

Abstract
Purpose
Immunotherapy has recently been shown to improve outcomes for advanced PD-L1-positive triple-negative breast cancer (TNBC) in the Impassion130 trial, leading to FDA approval of the first immune checkpoint inhibitor in combination with taxane chemotherapy. To further develop predictive biomarkers and improve therapeutic efficacy of the combination, interrogation of the tumor immune microenvironment before therapy as well as during each component of treatment is crucial. Here we use single-cell RNA sequencing (scRNA-seq) on tumor biopsies to assess immune cell changes from two patients with advanced TNBC treated in a prospective trial at predefined serial time points, before treatment, on taxane chemotherapy and on chemo-immunotherapy.
Methods
Both patients (one responder and one progressor) received the trial therapy, in cycle 1 nab-paclitaxel given as single agent, in cycle 2 nab-paclitaxel in combination with pembrolizumab. Tumor core biopsies were obtained at baseline, 3 weeks (after cycle 1, chemotherapy alone) and 6 weeks (after cycle 2, chemo-immunotherapy). Single-cell RNA sequencing (scRNA-seq) of both cancer cells and infiltrating immune cells isolated were performed from fresh tumor core biopsy specimens by 10x chromium sequencing.
Results
ScRNA-seq analysis showed significant baseline heterogeneity of tumor-infiltrating immune cell populations between the two patients as well as modulation of the tumor microenvironment by chemotherapy and immunotherapy. In the responding patient there was a population of PD-1high expressing T cells which significantly decreased after nab-paclitaxel plus pembrolizumab treatment as well as a presence of tissue-resident memory T cells (TRM). In contrast, tumors from the patient with rapid disease progression showed a prevalent and persistent myeloid compartment.
Conclusions
Our study provides a deep cellular analysis of on-treatment changes during chemo-immunotherapy for advanced TNBC, demonstrating not only feasibility of single cell analyses on serial tumor biopsies but also the heterogeneity of TNBC and differences in on-treatment changes in responder versus progressor.
Keywords: metastatic triple negative breast cancer, immunotherapy, single-cell RNA seq, nab-paclitaxel, anti-PD-1 antibody
Introduction
Immune checkpoint inhibitors such as the PD-1 inhibitor pembrolizumab and PD-L1 inhibitor atezolizumab have shown activity in early phase clinical trials of metastatic TNBC with durable responses in a subset of patients [1–4]. The combination treatment regimen of atezolizumab with nab-paclitaxel has been granted accelerated approval as first immunotherapy for TNBC patient. Furthermore, we and others have demonstrated that response rates and progression-free survival can be significantly enhanced when immune checkpoint inhibitors are combined with the chemotherapeutic nab-paclitaxel [5,6].
Taxane-based chemotherapy is considered a first-line standard of care in metastatic breast cancer [7,8]. It is reported that Taxane based chemotherapy exerts pleiotropic immune-modulating effects, and paclitaxel can promote dendritic cell maturation through a TLR4-dependent manner [9,10]. Nanoparticle albumin-bound-paclitaxel (nab-paclitaxel) is an albumin-bound formation of paclitaxel that was developed to avoid allergic reactions associated with intravenous administration of solvent-based (sb)-paclitaxel (polyethylated castor oil and polysorbate 80) [11]. Nab-paclitaxel can be administered without steroid premedication, which makes it an ideal partner for combination with immunotherapy.
To further improve outcomes in TNBC, optimal chemotherapy combination partners for immunotherapy and optimal sequencing of the therapies need to be defined based on their individual modulation of the tumor microenvironment, underscoring the need for biomarker analysis as well as longitudinal analyses of tumors on treatment.
Here we serially profiled tumors from two patients with easy to access metastatic TNBC who were treated with nab-paclitaxel chemotherapy initially followed by the combination of nab-paclitaxel and pembrolizumab. One patient had a partial response while the other patient showed rapid progression on therapy. We utilized single-cell RNA sequencing (scRNA-seq) of 10x genomics platform, which allowed us to barcode each single cell and sequence around 4000 single cells from each serial biopsy sample collected. We observed significant differences in tumoral immune infiltrates between responder and non-responder at baseline and on-treatment.
Methods
Study design and patients
Specimens were collected in a NYU IRB-approved clinical trial (NCT02752685) and patients gave written informed consent. The trial is an ongoing Phase 2 chemo-immunotherapy study in metastatic breast cancer with the primary objectives of safety and efficacy, as well as translational endpoints exploring the tumor immune microenvironment.
The patients chosen for this report were based on relatively easy access to tumor tissue for the serial biopsies (breast, lymph nodes, pleural effusion) and enrollment into the triple-negative cohort. Patients varied in extent of metastatic disease, biopsy site and prior treatment (one pre-treated, the other de novo metastatic) but both received nab-paclitaxel 100 mg/m2 on Days 1 and 8 of every 21-day cycle and pembrolizumab 200 mg on Day 1 of every 21-day cycle). In the first cycle pembrolizumab was omitted in order to conduct tissue analyses after treatment with nab-paclitaxel alone. Biopsies were obtained at baseline, after 1 cycle of nab-paclitaxel (between day 15–20 of cycle 1) and after the next cycle with combination nab-paclitaxel and pembrolizumab treatment (between day 15–20 of cycle 2).
Sample processing
Triple negative breast cancer immune cells were processed as previously described [12]. Briefly, fresh TNBC core biopsy tumor specimens were received in MACS Tissue Storage Solution (Cat #130-100-008) on ice and minced in 10 cm ultra-low attachment dishes (Cat # 3262, Corning), and digested with Rat tail collagenase IV (Cat # 17104019, Life Technologies) for 30 min. Red blood cells were lysed with ACK buffer (Cat # 420301, BioLegend) and single cells were obtained and stained with DAPI (Cat # 422801, Biolegend) and Calcein (Cat # 425201, Biolegend). The BD Aria III sorter was used with a 100 μm nozzle to sort live (DAPI- Calcein+) single cells.
Single-cell library construction and 10x chromium sequencing
The sorted cellular suspensions were loaded on a 10x Genomics Chromium instrument to generate single-cell gel beads in emulsion (GEMs). Approximately 4000 cells were loaded per channel. Single-cell RNA-Seq libraries were prepared using the following: Single Cell 3’ Reagent Kits v2: Chromium™ Single Cell 3’ Library & Gel Bead Kit v2, PN-120237; Single Cell 3’ Chip Kit v2 PN-120236 and i7 Multiplex Kit PN-120262″ (10x Genomics) as previously described [13], and following the Single Cell 3’ Reagent Kits v2 User Guide (Manual Part # CG00052 Rev A). Libraries were run on an Illumina HiSeq 4000 as 2 × 150 paired-end reads, one full lane per sample, for approximately >85% sequencing saturation.
