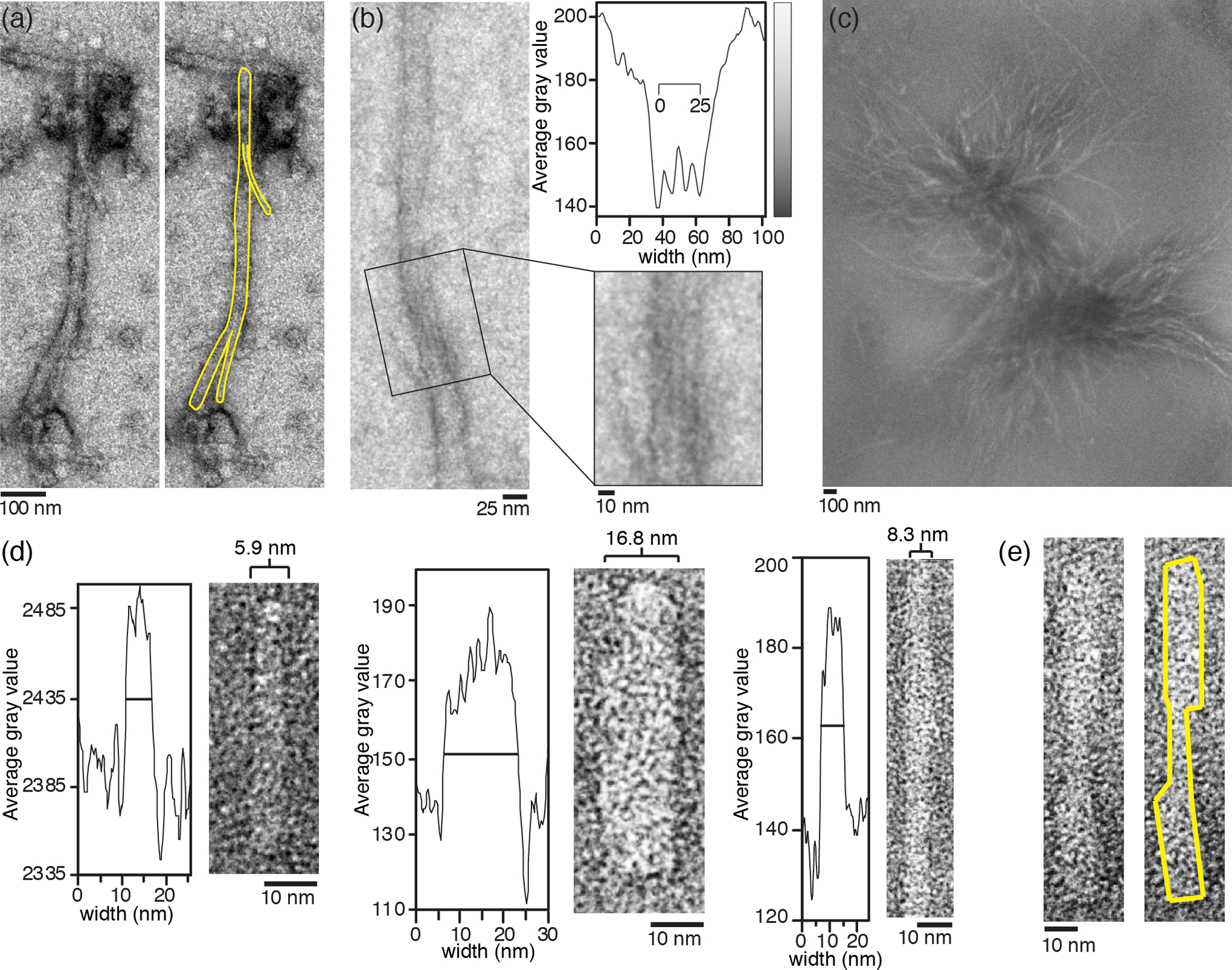
(a) Wide (> 16 nm) fibrils formed from 78.8 μM MBP-Q44-HttEx1; 170:1 FP:P. Left: fibril with two branch points (fibril border highlighted yellow). (b) Fibril area displaying striations that correspond to a supramolecular multifilament structure. High gray values mark regions with increased levels of staining. Top right: projected grey scale values summed along the fiber length, as a function of the fiber diameter, based on the aligned fiber section shown below. (c) Fibril bundle composed of intermediate (> 9 nm and < 16 nm) width fibrils, formed from 10.1 μM MBP-Q44-HttEx1; 510:1 FP:P. A band pass filter has been applied to the image to balance contrast. (d-e) Fibrils from a single sample of aggregated 28.6 μM MBP-Q44-HttEx1 (62.5:1 FP:P). (d) Although the majority of fibrils observed in this sample were categorized as narrow, TEM-based width analysis shows there is heterogeneity of fibril widths within the sample. Shown are aligned fiber sections and corresponding grey scale projections as in panel (b). (e) Fibril width can vary along the long axis of a single fibril.
Larger bundles of fibrils [Fig. 3 (c)] are observed in many of the samples containing ‘narrow’ and ‘intermediate’ fibrils but are not observed in any sample with a significant population of ‘wide’ fibrils. In our TEM data these fibril bundles often are extremely dark due to high levels of staining between and around bundled fibrils. Fig. 3 (c) includes an image processed to enhance the visibility of one such bundle (see Methods). The widths observed for individual fibrils within a bundle are consistent with the widths observed for the isolated narrow and intermediate fibrils. Since these fibril types feature more exposed flanking domains resulting in their fuzzy appearance (see above), it seems likely that this bundling may involve these flanking regions (see Discussion).
Impacts of temperature on cleavage and aggregation.
We wondered if our previously found effect of temperature on fibril polymorphism [36] may actually relate to an effect of temperature on the cleavage process, causing significant differences in the time-dependent monomer concentration. However, in direct comparisons, we find that the rate of monomer release by the protease employed (FXa) shows negligible differences at 22 and 37 °C [Fig. S3 (a, g, h), Fig. S5 (a)]. As an aside, in comparative measurements the rate of monomer release is sensitive to the length of the polyQ domain, with the cleavage of wild-type MBP-Q20-HttEx1 being significantly faster than for MBP-Q44-HttEx1 [Fig. S3 (e)]. To compare to the fibrils formed at 37 °C (above), we also allowed fibrils to form at 22 °C, at 10 – 45 μM and 625:1 – 35:1 FP:P. The average fibril width was consistently 15 – 16 nm, similar to the intermediate fibrils formed at 37 °C described above and in agreement with our prior work [36]. Thus, the concentration dependence of HttEx1 polymorphism described here is distinct from the temperature-induced effects described by us and others in prior work. Observed temperature effects on Q44-HttEx1 polymorphism are not attributable to differences in cleavage kinetics and monomer concentration.
Aggregation of ΔN15-Q44-HttEx1 fibrils
The httNT flanking domain accelerates and initiates the aggregation of HttEx1 [35, 50, 59], facilitating also alternative HttEx1 aggregation pathways that are not primarily driven by intermolecular interactions between polyQ domains [22, 50]. Given this key role, and the fact that both flanking domains stabilize HttEx1 supramolecular polymorphism [36], we set out to probe the impact of eliminating the httNT domain on Q44-HttEx1. To do so, rather than use FXa protease, we used an alternative trypsin cleavage protocol exploiting a trypsin cleavage site near the end of httNT [Fig. 4 (a)] [36, 56]. First, we performed a detailed analysis of the trypsin cleavage kinetics and cleavage products [Figs. S6–S7] at 22 °C, which revealed that trypsin preferentially cleaves MBP-Q44-HttEx1 at residue R[−1] (the last residue prior to HttEx1) to release Q44-HttEx1, but then sequentially and efficiently cleaves the httNT domain from the N-terminus to C-terminus at K6, K9, and K15 [Fig. S6]. Thus, we can rapidly generate ΔN15-Q44-HttEx1, which cannot assemble via an httNT-driven aggregation pathway [50]. We then monitored amyloid formation of Q44-HttEx1 and ΔN15-Q44-HttEx1 over time at 22 °C using a thioflavin T (ThT) dye that preferentially fluoresces when bound to amyloids [Fig. 4 (b)] [36, 53]. For normal Q44-HttEx1, a lag time of approximately 2 hours was observed prior to rapid polymerization that completed within 1 day [Fig. 4 (b, inset)]. In contrast, the lag time increased six fold and polymerization was completed within 3 days for the ΔN15-Q44-HttEx1 fibrils [Fig. 4 (b, inset)], supporting the known role of httNT in setting the amyloid kinetics [35, 50, 59]. Using SDS-PAGE and a previously-described HPLC-based sedimentation assay [36], monomer concentration was measured in parallel to the ThT assays at 22 °C. Complete monomer release was observed within 16 and 5 min for Q44-HttEx1 and ΔN15-Q44-HttEx1, respectively, [Fig. 4 (c)], well within the amyloid lag phases.
Fig. 4. Morphology of ΔN15-Q44-HttEx1 fibrils that lack most of httNT.
