

Abstract
Objective:
To report the rate of candidate actionable somatic mutations in patients with locally advanced and metastatic gastro-enteropancreatic (GEP) neuroendocrine tumors (NET) and of other genetic alterations that may be associated with tumorigenesis.
Methods:
A phase II mutation targeted therapy trial was conducted in patients with advanced well-differentiated G1/G2 GEP-NET. Mutations found in the mTOR pathway-associated genes led to treatment with the mTOR inhibitor everolimus, and were defined as actionable. Tumor deoxyribonucleic acid (DNA) from GEP-NET were sequenced and compared with germline DNA, using the OncoVAR-NET assay, designed for hybrid capture sequencing of 500 tumor suppressor genes and oncogenes. Somatic variants were called and copy-number (CN) variant analysis was performed.
Results:
Thirty patients (14 small-intestine, 8 pancreatic, 3 unknown primary NET, and 5 of other primary sites) harbored 37 lesions (4 patients had DNA of multiple lesions sequenced). Only 2 patients with sporadic NET (n = 26) had an actionable mutation leading to treatment with everolimus. Driver somatic mutations were detected in 18 of 30 patients (21/37 lesions sequenced). In the remaining samples without a driver mutation, CN alterations were found in 11/16 tumors (10/12 patients), including CN loss of chromosome (Chr) 18 (P<.05), CN gain of Chr 5, and loss of Chr 13. CN losses in Chr 18 were more common in patients without driver mutations detected. Pronounced genetic heterogeneity was detected in patients with multiple lesions sequenced.
Conclusion:
Genome-wide DNA sequencing may identify candidate actionable genes and lead to the identification of novel target genes for advanced well-differentiated GEP-NET.
INTRODUCTION
Neuroendocrine tumors (NET) are a heterogenous tumor group emerging from neuroendocrine cells, mainly in the gastro-intestinal and bronchopulmonary tracts. The incidence of NET has increased over the last three decades (1). The management of malignant and hormone-secreting NET consists of surgical resection as first line therapy, which is the only curative treatment modality in resectable NET. The second line consists of medical therapy and peptide-receptor radionuclide therapy (PRRT) for patients with unresectable disease that have been studied in large prospective studies. In the PROMID study, octreotide was found to have antiproliferative efficacy in advanced small intestine NET (2), while the CLARINET trial showed progression-free survival (PFS) benefit of treatment with lanreotide for patients with advanced gastro-enteropan-creatic (GEP)-NET (3). The RADIANT III and IV trials showed PFS benefit of treatment with everolimus, an inhibitor of the mammalian target of rapamycin (mTOR), in patients with GEP- and lung NET (4,5). In the SUN1111 trial, treatment with sunitinib, a multitarget tyrosine kinase inhibitor, improved PFS in patients with pancreatic NET (6). In addition to the medical therapy described above, the NETTER-1 trial reported the utility of PRRT with the DOTA-peptide DOTATATE labeled with 177Lutitium (177Lu-DOTATATE). Patients harboring advanced GEP-NET that were treated with 177Lu-DOTATATE had a prolonged PFS compared with the control group (7).
Great progress was also noted in the understanding of NET tumorigenesis both at the genetic and epigenetic levels. Karpathakis et al (8) performed a comprehensive integrated molecular analysis of small-intestine NET, showing not only that this group of tumors can be clustered into three distinct sub-groups based on their molecular profiles, but also that these clusters may be associated with the patient’s PFS (8). Substantial progress was also achieved in the understanding of the molecular characterization of pancreatic neuroendocrine tumors (PNET). Scarpa and colleagues performed whole-genome sequencing of a large number of PNET (9). In their pivotal work, the authors reported a surprisingly high rate of germline mutations that were detected in a sixth of presumably sporadic PNET. In addition, four subgroups of somatic alterations were defined according to the function of the mutated genes: chromatin remodeling, deoxyribonucleic acid (DNA) repair, mTOR signaling pathway, and telomere maintenance (9). These data further strengthen our knowledge on the high rate of ATRX and DAXX somatic mutations in PNET (10), supporting the role of alternate telomere lengthening in PNET tumorigenesis.
