

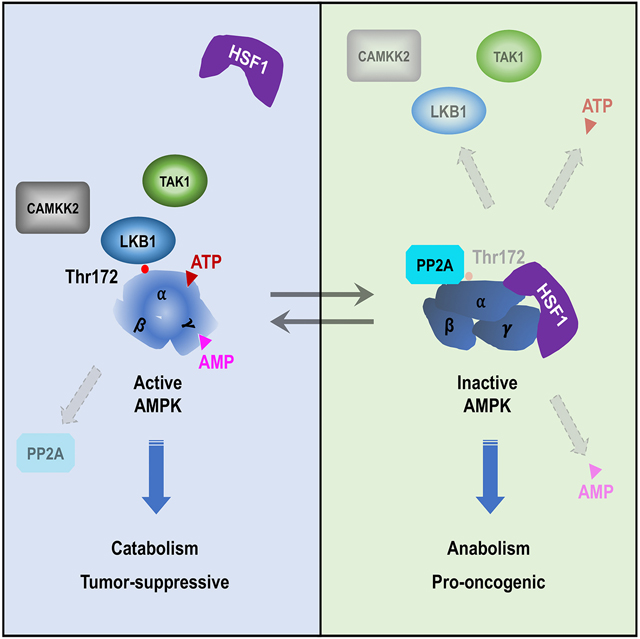
SUMMARY
Through transcriptional control of the evolutionarily conserved heat-shock or, proteotoxic stress, response, heat shock factor 1 (HSF1) preserves proteomic stability. Here, we show that HSF1, a physiological substrate for AMP-activated protein kinase (AMPK), constitutively suppresses this central metabolic sensor. By physically evoking conformational switching of AMPK, HSF1 impairs AMP binding to the γ subunits, enhances the PP2A-mediated de-phosphorylation and impedes the LKB1-mediated phosphorylation of Thr172, as well as retards ATP binding to the catalytic α subunits. These immediate and manifold regulations empower HSF1 to both repress AMPK under basal conditions and restrain its activation by diverse stimuli, thereby promoting lipogenesis, cholesterol synthesis and protein cholesteroylation. In vivo, HSF1 antagonizes AMPK to control body fat mass and drive the lipogenic phenotype and growth of melanomas, independently of its intrinsic transcriptional action. Thus, the physical AMPK-HSF1 interaction epitomizes a reciprocal kinase-substrate regulation whereby lipid metabolism and proteomic stability intertwine.
Keywords: AMPK, cholesteroylation, conformational switch, HSF1, lipogenesis, LKB1, oncogenesis, SHH, SREBP1
eTOC BLURB
Su et al. uncover an intriguing mutual antagonism whereby energy homeostasis and proteomic stability intertwine. While AMPK inactivates HSF1 via phosphorylation, HSF1 reciprocally suppresses AMPK by physically enforcing an inactive conformation. Thereby, HSF1 imposes a new layer of regulation on AMPK that impacts lipid metabolism, protein cholesteroylation, and tumor growth.
Graphical Abstract
INTRODUCTION
Heat shock factor 1 (HSF1) governs the heat-shock, or proteotoxic stress, response (HSR/PSR) transcriptionally, thereby safeguarding the proteome (Li et al., 2017). Contrary to its benefits against stress, HSF1 promotes oncogenesis (Dai et al., 2007; Jin et al., 2011; Min et al., 2007). The diverse underpinning mechanisms include mitigating oncogenes-induced senescence (Meng et al., 2010), strengthening oncogenic RAS signaling (Dai et al., 2012), promoting migration, invasion and epithelial-mesenchymal transition (Xi et al., 2012), and repressing tumor-suppressive amyloidogenesis (Tang et al., 2015).
We previously identified HSF1 as a physiological substrate for AMP-activated protein kinase (AMPK) (Dai et al., 2015). AMPK functions as heterotrimeric complexes, comprising a catalytic α subunit and two regulatory subunits, β and γ, which sense energy state via binding of glycogen and AMP/ADP/ATP, respectively (Carling, 2017; Hardie et al., 2012; Oakhill et al., 2012). The tumor suppressor LKB1/STK11, by phosphorylating α subunits on Thr172, activates AMPK (Hawley et al., 2003; Lizcano et al., 2004; Shaw et al., 2004; Woods et al., 2003). AMPK, in turn, phosphorylates effectors to repress anabolic but stimulate catabolic processes, thereby re-establishing energy homeostasis to survive energetic stress. AMPK represses lipogenesis, by inhibiting sterol regulatory element-binding transcription factor 1c (SREBP1c) and acetyl-CoA carboxylase (ACC) (Li et al., 2011; Munday et al., 1998), and impairs cholesterol (CHL) synthesis by inactivating hydroxymethylglutaryl-CoA reductase (HMGCR) (Clarke and Hardie, 1990). Upon activation by metabolic stressors, AMPK suppresses HSF1 via Ser121 phosphorylation (Dai et al., 2015).
Herein we report that HSF1 inhibits AMPK reciprocally. At the molecular level, HSF1 provokes conformational switching of AMPK via physical interactions. At the cellular level, HSF1 stimulates lipogenesis, CHL synthesis and protein cholesteroylation (CHLation). At the organismal level, HSF1 controls blood lipid levels and body fat mass. Moreover, this physical suppression of AMPK by HSF1 fosters the lipogenic phenotype and growth of melanomas in vivo.
RESULTS
Reciprocal regulations between the kinase-substrate pair AMPK and HSF1
Confirming our previous findings, active AMPK, but not LKB1, complexes phosphorylated HSF1 at Ser121 in vitro (Figures 1A and S1A). In melanoma cells, AMPKα1/2 knockdown diminished Ser121 phosphorylation but elevated inducible heat-shock proteins (Figure 1B), indicating HSF1 activation. Intriguingly, HSF1 was proposed to regulate mitochondrial biogenesis to sustain ATP levels, thereby suppressing AMPK (Qiao et al., 2017).
Figure 1: AMPK and HSF1 suppress each other reciprocally.

(A) In vitro phosphorylation of recombinant HSF1 by AMPKα1β1γ2. (B) Immunoblotting of HSF1 Ser121 phosphorylation and HSPs in A2058 cells stably expressing inducible scramble or AMPKα1/2-targeting shRNAs. (C) HSF1 dual reporter assays. (D and E) Immunoblotting of Thr172 phosphorylation and co-IP of transfected HSF1 and endogenous AMPKα in HEK293T cells. HC: heavy chain. WCL: whole cell lysates. (F) Immunoblotting of AMPKα, ACC, and Raptor phosphorylation in immortalized Rosa26-CreERT2; Hsf1fl/fl (Hsf1fl/fl, 4-OHT±) MEFs treated with 2μM CC overnight. (G) Immunoblotting of HSF1 in Hsf1fl/fl, 4-OHT+ MEFs reconstituted with lentiviral FLAG-HSF1. The signals of HSF1 were normalized against those of βActin, then all values were further normalized against that of HSF11–529. (H-J) ELISA quantitation of Thr172 phosphorylation in reconstituted Hsf1fl/fl, 4-OHT+ MEFs treated with 5mM phenformin, 500nM oligomycin or glucose starvation for 1hr. (K) Co-IP of endogenous AMPK and HSF1 in Hsf1fl/fl, 4-OHT− MEFs and immunoblotting of total and phospho-AMPKα in the supernatants before and after IP. (L) Co-IP of inactive AMPK and HSF1 in MEFs. Following depletion of p-AMPKα from cell lysates, the supernatant (the fourth lane) was IPed for HSF1. (M) Summary of the percentage of AMPK associated with and without HSF1. (N) The postulated model wherein HSF1, following binding to active AMPK as a substrate, constitutively interacts with and renders AMPK inactive.
Error bars are mean±SD, n=3 independent experiments, One-way ANOVA (C, G, H, I, and J) or Student’s t test (K and L, two-tailed). See also Figure S1.
Could HSF1 regulate AMPK directly? The paramount importance of Thr172 phosphorylation to AMPK activation prompted us to examine whether HSF1 affects this modification. To determine the necessity of HSF1 transcriptional activity, we overexpressed two transcription-deficient mutants, HSF11–323 lacking the C-terminal transactivation domain (AD) and HSF1324–529 lacking the N-terminal DNA-binding domain (DBD), in HEK293T cells (Figure 1C). HSF11–323, a dominant negative mutant, impaired endogenous HSF1 activity. Of interest, both mutants diminished Thr172 phosphorylation (Figure 1D). Moreover, HSF1S326A and HSF1S326D, showing diminished and enhanced transcriptional activity respectively (Figure S1B), reduced Thr172 phosphorylation as equally well as HSF1WT (Figure S1C). Thus, the transcriptional action of HSF1 is dispensable for AMPK suppression. Both HSF11–323 and HSF1324–529 co-precipitated with endogenous AMPK (Figure 1E), supporting a physical regulation.
In mouse embryonic fibroblasts (MEFs), Hsf1 deficiency activated AMPK, evidenced by elevated phosphorylation of AMPKα and its two substrates, ACC and RAPTOR (Gwinn et al., 2008; Munday et al., 1998) (Figure 1F). This effect was largely abolished by the AMPK inhibitor Compound C (CC) (Zhou et al., 2001) (Figure 1F), suggesting that HSF1 keeps AMPK inactive under non-stress conditions. To establish the physiological relevance of this transcription-independent regulation, we reconstituted the Hsf1-deficient (Hsf1fl/fl, 4-OHT+) MEFs with human FLAG-HSF1324–529 by lentiviral transductions. HSF1 is highly conserved between human and mouse. To circumvent the lack of monoclonal antibodies recognizing both HSF1 equally, we reconstituted the MEFs with human FLAG-HSF11–529 as a surrogate for Hsf1-proficient (Hsf1fl/fl, 4-OHT−) MEFs. Comparable levels of HSF11–529 and endogenous HSF1 resulted in similar AMPKα phosphorylation (Figures 1G–1J). Importantly, while the level of reconstituted FLAG-HSF1324–529 Low was only ∼12% of that of endogenous HSF1, AMPKα phosphorylation was comparable (Figures 1G–1J). Consistent with its low expression, the FLAG tag was only detected by enhanced ELISA and Proximity Ligation Assays (PLA) (Figures S1D and S1E), but not by immunoblotting and co-IP. Thus, these results confirm the transcription-independent AMPK suppression, without HSF1 overexpression. Reconstituted HSF1324–529 also dose-dependently blocked AMPK activation by phenformin or oligomycin treatment and glucose starvation (Figures 1H–1J). Unlike phenformin and oligomycin, glucose starvation activates AMPK without elevating AMP/ATP ratios (Zhang et al., 2017), suggesting an action of HSF1 operating beyond energy stress. Under basal conditions, endogenous HSF1 and AMPK readily co-precipitated in MEFs (Figure 1K). While IP of HSF1 depleted ∼66% of total cellular AMPKα, only ∼25% of phospho-AMPKα was depleted (Figure 1K), suggesting that more HSF1 interacts with non-phosphorylated, inactive AMPK. To confirm this, we first completely depleted phospho-AMPKα from cell lysates, followed by a second IP of HSF1. Indeed, non-phospho-AMPKα, accounting for ∼72% of total, was largely depleted by HSF1 IP (Figures 1L and S1F). Thus, this AMPK-HSF1 interaction is prevalent; moreover, under basal conditions a majority of AMPK associated with HSF1 is inactive (Figure 1M). Considering that active AMPK phosphorylates HSF1, we posited that following initial interactions as a substrate, HSF1 might deactivate AMPK and constitutively repress it (Figure 1N), a scenario distinct from conventional kinase-substrate interactions that are typically weak and transient. Of note, ∼34% of cellular AMPKα did not interact with HSF1 and HSF1 mutants suppressed AMPK dose-dependently, suggesting the abundance of cellular HSF1 as a limiting factor. To test this, we determined the AMPK:HSF1 molar ratios in HEK293T cells and MEFs (Figure S1G), which were estimated as 1.207±0.418 and 1.529±0.743 (mean±SD, n=3), respectively. This result suggests that HSF1 could maximally interact with ∼65% of total AMPK at a 1:1 stoichiometry in MEFs, in line with the IP results (Figure 1K). Hence, AMPK and HSF1, two key players in energy homeostasis and proteomic stability respectively, suppress each other reciprocally.