Alignment, barcode assignment and UMI counting
The Cell Ranger Single Cell Software Suite, version 1.3 was used to perform sample de-multiplexing, barcode ad UMI processing, and single-cell 3′ gene counting. A detailed description of the pipeline and specific instructions to run it can be found at: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger
Data analysis
All scRNAseq derived gene matrices were analyzed using Seurat R package v.2.3.4 [14]. Individual datasets first underwent a stringent filtering criterion to construct a matrix with relevant genes and cells. For a gene to be selected for downstream analysis, it had to be present in a minimum of three cells in the dataset. Similarly, for a cell to be selected, it had to have a minimum of 200 uniquely mapped genes. In addition, dead cells and cell doublets were regressed out by calculating metrics like mito.percentage (mito genes/nUMI) and unique genes mapped ratios (nGene/nUMI). These were different for each individual scRNAseq dataset usually with the upper limit of mito.percentage ranging from 0.05–0.1 and unique genes ranging from 6000–8000. Subsequent to these filtering steps, the dataset was ‘log normalized’ and scaled according to the default Z-scoring in the Seurat package. Briefly, using the Seurat R package we identified cell subpopulations by utilizing the most variable genes and the significant principal component directions that divided these variable genes into separate clusters as visualized by tSNE plots. DE genes were calculated using Wilcoxon rank sum test and the top 100 DE genes were calculated for individual cell subpopulations. We then calculated the most variable genes and principal component eigenvalues for the dataset on those variable genes. Finally, we identified the cell subpopulations using the ‘FindClusters’ Seurat function utilizing relevant PC space and a resolution ranging from 0.6–1.2. These subpopulations were visualized using tSNE plots and individual cell subpopulation specific differentially expressed (DE) genes were calculated by the ‘FindAllMarkers’ Seurat function.
For combining scRNAseq datasets before and after individual patient treatment, we utilized the canonical correlation analysis (CCA) approach [14]. Briefly, each individual dataset was filtered and scaled as described above. Highly variable genes were calculated for each and the top 1000–2000 common genes between both the datasets were extracted. CCA was then run using the ‘RunCCA’ Seurat function and CCA subspaces were calculated which were subsequently aligned by the ‘AlignSubspace’ Seurat function. Finally, we calculated cell subpopulations and performed downstream DE analysis on this integrated dataset. After this alignment, we derived DE data for individual cell subpopulations, overall DE genes for each individual treatment class and DE genes in cell subpopulations shared within different treatment classes.
Results
Increased lymphocytes within tumor infiltrating leukocytes (TILs) from a responder TNBC patient after nab-paclitaxel and pembrolizumab treatment
The responder patient (patient #1) is a 53-year old female who presented with de novo stage 4 TNBC with liver metastases. She had not received any systemic therapy and serial core biopsies were collected from the untreated breast primary. A baseline tumor core biopsy (#1a) was collected on the same day before treatment (day 0), the second one (#1b) after the nab-paclitaxel only cycle (day 16), and the third one (#1c) after nab-paclitaxel and pembrolizumab therapy (day 43, 14 days since cycle 2 starts). The patient experienced a partial response to treatment, maintained for one year.
Using unsupervised analysis, we identified a combined 14 different clusters from the three tumor samples from each of the three time points (Figure 1B). The clusters were visualized on tSNE plots as follows according to top 100 differentially expressed (DE) genes as previously published [15,16] with modifications: three clusters of tumor/epithelial cells (clusters 7, 10, 12) with KRT8, KRT18, KRT19, KRT7 expression; tumor infiltrating lymphocytes, including CD4+PD-1lo (cluster 0, signature gene IL7R, LTB, CD3E, CCR7), CD8+PD-1lo (cluster 1, CD8A, CD8B, IFN, GZMB), PD-1hi T cells (cluster 3, CD2, PDCD1, CD3D), T regulatory cells (Tregs, cluster 5, FOXP3, IL2RA, IL10RA) and B cells (cluster 2, CD79A, MS4A1); and tumor infiltrating myeloid cells, including NK cells (cluster 4, NKG7, KLRC1, GZMB, GNLY), CD14+ (cluster 6, CD14, LYZ), macrophages (cluster 8, CD68), CD16+ (cluster 9, FCGR3A), granulocytes/monocytes (cluster 11, FCN1, MNDA, HCK, LYZ, CD14, CD68) and plasmacytoid dendritic cells (pDC, cluster 13, LILRA4, IL3RA, CLEC4C) (Figure 1B, 1C, supplementary table 1).
Figure 1. Single-cell RNAseq (scRNAseq) from a responder TNBC patient (patient #1) undergo chemo-immunotherapy.