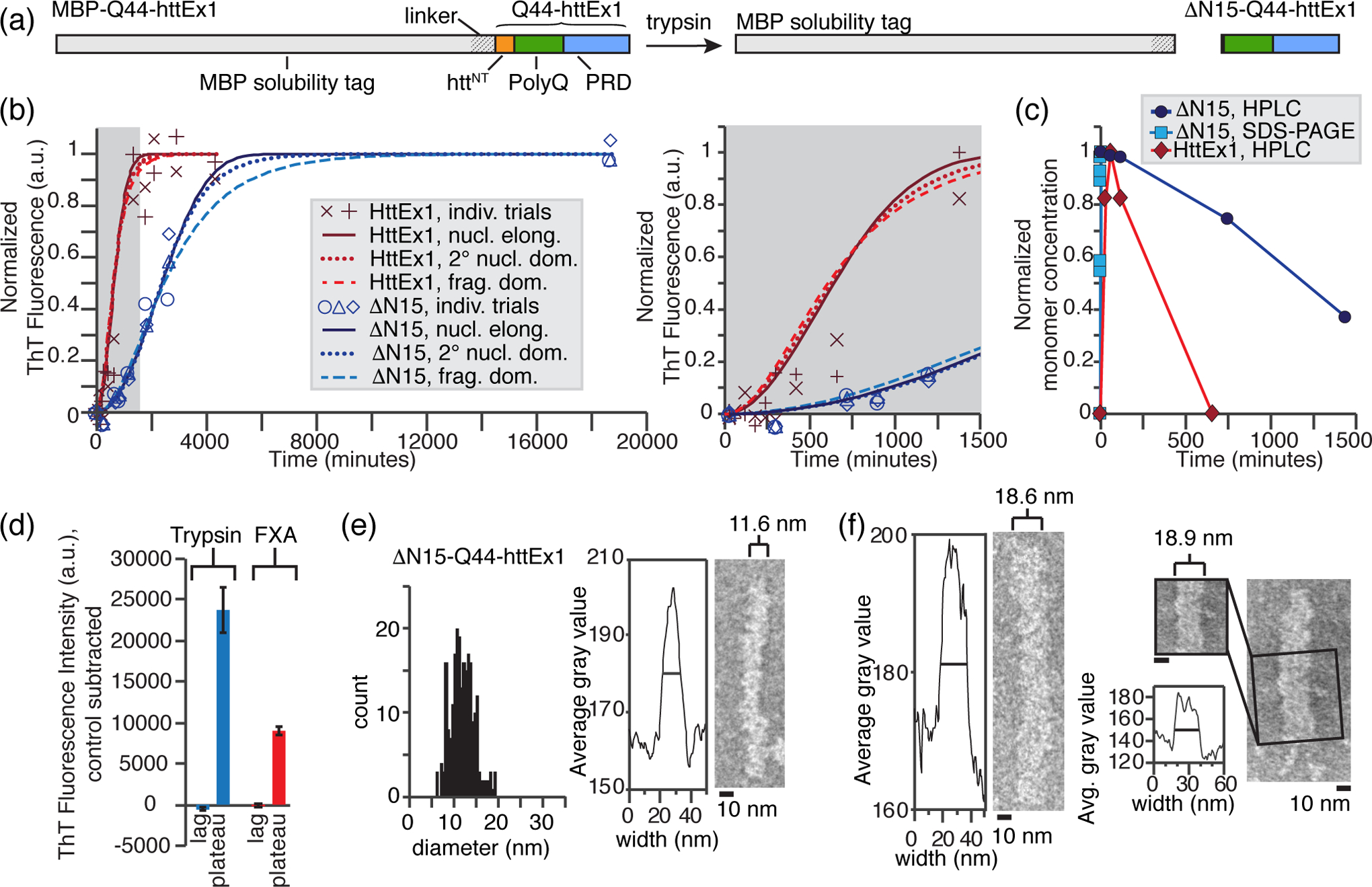
(a) Schematic diagram of the employed cleavage reaction, in which trypsin cleavage liberates ΔN15-Q44-HttEx1 after a rapid multistep cleavage process [Fig. S6]. (b) The formation of Q44-HttEx1 (red) and ΔN15-Q44-HttEx1 (blue) amyloid fibrils over time tracked by ThT fluorescence, normalized to the maximum fluorescence of the predicted curves (nucleated elongation) for each. Data is fit to nucleated elongation (solid), secondary nucleation dominated (dotted), and fragmentation dominated (dashed) aggregation kinetics models using AmyloFit [60]. Right: magnified inset of the lag phase. (c) Normalized Q44-HttEx1 (red) and ΔN15-Q44-HttEx1 (blue) monomer concentrations over time following cleavage by FXa and trypsin, respectively, measured by SDS-PAGE and HPLC [36]. The HPLC assays were performed in parallel to the ThT fluorescence assays. (d) Average ThT fluorescence in the lag and plateau phase for Q44-HttEx1 (red) and ΔN15-Q44-HttEx1 (blue). The average fluorescence of a control sample containing MBP-Q44-HttEx1 only was subtracted for each. The lag phase was measured at 50 and 60 minutes for ΔN15-Q44-HttEx1 (n = 3) and Q44-HttEx1 (n = 2), respectively. The plateau phase was measured at 4 and 3 days for ΔN15-Q44-HttEx1 (n = 3) and Q44-HttEx1 (n = 2) respectively. (e) Distribution of widths measured for ΔN15-Q44-HttEx1 fibrils, outfitted with uniform 13C,15N labels for ssNMR. The average width is between 11–12 nm (right), however a wide distribution of widths was observed (left). (f) Wide ΔN15-Q44-HttEx1 fibrils (~ 18 – 19 nm). Right: evidence of striations consistent with a supramolecular multifilament structure.
In order to gain insight into the possible effects the httNT domain has on the aggregation mechanism and the amyloid fibril kinetics, we fit the ThT results to different aggregation kinetics models [Fig. 4 (b)] using AmyloFit [60]. The aggregation kinetics of both Q44-HttEx1 and ΔN15-Q44-HttEx1 were found to be consistent with a typical primary nucleated elongation model (solid lines), although models dominated by secondary nucleation (dotted lines) and fragmentation (dashed lines) fit the data equally well [60]. Interestingly, the ThT fluorescence is ~2.5x higher in the plateau phase compared to cleavage with FXa protease [Fig. 4 (d)], even when seeding. We interpret this to indicate a structural difference in the trypsin-produced fibrils that changes the extent or mode of ThT dye binding to the fibril surface (see also Discussion) [61]. By TEM we observe that the ΔN15-Q44-HttEx1 fibrils predominantly have widths between 8–15 nm, with an average width of 11.6 nm [Fig. 4 (e)]. A small population of fibrils have widths exceeding 15 nm [Fig. 4 (f)]. The width again varies along the fiber axis in some fibrils, however at a much lower frequency than observed for Q44-HttEx1. Both isolated fibrils and bundled fibrils with frayed ends are observed. Striations were visible in a small number of fibrils [Fig. 4(e,f), far right]. Altogether, we observed that the missing httNT segment affects the fibril morphology and surface features, but does not appear to disrupt fibril bundling, providing further support for the prominent role of the PRD in those interfilament interactions [36]. However, branching events are not observed for the ΔN15 fibrils, potentially highlighting a role for the httNT domain in this phenomenon.
Comparison between Q44-HttEx1 and ΔN15-Q44-HttEx1 fibrils using MAS ssNMR
To gain more insights into the molecular consequences of omitting most of the httNT domain, we applied MAS ssNMR to the fibrils formed by uniformly 13C,15N labeled ΔN15-Q44-HttEx1 aggregated at 22 °C. 1D 13C cross-polarization (CP) ssNMR experiments were used to compare the rigid parts of Q44-HttEx1 and ΔN15-Q44-HttEx1 fibrils, both aggregated at 22 °C [Fig. 5 (a)]. The dominant peaks are very similar, but specific smaller peaks are missing in the ΔN15-Q44-HttEx1 fibrils. These peaks coincide with those signals that we previously attributed to the α-helical and partly immobilized httNT, providing an indirect confirmation of these prior assignments [35, 36]. The spectral differences are visualized in Fig. 5 (a, bottom), based on an overlay of the ΔN15-Q44-HttEx1 and Q44-HttEx1 spectra. In the aliphatic region several peaks are missing that correspond to the methyl groups in Met(1 and 8), Ala(2 and 10), Thr3 and Leu(4, 7, and 14), Cε of residues Lys(6, 9, and 15), and residue Glu5. There is also a noticeable decrease in the area of the broad peak observed in the backbone carbonyl (between 165 and 190 ppm) and Cα regions for ΔN15-Q44-HttEx1.
Fig. 5. MAS SSNMR comparison of ΔN15-Q44-HttEx1 and Q44-HttEx1 fibrils.
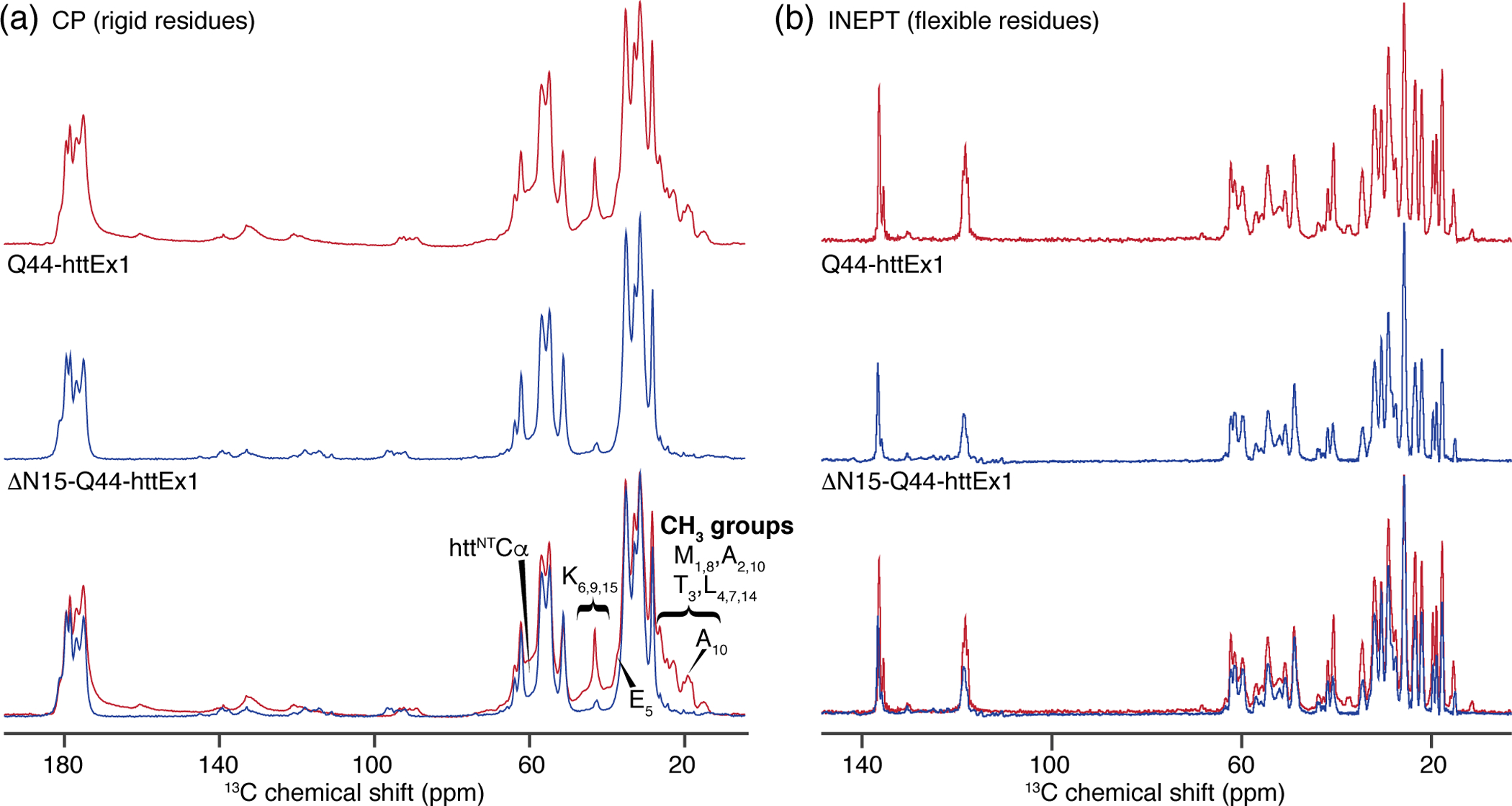
(a) Top: 1D cross polarization (CP) 13C spectrum of Q44-HttEx1 [36] and (middle) ΔN15-Q44-HttEx1, and (bottom) their overlaid comparison, normalized to the peak maxima for Q44-HttEx1. Spectral differences are consistent with the absence in the latter sample of the partly immobilized httNT seen in HttEx1 fibrils. (b) From top to bottom: 13C INEPT of Q44-HttEx1, ΔN15-Q44-HttEx1, and overlaid. The CP spectra feature signals from rigid and partly immobilized parts of the structure, while the INEPT data show only highly flexible residues. The spectra of Q44-HttEx1 are reprinted with permission from Lin et al. (2017) Nature Communications [36].