Given the growing number of treatment modalities available for unresectable NET, there is an increasing need for clinical tools that will enable the performance of precision medicine, by tailoring the treatment based on the molecular characteristics of each specific patient. The advances in high-throughput sequencing enable cost-efficacious and timely somatic DNA analysis for detecting driver mutations. This has in turn enabled great progress in mutation-targeted therapy in oncology in general (11–14), but still not in the management of NET.
In the current study, we analyzed data from a phase II mutation-targeted therapy in patients with unresectable well-differentiated GEP-NET, conducted in the National Cancer Institute. We detected driver somatic mutations using a panel of tumor-suppressor genes and oncogenes, and defined somatic mutations that were potentially actionable based on current knowledge and available drugs.
METHODS
This was a monocentric Phase II clinical trial (NCT02315625) of patients with nonresectable well-differentiated locally advanced and/or metastatic GEP-NET. Patients included were ≥18 years of age, with disease progression within 18 months before inclusion. Tumor tissue samples resected during surgery or obtained during tumor biopsy among patients that were not surgical candidates, were used for extracting somatic DNA. The patient’s tumor DNA was sequenced and compared with germline DNA extracted from peripheral blood lymphocytes, saliva, and/or histologically normal tissue adjacent to the tumor.
Investigations on human subjects were performed after approval by an institutional review board. Written informed consent was obtained from all study participants.
DNA Sequencing
The OncoVAR-NET assay was designed for GEP-NET tissue by hybrid capture sequencing to validate somatic mutations for 500 known tumor suppressor genes and oncogenes, including 15 target genes known to be associated with NET. Mutations in genes associated with the mTOR pathway (PTEN, PIK3CA, AKT1, MTOR, VHL, TSC1, TSC2, and NF1) were defined as actionable, and patients harboring these mutations were assigned to the everolimus study arm. Patients with mutations in other target genes (TP53, MEN1, FLT3, PDGFRA, ATM, KIT, and ATRX), or patients with no somatic mutation identified, were assigned to the sunitinib treatment arm. In the current analysis we report the extended gene panel sequencing results, while the analysis of the target genes and their clinical relevance will be analyzed after gathering a longer follow-up.
The sequencing methods were as previously described (15). Briefly, DNA was extracted using Qiagen Blood and Tissue DNA Extraction Kit (Qiagen Sciences, Inc., Germantown, MD), fragmented on a Covaris S1 sonicator (Covaris, Inc., Woburn, MA), followed by end-repair and phosphorylation. Blunt fragments were adenylated, ligated to Illumina Y-adapters (Illumina, Inc., San Diego, CA) and polymerase chain reaction (PCR)-amplified. Bait hybridization proceeded for 48 hours, followed by recovery of captured exome fragments by PCR. Captured exomes were sequenced on an Illumina HiSeq 2000 (Illumina).
The sequencing yielded a mean of 39.5 million reads per sample, of them 99.7% were mapped to target. The mean coverage per sample was 257×, with a minimal coverage of 2×, 20×, 50×, and 100×, in 98%, 93%, 84%, and 69% of the targets, respectively.
Bioinformatic Analysis
Data processing followed the “Best Practices” work-flow recommended by the Broad Institute (16). Sequencing reads were aligned to the human genome sequence (GRCh37), by the Burrows-Wheeler Aligner (17). The local realignment of sequencing reads was performed using the GATK suite from Broad Institute and duplicated reads were marked using Picard tools (18). Tumor samples were compared with normal DNA samples, using the somatic variant callers Strelka and Mutect-2 for single nucleotide variants and short insertion and deletions. Structural variants (>50 base pairs) were not reported in the current analysis due to low reliability when based on targeted sequencing data. Variant annotation and risk prediction were performed based on the National Center for Biotechnology Information dbSNP database using SnpEff (19).
Variants were further analyzed using an in-house developed R-based pipeline. Tumor DNA mutations were included only if they had an allele frequency of <1% in the ExAC database (Broad Institute, Cambridge, MA), were nonsynonymous, and had a minimal coverage of 30× for the reference germline DNA, 30× for the tumor DNA, and a variant allele fraction of 20% or more (Fig. 1). Oncoplot was produced using the Maftools package (20), based on data produced by the Mutect-2 variant caller, and the variants genomic distribution figure was produced using CRAVAT: cancer-related analysis of variants toolkit (21).