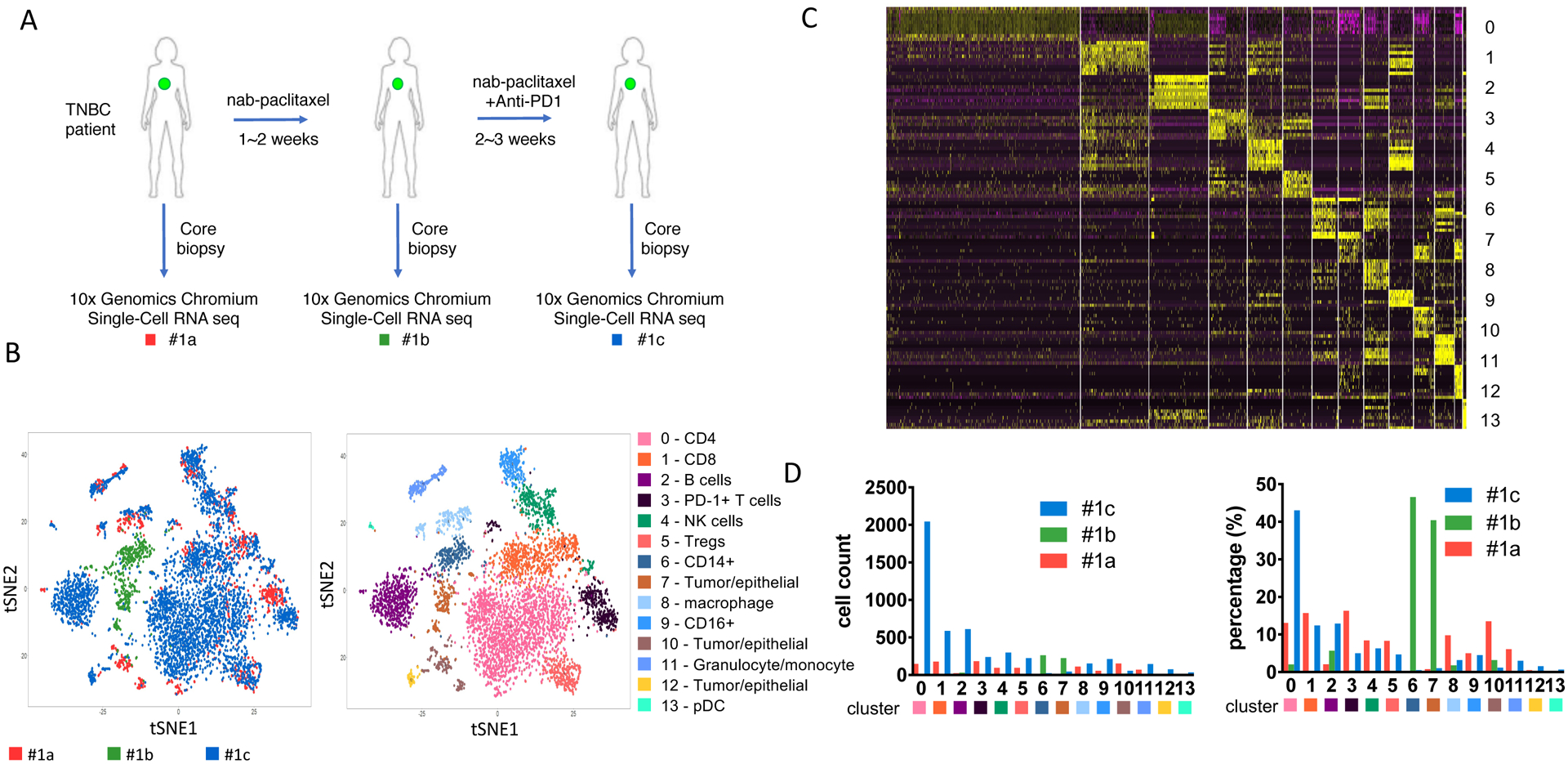
(A) Diagram of the core-biopsy samples collected from patient #1 during nab-paclitaxel and pembrolizumab (anti-PD1) treatment. Red, #1a, sample collected before treatment; green, #1b, sample collected after 1 cycle of nab-paclitaxel; blue, #1c, sample collected after treatment of nab-paclitaxel and pembrolizumab. (B) t-SNE analysis of the combined scRNAseq samples showed in panel A. Left panel, t-SNE plot of clustered cells overlayed with different color representing each TNBC patient biopsy. Right panel, clustering of different tumor or immune subpopulation with different color. (C) Heatmap of top differentially expressed (DE) genes from each cluster showed in panel B. (D) Cell number corresponding to each cluster from individual samples. Left panel, absolute cell count. Right panel, percentage of each population among each biopsy collected.
Among these 14 clusters of cells, there are a total of 970 immune cells from biopsy 1a, 317 cells from 1b and 4581 cells from 1c. The significant variation in specimen cellularity across the 3 time points is possibly due to the size/quality of the core biopsy and/or the impact of treatment. Due to the low number of cells at the 2nd time point, results have to be interpreted with caution, therefore we focus on immune cell changes between baseline and the 3rd time point. The most striking change on treatment was the significant expansion of CD4+PD-1low population (cluster 0) (Fig. 1D). This population represented 13.1% of cells at baseline (1a), which increased to 43.0% after pembrolizumab treatment (1c). Notably, cluster 0 showed heightened expression of CCR7, which marks circulating memory T cells that are activated [17,18]. CD8+ T cells (cluster 1) comprised 15.7% of cells at baseline (#1a) and remained relatively stable after pembrolizumab treatment at 12.4% (#1c) (Figure 1D). The PD-1+ T cells population (cluster 3) however, was greatly reduced from 16.3% at baseline (#1a) to 5.0% after treatment (#1c). This suggests that pembrolizumab can efficiently target intratumoral PD-1+ T cells. Induction of PD-1 on T cells is a hallmark of cancer, resulting in immune evasion [19]. Our data also suggest T cells from cluster 3 produce IFNγ and not only express high levels of PD-1, but also high levels of other co-inhibitory molecules including CTLA-4, LAG3 and TIGIT (Figure 1C). These exhausted T cells (Tex) have been reported to be a major target of PD-1 blockade and can predict clinical outcome in other cancers such as melanoma [20]. Consistent with reduced PD-1+ T cells and increased CD4+ T cells, the percentage of intratumoral Tregs is also reduced from baseline (8.3%) to 4.7% after the treatment (Figure 1D). CD14+ myeloid cells (cluster 6) were infrequent and remained relatively stable after treatment (0.1% to 0.5%, Figure 1D).
Tumor/epithelial cell frequencies (cluster 7, 10 and 12) significantly decreased with therapy (14.8% to 3.8%), consistent with the clinically observed tumor response (Figure 1D).
Persistent myeloid subpopulations and reduced T lymphocytes from a patient with disease progression after chemo-immunotherapy
The non-responder patient (patient #2) is a 51-year old female who presented with recurrent disease while receiving adjuvant chemotherapy (oral capecitabine, previously treated with neoadjuvant adriamycin, cyclophosphamide and paclitaxel) with pleural and lymph node metastases as well as malignant pleural effusions. The patient experienced rapid progression during study treatment, consistent with primary resistance to PD-1 blockade and died shortly after the study from progressive cancer. Both, pleural fluid (pf) and a biopsy of a nodal metastasis (ln) were obtained. Similar to the first patient, the tumor biopsy at the 2nd time point yielded limited cells, and thus was not suitable for analysis. At baseline, pleural fluid (#2a pf) and LN (#2a ln) were collected 2 days prior to treatment start. After nab-paclitaxel and pembrolizumab, both pleural fluid (#2c pf) and a LN core biopsy (#2c ln) were collected (day 31, 20 days since cycle 2 starts). The biopsy of #2c ln did not meet the quality control (QC) requirement and was not sent for scRNA seq analysis, all other three samples from patient #2 were further analyzed.