More insight is gained from 2D ssNMR spectra. The 2D 13C-13C DARR spectrum of ΔN15-Q44-HttEx1 [Fig. 6 (a, b)] consists overwhelmingly of Gln and Pro cross-peaks. As expected from the similarity of the 1D spectra, these peaks coincide perfectly with those of the polyQ amyloid core and the PPII-helical oligoPro segments of normal Q44-HttEx1 fibrils. In contrast, other cross-peaks previously assigned in the Q44-HttEx1 fibrils are absent. Most notably, these include signals from httNT residues, as expected. However, specific peaks from the C-terminal flanking domain are also absent. These include two peaks representing Val-103 along with additional alanine peaks and immobilized parts of the PRD in Q44-HttEx1 [36]. Thus, in absence of httNT these polyQ-proximal partly immobilized parts of the PRD become more mobile. Peaks representing proline residues with a random coil secondary structure (ProRC) are missing completely in the short-mixing DARR spectrum. They do occur in a proton-driven spin diffusion (PDSD) experiment with 250 ms of 13C-13C mixing [Fig. S8 (a)] designed to recouple longer-range or dynamically reduced dipolar couplings. Peaks of low intensity assigned to random coil alainines (AlaRC) are observed in the PDSD spectrum, however peaks assigned to V103 are not. We also performed 2D heteronuclear 13C-15N correlation spectra, shown in Fig. S8 (b). The NCACX and NCOCX spectra show rigid polyQ correlations that closely match the data seen previously for HttEx1 and other polyQ amyloid aggregates [40, 62]. No peaks from other amino acids are detected by NCACX or NCOCX. The lack of Pro peaks in these spectra is due to a combination of the lack of directly bonded N-1H in the Pro backbone, along with dynamics of the PRD (see below). As a workaround, we employed the 2D TEDOR experiment that is not dependent on 1H to 15N CP transfers, and thus works well for Pro residues [Fig. S8 (b); bottom]. Indeed, in this spectrum the Pro peaks are observed at a 15N frequency of 136 ppm, although the peaks require a longer TEDOR buildup time than expected for rigid residues [Fig. 6 (e)]. In summary, these ssNMR data reveal little to no difference in the rigid polyQ core of ΔN15-Q44-HttEx1 compared to Q44-HttEx1 [36, 40], but indicate dynamic changes in the immobilized oligoproline parts of the PRD, and a lack of httNT signals.
Fig. 6. 2D ssNMR analysis of ΔN15-Q44-HttEx1 and Q44-HttEx1 fibrils.
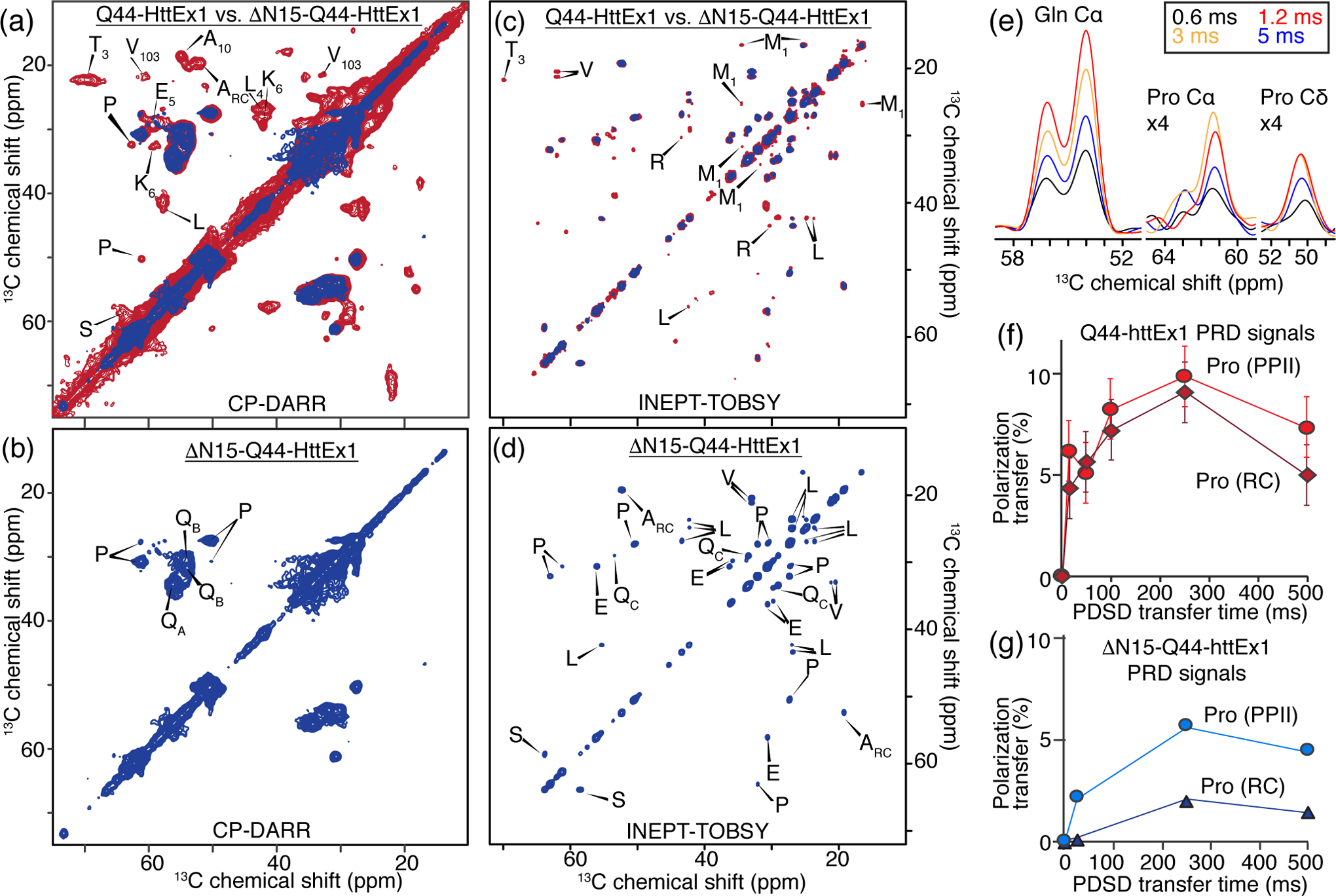
(a-b) Overlay of 2D CP/DARR ssNMR spectra for U-13C,15N ΔN15-Q44-HttEx1 (blue) and Q44-HttEx1 (red) [36], obtained at 13 kHz MAS and 8 ms DARR mixing. In these CP spectra, the signals from immobilized parts of the protein assembly are visible. Panel (b) shows assignment of preserved peaks, while (a) shows assignments for peaks absent in ΔN15-HttEx1 fibrils. (c-d) Analogous 13C-13C INEPT-TOBSY ssNMR spectra of the same samples, reflecting signals of flexible residues. (e) 1D slices from a series of 2D TEDOR spectra with marked mixing times, for Gln N-Cα and Pro N-Cα peaks. Differences in maximum transfer times indicate mobility differences between Gln, ProPPII and ProRC. (f) Normalized CP/PDSD buildup profiles for proline residues with random coil (RC; filled circles) and PPII-helical (filled diamonds) structure in PRD of Q44-HttEx1 [36], and (g) ΔN15-Q44-HttEx1 fibrils. Lower buildup curves are indicative of increased mobility, showing that the random-coil prolines are more dynamic than the PPII helices and that the PRD in ΔN15-Q44-HttEx1 fibrils retains higher mobility than in Q44-HttEx1 fibrils. Q44-HttEx1 spectra are reprinted with permission from Lin et al. (2017) Nature Communications [36].
Dynamics of the C-terminal flanking domain of ΔN15-Q44-HttEx1 fibrils
Polarization buildup curves from CP/PDSD spectra provided evidence of dynamic differences in the Q44-HttEx1 polymorphs [36]. We performed both 13C-13C PDSD and 13C-15N TEDOR variable-mixing time experiments [63] in order to analyze the mobility in ΔN15-Q44-HttEx1 fibrils [Fig. 6 (f, g)] [36]. Polarization transfer between directly bonded Cα-Cβ carbons is rapid in a rigid lattice, with maximum PDSD polarization transfer (10 – 20%) reached within 10 – 20 ms [36]. As before, such a fast buildup is observed for Cα-Cβ glutamine peaks within the amyloid core [36], indicating their crystal-like rigidity. However, the one-bond polarization buildup curves for PPII and ProRC residues in the PRD of ΔN15-Q44-HttEx1 experience a much slower buildup and lower peak maxima, indicating that segments of the PRD experience less rigidity than in HttEx1 [Fig. 6 (f, g)] [36]. A more dramatic difference was observed for two-bond Cα-Cγ polarization transfers, signifying that the ProRC residues experience more dynamics than those with a PPII structure. Analogous findings are obtained in the TEDOR 13C-15N recoupling experiments [63], where the N-Cα one-bond cross-peaks of the polyQ core and PRD are significantly different in their build-up timing [Fig. 6 (e)]. Combined, the MAS ssNMR results show that the PRD flanking domain of ΔN15-Q44-HttEx1 is more mobile that the polyQ core, even more so than their counterparts in Q44-HttEx1 fibrils.
Previously, we and others have noted the presence of a substantial segment of highly flexible residues in the C-terminal tail of aggregated HttEx1 [33, 36, 64]. These residues are detected using ssNMR experiments based on the INEPT scheme, which captures signals from residues with similar mobility to rapidly tumbling soluble protein [65]. In ΔN15-Q44-HttEx1 we again observe these signals with no notable changes in the positions of detected peaks, suggesting that the structural ensemble of these flexible residues is unchanged [Fig. 5 (b)]. The peak intensities are relatively similar, although there is a decrease in the peaks in the aromatic region (80 – 165 ppm), possibly due to a loss of one of the Phe residues within the missing httNT segment. 2D 13C-13C INEPT-based total through bond correlation spectroscopy (TOBSY) spectra showed more peaks for Q44-HttEx1 [Fig. 6 (c)] than in ΔN15-Q44-HttEx1 [Fig. 6 (d)] [36]. The peaks missing in ΔN15-Q44-HttEx1 are consistent with residues found in the httNT domain, along with signals assigned as Arg (which is not found in httNT). A small signal is observed that we previously assigned to M1 for Q44-HttEx1 [36], likely due to incomplete trypsin cleavage. In summary, these INEPT-based analyses show no evidence of a large change in the dynamics of the most flexible parts of the PRD flanking domains. Interestingly, they reveal (retrospectively) the presence of flexible residues in the httNT of the HttEx1 fibrils, coexisting with immobilized α-helical httNT (reaffirmed by the spectral differences in the CP spectra above). From these results we infer a subpopulation of proteins with a more disordered httNT, reminiscent of the findings reported by others on HttEx1 polymorphs that lack signals from the immobilized httNT [49, 64].