Fig. 1.
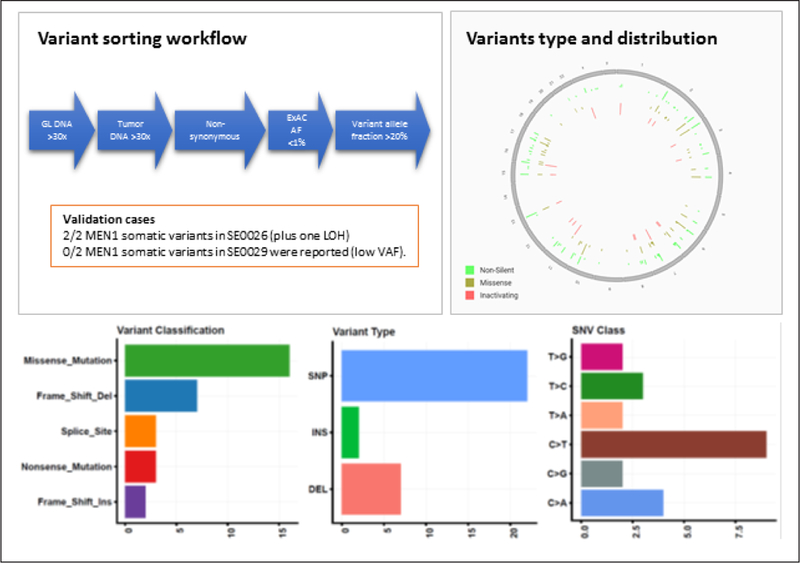
Variant sorting pipeline, variant genomic distribution, and characteristics. In order to avoid false positive calling of variants, stringent thresholds were used. We considered only variants with high coverage of both germline and tumor deoxyribonucleic acid (DNA) sequencing, and with a low frequency in the general population according to the ExAC database. The cutoff stringency is demonstrated by the sorting out of the MEN1 variants in patient SE0029, due to low variant allele frequency (VAF). The homogenous variant distribution throughout the genome, the high rate of missense mutation and single nucleotide polymorphisms (SNP), and the highest rate of nucleotide transition rather than transversion, all expected in this type of analysis, support the validity of the analysis. Patients SE0026 and SE0029 have germline MEN1 pathogenic variant. DEL = deletion; GL = germline; INS = insertion; LOH = loss of heterozygosity.
Analysis of copy number (CN) alteration based on the sequencing data, was performed on Nexus Copy Number 9.0 (BioDiscovery, Inc., El Segundo, CA). CN alteration was defined using a cutoff of 25% for differences between groups, and a P value threshold of <.05.
Analysis of Potential Actionable Genes
Driver somatic mutations were any variants with a high sequencing quality and coverage that were validated through direct inspection of the BAM file, since all of the genes sequenced were known oncogenes or tumor-suppressor genes. The list of genes in which driver somatic mutations were detected was compared with the OncoKB database (22) to identify potential actionable genes for GEP-NET. The evidence for the gene actionability were subgrouped according to the OncoKB classification into 4 levels: approved by the Food and Drug Administration (FDA; level 1), used in standard care (level 2), clinical evidence exists (level 3) and biological evidence exists (level 4).
Clinical Protocol
Patients were assigned to sunitinib or everolimus based on their somatic/germline mutations profile. Patients who had disease progression on either sunitinib or everolimus crossed over to the other drug. Treatment was continued until disease progression, unacceptable toxicity, or consent withdrawal. The full study protocol was reported previously (14).
RESULTS
The current analysis included 30 patients with tumor DNA sequencing available (mean age 53.7 ± 12.3 years, women). Fourteen of the patients harbored jejuno-ileal NET, 8 patients had pancreatic NET, 2 patients had duodenal NET, 2 patients had gastric NET, 1 patient had rectal NET, and 3 patients had metastatic NET of unknown primary site. A total of 37 lesions were sequenced (4 patients had multiple lesions sequenced). Twenty-six patients had sporadic NET, 3 patients had multiple endocrine neoplasia syndrome type 1 (MEN-1), and 1 had Cowden syndrome.