ScRNA seq analysis of patient #2 showed a very distinct distribution of immune cell infiltrates when compared to patient #1 (Figure 2A), with a total of 4746 cells from biopsy #2a pf, 107 cells from #2a ln and 1283 cells from #2c pf. Due to low cell number collected from #2a ln biopsy, we observed significant lower cell number and percentage of immune cells infiltrates comparing with baseline #2a pf sample. In patient #2, we identified 13 subpopulations, with significant dominance in myeloid cells (66.4% among all cells from baseline sample #2a pf), compared with 29.6% myeloid cells in patient #1 baseline sample (#1a). For #2a pf, the myeloid subpopulations include CD14+ monocytes (cluster 0, 1625 cells, 33.9%), CD68+ macrophages (cluster 2, 529 cells, 11.0%; cluster 7, 125 cells, 2.6%), three different DC populations (cluster 3, 508 cells, 10.5%; cluster 4, 323 cells 6.7%; cluster 11, 44 cells, 0.9%), and other myeloid subpopulations (cluster 8, 1 cell, 0%; cluster 10, 38 cells, 0.8%) (Figure 2B, 2C, supplementary table 2). Similar to what we observed in the analysis of patient #1, CD14+ cells do not respond to nab-paclitaxel plus pembrolizumab treatment, and patient #2, the CD14+ population expanded and increased to 50.8% post cycle 2 treatment (#2c pf). For macrophage clusters 2 and 7, it mainly showed M2 phenotype rather than M1 with expression of CSF1R, this is in contrast with Pt#1 whose infiltrates (cluster 8) show a M1 phenotype with CXCL9 and, CXCL10 and HLA-DR expression (supplementary table 1, 2) [21]. Our analysis also showed three different DC subpopulations in the baseline biopsy sample (#2a pf), including conventional DC (cluster 3, CD1C, HLA-DR, HLA-DQ, HLA-DP), plasmacytoid DC (cluster 4, LILRA4, CLEC4C, GZMB, IGJ) and BATF3 DC (cluster 11, CLEC9A, BTLA, BATF3, CD1C) (Figure 2B, supplementary table 2) [16,22]. Dendritic cells are important antigen-presenting cells, and BATF3 DC are critical for antigen cross-presentation to stimulate cytotoxic T cell immunity and effector T cell trafficking [22,23]. However, these three DC subgroups did not exert their immune surveillance function in the progressing patient and these DC subsets were found to be reduced after the treatment (#2c pf; cluster 3, 4.3%; cluster 4, 0.3%; cluster 11, 0.2%) (Figure 2A, 2C).
Figure 2. Persistent myeloid subpopulations from a metastatic TNBC patient progressed after chemo-immunotherapy.
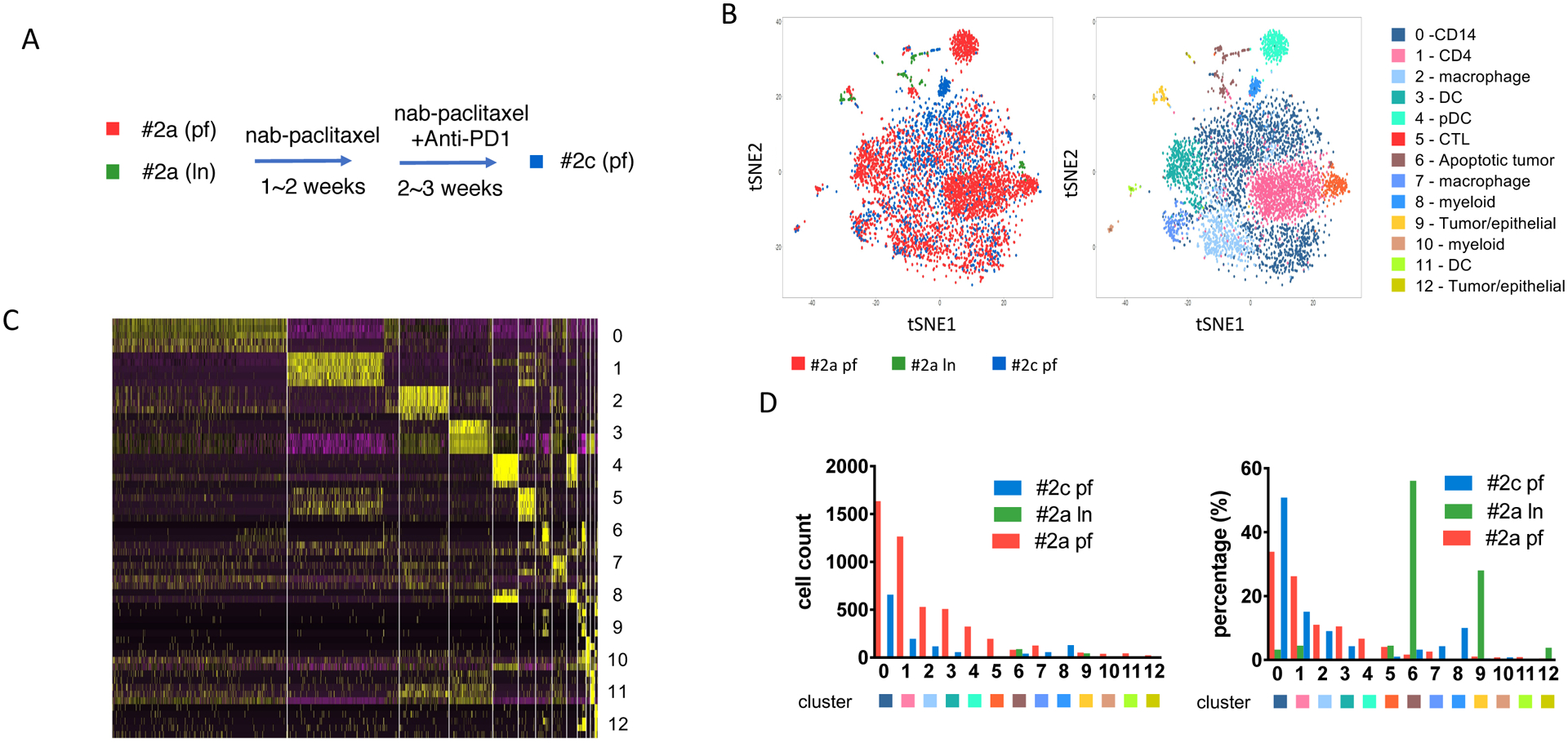
(A) Sample collection diagram from patient #2 before and after nab-paclitaxel and pembrolizumab (anti-PD1) treatment. Pleural fluid (PF) (#2a pf, red) and lymph node (LN) (#2a ln, green) samples collected before treatment; pleura fluid (#2c pf, blue) collected after treatment of nab-paclitaxel and pembrolizumab. (B) Immune infiltrates annotation shown as t-SNE plot of the combined samples collected from patient #2. The spatial location between each cell was calculated by principal component directions of most variable genes and visualized by t-SNE plot. Left panel, each dot represents one cell from each group. Red, #2a pf; green, #2a ln; blue, #2c pf. Right panel, annotation of total 13 clusters of cell populations from patient #2 biospies. Each subpopulation of cells visualized using tSNE plot and specific DE genes. (C) Heatmap of expression levels of top differentially expressed (DE) genes within each cell analyzed within each cluster identified corresponding to panel. (D) Cell number counts for each cluster identified. Left panel, each column represents total cell count for each cluster. Right panel, distribution of each cluster cell numbers from each sample as percentage in total cells.