HSF1 physically antagonizes AMPK via manifold mechanisms
To delineate the AMPK-interacting interfaces, we synthesized a HSF1 peptide library, comprising 22 non-overlapping peptides (24 amino acid each, Table S1). First, we screened for their AMPK binding in vitro. Nine peptides displayed AMPK binding (Figure 2A), representing two clusters located in the N- and C-terminal portion of HSF1, respectively. Once transfected into cells, 9 peptides diminished Thr172 phosphorylation (Figure 2B), among which 7 showed AMPK binding. Thus, these 22 short peptides are classified into four groups: 1) no or weak AMPK binding and without suppression (e.g. P1 and P4); 2) no or weak AMPK binding but with suppression (P11 and P12); 3) strong AMPK binding but without suppression (P8 and P10); and 4) strong AMPK binding and with suppression (P3, P5, P6, P17–20) (Figure 2C). We focused on the fourth group and included ones from the first and third group as negative controls. Binding of excessive substrates, such as HSF1 peptides, may disrupt AMPK complexes or alter subunit stoichiometry, leading to AMPK suppression. Countering this notion, SAMS peptides, a sensitive substrate (Davies et al., 1989), did not suppress AMPK (Figure 2D).
Figure 2: Individual HSF1 peptides suppress AMPK physically.

(A) Quantitation of the binding of HEK293T AMPKα to HSF1 peptides pre-coated on ELISA plates. Fold changes are presented as a heatmap. NP: no peptides. (B) ELISA quantitation of Thr172 phosphorylation in HEK293T cells transfected with 10μM peptides overnight. Fold changes are presented as a heatmap. (C) Summary of the HSF1 peptide library. (D) Quantitation of Thr172 phosphorylation in HEK293T cells transfected with 10μM SAMS peptides overnight. (E-H) Quantitation of Thr172 phosphorylation and AMP/ATP ratios in Hsf1fl/fl, 4-OHT± MEFs transfected with 10μM peptides overnight, followed by treatment of 1mM AMP for 5 min or 500nM oligomycin for 1 hr. (I) Co-IP of endogenous AMPK with biotinylated peptides in Hsf1fl/fl, 4-OHT+ MEFs. The ratios of α/β1/γ2 co-IPed with peptides were normalized against the one of B-P8-transfected cells. B: biotinylated. (J) Quantitation of mitochondrial mass in reconstituted Hsf1fl/fl, 4-OHT+ MEFs following transfection of 10nM siRNAs for 4 days by FACS using Mito-ID® red dye (data as geometric means of FL2-H). (K) Schematic depiction of the cause of elevated AMP/ATP ratio in Hsf1-deficient MEFs. (L and M) Visualization of interactions between biotinylated peptides and endogenous AMPK by PLA (red) in A2058 cells. Actin filaments were labeled with phalloidin-Alexa Fluor® 488 conjugates (green). NB: non-biotinylated. Scale bars: 10μm. (N) Immunoblotting of Thr172 phosphorylation in HEK293T cells transfected with indicated pLenti6 plasmids.
Three independent experiments are plotted individually in heatmaps. Error bars are mean±SD, n=3 independent experiments, One-way ANOVA. See also Figure S2 and Table S1.
Elevation of cellular AMP/ATP and/or ADP/ATP ratios is a prime cause of AMPK activation (Carling, 2017; Hardie et al., 2012). To determine whether HSF1 alters cellular energy state, we transfected 6 peptides, representing the first (P1), third (P8) and fourth (P5, P6, P17, and P19) group respectively, into Hsf1-proficient and –deficient MEFs. Compared to the negative controls (P1 and P8), P5, P6, P17, and P19 all diminished the elevated basal Thr172 phosphorylation in Hsf1-deficient MEFs (Figure 2E). Their corresponding scrambled peptides, as additional negative controls, failed to do so (Figures S2A and S2B). The heightened basal Thr172 phosphorylation in Hsf1-deficient MEFs was accompanied by an elevated AMP/ATP ratio (Figure 2F), detected by bioluminescence, a technique more sensitive than conventional chromatographic approaches (Khlyntseva et al., 2009). Naturally, this elevated AMP/ATP ratio would be presumed to cause AMPK activation.
Contrarily, our studies pinpointed an alternative mechanism. First, at basal conditions especially in Hsf1-deficient MEFs, P5, P6, P17, and P19 did not lower AMP/ATP ratios (Figures 2E–2H), dissociating AMPK suppression from energy state. Second, AMP treatment, directly elevating AMP/ATP ratios, stimulated Thr172 phosphorylation in MEFs transfected with control peptides; by contrast, P5, P6, P17, and P19 blocked this activation without lowering AMP/ATP ratios (Figures 2E and 2F). Third, the ATP synthase inhibitor oligomycin elevated AMP/ATP ratios universally; however, Thr172 phosphorylation was only induced in control peptide-transfected cells, again revealing the overriding effect of P5, P6, P17, and P19 (Figures 2G and 2H). Fourth, this suppression is not due to disruption of complexes or altered subunit stoichiometry. Compared to P8, which did not suppress AMPK, the ratio of α/β/γ subunits co-precipitated with biotinylated APMK-suppressing peptides remained unchanged (Figure 2I). Fifth, Hsf1-deficient MEFs showed reduced mitochondrial mass, which would be presumed to cause the elevated AMP/ATP ratio and AMPK activation; however, reconstituted HSF1324–529 fully rescued this defect transcription-independently (Figure S2C). This defect is due to AMPK activation, as it was fully rescued by Ampkα knockdown (Figures 2J and S2D). AMPK activation both promotes mitochondrial biogenesis and induces autophagy (Hardie et al., 2012). If reduced mitochondrial mass were the direct result of Hsf1 deficiency and the cause of AMPK activation, Ampkα knockdown would diminish mitochondrial mass further, owing to the loss of compensatory mitochondrial biogenesis (Figure 2K). This sharply contrasts with our findings. We reasoned that mitophagy, an autophagy-mediated mitochondrial degradation, might directly cause the mitochondrial defect. Indeed, two autophagy inhibitors, chloroquine and bafilomycin, fully rescued mitochondrial mass (Figures S2E). Thus, HSF1 acts downstream of cellular AMP/ATP, regulating AMPK independently of energy state. Furthermore, the elevated AMP/ATP ratio in Hsf1-deficient MEFs is the consequence, rather than the cause, of AMPK activation (Figure 2K).
In both Hsf1-proficient and -deficient MEFs, P5, P6, P17, and P19 did not alter the transcription of several genes implicated in mitochondrial biogenesis and regulated by HSF1 under fasting conditions (Figure S2F) (Qiao et al., 2017). To visualize peptide-AMPK interactions by PLA, we employed biotinylated peptides, which can be recognized by an anti-biotin antibody (Figure 2L). All antibodies were validated (Figure S2G). No PLA signals were observed in cells transfected with either non-biotinylated peptides or biotinylated scramble P6, demonstrating the specificity (Figure S2H). P5, P6, P8, P10, P17, and P19 all interacted with AMPK inside cells (Figure 2M). In support of the cruciality of interactions, HSF1 mutants lacking the interacting interfaces did not suppress Thr172 phosphorylation (Figure 2N).
Elevated cellular ADP or AMP binds to γ subunits to induce conformational changes in α subunits, both stimulating the LKB1-mediated phosphorylation and impeding the phosphatase-mediated de-phosphorylation of Thr172 to activate AMPK (Carling, 2017; Hardie et al., 2012; Oakhill et al., 2012). In addition, AMP can exert an allosteric activation (Hardie et al., 2012). While HSF1 peptides did not perturb energy state, it is unclear whether they interfered with AMP binding. To address this, we incubated recombinant active AMPKα1/β1/γ2 with mant-AMP, an adenosine analog intensifying its intrinsic fluorescence upon AMPKγ binding (Xiao et al., 2007). We first tested recombinant full-length HSF1 proteins. To provide physiological relevance, we applied a 2:1 AMPK:HSF1 molar ratio. Silver staining revealed that AMPKα1/β1/γ2 contained proteins other than the three core subunits under denaturing conditions; however, under native conditions the complex ran as a single band between the 242 and 480 KD markers (Figure S3A). This suggests that additional regulatory components were co-purified with AMPK core subunits from Sf9 cells, which is not surprising, given the intricate AMPK regulation in eukaryotes. Compared to the control GST and HSP90β, HSF1 impaired AMP binding (Figures 3A and 3B), congruent with its overriding effect on elevated AMP/ATP ratios. By contrast, the two classic substrates ACC1 and SREBP1 did not. Similarly, while P6 diminished mant-AMP binding, SAMS did not (Figure S3B), contradicting substrate binding as the cause. Unlike AMP, P6 blocked mant-AMP binding partially (Figure S3C), suggesting a non-competitive manner. As HSF1 can interact with HSP90 (Zou et al., 1998), a chaperone for AMPK (Zhang et al., 2012), HSF1 may act on AMPK via HSP90. However, our evidence contradicts this notion. First, AMPKα1/β1/γ2 contained no detectable HSP90 (Figure S3D). Second, HSP90 inhibition reduced Thr172 phosphorylation in Hsf1-deficient MEFs (Figure S3E). Contrary to HSF1, HSP90 maintains AMPK activation. Third, HSF1 peptides additively diminished Thr172 phosphorylation in the presence of HSP90 inhibition (Figure S3E), indicating a HSP90-independent mechanism.
Figure 3: HSF1 suppresses AMPK via manifold mechanisms.

(A and B) Quantitation of mant-AMP binding to recombinant AMPKα1β1γ2 incubated with HSF1. (C) De-phosphorylation of AMPKα1β1γ2 induced by HSF1 in vitro with and without 5μM OA. (D) Co-IP of endogenous AMPK and PP2A in HEK293T cells transfected with 10μM peptides overnight. The amounts of PP2A were normalized against that of α subunits, and the γ2/β1/α ratios were compared. A rabbit anti-PI3K Ab served as the IP control. (E) Quantitation of Thr172 phosphorylation in HEK293T cells transfected with 20nM siRNAs for 3 days, followed by transfection of pLenti6 plasmids for 2 days. (F) Impairment of the LKB1-mediated Thr172 phosphorylation of recombinant α1 by HSF1. (G) Co-IP of endogenous LKB1 and AMPKα in Hsf1fl/fl, 4-OHT± MEFs. (H and I) Effects of HSF1 on the α1-mediated SAMS phosphorylation and mant-ATP binding to α1. (J) CD spectral changes of AMPKα1β1γ2 induced by P6. SP6: scramble P6. (K) Following incubation with and without 10μM peptides at RT for 30 min, AMPKα1β1γ2 were IPed with anti-AMPKα Abs. Heat-denatured (HS) complexes served as a positive control. (L and M) Quantitation of Thr172 phosphorylation in HeLa cells treated with 10μM A23187 for 30 min or 500ng/ml recombinant TRAIL for 2 hr. (N) Schematic depiction of conformational switching of AMPK evoked by HSF1. In cells, AMPK either associate with or without HSF1, depending on the availability of HSF1. PPases: protein phosphatases.