Actionable Mutations
Onco VAR-NET sequence data was analyzed for 30 patients: 5/26 (19.2%) of the patients with sporadic NET had a somatic mutation in one of the 15 target genes. However, only 2 of them (2/26, 7.7%) were actionable mutations per the current study definition: a somatic PTEN in a patient with PNET, and a somatic NF1 mutation in a patient with small-intestine NET (SINET; Table 1). Among patients with hereditary NET, all 3 patients with MEN-1 had either loss of heterozygosity (LOH) or compound heterozygosity for MEN1 in the tumor DNA, 1 patient had somatic mutations in ATRX in 2 lesions (patient SE0029), and 1 patient had a somatic VHL mutation in addition to MEN1 LOH (patient SE0018) (23).
Table 1.
List of all Patients Included in the Clinical Study (n = 33) Including Thirty Patients Who Underwent Targeted Germline and Somatic Mutation Analysis by OncoVAR-NET
| Patient number |
Syndrome | Primary lesion |
Grade | Germline mutation |
Lesion number |
Somatic mutation data | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Hugo symbol |
HGVS.c | HGVS.p | Target gene |
Potentially actionable mutation |
||||||
| P3 | Unknowna | G2 | None | 1 | Chr2 | SF3B1 | c,1998G>C | p.Lys666Asn | |||
| 1 | Chr7 | TRRAP | c.3013C>T | p.Hisl005Tyr | |||||||
| P4 | PNET | G3 | None | ||||||||
| SE0001 | GNET | G2 | None | 1 | Chr1 | ARID1A | c.2402delG | p.Gly801fs | |||
| 1 | Chr11 | MEN1 | c.1542_1564del | p.514_522del | Yes | ||||||
| SE0002 | Cowden | PNET | Gl | PTEN c.697C>T; p.R233b |
1 | Chr10 | PTEN | LOH | |||
| SE0003 | SINET | G2 | None | 1 | ChrX | USP9X | c.4003C>T | p.Glnl335b | |||
| SE0004 | PNETb | Gl | None | 1 | Chr10 | PTEN | c.491_492+4delAGGTAA | p.Lysl64fs | Yes | ||
| 2 | Chr13 | IRS2 | c.2707A>G | p.Ser903Gly | |||||||
| SE0005 | SINET | Gl | None | ||||||||
| SE0006 | SINET | Gl | None | 1 | Chr1 | AKT3 | c. 1164–4A>G | ||||
| SE0007 | SINET | NA | None | ||||||||
| SE0010 | SINET | NA | None | ||||||||
| SE0011 | PNET | G2 | None | ||||||||
| SE0012 | RNET | G2 | None | 1 | Chr1 | PLEKHG5 | c.758C>T | p.Pro253Leu | |||
| 1 | Chr8 | FGFR1 | c,1731C>G | p.Asn577Lys | Yes | ||||||
| 1 | ChrX | USP9X | c.4071_4077delCTTTACT | p.Thrl359fs | |||||||
| SE0013 | SINET | G2 | None | 1 | Chr7 | MET | c.818C>A | p.Thr273Asn | Yes | ||
| 1 | Chr16 | CREBBP | c.953C>T | p.Ser318Leu | |||||||
| SE0014 | PNET | G2 | None | 1 | Chr3 | RAF1 | c.478C>T | p.Leul60Phe | |||
| SE0015 | SINET | Gl | None | ||||||||
| SE0016 | SINET | Gl | None | 1 | Chr11 | KMT2A | c.385T>G | p.Phel29Val | |||
| SE0017 | PNET | NA | None | ||||||||
| SE0018 | MENI | DNET | Gl | MEN1 c.402delC; p.Phel34fs |
1 | Chr3 | VHL | c.529A>T | p.Argl77b | Yes | |
| 1 | Chr11 | MEN1 | LOH | ||||||||
| SE0019 | Unknown | Gl | None | ||||||||
| SE0020 | SINET | G2 | None | ||||||||
| SE0021 | SINET | G2 | None | ||||||||
| SE0022 | SINET | NA | None | 1 | Chr2 | ERCC3 | c,1306G>T | p.Gly436Cys | |||