We identified two lymphoid subpopulations in the baseline sample (#2a pf) of patient #2, CD4+ T cells (cluster 1, CD2, IL7R, LTB, CCR7) and cytotoxic T lymphocytes (CTLs) (cluster 5, CD3E, GZMA). In contrast, patient #1 showed high expression of IFN and GZMB in the CD8+ population (cluster 1), key factors for cytotoxicity of CD8+ T cells. These two factors are not detectable in the top DE genes from patient #2 baseline sample (#2a pf). We did not identify a distinct PD-1 high T cell population or B cell population based on t-SNE plot analyzed. Instead, PD-1 low CD4+ and CD8+ were both reduced after treatment, as compared sample after treatment (#2c pf), for both CD4+ cells dropped from 26.2% at baseline (#2a pf) to 15.1% after the treatment (#2c pf), and CTL decreased from 4.1% at baseline (#2a pf) to 1.0% after the treatment (#2c pf). Taken together, our analysis of this non-responding patient shows that the combination treatment failed to unleash the T cells to activate anti-tumor immunity. Instead, the suppressive myeloid infiltrates, especially the CD14+ subpopulation, persisted and expanded at metastatic sites, leading to the progression of the tumor. It has been reported from a clinical trial of metastatic melanoma patients undergoing nivolumab treatment that high levels of CD14+HLA-DRloCD11b+ M-MDSC (monocytic-myeloid derived suppressor cells) before treatment are associated with poorer PFS and OS [24]. MDSCs have been shown to suppress immunity by blocking the activation and proliferation of CD8+ and CD4+ T cells whereas M1 macrophages have been shown to facilitate rejection of established tumors [25,26]. It has also been reported that MDSCs mediate local immunosuppression and the efficacy of checkpoint blockade in a CXCR2 dependent manner [27].
Distinct immune infiltrates composition of the two patients at both baseline and post treatment biopsies
To understand why these two patients responded differently to chemo-immunotherapy, despite the fact that they are both metastatic TNBC patients with high levels of immune infiltrates in the tumor, we cross compared these two patients at baseline before treatment (#1a) from patient #1 vs (#2a pf) from patient #2 and post nab-paclitaxel and pembrolizumab treatment (#1c) from patient #1 vs #2c pf) from patient #2). It is important to point out that biopsies in patient #1 were obtained from the breast cancer primary, whereas the sample of patient #2 used for this comparison was derived from malignant pleural effusion. This may limit the conclusions of our inter-patient comparison, whereas analyses presented above discussed intra-patient changes when serial biopsies were obtained from the same site.
As demonstrated by t-SNE plot at baseline, we observed that although at baseline both share the same cluster of both CD4+ (cluster 1) and CD8+ (cluster 3) populations, the cells from each patient are separated at different areas rather than evenly distributed within the annotated cluster, indicating that they exert distinct transcriptional profiling (Figure 3A, supplementary table 1). Further analysis of comparing the two tumors at baseline for significant gene expression fold change, the tumor of patient #1 shows an upregulation of FOS, JUNB, CD69 and RPS26 in CD4+ T cells, and an upregulation of RPS26, JUNB and RGS1 in CD8+ T cells (supplementary fig. 1, table 3). Both Fos and Jun proteins can form heterodimer and bind to AP-1 consensus sequences, and activate downstream IL-2 expression through NFAT, which will lead to T cell activation and proliferation [28,29]. In the meanwhile, co-stimulation is required for enhanced production of IL-2 and other cytokines provided by CD28 pathways [30], potentially indicating patient #1 is more prone to immune checkpoint blockade that inhibits co-inhibitory signaling. Increased CD69 in CD4+ cells from patient #1 at baseline indicates there are more antigen-experienced and activated T cells [31]. On the other hand, the myeloid infiltrates are mainly seen at baseline in the tumor of patient #2 (clusters 0, 2, 4, 5, 7 and 9). Unlike T lymphocyte infiltrates, myeloid infiltrates form both patients at baseline are evenly distributed rather than aggregate at different areas (Figure 3A).
Figure 3. Comparison between patient #1 and patient #2 biopsies.