A molecular model of the HttEx1 fibril architecture
The width of the observed single-filament Q44-HttEx1 fibrils appears to be approximately uniform. Since we would expect the narrow fibrils to be randomly oriented on the plane of the TEM grids, and with no indication of significant differences in width, it appears that the width and height of these filaments must be similar [Fig. 7 (a, left)]. Our current and prior data support the conclusion that wider fibril polymorphs are assembled from multiple narrow filaments adhered longitudinally, with the individual filaments mimicking the dimensions of the narrow fibrils, representing a type of supramolecular polymorphism [Fig. 7 (a, right)] [48]. With our new data in hand, we decided to revisit our structural model for the misfolded HttEx1 fibrils, to render our prior schematic model [36] more realistic. It was previously shown by ssNMR that the polyQ core of the Q44-HttEx1 fibrils (and other polyQ aggregates) features unusually long β-strands [40, 62, 64], in contrast to various recent structures of other disease-relevant amyloid fibrils [5, 66–68]. This point is also illustrated in Fig. S9 in the SI, which shows two such amyloid core architectures featuring compact architectures of short β-strand elements separated by β-arc bends or turns (“CASSA” motif [5]). In such amyloid structures neighboring amino acids often have different Ramachandran dihedral angles (Fig. S9e). This would result in characteristic peak patterns in NCOCX ssNMR spectra (Fig. S9c–h), as those spectra are based on polarization exchange between neighboring residues. Clearly, experimental polyQ NCOCX spectra are not consistent with an architecture with many short β-strands, and can only be explained by long and largely uninterrupted β-strands. In our detailed analysis of Q44-HttEx1 fibrils we previously concluded that the protofilament core featured β-strands of approximately 6.5 nm long, matching the TEM-observed width of the narrow fibril type [40]. Rather than CASSA motifs, the polyQ core is likely featuring a slab- or block-like architecture previously reported in other studies [69, 70]. Although not shown in our model, or easily seen in our TEM data, these polyQ-based fibrils may be twisted like other amyloids [71, 72].
Fig. 7. Model building of Q44-HttEx1 fibril structure and assembly.
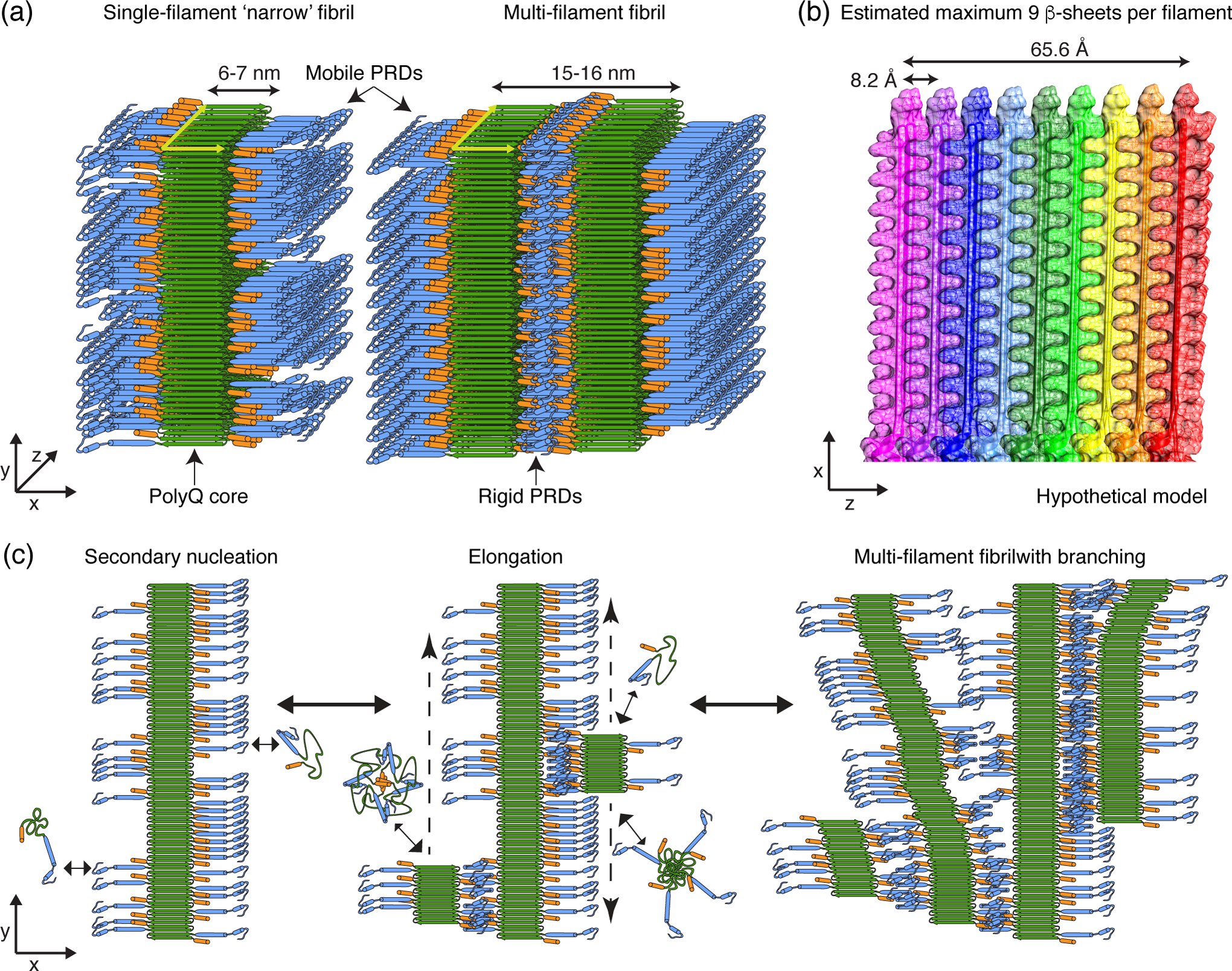
(a) Hypothetical model of a single-filament ‘narrow’ fibril and a multi-filament ‘wide’ fibril. (b) Based on TEM widths, each filament core contains several β-sheets with the depth of the sheet stack approximately the same as the β-strand length. Shown is a model of β-sheet stacking within a filament (see also Supplementary Fig. S10), with each sheet represented as a unique color, showing how approximately 9 stacked β-sheets make up a 6–7nm filament core.(c) Secondary nucleation events on the filament surface initiate the formation of protofilaments that can elongate in parallel (middle) or branch sideways while remaining associated with the template filament (right). Color coding in panels (a,c) is: green: polyQ b-strand core; blue: PRD, with blue cylinders being PPII helices; orange cylinder: httNT segment.
Our modeling of the Q44-HttEx1 fiber architecture will thus assume such a core architecture, in which the length of the β-strands closely approximates the filament width as seen in negative stain TEM [36]. Orthogonal to the β-strand long axis, the fiber diameter appears to be similar, and thus must be assembled from multiple β-sheets. In order to estimate the number of β-sheets within one protofilament, we can refer to the inter-β-sheet distances of 8.2 Å for HttEx1 fibrils, determined here by X-ray powder diffraction [Fig. S10 (a)] in agreement with previous work [36, 73]. To integrate the TEM, X-ray and ssNMR constraints, we built a structural model of a single filament using UCSF Chimera [Fig. 7 (b), S10 (c)] assuming a length and depth of the amyloid core of ~6.5 nm and torsion angle constraints [Fig. S10 (b)] that we previously determined [40, 74]. This model predicts a maximum of 9 sheets per filament, and features the interdigitating side chains identified in our prior MAS ssNMR studies [40]. The core is further stabilized by intra-sheet side-chain and backbone hydrogen bonds along with potential inter-sheet side-chain to backbone hydrogen bonds [Fig. S10 (c)]. This model would imply that most of the Gln residues are buried and distant from the solvent molecules. This observation is not only consistent with prior X-ray studies of polyQ peptides, but also with ssNMR relaxation and water-interaction measurements on HttEx1 and other polyQ aggregates [40, 62, 69, 75]. In summary, we constructed an improved model of the Q44-HttEx1 fibril base filaments, which provides a new perspective on their molecular architecture, with implications that will be examined below.
Discussion
Hierarchical supramolecular polymorphism of HttEx1 fibrils.
Supramolecular polymorphism has now been identified not only in the HD protein deposits, but also in amyloid structures formed in AD and other neurodegenerative diseases [36, 48, 76, 77]. We experimentally probed key parameters affecting this phenomenon in mutant HttEx1 aggregation. Across a range of different sample conditions we observed various indicators that HttEx1 fibrils contain hierarchical side-by-side assemblies of individual filaments, reflecting multiple levels of supramolecular assembly. The presence of protofilaments is seen by TEM as visible striations, fraying of fiber ends, branching events, and variations in width along the length of fibrils. Strikingly, the data point to a common width of 4 – 7 nm for the individual filaments, consistent with the Q44-HttEx1 protofilament architecture that we previously reported [36]. We observed a propensity for higher-order bundling of the narrow and intermediate fibrils, reminiscent of previous in vitro studies, and of HttEx1 inclusions in cells [31]. An intriguing observation is that the wide fibrils lacked the propensity for such higher order bundling. It is tempting to relate this to the disposition of disordered flanking segments on the surface of the filaments. These segments likely explain the fuzzy appearance of the narrow and intermediate fibrils (that are prone to bundling), but which is less apparent in the wider fibrils. In our prior work, we probed for solvent accessibility of the flanking domains in two temperature dependent polymorphs of Q44-httEx1 using a combination of MAS ssNMR and antibody binding assays [36]. We established that the httNT and PRD are partly buried and immobilized in the wider fibrils that had been formed at 22 °C compared to the narrower fibrils formed at 37 °C. In those studies, interactions among the flanking domains were proposed to be responsible for stabilizing the wider fibrils’ structure, with a particular role for the C-terminal PRD segment. The current data reinforce this conclusion. Even the removal of most of the httNT segment did not abolish the higher order assemblies, showing that httNT is not directly involved.
Protein concentration as a polymorphism controlling parameter.
Previous studies concluded that HttEx1 displays a temperature-dependent fibril polymorphism. Our current experiments identify protein concentration as another important determinant of fibril architecture. In particular, higher concentrations of Q44-HttEx1 monomer are necessary to induce the formation of the wide fibrils at 37 °C while lower concentrations result in distinct polymorphs, including narrower ones with more exposed flanking regions. It is worth noting that under disease conditions, the concentrations of HttEx1 are likely to be at or below the low end of the range studied here [78]. Thus, it is possible that the neuronal conditions favor the formation of narrower single-filament fibrils. Given that the flanking regions are well known to interact with protein partners and cellular membranes, their increased exposure on such fibrils may well be biologically relevant. Naturally, these same interactions may further influence the aggregation behavior and polymorphism, beyond what is seen here in vitro. It is important to note that the fibril width (in the nm range) is not directly related to the size of the μm-sized puncta or inclusions typically observed by fluorescence microscopy. This important point was recently visualized in cryo-electron tomography and super resolution studies [31, 79, 80].