| 1 | Chr18 | DCC | c.4111+6G>T | ||||||||
| SE0023 | SINET | Gl | None | ||||||||
| SE0024 | GNET | G2 | None | 1 | Chr3 | PBRM1 | c. 1134delC | p.Phe379fs | |||
| 1 | Chr18 | DCC | c,1779A>C | p.Glu593Asp | |||||||
| SE0025 | Missing data |
NA | Normal reference |
||||||||
| SE0026 | MEN1 | DNET | NA | MEN1 c.307delC; p.Leul03fs |
1 | Chr11 | MEN1 | c.1110delT | p.Phe370fs | Yes | |
| LN | 2 | Chr16 | CBFB | c.481C>T | p.Arg161Trp | ||||||
| 2 | Chr11 | MEN1 | LOH | ||||||||
| DNET | 3 | Chr11 | MEN1 | c. 1321T>A | p.Trp441Arg | Yes | |||||
| SE0027 | PNET | G2 | None | 1 | Chr7 | EGFR | c.560–8C>T | Yes | |||
| 1 | Chr13 | FOXO1 | c,1379G>T | p.Gly460Val | |||||||
| 1 | Chr11 | ATM | c.3661T>C | p.Trp1221Arg | Yes | ||||||
| SE0028 | SINET | NA | None | 1 | ChrX | TBX22 | c.350C>T | p.Ala117Val | |||
| Liver metastasis |
NA | ||||||||||
| SE0029 | MEN1 | PNET | G2 | MEN1 c.536A>G; p.E179G |
1 | ChrX | ATRX | c.6560_6570dupTTCGAGTTGTT | p.Asp2191fs | Yes | |
| 1 | Chr11 | MEN1 | LOH | ||||||||
| Gastric LN | 2 | Chr3 | SETD2 | c.2533T>C | p.Ser845Pro | Yes | |||||
| 2 | Chr11 | MEN1 | LOH | ||||||||
| Liver met | 3 | None | |||||||||
| Duodenal LN |
4 | None | |||||||||
| SE0030 | Unknown | Gl | None | Insufficient tumor DNA | |||||||
| SE0031 | None | 1 | Chr16 | TRAF7 | c.622delC | p.Arg208fs | |||||
| 1 | Chr11 | MEN1 | c.669+lG>C | Yes | |||||||
| SE0032 | None | 1 | Chr2 | LRP1B | c.6481C>T | p.Arg2161b | |||||
| 1 | Chr12 | CDKN1B | c.285dupC | p.Lys96fs | |||||||
| 1 | Chr17 | NF1 | c,1294delG | p.Val432fs | Yes | ||||||
| 1010012 | MEN1 | PNET | NA | NA | 1 | Insufficient tumor DNA | |||||
Abbreviations: DNA = deoxyribonucleic acid; DNET = duodenal neuroendocrine tumor; GNET = gastric neuroendocrine tumor; LN = lymph node; LOH = loss of heterozygosity; MEN1 = multiple endocrine neoplasia type 1; MET = metastases; NA = not applicable; NET = neuroendocrine tumor; PNET = pancreatic neuroendocrine tumor; RNET = rectal neuroendocrine tumor; SINET = small intestine neuroendocrine tumor.
Unknown: primary tumor was not located
In two samples of the same slide, the pathogenic variant was detected in one.
Nontarget Genes Analysis
The variants detected by the OncoVAR-NET assay, including variant sorting workflow, variant characteristics, and genomic distribution are detailed in Figure 1. Driver somatic mutations were identified in 18/30 patients (21/37 lesions) and mutation analysis identified 31 variants in these tumors (Fig. 2). Two tumor suppressor genes were found to be mutated in 2 different patients. DCC, located on chromosome (Chr) 18, had 2 nonsynonymous low-intermediate risk variants: a splice region variant, and a missense mutation in patients with gastric and SINET, respectively. USP9X, a gene involved in ubiquitination associated with pancreatic adenocarcinoma (24), had 2 high risk variants: nonsense and frameshift mutations, in patients with sporadic rectal and SINET, respectively.