AMPK was incubated with recombinant proteins at a 2:1 molar ratio in all experiments. Error bars are mean±SD, n=3 independent experiments, One-way ANOVA. See also Figure S3.
Only P6 blocked AMP binding, indicating additional mechanisms. Incubation of HSF1 with AMPKα1/β1/γ2 in vitro, surprisingly, diminished Thr172 phosphorylation (Figure 3C). Given the absence of ATP, ADP and AMP, this effect must be due to the co-purified phosphatase(s), stimulated by HSF1. Since PP2A de-phosphorylates Thr172 (Joseph et al., 2015), we asked whether the recombinant AMPK contained PP2A. Indeed, PP2A was detected (Figure S3F). Okadaic acid (OA), a potent PP2A inhibitor (Bialojan and Takai, 1998), both elevated basal Thr172 phosphorylation and blocked the effect of HSF1 completely (Figure 3C). The AMPK-suppressing, but not SAMS, peptides behaved similarly to HSF1 proteins (Figure S3G). In HEK293T cells, these peptides, but not the control and their scrambled counterparts, all heightened PP2A-AMPK interactions without disrupting the complexes or altering subunit stoichiometry (Figures 3D and S3H). Importantly, knocking down PP2A catalytic subunits both elevated basal Thr172 phosphorylation and mitigated the effect of HSF1 (Figures 3E and S3I). Thus, HSF1 exploits a PP2A-dependent de-phosphorylation mechanism, independent of adenosine nucleotides.
The evident suppression of Thr172 phosphorylation by HSF1 in PP2A-deficient cells suggests extra mechanisms. To determine whether HSF1 affects the LKB1-mediated phosphorylation, we performed in vitro LKB1 kinase assays using recombinant AMPKα1. Silver staining revealed that α1 ran as a single band (Figure S3J). Due to abundant basal phosphorylation, we de-phosphorylated α1 using recombinant PP2A. Following extensive phosphatase inactivation by a collection of inhibitors, including OA, the PP2C inhibitor sanguinarine chloride (SC) (Aburai et al., 2010), sodium fluoride (NaF), and sodium orthovanadate (Na3VO4), recombinant active LKB1-STRAD-MO25 complexes were added. Pre-incubation with HSF1, but not GST, HSP90β, ACC1 or SREBP1, mitigated the LKB1-meditated α1 phosphorylation (Figure 3F). Since no γ subunits were present, the AMP/ADP effect is excluded. Moreover, given the excessive inhibitors, this effect of HSF1 is independent of phosphatases. The AMPK-suppressing, but not SAMS, peptides exerted similar effects (Figures S3K-S3M). Importantly, Hsf1 deficiency heightened the LKB1-AMPKα interaction in MEFs (Figure 3G). Thus, HSF1 can impair the LKB1-mediated α phosphorylation, independently of ADP/AMP binding.
Can HSF1 act independently of Thr172 phosphorylation? AMP activates AMPK allosterically without inducing Thr172 phosphorylation (Gowans et al., 2013). HSF1, by impeding AMP binding, is expected to block this effect. During kinase assays, ATP can convert to AMP. To exclude the AMP effect, we performed in vitro AMPK kinase assays using recombinant α1 alone. With phosphatase inhibitors, ACC1, SREBP1, and HSF1 all inhibited the SAMS phosphorylation by α1 (Figure 3H), consistent with their status as substrates to compete for AMPK. Among the AMPK-suppressing peptides, only P6 and P17 inhibited SAMS phosphorylation (Figure S3N). Like SAMS, the Ser121-containing P6 was phosphorylated by AMPKα1; however, P5, P17 and P19 were not, revealing them as non-substrates (Figure S3O). Thus, the effect of P17 is not due to substrate competition. To test whether HSF1 impairs ATP binding, we incubated mant-ATP with α1 in the presence of phosphatase inhibitors. HSF1, but not ACC1 and SREBP1, blocked mant-ATP binding; as expected, HSP90β bound mant-ATP (Figure 3I). While the strong substrate SAMS did not impede ATP binding, the weak substrate P6 and non-substrate P17 both did (Figure S3P), refuting substrate binding as the cause. Unlike the ATP-competitive inhibitor CC, P6 impeded mant-ATP binding partially (Figure S3Q). Lastly, the recombinant HSF1 comprised transcription-inactive monomers predominantly (Figure S3R). Thus, HSF1 employs a fourth mechanism—blocking ATP binding to the active site, independently of substrate binding.
These diverse impacts suggest that HSF1 may induce global conformational changes of AMPK. Revealed by the circular dichroism (CD) spectroscopy, compared to its scrambled counterpart, P6 caused a reduction in α helix but an increase in β-sheet structure in AMPKα1/β1/γ2 (Figure 3J). Again, P6 did not disrupt the complex or alter subunit stoichiometry (Figure 3K). Distinct from the control peptides, P5, P6, P17 and P19 all displayed a negative peak around 200nm in CD spectra (Figures S3S), a characteristic of structural disorder (Chemes et al., 2012).
These immediate and manifold regulations predict that HSF1 can suppress AMPK irrespective of stimulatory signals. Indeed, in HeLa cells, deficient for LKB1 (Shaw et al., 2004), P5, P6, P17 and P19 all suppressed Thr172 phosphorylation induced by A23187 (Figure 3L), a calcium ionophore activating AMPK via CaMKK2 (Hawley et al., 2005), or by the apoptosis inducer TRAIL (Figure 3M), which activates AMPK via TAK1 (Herrero-Martin et al., 2009). Thus, HSF1 blocks AMPK activation by all three known upstream kinases transducing distinct stimuli. In aggregate, our studies support an unexpected multimechanistic inhibition of AMPK by HSF1 via induction of conformational switch (Figure 3N).
HSF1 represses AMPK to stimulate lipogenesis
AMPK represses lipogenesis and CHL synthesis. SREBP1/2 regulate key players in both processes transcriptionally, including ACC1, fatty acid synthase (FASN), low-density lipoprotein receptor (LDLR), and HMGCR (Horton et al., 2002). AMPK inactivates SREBP1c/2 via phosphorylation (Li et al., 2011). Moreover, it phosphorylates and inhibits ACC1 and HMGCR, two rate-limiting enzymes for lipogenesis and CHL synthesis respectively (Clarke and Hardie, 1990; Munday et al., 1998).
In A2058 cells, inducible HSF1 knockdown elevated SREBP1c Ser372 phosphorylation, diminished ACC1, FASN, LDLR and HMGCR mRNAs, and impaired the nuclear translocation and genomic DNA binding of SREBP1, revealed by PLA (Dai et al., 2015), all of which were reversed by either genetic or pharmacological AMPK inhibition (Figures 4A–4E and S4A-S4E). Therefore, HSF1 promotes SREBP1 transcriptional activation primarily via AMPK suppression.
Figure 4: HSF1 heightens lipogenesis through AMPK suppression.

(A) Immunoblotting of p-SREBP1c in A2058 cells stably expressing inducible HSF1-targeting shRNAs with and without AMPKα1/2 knockdown. P: precursor; M: mature. (B) Quantitation of mRNA levels of lipogenic genes in A2058 cells described in (A), by qRT-PCR. Fold changes are presented as a heatmap (three independent experiments plotted individually). (C) SREBP1 nuclear translocation in A2058 cells described in (A). LDH and Lamin A/C are the cytoplasmic and nuclear marker, respectively. (D and E) Visualization of SREBP1 binding to genomic DNAs by PLA (D, scale bars: 10μm) and further quantitation by FACS in A2058 cells described in (A) (E, data as geometric means of FL2-H). (F) Quantitation of cellular lipid content by Nile red staining in HEK293T cells transfected with shRNAs.
Error bars are mean±SD, n=3 independent experiments, One-way ANOVA. See also Figure S4.
Congruently, HSF1 knockdown diminished cellular neutral lipid content, detected by Nile red staining (Greenspan et al., 1985); and, concurrent AMPKα1/2 knockdown fully rescued this defect (Figure 4F). CC also increased cellular lipid content, largely via AMPK suppression (Figure S4F). Contrary to HSF1 deficiency, HSF1324–529 overexpression promoted SREBP1 DNA binding and elevated mRNAs of lipogenic genes, actions largely dependent on AMPK (Figures S4G and S4H). Together, our findings pinpoint a key causative role of AMPK in the HSF1-controlled lipogenesis.
HSF1 represses AMPK to enhance CHL synthesis and protein CHLation
HSF1 deficiency diminished cellular CHL; conversely, HSF1 peptides elevated CHL (Figures 5A and 5B). We were particularly intrigued by how lipids impact cellular proteome. Lipidation affects the stability, localization, and activities of target proteins; and, several types of protein lipidation, including farnesylation, myristylation, palmitoylation, and CHLation, have been demonstrated (Nadolski and Linder, 2007; Resh, 2012).
Figure 5: HSF1 controls CHL synthesis and protein CHLation via AMPK.

(A and B) Quantitation of cellular CHL in A2058 cells and MEFs following HSF1 depletion or in A2058 cells transfected with 10μM HSF1 peptides twice in 7 days. (C) Establishment and validation of SHH CHLation sandwich ELISA. SFM: serum-free medium. (D and E) Quantitation of SHH-N CHLation in A2058 cells with inducible HSF1 knockdown for 6 days with either AMPKα knockdown or 50μM CHL supplementation in the medium for 24 hr. Secreted CHLated SHH-N were normalized against total SHH-N present in the medium. (F) Quantitation of SHH-N CHLation in Hsf1-deficient MEFs reconstituted with HSF1324–529. (G) Quantitation of SHH mRNAs in cells deficient for HSF1. (H) ChIP analysis of HSF1 binding to the SHH promoter in A2058 cells expressing inducible HSF1-targeting shRNAs. Alphoid DNA loci served as a negative control. (I) Left panel: visualization of GLI1 binding to genomic DNAs by PLA (red) in parental A2058 cells stimulated for 4 hr by the conditioned media collected from A2058 cells expressing inducible scramble or HSF1-targeting shRNAs. Scale bars: 10μm. Right panel: Quantitation of GLI1-gDNA binding PLA by FACS (data as geometric means of FL2-H).
Error bars are mean±SD, n=3 independent experiments, One-way ANOVA (A-I) or Student’s t test (A, C, and G, two-tailed). See also Figure S5.
To date, only members of the hedgehog signaling pathway are known to undergo CHLation, among which the most prominent example is sonic hedgehog (SHH) (Jeong and McMahon, 2002; Porter et al., 1996; Xiao et al., 2017). To quantitate CHLated SHH N-terminus (SHH-N), we developed a sandwich ELISA by combining a capturing anti-SHH-N and a detecting anti-CHL antibody (Figure 5C). The specificity of this anti-CHL antibody was verified by both a dose-dependent recognition of CHL in vitro and diminished signals in cells treated with methyl-β-cyclodextrin, a chemical commonly used to deplete cellular CHL (Zidovetzki and Levitan, 2007) (Figures S5A and S5B). While not detecting free CHL, our ELISA detected CHLated, but not non-CHLated, recombinant SHH-N proteins (Figure 5C). Correlated with depleted cellular CHL was a marked reduction in CHLated SHH-N secreted to the culture medium by HSF1-deficient A2058 cells; and, this defect was largely rescued by AMPKα knockdown (Figure 5D). Also, we uncovered a reduction in total secreted SHH-N (Figure S5C). Nonetheless, normalization of CHLated against total SHH-N still reveals impaired CHLation (Figure 5D). Addition of CHL to the culture media also rescued this impaired CHLation (Figure 5E), indicating depletion of cellular CHL as a direct cause. This impairment is not due to defective secretion, as the normalized intracellular CHLated SHH-N was also diminished (Figure S5D). This HSF1-regulated CHLation was observed in MEFs as well (Figures S5E and S5F). Of note, this defective CHLation in Hsf1-deficient MEFs was fully rescued by reconstituted HSF1324–529 (Figure 5F). Interestingly, SHH is a direct transcriptional target of HSF1 (Figures 5G and 5H).