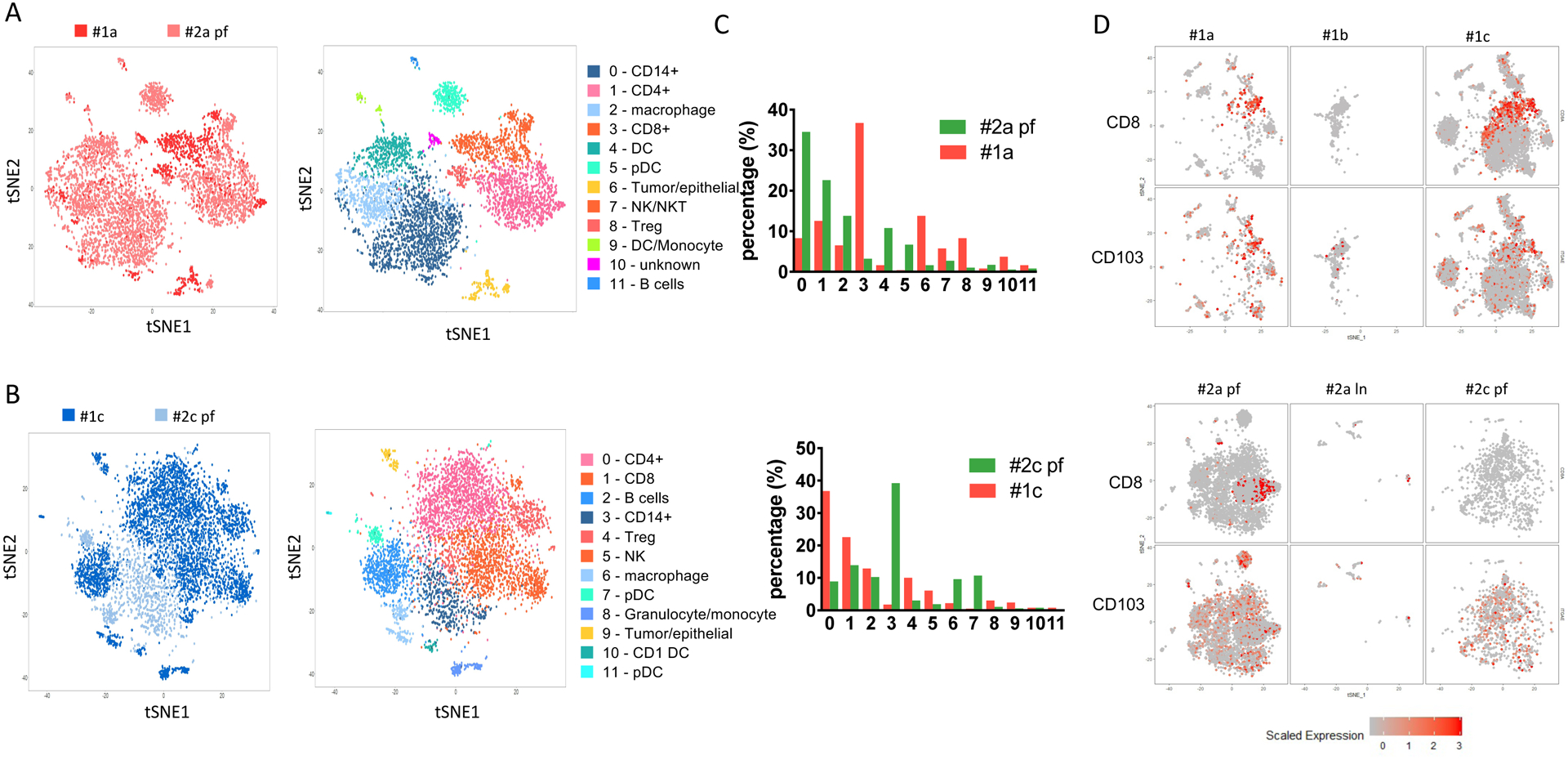
(A) Comparison of baseline samples between patient #1 (#1a) and patient #2 (#2a pf) collected before the treatment. Left panel, combined tSNE plot analysis of #1a and #2a pf. Red, #1a; pink, #2a pf. Right panel, annotation of tSNE plot for each cell subpopulations. (B) Comparison of after treatment samples from patient #1 (#1c, dark blue) and patient #2 (#2c pf, light blue). The tSNE plots are colored by either cell group (left panel) or subpopulation annotation (right panel). (C) Cell number counts for each cluster identified and shown by absolute cell count (upper panel) or percentage (lower panel). (D) TRM infiltration in each sample collected from patient #1 and patient #2. CD8 and CD103 (ITGA) expression levels on each cell from individual samples collected from patient #1 (upper panel) and patient #2 (lower panel).
When we compared the two post treatment biopsies of responding patient #1 (#1c) and nonresponding patient #2 (#2c pf), the remaining CD4+ and CD8+ T cells do not separate in the analysis of both tumors, indicating similarity at the transcriptional level (Figure 3B). This is further confirmed by volcano plot analysis of comparing both CD4+ and CD8+ T cells between the post-treatment samples (#1c and #2c pf), no major difference of upregulated genes is observed (Supplementary fig. 2, table 4). On the other hand, genes expression of myeloid cell markers, such as CD14 and CD68, are decreased in the non-responding patient (#2c pf). Whether this has biological meaning, remains to be explored. Meanwhile, #1c has a much higher percentage of T cell infiltrates when compared with #2c pf, including percentages of CD4+ (36.8% vs 8.9%), CD8+ (22.6% vs 13.9%) and Treg (10% vs 3%) (Figure 3C). This is in contrast to the observed lower frequency of CD4+ cells from the responding patient at baseline biopsy sample #1a (12.6%) when compared to the with the frequency of CD4+ cells in the non-responding patient sample #2a pf (22.6%).
The CD8+ T cell population of the responding patient at baseline (#1a) showed characteristics of resident memory T cells (TRM, displaying the phenotype of CD8+CD103+) and this population was further expanded post cycle 2 treatment (#1c). This contrasts with non-responder patient#2 samples, where no TRM were detected before (#2a pf) or after treatment (#2c pf) (Figure 3D). CD8+ TRM tumor infiltration has been previously demonstrated in breast cancer patients, where the TRM gene signature was significantly associated with improved patient survival in TNBC patients [32]. Whether TRM infiltration in TNBC tumor correlates with response to anti-PD-1 treatment, remains to be explored.
Conclusions
Here, we demonstrate feasibility of profiling serial biopsy samples from TNBC patients that undergo chemoimmunotherapy using scRNA sequencing technology. By assessing subsets of tumor-infiltrating immune cells on a single cell level, we show a significantly different composition in the pre-treatment tumor microenvironment as well as on-treatment changes associated with response to PD-1 blockade. The main differences observed at baseline included the presence of IFN+ and GZMB+ CD8+ T cells as well as TRM in the responding patient versus the presence of a dominant myeloid infiltrate along with absence pf PD-1high T cells in the non-responder. This is consistent with prior reports that TRM in breast cancer are associated with improved prognosis [32] and that tumor-infiltrating lymphocytes predict response to immunotherapy[33]. The most striking difference on-treatment was the significant expansion of CD4+PD-1low population and decrease in PD-1+ T cells population in the responder patient versus the expansion of myeloid cell populations in the nonresponder.
A limitation of our results is the small sample size of two patients as well as the difference in prior treatments which is a recognized prognostic factor, as patients with de novo metastatic disease (such as patient #1) have a greater chance of response to therapy versus patients with pretreated/chemotherapy resistant disease (such as patient #2) [5]. Furthermore, the different biopsy sites between the two patients limit cross-baseline comparison as the immune infiltrate can vary across primary and different metastatic sites [33]. However, while only baseline and post chemo-immunotherapy samples yielded adequate cell numbers for analyses, serial biopsies for each patient were taken from the same site which allows intra-patient comparison of pre- and post-treatment tumor samples.