A possible rationale for the observed concentration dependence is that prior work has identified at least two parallel competing aggregation mechanisms with different dependences on the monomer concentration [50, 52]. At high concentrations, direct interactions among the expanded polyQ domains were reported to drive the aggregation process, with the flanking domains playing a secondary role. At lower concentrations, the httNT domains drive aggregation by first undergoing non-amyloidogenic self-assembly, causing locally high concentrations that then trigger amyloidogenic interactions among the polyQ segments. Consistent with this second mechanistic model, we observe that the ΔN15-Q44-HttEx1 monomers aggregate at a slower rate than Q44-HttEx1.
Role of httNT in HttEx1’s aggregation mechanisms.
In many ways the ΔN15-Q44-HttEx1 fibrils are morphologically similar to normal HttEx1. They also appear capable of higher order fiber bundling, showing that httNT is not required for these types of interfilament interactions. Yet, there are a number of interesting differences. A clear difference in ThT fluorescence (for the same amount of protein) indicates a change in the binding of this amyloid-binding dye to the two kinds of fibril, with potential implications for our understanding of polymorph-specific dyes and contrast agents [61]. MAS ssNMR detected no changes in the polyQ amyloid core structure relative to that of normal HttEx1 fibrils [36, 40, 64], despite this technique being highly sensitive to such structural changes. Thus, differences in the (weight-normalized) ThT signal must stem from changes in the disposition or accessibility of the polyQ-based amyloid core surface, with an increased number of accessible binding sites present for the same fibril mass. The ΔN15-Q44-HttEx1 fibrils have an average width of 11.6 nm, unlike any of the fibril types observed for Q44-HttEx1, exceeding the width of the ‘narrow’ fibrils that form at low HttEx1 concentrations. Relative to our model of the Q44-HttEx1 fibril architecture [Fig. 7], the average width of fibrillar ΔN15-Q44-HttEx1 is approximately 4 nm less than the average width of the 22 °C Q44-HttEx1 fibrils. It remains unclear whether this is due to a closer inter-protofilament distance enabled by the lack of httNT domain, or a change in the filament architecture or the number of β-sheets within a filament. Nonetheless, both Q44-HttEx1 and ΔN15-Q44-HttEx1 fibrils formed with kinetics that were consistent with a nucleated elongation model, including secondary nucleation and fragmentation events [Fig. 4 (b)]. One notable difference was that there is an absence of apparent branching in the ΔN15-Q44-HttEx1 fibrils even though we do see side-by-side bundling. A recent study has argued that branching by HttEx1 is due to secondary nucleation on the surface of existing fibrils [51]. Thus, our observations may indicate a role for the httNT segment in such a secondary nucleation process. If so, the interactions that still stabilize side by side bundling of (ΔN15-Q44-HttEx1) filaments and protofilaments must be distinct from the interactions that mediate this nucleation and branching process.
Structural architecture of HttEx1 deposits and associated biological implications
We observe that protein monomer concentration plays a key role in dictating the fibril morphology. An interesting question relates to the molecular underpinnings of the observed morphological features. MAS ssNMR chemical shifts are highly sensitive to structural differences, but no MAS ssNMR study has yet identified any significant change in the polyQ signature motif [35, 36, 40, 49, 62, 64, 81], contradicting suggestions of qualitative differences in polyQ architecture proposed from other structural techniques [18, 82]. In our data the main differences between polymorphs occur on the fibril surfaces, in terms of the exposure of non-polyQ flanking domains and the ThT-accessible amyloid core surface.
In Fig. 7 we presented our new model for the HttEx1 fibril structure, which explains not only the ssNMR-detected exposure and burial of respective HttEx1 domains, but also fits the TEM-observed protofilament dimensions. A key feature of our structural model is that the HttEx1 fibril surface combines regions of exposed polyQ amyloid core with regions covered with partly disordered non-amyloid flanking domains. Both aspects of the fibril surface have potentially important implications for the aggregates’ biological properties. Amyloid fibril surfaces are increasingly thought to provide a molecular context that enhances the conversion of (soluble) monomers to amyloidogenic nuclei through secondary nucleation events [Fig. 7 (c, left)] [4]. In HD and other amyloid diseases, there is also an increasing realization that the domains that form a “fuzzy” coat around an amyloid fibril core may be of particular disease relevance by contributing to toxic effects exerted by cytotoxic aggregates [34–36, 42–44]. The httNT segment is thought to interact with cellular membranes and chaperones [45], and houses the potential ubiquitination sites of HttEx1 [83]. The C-terminal flanking domains are similarly implicated in interactions with membranes and other cellular proteins [12, 84, 85]. Recognition of exposed flanking domains may enable the targeting of deposited HttEx1 (e.g. for degradation), but their interactions may also have a detrimental effect by recruiting and trapping essential cellular components. In the HttEx1 polymorphs these domains (especially the PRD) mediate the supramolecular self-assembly into side-by-side multifilament fibrils and higher order bundles. Thus, the exposed flanking domains may be in competition with cellular components and with each other in forming dense filament clusters. This may underpin the reported lower levels of toxicity for large HttEx1 aggregates detected by (confocal) microscopy [86].
Mechanistic underpinnings of fibril surface variability and supramolecular polymorphism.
How may these structural variations originate during fibril assembly? The structure of the aggregation end products is dictated by the misfolding and aggregation pathway that led to their formation. As such, we conclude that the dominant aggregation pathway of HttEx1 must be concentration dependent. This finding is reminiscent of prior work that pointed to a toggling between polyQ- and httNT-driven pathways, which was concentration dependent [50]. At low concentrations, the aggregation initiating effect of httNT was dominant, while at higher concentrations aggregation was driven by the expanded polyQ domain. The httNT-driven and polyQ-driven aggregation mechanisms likely facilitate different core and flanking domain organization [50]. As illustrated schematically in Fig. S11, the fibrils may end up with different degrees of symmetry or clustering in the distribution of flanking domains. For example, the ΔN15-Q44-HttEx1 fibrils may have a more symmetric distribution [Fig. S11 (b)], due to the lack of httNT-driven aggregation. The flanking domain distribution would likely modulate how surface-nucleated filaments grow [Fig. S11 (a)], change the interfilament distances, and affect the propensity for multifilament fibrils to form. The cellular environment is likely to contribute additional complicating factors that can impose a bias on the relative alignment of the flanking domains or otherwise change the architecture of the resulting aggregates. Membrane interactions involving either of the flanking domains have been reported, which would likely change their role in the aggregation process [85, 87, 88]. Similarly, one can expect effects from chaperones that bind the flanking domains and various post-translational modifications [44, 47, 89]. The exact molecular consequences of these intracellular interactions are as yet incompletely understood and warrant further study.
Potential implications for mutant huntingtin at low concentration in vivo.
The concentration-dependent formation of aggregates with different morphologies, dynamics and surface characteristics may have important implications for our understanding of the fate of mutant huntingtin in a cellular context. Based on currently available data, the concentration of mutant Htt (and its fragments) in vivo is measured to be much lower than the values in typical use in mechanistic studies using purified proteins [78]. It is interesting to note the distinct behavior seen at very low effective monomer concentrations produced by the very slow release of monomer at highly substoichiometric FXa concentrations [Fig. S2 (i, l)]. We might have expected narrow fibrils composed of one filament each, but instead observe intermediate-width fibrils with an otherwise similar appearance to those formed at 22 °C [36]. Thus, it may be that very slow aggregation or extremely low monomer concentrations trigger yet another dominant aggregation pathway. Understanding the structural and mechanistic aspects of HttEx1 misfolding and aggregation under ultralow HttEx1 concentrations may be an important direction for future studies.
Conclusion
Together, our results indicate that HttEx1 polymorphism is driven not only by temperature, but also by other parameters including the concentration of monomer in solution. Our findings provide further support for the conclusion that the polymorphism of HttEx1 (with and without the crucial httNT segment) produces filaments with a common polyQ amyloid core, which arrange into different supramolecular arrangements depending on their misfolding pathway. Interestingly, despite initiating and accelerating HttEx1 aggregation, the httNT domain is not necessary for the interfilament interactions but does appear to be important for initiating fibril branching. Naturally, cellular conditions feature numerous complicating factors, including aggregation-modulating (sub)cellular membranes, chaperones and htt post-translational modifications [85, 89–91]. These factors may further modulate the misfolding process and warrant further investigation.
Materials and Methods
Protein expression and purification
The MBP-fusion protein MBP-Q44-HttEx1, featuring mutant HttEx1 with 44 consecutive glutamine residues within the polyQ domain, was subcloned into a pMALc2x plasmid by Genscript (Piscataway, NJ) as previously described [36, 92]. Overexpression in E. coli BL21(DE3)pLysS cells (Invitrogen, Grand Island, NY) was done at natural isotopic abundance for most samples, using a protocol optimized for isotopic labeling [36]. Samples intended for MAS ssNMR were uniformly 13C,15N labeled with 13C D-glucose and 15N ammonium chloride (SKU CLM-139–10 and NLM-467–10, Cambridge Isotopes, Andover, MA) during overexpression. Cells were harvested by pelleting at 7,000 g and resuspended in PBS buffer (SKU BP399–4, Fisher BioReagents, Pittsburgh, PA). 1 mM phenylmethanesulfonyl fluoride (PMSF) (SKU 215740050, Acros Organics, New Jersey, USA) was added to the resuspended cells just prior to lysis and replenished every 0.5 hour as necessary. The cells were lysed by microfluidization using an M-110Y high-pressure pneumatic high shear fluid processor (Microfluidics, Newton, MA) until the transparency of the lysate ceased changing as determined visually. Debris was removed by centrifugation at 38,720g for 1 hour using a Sorvall RC 3C Plus centrifuge (Thermo Scientific, Waltham, MA) and subsequently by filtering over Nalgene Rapid-Flow (Thermo Scientific, Waltham, MA) and/or Millex-GP syringe-driven (Millipore Sigma, Burlington, MD) 0.22 μm PES membranes. The soluble fusion protein was purified using a HisTrap HP nickel column (GE Healthcare, Uppsala, Sweden) with an imidazole (SKU I5513–100G, Sigma, St. Louis, MO) gradient. The purified protein was exchanged into an imidazole-free PBS buffer over an Amicon Ultra centrifugal filter (MilliporeSigma, Burlington, MA) with a regenerated cellulose membrane (10,000 MWCO). The concentration of the fusion protein was determined by its average absorbance (n = 3) at 280 nm, which was measured in a 100 μL quartz cuvette with a 1 cm path length by a DU 800 UV-visible spectrophotometer (Beckman Coulter, Brea, CA). The extinction coefficient of the fusion protein is estimated to be 66350 M−1 cm−1 (determined using the ProtParam tool by ExPASy [93]). The protein purity, molecular weight, and isotopic labeling yield were verified by ESI-TOF MS and SDS-PAGE, as described below [34].