CHLation is critical to the biological activities of SHH (Jeong and McMahon, 2002). The genomic DNA binding of GLI1, a key downstream effector (Evangelista et al., 2006), can be quantitated by PLA to indicate SHH activity (Figure S5G). To activate SHH signaling, we stimulated parental A2058 cells with conditioned media collected from A2058 cells expressing either scramble or HSF1-targetting shRNAs. The media conditioned from HSF1-deficient cells induced less GLI1 DNA binding (Figures 5I and S5H), suggesting impaired paracrine SHH signaling owing to HSF1 deficiency. Thus, these results uncover a previously unrecognized role of HSF1 in controlling protein CHLation. Moreover, HSF1 regulates SHH signaling, both transcriptionally and posttranslationally.
HSF1 controls organismal lipid metabolism via AMPK
AMPK is activated in Hsf1−/− mouse tissues (Figures 6A and 6B). Compared to age-matched Hsf1+/+ mice, strikingly, Hsf1−/− mice displayed reduced body fat mass (Figure 6C). However, Hsf1−/− mice exhibit no changes in food intake and locomotor activity, but a reduction in total body oxygen consumption (Ma et al., 2015). Thus, this reduced body fat mass is not due to imbalanced energy intake and expenditure. Rather, it may be contributed by impaired lipogenesis and CHL synthesis, owing to intrinsic AMPK activation. To test this, we treated mice with the AMPK inhibitor CC, which markedly rescued the diminished body fat mass and blood lipids in Hsf1−/− mice (Figures 6C–6E).
Figure 6: HSF1 controls organismal lipid metabolism via AMPK.

(A and B) Immunoblotting of p-AMPKα in mouse tissues, 2 mice each genotype. (C) NMR body composition analyses of Hsf1+/+ and Hsf1−/− mice before and after i.p. injection of CC (n=7). (D and E) Plasma lipid levels of the same mice described in (C) (n=6 or 7). (F) Immunoblotting of HSF1 in tissues of Hsf1fl/Δ and Meox2-Cre; Hsf1fl/Δ mice (left). NMR body composition analyses of these mice before and after i.p. injection of CC (right, n=10 or 12). (G) Immunoblotting of HSF1 and AMPKα in mouse livers following hydrodynamic injections of scramble or Ampkα1/2-targeting shRNAs, 2 mice each group. (H) NMR composition analyses of livers from mice injected with scramble or Ampkα1/2-targeting shRNAs (n=5).
Error bars are mean±SD, unpaired and paired Student’s t test (C-F, two-tailed) or One-way ANOVA (H). See also Figure S6.
Due to the embryonic lethality caused by placental defects on inbred genetic backgrounds, Hsf1−/− mice are maintained on a mixed background (Xiao et al, 1999). To exclude potential genetic modifiers, we employed Hsf1fl/fl mice on the inbred C57BL/6J background (Su et al., 2016). To retain placental Hsf1 expression but delete it in embryonic tissues, we utilized C57BL/6J Meox2-cre mice (Tallquist and Soriano, 2000). By crossing Hsf1+/Δ; Meox2-cre mice with Hsf1fl/fl mice, we generated Hsf1fl/Δ mice with and without Meox2-cre. Due to mosaic Cre expression (Tallquist and Soriano, 2000), Hsf1 was deleted incompletely (Figure 6F). Nonetheless, these Hsf1fl/Δ; Meox2-cre mice still exhibited reduced body fat mass compared to Hsf1fl/Δ littermates, which was rescued by CC (Figure 6F), confirming the specific role of HSF1 in controlling body fat mass.
While its lipogenic effect was largely AMPK-dependent (Figure S4), CC can affect other targets (Bain et al., 2007). To exclude potential off-target effects in vivo, we depleted Ampkα1/2 in mouse livers via hydrodynamic shRNA delivery (Suda and Liu, 2007). Hsf1fl/Δ; Meox2-cre mice showed elevated Thr172 phosphorylation and reduced fat mass in livers; importantly, in vivo delivery of Ampkα1/2-targeting shRNAs restored their liver fat mass to a level comparable to that of Hsf1fl/Δ littermates injected with scramble shRNAs (Figures 6G and 6H), revealing a key causative role of AMPK activation. Moreover, hepatocyte-specific Hsf1 deletion using Alb-cre mice phenocopied the diminished liver fat mass (Figure S6A), confirming the liver-autonomous effect of HSF1. This liver-targeted Ampkα1/2 knockdown even marginally elevated body fat mass in Hsf1fl/Δ; Meox2-cre mice (Figure S6B), in line with a central role of livers in lipid metabolism. Thus, HSF1, at least in part via AMPK, controls organismal lipid metabolism.
HSF1 drives the lipogenic phenotype and growth of melanomas via AMPK suppression
Cancer cells rely on lipids for their rapid growth (Menendez and Lupu, 2007; Beloribi-Djafaflia et al., 2016). Given its potent pro-oncogenic role (Dai and Sampson, 2016), we asked whether HSF1 promotes lipogenesis and CHL synthesis in cancer.
To this end, we employed a human melanoma xenograft model wherein A2058 cells stably overexpressing either LacZ or HSF11–529 were transplanted into NOD/SCID mice. HSF11–529 overexpression accelerated in vivo melanoma growth (Dai et al., 2015). Congruent with suppressed AMPK signaling (Figure S7A), HSF11–529 overexpression increased the fat mass, CHL level, and SHH-N CHLation in melanomas (Figures 7A-7C). To address whether HSF1 promotes oncogenesis via AMPK suppression independently of transcription, we transduced A2058 cells stably expressing scramble or AMPKα1/2-targeting shRNAs with lentiviral HSF1324–529 (Figure S7B). In vitro, AMPKα1/2 deficiency, HSF1324–529 expression, and the combination all stimulated cell proliferation comparably (Figure S7C). In vivo, HSF1324–529 expression alone accelerated melanoma growth (Figures 7D and 7E), revealing a transcription-independent pro-oncogenic role of HSF1 unrecognized previously. Importantly, whereas AMPKα1/2 deficiency accelerated the growth of LacZ-expressing melanomas, it failed to further accelerate the growth of HSF1324–529-expressing melanomas (Figure 7D). Compared to AMPKα1/2 deficiency, HSF1324–529 expression drove a faster tumor growth, accompanied by deteriorated body conditions of the hosts (Figures 7D and S7D), suggesting that the pro-oncogenic effect of HSF1324–529 is partly AMPK-dependent. Interestingly, this mutant sequestrated JNK apart from mTORC1 to enhance protein translation (Su et al., 2016), a potential AMPK-independent mechanism. AMPKα1/2 deficiency or HSF1324–529 expression individually increased CHL and SHH-N CHLation in melanomas to comparable levels; however, the combination did not elicit an additive effect (Figures S7E and 7F). Thus, the lipogenic effect of HSF1324–529 is largely AMPK-dependent. Of note, enhanced CHL synthesis is causally linked to intestinal tumorigenesis (Wang et al., 2018). In support of a role of CHL in fostering malignant growth, intra-tumoral levels of CHL and CHLated SHH-N were positively correlated with melanoma growth rates (Figures S7F and S7G). Lastly, in diverse human cancers, increased HSF1 mRNA levels are correlated with diminished AMPKα Thr172 phosphorylation (data generated by The Cancer Genome Atlas Research Network, Figure 7G). Together, these results suggest that HSF1, via AMPK suppression, reprograms lipid metabolism and protein lipidation to support malignancy.
Figure 7: HSF1 drives the lipogenic phenotype and accelerates the growth of melanomas via AMPK.

(A) NMR composition analyses of xenografted A2058 melanomas stably expressing either LacZ or HSF11–529 (n=6). (B) Immunostaining of CHL in these melanomas. Scale bars: 20μm. (C) Quantitation of normalized SHH-N CHLation in these melanomas (n=5). (D) Subcutaneous xenograft of modified A2058 cells in NOD/SCID mice. Tumor volumes are expressed as fold changes (n=9 or 10). Tumor growth curves were fitted to exponential growth models to derive tumor-doubling time (DT) and growth rate constants (K). K values were compared. (E) Immunoblotting of p-AMPKα in melanomas, 3 tumors per group. (F) Quantitation of normalized CHLated SHH-N in melanomas (n=9 or 10). (G) Tukey boxplots showing an inverse correlation between HSF1 mRNA expression and AMPKα Thr172 phosphorylation RPPA scores in diverse human cancers. Patients’ samples were stratified according to the median values of HSF1 mRNA expression Z-scores.
Error bars are mean±SD, except (D, mean±SEM), Student’s t test (two-tailed for A and C, one-tailed for G) or One-way ANOVA (D and F). See also Figure S7.
DISCUSSION
Reciprocal regulation between the kinase-substrate pair AMPK and HSF1
AMPK inactivates HSF1, causing proteomic instability and tumor suppression (Dai et al., 2015). Now, we uncover an intriguing mutual antagonism between AMPK and HSF1 whereby energy homeostasis and proteomic stability intertwine. This reciprocal AMPK suppression may illustrate a previously unappreciated substrate-mediated kinase regulation. Apparently, not all substrates are equal. Among the three tested substrates, only HSF1 can do so. Correlated with this suppression is an abundant and constitutive HSF1-AMPK interaction in vivo, which is beyond the substrate binding and distinct from the conventional kinase-substrate interaction. Thus, it remains unclear how widespread reciprocal regulation is between kinase-substrate pairs or, more broadly, interacting partners.
HSF1, by enforcing an inactive conformation, is a direct antagonist of AMPK
Our in vitro studies, using recombinant proteins and synthetic peptides, provide the direct evidence showing the physical regulation of AMPK by HSF1. The peptide library empowers us to dissect the physical AMPK suppression in high resolution. Two prime interfaces on HSF1 were identified, supporting the constitutive HSF1-AMPK interaction in vivo. While P3, P5 and P6 encompass part of the N-terminal DBD, P17-P20 comprise half of the C-terminal AD. Thus, this AMPK-HSF1 interaction may constitute a new HSF1-repressing mechanism.
Excessive exogenous substrates compete with endogenous ones for AMPK phosphorylation. However, under this scenario AMPK remains active, merely unable to access endogenous substrates. Demonstrating this, transfection of SAMS peptides into cells did not diminish AMPK phosphorylation; by contrast, HSF1 peptides did, revealing a direct impact on AMPK per se. Since P6 contains Ser121, this substrate binding to the active site may physically hinder the Thr172 phosphorylation by upstream kinases and block ATP binding. Yet, our findings collectively refute this notion: 1) SAMS, ACC1 and SREBP1, all known substrates binding to the same active site, did not suppress AMPK, indicating that substrate binding is at least insufficient and suggesting that P6 interacts with non-active sites as well; 2) the non-substrate P5, P17 and P19 impaired the LKB1-mediated phosphorylation. P17 also mitigated ATP binding, indicating that substrate binding is not necessary and pinpointing binding to non-active sites as the primary cause; and 3) P6 impaired both ATP binding to α subunits and AMP binding to γ subunits. However, these two binding sites are not in proximity (Xiao et al, 2011). P6, unlikely, hinders both binding physically.