It is of great need to generate a robust system that can be used for core biopsy samples, to help identify predictive biomarkers for immunotherapies[33]. Due to the scarce number of the cells obtained in these samples, the conventional method of flow cytometry and other routine immunology techniques remain challenging for serial biopsies. Through the unbiased transcriptome analysis of different subpopulations of immune cells, we are able to identify candidate genes that may be critical for the outcome of the patient in response to treatment. This is in contrast with flow cytometry analysis which only has limited markers that can be tested in each sample.
It is not always feasible to perform scRNA seq for TNBC patients or other clinical samples, due to its complex requirement for timing and the techniques. Instead, standard immunohistochemistry is more widely used for specimens obtained. As a newly emerged method that provides much more informative data for the immune composition of the tumors, scRNA seq holds the promise of providing indications for biomarkers and therapeutic targets to guide future immunotherapy strategies when properly used.
Supplementary Material
Supplementary table 2. Signature genes of each cluster from patient #2
Supplementary table 1. Signature genes of each cluster from patient #1
Supplementary Figure 1. Volcano plot of gene expression level comparison of baseline samples between patient #1 (#1a) and #2 (#2a) before treatment.
Supplementary Figure 2. Comparison of between patient #1 (#1c) and #2 (#2c pf) after treatment of nab-paclitaxel and pembrolizumab.
Supplementary table 4. Differentially expressed genes between #1c and #2c pf within CD4+ an CD8+ T cells
Supplementary table 3. Differentially expressed genes between #1a and #2a within CD4+ an CD8+ T cells
Acknowledgments
We would like to thank the patients participating in the trial. We would also like to thank staff at the Clinical Trial Office (CTO) of NYU Langone’s Perlmutter Cancer Center and the Center for Biospecimen Research and Development (CBRD) at NYU Langone Health for supporting this research.
Funding
This work has been supported by the NIH CA219670, CA216188, CA205150 and CA195740 (K.K.W.) as well as P30CA016087 (for the Center for Biospecimen Research and Development and Genome Technology Core at NYU Langone Health). CMP was supported by funds from Breast Cancer Research Foundation, NCI Breast SPORE program (P50-CA58223), and RO1-CA148761.
Conflict of interests
C.M.P is an equity stockholder and consultant, and Board of Director Member, of BioClassifier LLC and GeneCentric Diagnostics. C.M.P is also listed an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. M.K. receive research support from Merck, Agenus and AgenTus. M.K. is a consultant for XCella Biosciences and Agenus. K.K.W. is a founder and equity holder of G1 Therapeutics. K.K.W. has sponsored Research Agreements with MedImmune, Takeda, TargImmune and BMS. K.K.W. has consulting & sponsored research agreements with AstraZeneca, Janssen, Pfizer, Novartis, Merck, Ono, Array. S.A. receives research funding to institution from Genentech, Merck, BMS, Amgen, Novartis and Celgene and is uncompensated consultant or steering committee member for BMS, Genentech and Merck.
Footnotes
Availability of the data and materials
The datasets used and/or analyzed during current study are available from the corresponding authors upon request
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The protocol was approved by the NYU IRB for the following studies a) Phase 2 nab-paclitaxel plus pembrolizumab in HER2-negative breast cancer (15–00441, ClinicalTrials.gov: NCT02752685), b) the NYU institutional universal collection protocol (16–00122) and c) Dissecting the Immunobiology of Breast Cancer protocol (S17–01382).
References
- 1.Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L (2016) Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 34 (21):2460–2467. doi: 10.1200/JCO.2015.64.8931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, Kuter I, Nanda R, Cassier PA, Delord JP, Gordon MS, ElGabry E, Chang CW, Sarkar I, Grossman W, O’Hear C, Fasso M, Molinero L, Schmid P (2019) Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol 5 (1):74–82. doi: 10.1001/jamaoncol.2018.4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, Cescon DW, Iwata H, Campone M, Nanda R, Hui R, Curigliano G, Toppmeyer D, O’Shaughnessy J, Loi S, Paluch-Shimon S, Tan AR, Card D, Zhao J, Karantza V, Cortes J (2018) Pembrolizumab Monotherapy for Previously Treated Metastatic Triple-Negative Breast Cancer: Cohort A of the Phase 2 KEYNOTE-086 Study. Ann Oncol. doi: 10.1093/annonc/mdy517 [DOI] [PubMed] [Google Scholar]
- 4.Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, Liu MC, Iwata H, Ryvo L, Wimberger P, Rugo HS, Tan AR, Jia L, Ding Y, Karantza V, Schmid P (2018) Title: Pembrolizumab Monotherapy for Previously Untreated, PD-L1-Positive, Metastatic Triple-Negative Breast Cancer: Cohort B of the Phase 2 KEYNOTE-086 Study. Ann Oncol. doi: 10.1093/annonc/mdy518 [DOI] [PubMed] [Google Scholar]
- 5.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, Investigators IMT (2018) Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379 (22):2108–2121. doi: 10.1056/NEJMoa1809615 [DOI] [PubMed] [Google Scholar]
- 6.Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, Gallardo C, Lipatov O, Barrios CH, Holgado E, Iwata H, Masuda N, Torregroza Otero M, Gokmen E, Loi S, Guo Z, Zhao J, Aktan G, Karantza V, Schmid P (2020) KEYNOTE-355: Randomized, double-blind, phase III study of pembrolizumab + chemotherapy versus placebo + chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer. Journal of Clinical Oncology 38 (15_suppl):1000–1000. doi: 10.1200/JCO.2020.38.15_suppl.1000 [DOI] [PubMed] [Google Scholar]
- 7.Cardoso F, Harbeck N, Fallowfield L, Kyriakides S, Senkus E, Group EGW (2012) Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 23 Suppl 7:vii11–19. doi: 10.1093/annonc/mds232 [DOI] [PubMed] [Google Scholar]
- 8.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology — breast cancer (2018). V1 [DOI] [PubMed] [Google Scholar]