Fibril formation
MBP-Q44-HttEx1 was cleaved to release Q44-HttEx1 by treating with Factor Xa (FXa) protease (SKU PR-V5581, Promega, Madison, WI), or cleaved to release ΔN15-Q44-HttEx1 by treating with N-tosyl-L-phenylalanine chloromethyl ketone treated trypsin lyophilized powder from bovine pancreas (SKU T1426 from Sigma-Aldrich, St. Louis, MO) re-dissolved in PBS buffer with 3 μM HCl. The progression of protein cleavage was monitored by SDS-PAGE (Bio-Rad Mini-Protein Precast TGX Gels 12%) [36] or by ESI-TOF MS as described below. For TEM studies, 10.1 – 98.9 μM MBP-Q44-HttEx1 was treated with FXa at fusion protein to protease molar ratios of 1500:1 – 4.25:1 FP:P as indicated in Fig. 2 and Fig. S2, and the mixture was incubated at 37 °C for at least 1 day to allow for Q44-HttEx1 aggregation. For ΔN15-Q44-HttEx1, 11.6 μM MBP-Q44-HttEx1 was cleaved by trypsin at 3:1 FP:P and incubated for at least 3 days during aggregation. Unless otherwise indicated, in all ΔN15-Q44-HttEx1 preparations the trypsin cleavage reaction was quenched after 10 minutes with the addition of 26 nM PMSF.
Transmission electron microscopy
Fibrils were pelleted at 2880 g for 20–30 minutes and washed with PBS buffer three times to remove free MBP and un-aggregated monomers and oligomers. Aliquots of fibril suspensions were diluted into PBS buffer and 5 μL of the diluted suspension were deposited and adsorbed onto freshly glow-discharged 400 mesh carbon-coated copper grids (SKU FCF400CU50, Electron Microscopy Sciences, Hatfield, PA) for 0.5 – 2 minutes. Excess buffer was removed by blotting. The negative staining agent used was 1% (w/v) uranyl acetate (UA) (SKU 22400, Electron Microscopy Sciences, Hatfield, PA), as indicated. Stain was applied to the grids for approximately 5 seconds before blotting, and then grids were air dried for 5 minutes. Images of the negatively-stained samples were obtained on a Tecnai T12 TEM (FEI, Hillsboro, OR) operating at 120 kV and equipped with an UltraScan 1000 CCD camera (Gatan, Pleasanton, CA). Fibril widths were measured transverse to each fiber axis using the straight Free-Hand tool of ImageJ [94]. Three measurements were obtained per fibril, except in fibrils where the width varied significantly, in which case three measurements were obtained at the widest and narrowest points of the fibril as well as in an intermediate area, and the average of each was included in the analysis. In select cases, fibril widths were verified on isolated and vertically aligned fibrils using the Plot Profile tool of ImageJ, which plots the average gray scale intensity values across a rectangular region of an image [94], and the width of the resulting peak was measured at its estimated half-height. Fourier transform band pass filters were applied to images of fibril bundles without saturation, with 5% tolerance of direction, and without stripe suppression or autoscaling using ImageJ in order to aid in the visibility of individual fibrils within the bundles, as indicated in Fig. 3 (b) [94].
Cleavage kinetics assays by SDS-PAGE and ESI-TOF MS
The kinetics of monomer release by FXa or trypsin cleavage on MBP-Q44-HttEx1 was monitored by SDS-PAGE for samples indicated in Figs. S3, S6, S7. Following the addition of protease to MBP-Q44-HttEx1 as described above, aliquots (15 μL) were removed from the reaction mixture at the indicated time points and mixed with an equal volume of SDS-PAGE loading dye to terminate the proteolysis reaction. An SDS-PAGE gel (Bio-Rad Mini-Protein Precast TGX Gels 12%) stained with Coomassie R-250 dye was analyzed by the Gel analyzer tool in ImageJ [94] in order to estimate the ng of uncleaved MBP-Q44-HttEx1 material present at each time point. Michaelis-Menten kinetics were calculated from the initial cleavage rates using Graphpad Prism version 7 for Mac (Graphpad Software, La Jolla, CA). Kinetics of cleavage were also measured in samples with 1 molar ratios of MBP-Q44-HttEx1 to either FXa or trypsin protease, as indicated in Figs. S3, S6, S7. In these samples, aliquots of a few μL were removed from the reaction mixture and added to formic acid (SKU A117–50, Fisher Chemical, Pittsburgh, PA) to quench the proteolysis reaction at 30 second, 1 minute, 5 minute, and 6 hour time points. ESI-TOF MS was used to analyze the kinetics of cleavage and identify cleavage products, as described below.
Thioflavin T fluorescence assay
Thioflavin T (ThT) fluorescence assays were performed as described previously [36]. Briefly, FXa or trypsin was added to 11.6 μM MBP-Q44-HttEx1 at 5:1 and 3:1 FP:P, respectively. The samples were mixed with a vortex for 15 seconds and then allowed to aggregate at 22 °C for several days. At various timepoints, the aggregates were resuspended using a vortex and 40 μL aliquots of the suspensions were immediately diluted into ThT (SKU 156877, MP Biomedicals LLC, Solon, OH) stock solution (250 μM ThT, 0.02% w/v NaN3, 1x PBS) for a total volume of 400 μL and protein concentration of 1.27 nM. Samples were excited at 445 nm using a 2 nm excitation slit on a FluoroMax-4 spectrofluorometer (Horiba; Kyoto, Japan). Emission was recorded at 489 nm through a 4 nm emission slit. Two measurements were obtained for each sample per timepoint. Curves were fit to nucleated elongation, secondary nucleation dominated, and fragmentation dominated models using AmyloFit over 50 basin hops [60].
HPLC-based sedimentation assay
As described previously [36], an HPLC-based sedimentation assay was used in parallel to the ThT assays in order to accurately monitor monomer concentrations over time. Briefly, aggregates were removed from 15 μL aliquots of the aggregation mixture by centrifugation at 20,800g for 15 min. The supernatant (5 μL) was subsequently diluted 2x into formic acid and loaded onto an Agilent Zorbax SB-C8 4.6 × 50 mm HPLC column with a 1.8 μm particle size using an analytical HPLC (Agilent Technologies). The monomer was eluted over a 15 – 35% C2H3N/H2O gradient (0.05% TFA, 37 °C). The elution was monitored by absorbance at 215 nm (A215), and monomer concentrations were determined by integrating the A215 peaks using ChemStation (Agilent Technologies).
Potential trypsin cleavage products of MBP-Q44-HttEx1
ExPASy’s PeptideCutter tool [93] was used to determine 42 possible trypsin cleavage sites within MBP-Q44-HttEx1, excluding 3 sites (K[−142]P[−141], R[−101]P[−100], and K[104]P[105]) with 19.3%, 29.3% and 23.8% probabilities of cleavage. These residue numbers are relative to the numbering for Q44-HttEx1 following FXa cleavage of MBP-Q44-HttEx1. The 42 potential cleavage sites were marked onto the predicted structure of MBP based on the structure of fusion protein MBP-L30 (PBD ID: 1NMU) using UCSF Chimera [Fig. S7] [95]. The molecular weights of all fragments were calculated using ExPASy’s ProtParam tool [93] assuming 42 active cleavage sites. The molecular weights of all possible fragments were calculated by combining the molecular weights of individual fragments and adding 18 Da (the molecular weight of water) for each peptide bond formation. A total of 945 possible cleavage products were identified, 760 of which have molecular weights above 5000 Da.
Mass Spectrometry
The mass of the MBP-Q44-HttEx1 fusion protein (56.685 kDa) was confirmed within ± 1 Da by ESI-TOF MS using a MaXis II ESI-QTOF mass spectrometer (Bruker Daltonics, Billerica, MA). Mass spectra were deconvoluted by maximum entropy using Compass Data Analysis Hystar 4.1 software (Bruker Daltonics, Billerica, MA), with lower and upper limits of 5000 and 100,000 m/z. MBP-Q44-HttEx1 was deemed usable for experiments if the MS deconvolution curve indicated high purity without any indication of prior proteolytic cleavage. To identify the trypsin cleavage products of MBP-Q44-HttEx1, the fusion protein was cleaved with trypsin in 7 independent trials and the mass spectra were obtained from 5 μL aliquots of the reaction mixture quenched with formic acid within the first six hours following the addition of trypsin. PMSF was not added to any of the reaction mixtures. All peaks observed in the resulting deconvoluted mass spectra were screened against the expected molecular weights of 760 possible cleavage products with MW > 5000 DA within ± 2 Da. An additional 4 independent trials were analyzed after 1 day of aggregation.
Magic Angle Spinning solid state NMR spectroscopy
Isotopically labeled samples were prepared and then packed by pelleting a hydrated suspension of purified protein aggregates into 3.2 mm zirconia thin wall MAS rotors (Bruker Biospin, Billerica, MA). This sedimentation process was done using a home-built ultracentrifugal packing device under centrifugation at ~ 130,000 g in a Beckman Coulter Optima L-100 XP ultracentrifuge equipped with a SW-32 Ti rotor [96]. For ΔN15-Q44-HttEx1, 7 mg of protein was packed into the MAS rotor. Caps were sealed to the rotor with epoxy glue to ensure stable sample hydration. Samples were studied by MAS ssNMR in an unfrozen state. 1D 13C and 2D 13C-13C MAS ssNMR experiments were performed using a wide-bore Bruker Avance I NMR spectrometer operating at a 1H Larmor frequency of 600 MHz (14.1 Tesla) and equipped with a 3.2 mm MAS probe with an HCN ‘EFree’ coil (Bruker Biospin). 2D 15N-13C NCOCX, 2D 15N-13C NCACX and 2D 15N-13C TEDOR spectra [63, 97] were acquired on a Bruker AVANCE NEO 600 MHz (14.1 T) spectrometer, using a 3.2 mm EFree HCN MAS Probe from Bruker Biospin. Immobilized and rigid parts of the fibrils were studied using 1D 13C and 2D 13C-13C cross polarization (CP) based experiments at 12.5 kHz MAS. 13C-13C correlations were obtained from 2D experiments with a ramped 1H-13C CP step and 8 ms dipolar-assisted rotational resonance (DARR) 13C-13C mixing [98]. Dipolar-recoupling curves based on 13C-13C 2D Proton-Driven Spin Diffusion (PDSD) experiments were acquired with mixing times of 0, 25, 250, and 500 ms. The volumes of 2D peaks were integrated using the Sparky NMR software package and normalized to the corresponding peak volume at the diagonal in the PDSD experiment that employed 0 ms mixing [36]. Flexible parts of the fibrils were identified from 1D J-coupling-based 13C spectra which were acquired using rotor-synchronized refocused insensitive nuclei enhanced polarization transfer (INEPT) 1H-13C transfers at 8.333 kHz MAS rate. 2D spectra showing 13C-13C correlations between highly mobile carbons were obtained by combining refocused INEPT 1H-13C transfers with the P913 total through bond correlation spectroscopy (TOBSY) pulse sequence [99]. For the 13C 1D experiments, two-pulse phase modulation (TPPM) 1H decoupling (typically at 83 kHz) was applied during acquisition, and for all 2D 13C-13C experiments the same 1H decoupling was applied during evolution and acquisition [100]. 2D NCACX, NCOCX and TEDOR spectra were acquired at 13kHz MAS and 275K temperature, using 100kHz TPPM proton decoupling, 3s recycle delay and 256 scans per datapoint. For the NCOCX and NCACX experiments, the first step was a CP-MAS transfer from 1H to 15N with a 70–100% ramp on 1H, using rf fields of approx. 75 (1H) and 50 (15N) kHz, and optimized contact time of 1ms. After the first CP step, the magnetization was transferred from 15N to 13C using a 90–100% ramp on 13C, and selective transfer was achieved by setting the carrier frequency to be on-resonance with either C’ or Cα and with optimized contact time of 5 and 2ms respectively. For all transfer pulses, a two-dimensional grid search was used to optimize the power level of the 13C and 15N channels. The homonuclear 13C mixing via DARR was set to 50ms for the NCACX and NCOCX experiments. 2D TEDOR experiments were recorded using a 5μs 90° carbon pulse, 2.5μs 90° proton pulse, 5μs 90° nitrogen pulse, and TEDOR block total durations of 0.6, 1.2, 3, 5 and 8ms. Spectra were acquired with Bruker Topspin, processed in NMRPipe [101], and analyzed with the CcpNmr Analysis program version 2.4 developed by the Collaborative Computation Project for the NMR community (CCPN). The chemical shifts of 13C and 15N were indirectly referenced to 4,4-dimethyl-4-silapentane-1–1 sulfonic acid and liquid ammonia based on external measurements of the 13C signals of adamantane [102]. Experimental details for each MAS ssNMR experiment are available in Table S4.