Instead, our evidence supports induction of conformational switching by HSF1 as a root cause. Of interest is the α-to-β transition provoked by HSF1, a structural change implicated in protein deformation and aggregation (Ding et al., 2003; Qin and Buehler, 2010). Collectively, our findings support a model wherein HSF1 interaction instigates the switching of AMPK to an inactive conformation, which favors de-phosphorylation but disfavors phosphorylation as well as binding of AMP and ATP. Thereby, HSF1 renders AMPK unreceptive to activation; contrarily, without HSF1 association, AMPK is primed for activation. Our findings further suggest that under non-stress conditions, a large part of cellular AMPK interacts with inactive, monomeric HSF1, constituting a mutually repressive complex. And, the other small part is free of HSF1 and available for activation. Hence, by dictating how much cellular AMPK is activable, HSF1 imposes a new layer of regulation overriding AMPK stimuli. This model predicts that conditions altering this HSF1-AMPK interaction would modulate both activities. Upon activation by metabolic stress, AMPK recruits HSF1 as a substrate, both preventing HSF1 DNA binding and, as a negative feedback, restraining its own activation. Contrarily, heat shock diminishes Ser121 phosphorylation (Dai et al., 2015), suggesting disruption of this HSF1-AMPK interaction, which releases HSF1 for DNA binding and is expected to prime AMPK activation. This is consistent with the AMPK activation by heat shock in rat hepatocytes (Corton et al., 1994). AMPK inactivation by heat shock, however, has been reported in other cell types (Dai et al., 2015; Kodiha et al., 2011). This discrepancy may be partly due to the direct impacts of heat on AMPK, as γ subunits could misfold and aggregate under heat shock (Dai et al., 2015). Future work is needed to elucidate how this HSF1-AMPK interaction responds to heat stress and controls the HSR/PSR.
Our results do not exclude indirect AMPK-regulating mechanisms in vivo. In fact, the elevated AMP/ATP ratio in Hsf1-deficient MEFs could exacerbate its activation. Substrate competition may be another. When overexpressed, the direct and indirect effects of HSF11–529 cannot be distinguished. By contrast, HSF1 peptides, except P6, are non-substrates, enabling the separation. Of note, HSF1324–529 is a non-substrate and does not interfere with endogenous HSF1.
Control of lipid metabolism and protein CHLation by HSF1
Our studies reveal a remarkable lipogenic action of HSF1, extending its biological impacts beyond the proteome. By influencing protein folding, activity and turnover, posttranslational modifications impact the proteome. The role of HSF1 in SHH CHLation is unexpected; thus, controlling protein lipidation may be a new means for HSF1 to govern proteomic stability.
HSF1-mediated AMPK suppression in human pathologies
The physical AMPK regulation suggests implications of HSF1 in obesity, insulin resistance and type 2 diabetes.
Moreover, a growing body of evidence has pinpointed HSF1 as a powerful, multifaceted pro-oncogenic factor. Herein illuminated is a new underlying mechanism----stimulation of anabolism, in particular lipogenesis and protein lipidation. Cancer cells produce ample lipids to fuel their malignant growth; hence, this lipogenic effect of HSF1 fully aligns with its pro-oncogenic role in general. Moreover, by stimulating SHH expression and CHLation, HSF1 heightens the oncogenic SHH signaling in particular (Evangelista et al., 2006). Of note, at the physiological level HSF1 only suppresses part of cellular AMPK. While this regulation can elude the tumor-suppressive effects of AMPK hyper-activation, it retains the benefits of mild AMPK activation, such as adaptation to metabolic stress, which may be critical to tumor cell survival (Hardie, 2015). To date, almost all biological functions of HSF1 have been attributed to its transcriptional activity; by contrast, our study demonstrates that HSF1 can promote oncogenesis transcription-independently. Thus, therapeutic targeting of HSF1 should consider its both transcription-dependent and -independent actions.
Contrary to fostering malignancy, AMPK suppression may be beneficial under other pathological conditions, including ischemic stroke and neurodegenerative disorders. Previous studies have demonstrated neuronal AMPK activation under stroke, and AMPK inhibition resulted in neuroprotection in mice subjected to stroke (Li et al., 2007; McCullough e t al., 2005). Moreover, AMPK activation is implicated in Huntington’s and Alzheimer’s disease (Ju et al. 2011; Mairet-Coello et al., 2014). Thus, this HSF1-dependent AMPK suppression may broadly impact human physiology and pathology.
STAR★METHODS
Experimental Model and Subject Details
Cells
HeLa and A2058 cells were purchased from ATCC and HEK293T cells were purchased from GE Dharmacon. They were authenticated recently by ATCC. Immortalized Rosa26-CreERT2; Hsf1fl/fl MEFs (male) were described previously (Su et al., 2016). All cell cultures were maintained in DMEM supplemented with 10% HyClone™ bovine growth serum and 100μg/ml normocin™. These cell lines have been routinely tested for mycoplasma contamination using MycoAlert™ Mycoplasm Detection kits.
Organisms/Strains
Meox2-Cre and Alb-Cre mice on the C57BL/6J background and NOD.CB17-Prkdcscid/J (NOD/SCID) mice were obtained from The Jackson Laboratory. Hsf1−/− mice (129SvJ/BALB/cJ) were a generous gift from Dr. Ivor Benjamin and described previously (Dai et al. 2007; Su et al., 2016). Hsf1fl/fl mice on the C57BL/6J background were described previously (Su et al., 2016). All mouse experiments were approved by institutional Animal Care and Use Committees.
Method Details
In vitro kinase assays
All kinase assays were performed in 200μl kinase buffer supplemented with 200μM ATP and various phosphatase and protease inhibitors, containing 10μM Na3VO4, 10μM NaF, 5μM okadaic acid (OA), 10μM sanguinarine chloride (SC), and 1x Halt™ Protease Inhibitor Cocktail. Reactions were incubated at 30°C for 15–30 min. The LKB1 kinase buffer comprises 50mM HEPES, 50mM NaCl, 5mM MgCl2, and 1mM DTT, pH 7.0. The AMPK kinase buffer comprises 25mM MOPS, 12.5mM β-glycerophosphate, 25mM MgCl2, 5mM EGTA, 2mM EDTA, and 0.25mM DTT, pH 7.2. For the kinase assays using various recombinant proteins, all recombinant proteins were pre-dialyzed against 1xPBS using Slide-A-Lyzer MINI Dialysis Devices, 7K MWCO, 0.1ml.
For HSF1 de-phosphorylation, 200ng recombinant HSF1 proteins were first incubated with 100 units of lambda phosphatases at 30°C for 1 hr. Following inactivation with phosphatase inhibitors and protease inhibitors at RT for 30 min, HSF1 proteins were incubated with 75ng recombinant AMPKα1β1γ2 proteins in the presence of 200μM ATP at 30°C for 30 min.
For AMPKα1 de-phosphorylation, 200ng recombinant AMPKα1 proteins were first incubated with 0.1 mU/ml bovine kidney PP2A1 proteins at 30°C for 30 min with 600rpm shaking, followed by incubation with the mixture of phosphatase and protease inhibitors at RT for 5 min to inactivate PP2A. In assays including HSF1 peptides, 10μM peptides were first incubated with AMPK at RT for 30 min.
Detection of AMPK binding by ELISA
10μM HSF1 peptides in 100μl PBS were coated on an ELISA microplate at 4°C overnight. The plates were blocked with 1% BSA in PBS at RT for 30 min, followed by incubation with 100μg HEK293T cell lysates at 4°C overnight. After washing with PBS for 3 times, each well was incubated with a mouse monoclonal anti-AMPKα1/2 antibody (1:1000 diluted in the blocking buffer) at 4°C overnight. Following washing, the wells were incubated with an anti-mouse IgG (H+L)-HRP conjugate (1:1000 diluted in the blocking buffer) at RT for 1 hr. Signals were developed using the 1-Step™ Ultra TMB-ELISA Substrate Solution.
Measurement of cellular ATP and AMP levels
Cellular AMP levels were measured using AMP-Glo™ Assay Kit and cellular ADP/ATP levels were measured by EnzyLight™ ADP Assay Kit. All cells were first lysed in the assay buffer from the ADP Assay Kits. After protein quantitation, equal amounts of cell lysates were used for absolute quantitation of ATP/ADP and AMP levels based on the standard curves. AMP, ADP and ATP standards were all dissolved in the assay buffer.
Proximity Ligation Assay (PLA)
The PLA procedure was described in detail previously (Dai et al., 2015; Su et al., 2016). Duolink® In Situ PLA® anti-rabbit Plus probes, anti-mouse Minus probes, and Detection Reagents Red and Green were all purchased from Sigma-Aldrich. For detection of AMPK-peptide interactions, rabbit anti-AMPKα1 Abs (1:100 dilution), rabbit anti-AMPKβ1+β2 monoclonal Ab clone E.427.6 (1:100 dilution), or rabbit anti-AMPKγ1+2+3 polyclonal Abs (1:100, dilution) were combined with mouse monoclonal anti-biotin Ab-2 BTN.4 (1:100 dilution). For detection of genomic DNA interactions, rabbit anti-GLI1 Abs (1:200 dilution) or rabbit anti-SREBP1 Abs (1:200 dilution) were combined with mouse monoclonal anti-dsDNA Abs HYB331–01 (1:100 dilution). The detailed procedure was described previously (Dai et al., 2015). Nuclei were counterstained with Hoechst 33342. PLA signals were documented by a Zeiss LSM780 confocal microscope or quantitated by a BD FACSCalibur™ flow cytometer.
Immunofluorescence
For antibody validations, fixed cells were incubated with primary antibodies diluted in 5% normal goat serum as described in the PLA experiments at 4°C overnight, followed by incubation with donkey anti-rabbit or anti-mouse IgG (H+L) CF594 conjugates, highly cross-absorbed (1:100 dilution) at RT for 1 hr. For melanoma staining, sections were incubated with rabbit anti-cholesterol Abs (1:100 dilution) at 4°C overnight, followed by incubation with donkey anti-rabbit IgG (H+L) CF594 conjugates (1:200 dilution) at RT for 1 hr. Fluorescent signals were captured using a Zeiss LSM780 confocal microscope and images of different fluorescence channels were overlaid using the ImageJ software.
Mant-AMP and mant-ATP binding assays
All recombinant proteins were pre-dialyzed against 1x PBS. AMPK(A1/B1/G2) proteins (75 or 100ng) or 50ng AMPKα1 proteins were diluted in the kinase buffer supplemented with 10μM Na3VO4, 10μM NaF, 5μM OA, 10μM SC, and 1x Halt™ Protease Inhibitor Cocktail. Following incubation at RT for 5 min, either 10μM peptides or recombinant proteins (AMPK/protein molar ratio 2:1) were added and incubated at RT for 30 min. Following addition of 100nM mant-AMP or 15μM mant-ATP to the mixtures at RT for 5 min, fluorescence was measured at Ex/Em 355/448 using a CLARIOstar microplate reader (BMG LABTECH).
Circular dichroism spectroscopy
Recombinant active AMPKα1β1γ2 complexes (20μg) were first dialyzed against 18.8mM sodium phosphate buffer pH 7.4 using Slide-A-Lyzer MINI Dialysis Devices, 7K MWCO, 0.1ml at 4°C for 48 hr with stirring. The buffer was changed 5 times during dialysis. The quality of dialyzed samples was first examined by UV-Vis spectroscopy. Dialyzed 20μg AMPKα1β1γ2 was pre-incubated with 10μM SP6 or P6 at RT for 30 min, followed by CD spectroscopy. The CD spectra were measured using an AVIV Model 420SF CD spectrometer (AVIV Biomedical), scanning wavelength from 240nm to 190nm with 5 sec/nm for 5 times. The spectrum of the sodium phosphate buffer was subtracted from the spectra of tested samples. The spectrum of P6 alone was subtracted from the spectrum of combined AMPK and P6. To calculate molar ellipticity, the following parameters (AMPK aa1345, 3.24×10−7 mole/L, 0.1mm cell pathlength) were used. The curves were re-plotted using GraphPad Prism 7. To determine the secondary structure fractions, the CDPro software package was used to analyze the CD spectra.