- 9.Emens LA, Middleton G (2015) The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res 3 (5):436–443. doi: 10.1158/2326-6066.CIR-15-0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfannenstiel LW, Lam SS, Emens LA, Jaffee EM, Armstrong TD (2010) Paclitaxel enhances early dendritic cell maturation and function through TLR4 signaling in mice. Cell Immunol 263 (1):79–87. doi: 10.1016/j.cellimm.2010.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schettini F, Giuliano M, De Placido S, Arpino G (2016) Nab-paclitaxel for the treatment of triple-negative breast cancer: Rationale, clinical data and future perspectives. Cancer Treat Rev 50:129–141. doi: 10.1016/j.ctrv.2016.09.004 [DOI] [PubMed] [Google Scholar]
- 12.Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, He MX, Walker W, Zhang G, Tian T, Cheng C, Wei Z, Palakurthi S, Bittinger M, Vitzthum H, Kim JW, Merlino A, Quinn M, Venkataramani C, Kaplan JA, Portell A, Gokhale PC, Phillips B, Smart A, Rotem A, Jones RE, Keogh L, Anguiano M, Stapleton L, Jia Z, Barzily-Rokni M, Canadas I, Thai TC, Hammond MR, Vlahos R, Wang ES, Zhang H, Li S, Hanna GJ, Huang W, Hoang MP, Piris A, Eliane JP, Stemmer-Rachamimov AO, Cameron L, Su MJ, Shah P, Izar B, Thakuria M, LeBoeuf NR, Rabinowits G, Gunda V, Parangi S, Cleary JM, Miller BC, Kitajima S, Thummalapalli R, Miao B, Barbie TU, Sivathanu V, Wong J, Richards WG, Bueno R, Yoon CH, Miret J, Herlyn M, Garraway LA, Van Allen EM, Freeman GJ, Kirschmeier PT, Lorch JH, Ott PA, Hodi FS, Flaherty KT, Kamm RD, Boland GM, Wong KK, Dornan D, Paweletz CP, Barbie DA (2018) Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 8 (2):196–215. doi: 10.1158/2159-8290.CD-17-0833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8:14049. doi: 10.1038/ncomms14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36 (5):411–420. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, Wang S, Rabasha B, Liu D, Zhang G, Margolais C, Ashenberg O, Ott PA, Buchbinder EI, Haq R, Hodi FS, Boland GM, Sullivan RJ, Frederick DT, Miao B, Moll T, Flaherty KT, Herlyn M, Jenkins RW, Thummalapalli R, Kowalczyk MS, Canadas I, Schilling B, Cartwright ANR, Luoma AM, Malu S, Hwu P, Bernatchez C, Forget MA, Barbie DA, Shalek AK, Tirosh I, Sorger PK, Wucherpfennig K, Van Allen EM, Schadendorf D, Johnson BE, Rotem A, Rozenblatt-Rosen O, Garraway LA, Yoon CH, Izar B, Regev A (2018) A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175 (4):984–997 e924. doi: 10.1016/j.cell.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N (2017) Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356 (6335). doi: 10.1126/science.aah4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell JJ, Murphy KE, Kunkel EJ, Brightling CE, Soler D, Shen Z, Boisvert J, Greenberg HB, Vierra MA, Goodman SB, Genovese MC, Wardlaw AJ, Butcher EC, Wu L (2001) CCR7 expression and memory T cell diversity in humans. J Immunol 166 (2):877–884 [DOI] [PubMed] [Google Scholar]
- 18.Bromley SK, Thomas SY, Luster AD (2005) Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol 6 (9):895–901. doi: 10.1038/ni1240 [DOI] [PubMed] [Google Scholar]
- 19.Sharpe AH, Pauken KE (2018) The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 18 (3):153–167. doi: 10.1038/nri.2017.108 [DOI] [PubMed] [Google Scholar]
- 20.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, D’Andrea K, Pauken KE, Herati RS, Staupe RP, Schenkel JM, McGettigan S, Kothari S, George SM, Vonderheide RH, Amaravadi RK, Karakousis GC, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, Wherry EJ (2017) T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545 (7652):60–65. doi: 10.1038/nature22079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D (2017) Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer 5 (1):53. doi: 10.1186/s40425-017-0257-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S, Dai D, Horton B, Gajewski TF (2017) Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31 (5):711–723 e714. doi: 10.1016/j.ccell.2017.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322 (5904):1097–1100. doi: 10.1126/science.1164206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, Kroeger J, Richards A, McCormick L, Moberg V, Cronin H, Zhao X, Schell M, Chen YA (2016) Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res 4 (4):345–353. doi: 10.1158/2326-6066.CIR-15-0193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9 (3):162–174. doi: 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 174 (2):636–645 [DOI] [PubMed] [Google Scholar]
- 27.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL (2014) Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med 6 (237):237ra267. doi: 10.1126/scitranslmed.3007974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foletta VC, Segal DH, Cohen DR (1998) Transcriptional regulation in the immune system: all roads lead to AP-1. J Leukoc Biol 63 (2):139–152 [DOI] [PubMed] [Google Scholar]
- 29.Jain J, McCaffrey PG, Miner Z, Kerppola TK, Lambert JN, Verdine GL, Curran T, Rao A (1993) The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature 365 (6444):352–355. doi: 10.1038/365352a0 [DOI] [PubMed] [Google Scholar]
- 30.Rincon M, Flavell RA (1994) AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO J 13 (18):4370–4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swat W, Dessing M, von Boehmer H, Kisielow P (1993) CD69 expression during selection and maturation of CD4+8+ thymocytes. Eur J Immunol 23 (3):739–746. doi: 10.1002/eji.1830230326 [DOI] [PubMed] [Google Scholar]
- 32.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajah AS, Gyorki DE, Thornton CM, Beavis PA, Fox SB, Kathleen Cuningham Foundation Consortium for Research into Familial Breast C, Darcy PK, Speed TP, Mackay LK, Neeson PJ, Loi S (2018) Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24 (7):986–993. doi: 10.1038/s41591-018-0078-7 [DOI] [PubMed] [Google Scholar]
- 33.Wein L, Savas P, Luen SJ, Virassamy B, Salgado R, Loi S (2017) Clinical Validity and Utility of Tumor-Infiltrating Lymphocytes in Routine Clinical Practice for Breast Cancer Patients: Current and Future Directions. Front Oncol 7:156. doi: 10.3389/fonc.2017.00156 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 2. Signature genes of each cluster from patient #2
Supplementary table 1. Signature genes of each cluster from patient #1
Supplementary Figure 1. Volcano plot of gene expression level comparison of baseline samples between patient #1 (#1a) and #2 (#2a) before treatment.
Supplementary Figure 2. Comparison of between patient #1 (#1c) and #2 (#2c pf) after treatment of nab-paclitaxel and pembrolizumab.
Supplementary table 4. Differentially expressed genes between #1c and #2c pf within CD4+ an CD8+ T cells
Supplementary table 3. Differentially expressed genes between #1a and #2a within CD4+ an CD8+ T cells