X-ray powder diffraction
Fibrils were pelleted by ultracentrifugation at 100,000 g for 4 hours (Beckman Coulter Optima MAX Ultracentrifuge, with TLA 120.2 rotor) and then the supernatant was removed. The hydrated pellet was packed into a glass capillary (0.7 mm) using a spatula and PBS buffer was inserted on both ends of the capillary by syringe. Capillaries were then sealed with capillary wax. A Rigaku FR-E generator (2kW, spot size 0.07 mm) with a source wavelength of 1.541780 Å was placed 65 mm from the sample and diffraction data was collected on a Rigaku Saturn 944 CCD camera (Tokyo, Japan) operating at −45 °C. The sample remained at room temperature during data collection. Diffraction datasets were analyzed in Structure Studio v. 2.2.3 r1 (Rigaku).
Structural model building
A model β-hairpin structure of a Q44-HttEx1 monomer was built in UCSF Chimera version 1.13.1 using partial structural constraints obtained previously by MAS NMR [36, 40]. Hydrogens were added to the monomer structure using Dock Prep and charges were assigned using an AMBER ff14SB force field. The monomer structure was duplicated and the duplicate was docked to the original using a Dock Prep method, which considers both steric interactions and H-bonds, to create a two-molecule sheet structure. The two-molecule structure was subsequently iteratively duplicated and docked using Dock Prep to create an 18-mer composed of nine two-molecule sheets. Each structure underwent an iterative energy minimization process involving a total of 12,000 steepest descent steps of 0.02 Å and 1000 conjugate gradient steps of 0.02 Å.
Supplementary Material
Highlights.
Mutant huntingtin exon 1 displays hierarchical supramolecular polymorphism.
Fibril bundling is polymorph dependent, mediated by mobile segments on the surface.
N- and C-terminal flanking domains have distinct roles in the assembly mechanism.
Protein concentration is identified as a driver of huntingtin polymorphism.
Competing mechanisms provide a rationale for polymorphic behavior.
Acknowledgements
We thank Drs. Mingyue Li and Abhishek Mandal for helpful discussions related to experimental design and data analysis. We also acknowledge Audrey Valentine and James Nassur for their contributions to preliminary experiments towards this manuscript. We thank Michael Delk for technical assistance with the NMR spectrometers, Dr. Jinwoo Ahn, Dr. Rieko Ishima, and Christine Monnie for their help and use of the mass spectrometer, and Dr. Alexander Makhov for his help and the use of the electron microscope facility. This work was enabled by funding from the University of Pittsburgh, National Institutes of Health (NIH grants R01 GM112678 to P.C.A.v.d.W., T32 GM088119 to J.C.B., R01 CM11642 S1 to Dr. Jinwoo Ahn and Dr. Rieko Ishima), the Achievement Rewards for College Scientists (ARCS) Foundation (J.C.B.), grant UL1 RR024153 from the National Center for Research Resources (NCRR), and a grant from the CampagneTeam Huntington (P.C.A.v.d.W.).
Abbreviations
- HD
Huntington’s disease
- htt
huntingtin
- httNT
huntingtin N-terminal domain
- polyQ
polyglutamine
- PRD
proline rich domain
- HttEx1
huntingtin exon 1
- Q44-HttEx1
huntingtin exon 1 with 44 residues in the polyQ domain
- MBP-Q44-HttEx1
fusion protein of maltose binding protein and Q44-HttEx1
- ΔN15-Q44-HttEx1
Q44-HttEx1 lacking the first 15 residues of the httNT domain
- FXa
Factor Xa
- FP:P
fusion protein to protease molar ratio
- MAS ssNMR
magic angle spinning solid state nuclear magnetic resonance
- CP
cross polarization
- DARR
dipolar assisted rotational resonance
- PDSD
proton-driven spin diffusion
- INEPT
insensitive nuclei enhanced by polarization transfer
- TOBSY
through bond correlation spectroscopy
- TPPM
two-pulse phase-modulated decoupling
- TEM
transmission electron microscopy
- ESI-TOF MS
electrospray ionization time-of-flight mass spectrometry
Footnotes
Accession numbers
PDB ID: 1NMU
Declarations of interest: none.
References
- [1].Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington Disease. Nat Rev Dis Primers. 2015;1:15005. [DOI] [PubMed] [Google Scholar]
- [2].Mangiarini L, Sathasivam K, Seller M, Cozens BA, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. [DOI] [PubMed] [Google Scholar]
- [3].DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. [DOI] [PubMed] [Google Scholar]
- [4].Chiti F, Dobson CM. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem. 2017;86:27–68. [DOI] [PubMed] [Google Scholar]
- [5].van der Wel PCA. Insights into protein misfolding and aggregation enabled by solid-state NMR spectroscopy. Solid State Nucl Magn Reson. 2017;88:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Matlahov I, Van der Wel PCA. Conformational Studies of Pathogenic Expanded Polyglutamine Protein Deposits from Huntington’s Disease. Exp Biol Med. 2019;244:1584–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ruggeri FS, Vieweg S, Cendrowska U, Longo G, Chiki A, Lashuel HA, et al. Nanoscale studies link amyloid maturity with polyglutamine diseases onset. Sci Rep. 2016;6:31155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ast A, Buntru A, Schindler F, Hasenkopf R, Schulz A, Brusendorf L, et al. mHTT Seeding Activity: A Marker of Disease Progression and Neurotoxicity in Models of Huntington’s Disease. Mol Cell. 2018;71:675–88.e6. [DOI] [PubMed] [Google Scholar]
- [9].Pearce MMP, Kopito RR. Prion-Like Characteristics of Polyglutamine-Containing Proteins. CSH Perspect Med. 2018;8:a024257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Drombosky KW, Rode S, Kodali R, Jacob TC, Palladino MJ, Wetzel R. Mutational analysis implicates the amyloid fibril as the toxic entity in Huntington’s disease. Neurobiol Dis. 2018;120:126–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp Neurol. 2012;238:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qin Z-H, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR, et al. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci. 2004;24:269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sun C-S, Lee C-C, Li Y-N, Yao-Chen Yang S, Lin C-H, Chang Y-C, et al. Conformational switch of polyglutamine-expanded huntingtin into benign aggregates leads to neuroprotective effect. Sci Rep. 2015;5:14992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tycko R Amyloid polymorphism: structural basis and neurobiological relevance. Neuron. 2015;86:632–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yoshimura Y, Lin Y, Yagi H, Lee Y-H, Kitayama H, Sakurai K, et al. Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc Natl Acad Sci USA. 2012;109:14446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Boatz JC, Whitley MJ, Li M, Gronenborn AM, Van der Wel PCA. Cataract-associated P23T γD-crystallin retains a native-like fold in amorphous-looking aggregates formed at physiological pH. Nat Commun. 2017;8:15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aguzzi A, Heikenwaelder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–61. [DOI] [PubMed] [Google Scholar]
- [18].Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci USA. 2009;106:9679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kodali RB, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struc Biol. 2007;17:48–57. [DOI] [PubMed] [Google Scholar]
- [20].Habchi J, Chia S, Limbocker R, Mannini B, Ahn M, Perni M, et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc Natl Acad Sci USA. 2017;114:E200–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet–rich amyloid fibrils. Nat Chem Biol. 2012;8:93–101. [DOI] [PubMed] [Google Scholar]
- [22].Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon I-JL, et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nagy-Smith K, Moore E, Schneider J, Tycko R. Molecular structure of monomorphic peptide fibrils within a kinetically trapped hydrogel network. Proc Natl Acad Sci USA. 2015;112:9816–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Walker LC, Levine H. Corruption and spread of pathogenic proteins in neurodegenerative diseases. J Biol Chem. 2012;287:33109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci USA. 2014;111:E4376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Paravastu AK, Leapman RD, Yau W-M, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lu J-X, Qiang W, Yau W-M, Schwieters CD, Meredith SC, Tycko R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell. 2013;154:1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, et al. Atomic Resolution Structure of Monomorphic Aβ 42 Amyloid Fibrils. J Am Chem Soc. 2016;138:9663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kodali RB, Williams AD, Chemuru S, Wetzel R. Aβ(1–40) forms five distinct amyloid structures whose β-sheet contents and fibril stabilities are correlated. J Mol Biol. 2010;401:503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shahmoradian SH, Galaz-Montoya JG, Schmid MF, Cong Y, Ma B, Spiess C, et al. TRiC’s tricks inhibit huntingtin aggregation. eLife. 2013;2:e00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bäuerlein FJB, Saha I, Mishra A, Kalemanov M, Martínez-Sánchez A, Klein R, et al. In Situ Architecture and Cellular Interactions of PolyQ Inclusions. Cell. 2017;171:179–87.e10. [DOI] [PubMed] [Google Scholar]
- [32].Shen K, Calamini B, Fauerbach JA, Ma B, Shahmoradian SH, Serrano Lachapel IL, et al. Control of the structural landscape and neuronal proteotoxicity of mutant Huntingtin by domains flanking the polyQ tract. eLife. 2016;5:1–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Caulkins BG, Cervantes SA, Isas JM, Siemer AB. Dynamics of the Proline-Rich C-Terminus of Huntingtin Exon-1 Fibrils. J Phys Chem B. 2018;122:9507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hoop CL, Lin H-K, Kar K, Hou Z, Poirier MA, Wetzel R, et al. Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance. Biochemistry. 2014;53:6653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PCA. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J Am Chem Soc. 2011;133:4558–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin H-K, Boatz JC, Krabbendam IE, Kodali R, Hou Z, Wetzel R, et al. Fibril polymorphism affects immobilized non-amyloid flanking domains of huntingtin exon1 rather than its polyglutamine core. Nat Commun. 2017;8:15462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Choudhury KR, Bhattacharyya NP. Chaperone protein HYPK interacts with the first 17 amino acid region of Huntingtin and modulates mutant HTT-mediated aggregation and cytotoxicity. Biochem Biophys Res Commun. 2015;456:66–73. [DOI] [PubMed] [Google Scholar]
- [38].Monsellier E, Redeker V, Ruiz-Arlandis G, Bousset L, Melki R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J Biol Chem. 2015;290:2560–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R. Structural Features and Domain Organization of Huntingtin Fibrils. J Biol Chem. 2012;287:31739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hoop CL, Lin H-K, Kar K, Magyarfalvi G, Lamley JM, Boatz JC, et al. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin-based polyglutamine core. Proc Natl Acad Sci USA. 2016;113:1546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bhattacharyya AM, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, et al. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–35. [DOI] [PubMed] [Google Scholar]