Quantitation of total and cholesteroylated SHH-N by ELISA
The Human Sonic Hedgehog/Shh N-Terminus Quantikine ELISA Kit was used and adapted to detect total and cholesteroylated SHH-N, respectively. Twenty-four hours before the assays, cells were changed to fresh medium and/or treated with 20μg/ml cholesterol. The collected media were centrifuged at the maximum speed using a desktop Eppendorf centrifuge to remove cell debris. Either 100μl medium, 100μg cell lysates or 50μg melanoma lysates diluted in the assay diluent (total 200μl volume) were added to each well on a microplate that had already been coated with a monoclonal anti-SHH-N antibody, and incubated at RT for 2 hr. For detection of total SHH-N, 200μl another monoclonal anti-SHH-N antibody that was conjugated to HRP were added to the wells and incubated at RT for 2 hr. For detection of cholesteroylated SHH-N, 200μl anti-CHL antibody (1:1000 dilution) was added to the wells and incubated at 4°C overnight. Following washing 3 times, 200μl anti-rabbit IgG (H+L)-HRP conjugates (1:1000 dilution) were added and incubated at RT for 2 hr.
Recombinant human SHH-N proteins without cholesteroylation from this ELISA kit and cholesteroylated human SHH-N proteins were used for validation. First, this ELISA was tested with serum-free medium supplemented with and without 50μM free CHL. Next, equal amounts of recombinant SHH-N proteins with and without cholesteroylation were captured on the plates, followed by detection with either anti-CHL Abs or another anti-SHH-N Ab conjugated with HRP from the Shh N-Terminus Quantikine ELISA Kit.
Image analysis
Immunoblotting signals and western blots were quantitated by the ImageJ software.
Animal studies
For in vivo pharmacological inhibition of AMPK, Hsf1+/+ and Hsf1−/− mice (7–8 weeks of age) or Hsf1fl/Δ and Meox2-Cre; Hsf1fl/Δ mice (2–3 months of age) were i.p. injected with 2mg/kg/day CC for 10 days. In each treatment group, both male and female mice were included. Before and after CC treatment, blood samples were collected, and body composition was analyzed.
Liver-targeted in vivo shRNA delivery in Hsf1fl/Δ and Meox2-Cre; Hsf1fl/Δ mice (2–3 months of age, both males and females) was achieved via hydrodynamic injection. For each mouse 40μg of plasmids, either Scramble alone or 1:1 combined Ampkα1 and α2 shRNAs, were incubated with 5μl of In Vivo-JetPEI® transfection reagent in 200μl sterile 5% glucose solution at RT for 15min. Before injections, DNA/in vivoJetPEI® complexes were diluted in sterile 5% glucose and total volumes equaled to 10% of mouse body weights. The whole injection solutions were delivered into tail veins at a constant rate within 5–7 seconds. Mice were injected 5 times at 3-day intervals.
For the melanoma xenograft study, 1 ×106 modified A2058 cells were s.c. injected into the left flanks of 9-week-old male NOD.CB17-Prkdc<scid>/J (NOD/SCID) mice. Tumor sizes were measured weekly using a caliper by an animal technician who was blinded to this study. Tumor volumes were calculated following the formula [(width)2 × length] ÷ 2.
Nuclear magnetic resonance (NMR)-based body and tumor composition analysis
Body, liver and tumor composition were measured using an EchoMRI-130™ whole-body composition analyzer (EchoMRI LLC) according to manufacturer’s instructions.
HSF1 peptide library
The HSF1 peptide library and biotinylated peptides were synthesized by GenScript Custom Peptide Synthesis Service. The amino acid sequences of individual peptides are listed in Table S1. For biotinylation, a biotin moiety was conjugated to the N-terminus of each peptide during synthesis. Peptides were dissolved in 0.01N NaOH to make 1mM stocks.
Plasmid construction
The plasmids used in this study, including pLenti6-LacZ, pHSE-SEAP, pCMV-Gaussia luciferase, pLenti6FLAG-HSF11–529 plasmid were described previously (Su et al., 2016). pBabe-LacZ, pBabe-FLAG-HSF1WT, pBabe-FLAG-HSF1S326A, and pLenti6-FLAG-HSF1S326D were described previously (Tang et al., 2015; Su et al., 2016). pLenti6-FLAG-HSF11–323, pLenti6-FLAG-HSF1324–529 and pLenti6-FLAG-HSF1Δ49–144, Δ385–480 plasmids were constructed from pLenti6-FLAG-HSF11–529 using a Q5® Site-Directed Mutagenesis Kit.
Conditional deletion of Hsf1 and glucose starvation in MEFs
To delete Hsf1, immortalized Rosa26-CreERT2; Hsf1fl/fl MEFs were pre-treated with ethanol or 1μM (Z)-4Hydroxytamoxifen (4-OHT) for 7 days. For glucose starvation, MEFs were cultured in glucose-, serum-free medium supplemented with 4mM L-glutamine and 1mM sodium pyruvate.
Quantitation of AMPKα Thr172 phosphorylation
AMPKα phosphorylation was quantitated by ELISA using the PathScan® Phospho-AMPKα (Thr172) Sandwich ELISA Antibody Pair, 100μg cell lysates per well. Signals were developed using the 1-Step™ Ultra TMB-ELISA Substrate Solution.
Co-IP of endogenous AMPK complexes and HSF1 peptides
Following transfection of 10μM biotinylated peptides overnight in Hsf1-deficient (Hsf1fl/fl, 4-OHT+) MEFs, biotinylated peptides were precipitated by streptavidin beads. The levels of biotinylated peptides in cell lysates were quantitated by direct ELISA using streptavidin-HRP.
Concurrent knockdown of HSF1 and AMPKα in A2058 cells
A2058 cells stably expressing inducible scramble or HSF1-targeting shRNAs were first treated with 25μg/ml doxycycline for 2 days, followed by transfection of 10nM control or AMPKα1/2-targeting siRNAs using using jetPRIME® transfection reagents for 4 days. These transfected cells were simultaneously treated with doxycycline for another 4 days to induce shRNA expression. For inducible HSF1 knockdown alone, A2058 cells were treated with doxycycline continuously for 6 days.
In vitro phosphorylation of SAMS peptides by AMPKα1
First, 10μM biotinylated SAMS peptides were incubated on a Pierce™ Streptavidin Coated Plate overnight. Recombinant active AMPKα1 was pre-incubated with individual recombinant proteins (2:1 molar ratio) at RT for 30 min. The protein mixtures were then added to the SAMS peptide-coated microplates in the presence of 5mM ATP and various phosphatase inhibitors and incubated at 30 ° C for 1 hr. Phosphorylation of SAMS peptides was quantitated by ELISA using a mouse anti-phosphoserine/threonine Ab and secondary anti-mouse IgG HRP conjugates.
Nile red staining
Following transfection of shRNA plasmids for 4 days, HEK293T cells grown in 96-well plates were fixed and stained with 20μg/ml Nile red dissolved in 100% alcohol at RT for 5 min. Green fluorescence intensities of lipid droplets were measured at Ex/Em 485/535nm. Subsequently, each well was stained with Hoechst 33342 for 1 min and measured at Ex/Em 358/461nm to quantitate cell numbers. For each well, the fluorescence intensity of Nile red was normalized against that of Hoechst 33342.
Immunoblotting and Immunoprecipitation
Whole-cell protein extracts were prepared in cold cell-lysis buffer (100 mM NaCl, 30 mM Tris-HCl pH 7.6, 1% Triton X-100, 20 mM sodium fluoride, 1 mM EDTA, 1 mM sodium orthovanadate, and 1x Halt™ protease inhibitor cocktail). Proteins were transferred to nitrocellulose membranes. Following incubation with the blocking buffer (5% non-fat milk in 1x TBS-T) for 2 hr at RT, membranes were incubated with primary antibodies (1:1,000 dilution in the blocking buffer) overnight at 4°C. After washing with 1xTBS-T for 3 times, membranes were incubated with peroxidase-conjugated secondary antibodies (1: 2,500 dilution in the blocking buffer) at RT for 1 hr. Signals were detected using SuperSignal West chemiluminescent substrates.
For IP, 1mg whole cell lysates were incubated with anti-AMPKα (23A3) sepharose beads, anti-AMPKα (23A3) Ab, or phospho-AMPKα Thr172 (40H9) Ab, 2μg rabbit anti-HSF1 Abs H-311 or 10μl rabbit monoclonal anti-FLAG antibodies at 4°C overnight. Either normal rabbit IgG or rabbit anti-PI3K p110α (C73F8) were used as the negative controls. Protein G MagBeads were used to precipitate primary Abs. After washing with the lysis buffer for 3 times, beads were boiled in 1x loading buffer for 5 min before loading on SDS-PAGE.
Real-time quantitative RT-PCR
Total RNAs were extracted using RNA STAT-60 reagent, and RNAs were used for reverse transcription using a Verso cDNA Synthesis kit. Equal amounts of cDNA were used for quantitative RCR reaction using a DyNAmo SYBR Green qPCR kit. Signals were detected by an Agilent Mx3000P qPCR System (Agilent Genomics). ACTB was used as the internal control. The sequences of individual primers for each gene are listed in Table S2.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed using rabbit anti-HSF1 Abs H-311 or normal rabbit IgG, as described previously (Tang et al., 2015; Su et al., 2016). The sequences of individual primers for CHIP-qPCR are listed in Table S2.
shRNA and siRNA knockdown
The sequences of individual shRNAs and siRNAs are listed in Table S3. shRNAs were transfected at 1μg using Turbofect transfection reagents for 3 days. In general, siRNAs were transfected at 10nM using jetPRIME® transfection reagents for 4 days.
Transfection and HSF1 dual reporter assay
Plasmids were transfected with TurboFect transfection reagents. HEK293T cells were co-transfected with heat shock element (HSE)-secreted embryonic alkaline phosphatase (SEAP), CMV-Gaussia luciferase (GLuc) reporter plasmids, along with various indicated plasmids. After 48 hr, reporter activities in culture media were measured. SEAP and luciferase activities in culture supernatants were quantitated using a NovaBright™ Phospha-Light™ EXP Assay Kit for SEAP and a Pierce™ Gaussia Luciferase Glow Assay Kit, respectively. Luminescence signals were measured by a CLARIOstar microplate reader (BMG LABTECH). SEAP activities were normalized against GLuc activities.
Cholesterol Assays
Cellular cholesterol levels were quantitated using an Amplex™ Red Cholesterol Assay Kit. For lipids extraction, equal numbers of cells were incubated with 200μl chloroform-methanol mixture (2:1v/v) and vortexed. Following centrifugation in an Eppendorf microcentrifuge at the maximal speed, the organic layers were transferred to a new tube and air dried in a chemical fume hood. Cholesterol levels were quantitated according to the manufacturer’s instructions.
Blood plasma chemistry
Mouse blood plasma lipids were measured using a Beckman Coulter Synchron CX5 Chemistry Analyzer (Beckman Coulter Inc.)
Cell viability assays
Cell viability was measured in 96-well microplate format using CellTiter-Blue® Cell Viability Assay. Fluorescent signals were measured by a CLARIOstar microplate reader (BMG LABTECH).
Quantification and Statistical Analysis
All experiments, except the animal studies, immunostaining for antibody validation and human data, have been repeated at least three times. All statistical analyses were performed using Prism 7.0 or 8.0 (GraphPad software). Detailed statistical methods were described in the figure legends. Statistical significance: n.s.: not significant, p>0.05; *p< 0.05; **p<0.01; ***p<0.001.
Data and Software Availability
Original images and western blot data are deposited in Mendeley
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-HSF1 monoclonal antibody [EP1710Y] | Abcam | Cat# ab52757, RRID: AB_880518 |
| Rabbit anti-LDH monoclonal antibody [EP1563Y] | Abcam | Cat# 1980-1, RRID: AB_991755 |
| Mouse anti-AMPKα1/2 monoclonal antibody | Bio-Rad | Cat# MCA2673GA; RRID: AB_1604623 |
| Donkey Anti-Mouse IgG (H+L) Highly Cross-Adsorbed Antibody, CF594 Conjugated | Biotium | Cat# 20115, RRID: AB_10559815 |
| Donkey Anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Antibody, CF594 Conjugated | Biotium | Cat# 20152, RRID: AB_10563030 |
| Rabbit anti-AMPK (23A3) monoclonal Antibody | Cell Signaling Technology | Cat# 2603, RRID: AB_490795 |
| Rabbit anti-phospho-Raptor (Ser792) polyclonal antibody | Cell Signaling Technology | Cat# 2083, RRID: AB_2249475 |
| Mouse anti Lamin A/C (4C11) monoclonal antibody | Cell Signaling Technology | Cat# 4777, RRID: AB_10545756 |
| Rabbit anti-LKB1 (D60C5) monoclonal antibody | Cell Signaling Technology | Cat# 3047, RRID: AB_2198327 |
| Mouse anti-DYKDDDDK (FLAG) Tag (9A3) monoclonal antibody | Cell Signaling Technology | Cat# 8146, RRID: AB_10950495 |
| Rabbit anti-phospho-SREBP-1c (Ser372) polyclonal antibody | Cell Signaling Technology | Cat# 9874, RRID: AB_10949508 |
| Rabbit anti-PP2A C Subunit polyclonal antibody | Cell Signaling Technology | Cat# 2038, RRID: AB_2169495 |
| Rabbit anti-phospho-AMPKα, (Thr172) (40H9) monoclonal antibody | Cell Signaling Technology | Cat# 2535, RRID: AB_331250 |
| Rabbit anti-DYKDDDDK (FLAG) Tag (D6W5B) monoclonal antibody | Cell Signaling Technology | Cat# 14793, RRID: AB_2572291 |
| Rabbit anti-GST (91G1) monoclonal antibody | Cell Signaling Technology | Cat# 2625, RRID: AB_490796 |
| Rabbit anti-phospho-Acetyl-CoA Carboxylase (Ser79) polyclonal antibody | Cell Signaling Technology | Cat# 3661, RRID: AB_330337 |
| Rabbit anti-PI3 Kinase p110α (C73F8) monoclonal antibody | Cell Signaling Technology | Cat# 4249, RRID: AB_2165248 |
| Rabbit anti-Raptor (24C12) monoclonal antibody | Cell Signaling Technology | Cat# 2280, RRID: AB_561245 |
| Rabbit anti-Acetyl-CoA Carboxylase (C83B10) monoclonal antibody | Cell Signaling Technology | Cat# 3676, RRID: AB_2219397 |
| Mouse anti-Phosphoserine / threonine monoclonal antibody | ECM Biosciences | Cat# PM3801, RRID: AB_1944434 |
| Rabbit anti-AMPKα1 monoclonal antibody | GeneTex | Cat# GTX61229; RRID: AB_10620245 |
| DYKDDDDK (FLAG) Tag antibody-HRP | GenScript | Cat# A01428, RRID: AB_1720817 |
| β-Actin antibody-HRP | GenScript | Cat# A00730, RRID: AB_914100 |
| Peroxidase-AffiniPure goat anti-mouse IgG (H + L) antibody | Jackson ImmunoResearch Labs | Cat# 115-035-003, RRID: AB_10015289 |
| Peroxidase-AffiniPure goat anti-rabbit IgG (H+L) antibody | Jackson ImmunoResearch Labs | Cat# 111-035-003, RRID: AB_2313567 |
| Peroxidase-AffiniPure goat anti-rat IgG (H+L) antibody | Jackson ImmunoResearch Labs | Cat# 112-035-003, RRID: AB_2338128 |
| Rabbit anti-SREBP1 polyclonal antibody | Novus Biologicals | Cat# NB100-2215; AB_10002406 |
| Mouse anti-dsDNA (HYB331-01) monoclonal antibody | Santa Cruz Biotechnology | Cat# sc-58749; AB_783088 |
| Rabbit anti-HSF1 (H-311) polyclonal antibody | Santa Cruz Biotechnology | Cat# sc-9144, RRID: AB_2120276 |
| Rat anti-HSF1 (10H8) monoclonal antibody | Santa Cruz Biotechnology | Cat# sc-13516, RRID: AB_627751 |
| Mouse anti-biotin Ab-2 (BTN.4) monoclonal antibody | Thermo Fisher Scientific | Cat# MA511251; RRID: AB_10980856 |
| Rabbit anti-AMPKβ1+2 (E.427.6) monoclonal antibody | Thermo Fisher Scientific | Cat# MA515090; RRID: AB_10981956 |
| Rabbit anti-AMPKγ1+2+3 polyclonal antibody | Thermo Fisher Scientific | Cat# PA536314; RRID: AB_2553469 |
| Rabbit anti-GLI1 polyclonal antibody | Thermo Fisher Scientific | Cat# PA5-32206; RRID: AB_2549679 |
| Mouse anti-SREBP1 (2A4) monoclonal antibody | Thermo Fisher Scientific | Cat# MA5-16124, RRID: AB_11152118 |
| Rabbit anti-cholesterol polyclonal antibody | USBiological | Cat# 139711 |
| ImmPRESS® Excel Amplified HRP Polymer Staining Kit (Goat anti-rabbit IgG and horse anti-goat IgG HRP polymers) | Vector Laboratories | Cat# MP-7601, RRID: AB_2336533 |
| Bacterial and Virus Strains | ||
| NEB® 5-α Competent E. coli (High Efficiency) | New England Biolabs | Cat# C2987 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bafilomycin A1 | Axxora LLC | Cat# LKT-B0025 |
| Chloroquine diphosphate | Axxora LLC | Cat# LKT-C2950 |
| Mant-AMP | Axxora LLC | Cat# JBS-NU-236S |
| Recombinant active human AMPKα1/PRKAA1 proteins | Cell Sciences | Cat# CRA019 |
| AMPKα (23A3) Rabbit monoclonal antibody (Sepharose® Bead Conjugate) | Cell Signaling Technology | Cat# 6707 |
| Recombinant human His-HSF1 proteins | Enzo Life Sciences | Cat# ADI-SPP-900 |
| Recombinant human HSP90β proteins | Enzo Life Sciences | Cat# ALX-201-147-C025 |
| HSF1 peptides (see Table S1: HSF1 peptide sequences) | GenScript | Custom order |
| Puromycin | InvivoGen | Cat# ant-pr-1 |
| 17-DMAG | LC Laboratories | Cat# D-3440 |
| Recombinant human SREBP1 proteins | Lifespan BioSciences, Inc | Cat# LS-G56019 |
| (Z)-4-Hydroxytamoxifen | LKT Laboratories | Cat# H9711 |
| Recombinant bovine kidney PP2A1 proteins | Millipore | Cat# 53-950-8 |
| Lambda phosphatases | New England Biolabs | Cat# P0753S |
| In Vivo-JetPEI® transfection reagent | PolyPlus-transfection | Cat# 201 |
| CellTiter-Blue® cell viability assay kit | Promega | Cat# G8081 |
| Recombinant cholesteroylated human SHH-N proteins | R&D Systems | Cat# 8908-SH-005/CF |
| AMP | Research Product International | Cat# A11240 |
| ATP | Research Product International | Cat# A30030 |
| Dorsomorphin dihydrochloride (Compound C) | Santa Cruz Biotechnology | Cat# sc-361173 |
| A23187 | Sigma-Aldrich | Cat# C7522 |
| Cholesterol-water soluble | Sigma-Aldrich | Cat# C4951 |
| Duolink® In Situ PLA anti-mouse Minus probes | Sigma-Aldrich | Cat# DUO92004 |
| Duolink® In Situ PLA anti-rabbit Plus probes | Sigma-Aldrich | Cat# DUO92002 |
| Duolink® In Situ PLA Detection Reagents Green | Sigma-Aldrich | Cat# DUO92014 |
| Duolink® In Situ PLA Detection Reagents Red | Sigma-Aldrich | Cat# DUO92008 |
| Recombinant active human LKB1/MO25/STRAD proteins | Sigma-Aldrich | Cat# SRP0246 |
| Recombinant human ACC1 proteins | Sigma-Aldrich Corporation | Cat# A6986 |
| Recombinant GST proteins | SignalChem Lifesciences | Cat# G52-30U |
| RNA STAT-60 reagent | Tel-Test, Inc. | Cat# RNA STAT-60 |
| ActinGreen™ 488 ReadyProbes® Reagent | Thermo Fisher Scientific | Cat# R37110 |
| Alfa Aesar™ Okadaic acid | Thermo Fisher Scientific | Cat# NC0314406 |
| Amplex™ Red Cholesterol Assay Kit | Thermo Fisher Scientific | Cat# A12216 |
| Apexbio Technology LLC sanguinarine chloride | Thermo Fisher Scientific | Cat# 50-115-2424 |
| DSP crosslinker | Thermo Fisher Scientific | Cat# PI22585 |
| Gibco™ recombinant human TRAIL proteins | Thermo Fisher Scientific | Cat# PHC1634 |
| Halt™ Protease Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78430 |
| Mant-ATP | Thermo Fisher Scientific | Cat# M12417 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Thermo Fisher Scientific | Cat# PI78835 |
| Nile Red | Thermo Fisher Scientific | Cat# N1142 |
| Oligomycin | Thermo Fisher Scientific | Cat# AAJ61898MA |
| Pierce™ High Sensitivity Streptavidin-HRP | Thermo Fisher Scientific | Cat# 21134 |
| Recombinant human AMPK(A1/B1/G2) proteins | Thermo Fisher Scientific | Cat# PV6238 |
| Selleck Chemical LLC phenformin | Thermo Fisher Scientific | Cat# 50-579-2 |
| Critical Commercial Assays | ||
| EnzyLight® ADP Assay Kit | BioAssay Systems | Cat# EADP-100 |
| PathScan® Phospho-AMPKα (Thr172) Sandwich ELISA Antibody Pair | Cell Signaling Technology | Cat# 7955 |
| Mito-ID® red detection kit | Enzo Life Sciences | Cat# ENZ-51007 |
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs Inc. | Cat# E0552S |
| JetPRIME® transfection reagents | PolyPlus-transfection® SA | Cat# 114 |
| AMP-Glo® Assay Kit | Promega | Cat# V5011 |
| Human Sonic Hedgehog/Shh N-Terminus Quantikine ELISA Kit | R&D Systems | Cat# DSHH00 |
| DyNAmo SYBR Green qPCR kit | Thermo Fisher Scientific | Cat# F416XL |
| Thermo Scientific™ Pierce™ Silver Stain Kit for Mass Spectrometry | Thermo Fisher Scientific | Cat# 24600 |
| Verso cDNA Synthesis kit | Thermo Fisher Scientific | Cat# AB1453A |
| Deposited Data | ||
| Raw images of western blots and original image files | Mendeley Data | http://doi.org/10.17632/bpr9h3gpfn.1 |
| Experimental Models: Cell Lines | ||
| A2058 | ATCC | Cat# CRL-11147 |
| HeLa | ATCC | Cat# CCL-2 |
| HEK293T | GE Dharmacon | Cat# HCL4517 |
| Immortalized Rosa26-CreERT2; Hsf1fl/fl MEFs | Su et al., 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Hsf1fl/fl mice (C57BL/6J) | Su et al., 2016 | N/A |
| Hsf1−/− mice (129SvJ/BALB/cJ) | Xiao et al., 1999 | N/A |
| Alb-Cre mice (C57BL/6J) | The Jackson Laboratory | IMSR Cat# JAX:003574; RRID:IMSR_JAX:003574 |
| Meox2-Cre mice (C57BL/6J) | The Jackson Laboratory | IMSR Cat# JAX:003755; RRID:IMSR_JAX:003755 |
| NOD.CB17-Prkdcscid/J mice (NOD/SCID) | The Jackson Laboratory | IMSR Cat# JAX:001303; RRID:IMSR_JAX:001303 |
| Oligonucleotides | ||
| qRT-PCR primers (see Table S2: Primer sequences) | IDT | N/A |
| ChIP qPCR primers (see Table S2: Primer sequences) | IDT | N/A |
| Mutagenesis and sequencing primers (see Table S2: Primer sequences) | IDT | N/A |
| The target sequences of siRNAs and shRNAs (see Table S3) | N/A | N/A |
| Recombinant DNA | ||
| pLenti6-LacZ | Su et al., 2016 | N/A |
| pHSE-SEAP | Clontech Laboratories | Cat# 631910 |
| pCMV-Gaussia luciferase | Thermo Fisher Scientific | Cat# 16147 |
| pLenti6-FLAG-HSF11–529 | This paper | N/A |
| pBabe-FLAG-HSF11–323 | This paper | N/A |
| pBabe-FLAG-HSF1324–529 | This paper | N/A |
| pLenti6-FLAG-HSF1324–529 | This paper | N/A |
| pBabe-LacZ | Su et al., 2016 | N/A |
| pBabe-FLAG-HSF11–529 | Gift from Robert Kingston | Addgene #1948 |
| pBabe-FLAG-HSF1S326A | Tang et al., 2015 | N/A |
| pLenti6-FLAG-HSF1S326D | Tang et al., 2015 | N/A |
| pLenti6-FLAG-HSF1Δ49–144,Δ385–480 | This paper | N/A |
| Software and Algorithms | ||
| CDPro | CDPro software | https://sites.bmb.colostate.edu/sreeram/CDPro/ |
| FlowJo | FlowJo, LCC | https://www.flowjo.com/ |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Prism 7 and 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Protein G MagBeads | GenScript | Cat# L00274 |
| HyClone™ bovine growth serum | HyClone Laboratories | Cat# SH30541.03IR |
| Normocin™ | Invivogen | Cat# ant-nr-1 |
| DMEM 4.5g/L Glucose w/ L-Glutamine | Lonza | Cat# 12-604Q |
| MycoAlert™ Mycoplasm Detection kits | Lonza | Cat# LT07-418 |