- [42].Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr Trends Neurol. 2011;5:65–78. [PMC free article] [PubMed] [Google Scholar]
- [43].Duennwald ML, Jagadish S, Muchowski PJ, Lindquist SL. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci USA. 2006;103:11045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, et al. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol. 2009;16:1279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Arndt JR, Chaibva M, Legleiter J. The emerging role of the first 17 amino acids of huntingtin in Huntington’s disease. Biomolecular Concepts. 2015;6:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cariulo C, Azzollini L, Verani M, Martufi P, Boggio R, Chiki A, et al. Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc Natl Acad Sci USA. 2017;10:201705372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].DeGuire SM, Ruggeri FS, Fares M-B, Chiki A, Cendrowska U, Dietler G, et al. N-terminal Huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. J Biol Chem. 2018;293:18540–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;56:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Isas JM, Langen A, Isas MC, Pandey NK, Siemer AB. Formation and Structure of Wild Type Huntingtin Exon-1Fibrils. Biochemistry. 2017;56:3579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jayaraman M, Mishra R, Kodali R, Thakur AK, Koharudin LMI, Gronenborn AM, et al. Kinetically competing huntingtin aggregation pathways control amyloid polymorphism and properties. Biochemistry. 2012;51:2706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wagner AS, Politi AZ, Ast A, Bravo-Rodriguez K, Baum K, Buntru A, et al. Self-assembly of mutant huntingtin exon-1 fragments into large complex fibrillar structures involves nucleated branching. J Mol Biol. 2018;430:1725–44. [DOI] [PubMed] [Google Scholar]
- [52].Newcombe EA, Ruff KM, Sethi A, Ormsby AR, Ramdzan YM, Fox A, et al. Tadpole-like Conformations of Huntingtin Exon 1 Are Characterized by Conformational Heterogeneity that Persists regardless of Polyglutamine Length. J Mol Biol. 2018;430:1442–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci USA. 1999;96:4604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Duim WC, Chen B, Frydman J, Moerner WE. Sub-Diffraction Imaging of Huntingtin Protein Aggregates by Fluorescence Blink-Microscopy and Atomic Force Microscopy. ChemPhysChem. 2011;12:2387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–72. [DOI] [PubMed] [Google Scholar]
- [56].Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–58. [DOI] [PubMed] [Google Scholar]
- [57].Dahlgren PR, Karymov MA, Bankston J, Holden T, Thumfort P, Ingram VM, et al. Atomic force microscopy analysis of the Huntington protein nanofibril formation. Nanomedicine. 2005;1:52–7. [DOI] [PubMed] [Google Scholar]
- [58].Lewandowski JR, van der Wel PCA, Rigney M, Grigorieff N, Griffin RG. Structural complexity of a composite amyloid fibril. J Am Chem Soc. 2011;133:14686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wetzel R Physical Chemistry of Polyglutamine: Intriguing Tales of a Monotonous Sequence. J Mol Biol. 2012:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Meisl G, Kirkegaard JB, Arosio P, Michaels TCT, Vendruscolo M, Dobson CM, et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat Protoc. 2016;11:252–72. [DOI] [PubMed] [Google Scholar]
- [61].Wetzel R, Chemuru S, Misra P, Kodali R, Mukherjee S, Kar K. An Aggregate Weight-Normalized Thioflavin-T Measurement Scale for Characterizing Polymorphic Amyloids and Assembly Intermediates. Methods Mol Biol. 2018;1777:121–44. [DOI] [PubMed] [Google Scholar]
- [62].Schneider R, Schumacher MC, Mueller H, Nand D, Klaukien V, Heise H, et al. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J Mol Biol. 2011;412:121–36. [DOI] [PubMed] [Google Scholar]
- [63].Jaroniec CP, Filip C, Griffin RG. 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly (13)C,(15)N-labeled solids. J Am Chem Soc. 2002;124:10728–42. [DOI] [PubMed] [Google Scholar]
- [64].Isas JM, Langen R, Siemer AB. Solid-State Nuclear Magnetic Resonance on the Static and Dynamic Domains of Huntingtin Exon-1 Fibrils. Biochemistry. 2015;54:3942–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Matlahov I, van der Wel PCA. Hidden motions and motion-induced invisibility: Dynamics-based spectral editing in solid-state NMR. Methods. 2018;148:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, et al. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell. 2017;171:615–27.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Schütz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, et al. Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew Chem Int Ed. 2015;54:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sharma D, Shinchuk LM, Inouye H, Wetzel R, Kirschner DA. Polyglutamine homopolymers having 8–45 residues form slablike β-crystallite assemblies. Proteins. 2005;61:398–411. [DOI] [PubMed] [Google Scholar]
- [70].Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci USA. 2002;99:17014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kurouski D, Kar K, Wetzel R, Dukor RK, Lednev IK, Nafie LA. Levels of supramolecular chirality of polyglutamine aggregates revealed by vibrational circular dichroism. FEBS Lett. 2013;587:1638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Periole X, Huber T, Bonito-Oliva A, Aberg KC, van der Wel PCA, Sakmar TP, et al. Energetics Underlying Twist Polymorphisms in Amyloid Fibrils. Journal of Physical Chemistry B. 2018;122:1081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid β-peptide of amyloid plaques. Proc Natl Acad Sci USA. 2002;99:5596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- [75].Sikorski P, Atkins E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecules. 2005;6:425–32. [DOI] [PubMed] [Google Scholar]
- [76].Iadanza MG, Silvers R, Boardman J, Smith HI, Karamanos TK, Debelouchina GT, et al. The structure of a β2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat Commun. 2018;9:4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Röder C, Vettore N, Mangels LN, Gremer L, Ravelli RBG, Willbold D, et al. Atomic structure of PI3-kinase SH3 amyloid fibrils by cryo-electron microscopy. Nat Commun. 2019;10:884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, Miller JRC, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J Clin Invest. 2015;125:1979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sahl SJ, Weiss LE, Duim WC, Frydman J, Moerner WE. Cellular Inclusion Bodies of Mutant Huntingtin Exon 1 Obscure Small Fibrillar Aggregate Species. Sci Rep. 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Duim WC, Jiang Y, Shen K, Frydman J, Moerner WE. Super-resolution fluorescence of huntingtin reveals growth of globular species into short fibers and coexistence of distinct aggregates. ACS Chem Biol. 2014;9:2767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kar K, Hoop CL, Drombosky KW, Baker MA, Kodali R, Arduini I, et al. β-hairpin-mediated nucleation of polyglutamine amyloid formation. J Mol Biol. 2013;425:1183–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Perevozchikova T, Stanley CB, McWilliams-Koeppen HP, Rowe EL, Berthelier V. Investigating the structural impact of the glutamine repeat in huntingtin assembly. Biophys J. 2014;107:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Juenemann K, Jansen AHP, van Riel L, Merkx R, Mulder MPC, An H, et al. Dynamic recruitment of ubiquitin to mutant huntingtin inclusion bodies. Sci Rep. 2018;8:1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gao Y-G, Yang H, Zhao J, Jiang Y-J, Hu H-Y. Autoinhibitory Structure of the WW Domain of HYPB/SETD2 Regulates Its Interaction with the Proline-Rich Region of Huntingtin. Structure. 2014;22:378–86. [DOI] [PubMed] [Google Scholar]
- [85].Burke KA, Kauffman KJ, Umbaugh CS, Frey SL, Legleiter J. The interaction of polyglutamine peptides with lipid membranes is regulated by flanking sequences associated with huntingtin. J Biol Chem. 2013;288:14993–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7:925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tao M, Pandey NK, Barnes R, Han S, Langen R. Structure of Membrane-Bound Huntingtin Exon 1 Reveals Membrane Interaction and Aggregation Mechanisms. Structure/Folding and Design. 2019;0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16:2600–15. [DOI] [PubMed] [Google Scholar]
- [89].Gu X, Greiner ER, Mishra R, Kodali RB, Osmand AP, Finkbeiner S, et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Darrow MC, Sergeeva OA, Isas JM, Galaz-Montoya JG, King JA, Langen R, et al. Structural Mechanisms of Mutant Huntingtin Aggregation Suppression by the Synthetic Chaperonin-like CCT5 Complex Explained by Cryoelectron Tomography. J Biol Chem. 2015;290:17451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Chiki A, DeGuire SM, Ruggeri FS, Sanfelice D, Ansaloni A, Wang Z-M, et al. Mutant Exon1 Huntingtin Aggregation is Regulated by T3 Phosphorylation-Induced Structural Changes and Crosstalk between T3 Phosphorylation and Acetylation at K6. Angewandte Chemie International Edition. 2017;56:5202–7. [DOI] [PubMed] [Google Scholar]
- [92].Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 2002;277:41032–7. [DOI] [PubMed] [Google Scholar]
- [93].Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Chao JA, Prasad GS, White SA, Stout CD, Williamson JR. Inherent Protein Structural Flexibility at the RNA-binding Interface of L30e. J Mol Biol. 2003;326:999–1004. [DOI] [PubMed] [Google Scholar]
- [96].Mandal A, Boatz JC, Wheeler TB, van der Wel PCA. On the use of ultracentrifugal devices for routine sample preparation in biomolecular magic-angle-spinning NMR. J Biomol NMR. 2017;67:165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Baldus M, Petkova AT, Herzfeld J, Griffin RG. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol Phys. 1998;95:1197–207. [Google Scholar]
- [98].Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–7. [Google Scholar]
- [99].Baldus M, Meier BH. Total correlation spectroscopy in the solid state. The use of scalar couplings to determine the through-bond connectivity. J Magn Reson A. 1996;121:65–9. [Google Scholar]
- [100].Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–8. [Google Scholar]
- [101].Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- [102].Harris RK, Becker ED, De Menezes SM, Granger P, Hoffman RE, Zilm KW. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Magn Reson Chem. 2008;46:582–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.