| Streptavidin Magnetic Beads | New England Biolabs | Cat# S1420S |
| 1-Step™ Ultra TMB-ELISA Substrate Solution | Thermo Fisher Scientific | Cat# 34028 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H1399 |
| Normal goat serum | Thermo Fisher Scientific | Cat# OB006001 |
| NovaBright™ Phospha-Light™ EXP Assay Kit for SEAP | Thermo Fisher Scientific | Cat# N10578 |
| Pierce™ Gaussia Luciferase Glow Assay Kit | Thermo Fisher Scientific | Cat# 16160 |
| Pierce™ Streptavidin Coated Plates, Clear, 96-Well | Thermo Fisher Scientific | Cat# 15124 |
| Slide-A-Lyzer MINI Dialysis Devices, 7K MWCO, 0.1ml | Thermo Fisher Scientific | Cat# 69560 |
| SuperSignal West Pico Plus chemiluminescent substrates | Thermo Fisher Scientific | Cat# 34580 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat# 34096 |
| TurboFect transfection reagents | Thermo Fisher Scientific | Cat# R0531 |
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to the Lead Contact Chengkai Dai (chengkai.dai@nih.gov).
HIGHLIGHTS.
The kinase-substrate pair AMPK and HSF1 constitute a mutually repressive complex
By physically evoking a conformational switch, HSF1 is a potent antagonist of AMPK
HSF1, via AMPK suppression, governs lipid metabolism and protein cholesteroylation
This AMPK repression is a transcription-independent pro-oncogenic mechanism of HSF1
ACKNOWLEDGMENTS
We would like to thank Holly Morris for her technical support for the animal care and studies, Sergey Tarasov and the Biophysics Resource (BR) for their assistance with the CD study, the Optical Microscopy and Image Analysis lab (OMAL) for their assistance with the confocal microscopy studies. This work was supported by the grant from NIH to C.D. (R21CA184704) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
CONFLICTS OF INTEREST
The authors have declared no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aburai N, Yoshida M, Ohnishi M, Kimura K (2010) Sanguinarine as a potent and specific inhibitor of protein phosphatase 2C in vitro and induces apoptosis via phosphorylation of p38 in HL60 cells. Biosci Biotechnol Biochem 74: 548–52 [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P (2007) The selectivity of protein kinase inhibitors: a further update. Biochem J 408: 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloribi-Djefaflia S, Vasseur S, Guillaumond F (2016) Lipid metabolic reprogramming in cancer cells. Oncogenesis 5: e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialojan C, Takai A (1988) Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J 256: 283–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D (2017) AMPK signalling in health and disease. Curr Opin Cell Biol 45: 31–37 [DOI] [PubMed] [Google Scholar]
- Chemes LB, Alonso LG, Noval MG, de Prat-Gay G (2012) Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods Mol Biol 895: 387–404 [DOI] [PubMed] [Google Scholar]
- Choi DC, Chae YJ, Kabaria S, Chaudhuri AD, Jain MR, Li H, Mouradian MM, Junn E (2014) MicroRNA-7 protects against 1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J Neurosci 34: 12725–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY (2014) A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515: 274–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PR, Hardie DG (1990) Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J 9: 2439–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP (2001) AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci 17: 45–58 [DOI] [PubMed] [Google Scholar]
- Dai C (2018) The heat-shock, or HSF1-mediated proteotoxic stress, response in cancer: from proteomic stability to oncogenesis. Philos Trans R Soc Lond B Biol Sci 373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Sampson SB (2016) HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol 26: 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L, Lindquist S (2012) Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 122: 3742–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130: 1005–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, Dai C (2015) Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J 34: 275–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Carling D, Hardie DG (1989) Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem 186: 123–8 [DOI] [PubMed] [Google Scholar]
- Ding F, Borreguero JM, Buldyrey SV, Stanley HE, Dokholyan NV (2003) Mechanism for the alpha-helix to beta-hairpin transition. Proteins 53: 220–8 [DOI] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD (2007) Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, Tian H, de Sauvage FJ (2006) The hedgehog signaling pathway in cancer. Clin Cancer Res 12: 5924–8 [DOI] [PubMed] [Google Scholar]
- Gowans GJ, Hawley SA, Ross FA, Hardie DG (2013) AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18: 556–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan P, Mayer EP, Fowler SD (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 100: 965–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2015) Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin Cancer Res 21: 3836–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG (2005) Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2: 9–19 [DOI] [PubMed] [Google Scholar]
- Herrero-Martin G, Hoyer-Hansen M, Garcia-Garcia C, Fumarola C, Farkas T, Lopez-Rivas A, Jaattela M (2009) TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J 28: 677–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, McMahon AP (2002) Cholesterol modification of Hedgehog family proteins. J Clin Invest 110: 591–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Moskophidis D, Mivechi NF (2011) Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab 14: 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph BK, Liu HY, Francisco J, Pandya D, Donigan M, Gallo-Ebert C, Giordano C, Bata A, Nickels JT Jr., (2015) Inhibition of AMP Kinase by the Protein Phosphatase 2A Heterotrimer, PP2APpp2r2d. J Biol Chem 290: 10588–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, Sun CP, Tao MH, Tu PH, Chang C, Dickson DW, Chern Y (2011) Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol 194: 209–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Labbadia J, Morimoto RI (2017) Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol 27: 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD (2007) Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke 38: 2992–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13: 376388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23: 833–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F (2013) The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron 78: 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV (2005) Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem 280: 20493–502 [DOI] [PubMed] [Google Scholar]
- Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7: 763–77 [DOI] [PubMed] [Google Scholar]
- Meng L, Gabai VL, Sherman MY (2010) Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 29: 5204–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF (2007) Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 26: 5086–97 [DOI] [PubMed] [Google Scholar]
- Munday MR, Campbell DG, Carling D, Hardie DG (1988) Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem 175: 331–8 [DOI] [PubMed] [Google Scholar]
- Nadolski MJ, Linder ME (2007) Protein lipidation. FEBS J 274: 5202–10 [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Scott JW, Kemp BE (2012) AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol Metab 23: 125–32 [DOI] [PubMed] [Google Scholar]
- Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H (2008) Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem 283: 3979–87 [DOI] [PubMed] [Google Scholar]
- Porter JA, Young KE, Beachy PA (1996) Cholesterol modification of hedgehog signaling proteins in animal development. Science 274: 255–9 [DOI] [PubMed] [Google Scholar]
- Qiao A, Jin X, Pang J, Moskophidis D, Mivechi NF (2017) The transcriptional regulator of the chaperone response HSF1 controls hepatic bioenergetics and protein homeostasis. J Cell Biol 216: 723–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Buehler MJ (2010) Molecular dynamics simulation of the alpha-helix to beta-sheet transition in coiled protein filaments: evidence for a critical filament length scale. Phys Rev Lett 104: 198304 [DOI] [PubMed] [Google Scholar]
- Resh MD (2012) Targeting protein lipidation in disease. Trends Mol Med 18: 206–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset CI, Leiper FC, Kichev A, Gressens P, Carling D, Hagberg H, Thornton C (2015) A dual role for AMP-activated protein kinase (AMPK) during neonatal hypoxic-ischaemic brain injury in mice. J Neurochem 133: 242–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101: 3329–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KH, Cao J, Tang Z, Dai S, He Y, Sampson SB, Benjamin IJ, Dai C (2016) HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat Cell Biol 18: 527–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Liu D (2007) Hydrodynamic gene delivery: its principles and applications. Mol Ther 15: 2063–9 [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P (2000) Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26: 113–5 [DOI] [PubMed] [Google Scholar]
- Tang Z, Dai S, He Y, Doty RA, Shultz LD, Sampson SB, Dai C (2015) MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160: 729–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasca CI, Dal-Cim T, Cimarosti H (2015) In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol Biol 1254: 197–210 [DOI] [PubMed] [Google Scholar]
- Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, Alrefai WA, Ford DA, Tontonoz P (2018) Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 22: 206–220 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: 2004–8 [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 16: 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi C, Hu Y, Buckhaults P, Moskophidis D, Mivechi NF (2012) Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J Biol Chem 287: 35646–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ (2007) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449: 496–500 [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472: 230–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Tang JJ, Peng C, Wang Y, Fu L, Qiu ZP, Xiong Y, Yang LF, Cui HW, He XL, Yin L, Qi W, Wong CC, Zhao Y, Li BL, Qiu WW, Song BL (2017) Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol Cell 66: 154–162 e10 [DOI] [PubMed] [Google Scholar]
- Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ (1999) HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J 18: 5943–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, Wu YQ, Li TY, Ye Z, Lin SY, Yin H, Piao HL, Hardie DG, Lin SC (2017) Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I (2007) Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta 1768: 1311–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94: 471–80 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original images and western blot data are deposited in Mendeley