Fig. 2.
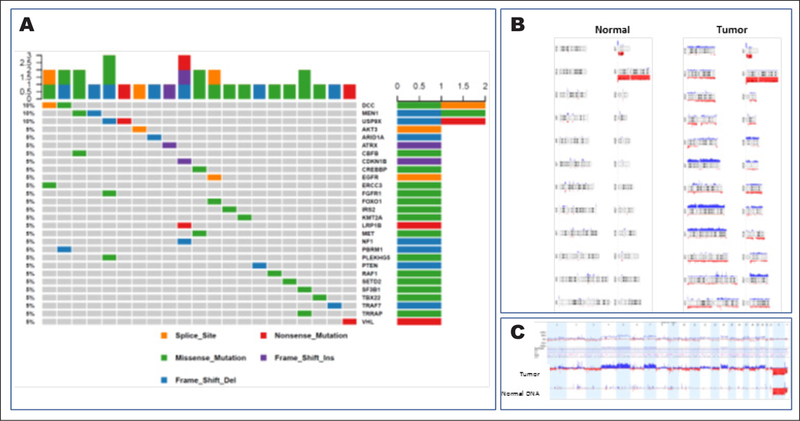
Oncoplot of somatic variants identified in patients with locally advanced and/or metastatic gastro-enteropancreatic neuroendocrine tumors. The oncoplot describes the pathogenic variants detected in 21/37 tumors sequenced by the Mutect variant caller, in 18/30 patients included in the deoxyribo-nucleic acid (DNA) sequencing analysis. Each column represents one tumor. The three genes at the top were mutated in two tumors. A, Two genes with pathogenic variants detected here were rarely reported previously in the context of neuroendocrine tumors: DCC and USP9X. B, Normal DNA has no copy-number (CN) alterations, whereas various regions of CN gain (blue) or loss (red) were detected in the tumor DNA. C, Subtraction analysis of CN alterations between the tumor and the germline DNA demonstrated copy number gain in chromosomes 4 and 5, and copy number loss in chromosome 18.
Copy-Number Alterations
CN alterations were found in 11/16 tumors (10/12 patients) in which no driver somatic mutation was detected. Comparison between CN of these tumors and the corresponding normal DNA revealed CN loss in Chr 18 (25% difference; P<.05), CN gain in Chr 5, and a trend of Chr 13 loss (P<.1). CN losses in Chr 18 were more common in patients without driver mutations compared to those with driver mutations with borderline statistical significance (P<.1).
Tumor Genetic Heterogeneity
Four patients had multiple tumor DNA analyzed. In one patient (SE004), two samples of the same slide were analyzed. In one, a somatic deleterious PTEN mutation was detected, whereas in the other no pathogenic variant was found. In three other patients, multiple lesions were analyzed (2, 3 and 4 lesions, each). Among those patients, intertumor heterogeneity was detected in each patient (Table 1).
In addition to the heterogeneity in terms of tumor DNA variants, patient SE0029 had intertumoral heterogeneous CN alterations, and this was concordant with the somatic variant intertumor heterogeneity detected in the lesions (Fig. 3).
Fig. 3.

Genetic heterogeneity between various lesions of one patient with a germline mutation in the MEN1 gene, reflected both in deoxyribonucleic acid (DNA) sequencing and copy number alteration patterns. Germline DNA, extracted from saliva, showing heterogenous MEN1 mutation. The unaffected allele is lost in the pancreatic neuroendocrine tumor (PNET) and the gastric lymph node (LN). In these two lesions, pathogenic variants were detected in the ATRX gene (insertion) and the SETD2 gene (missense), respectively. In addition, an identical copy-number alteration signature is demonstrated for these two lesions. All together, these findings demonstrate the clonality of these two lesions. The other lesions, liver metastasis and duodenal LN, show only the MEN1 heterogenous variant, which may point toward an undetected driver mutation or epimutation, but show again the clonality. The heterogeneity between the two pairs of tumors is also evident.
Potential Novel Actionable Genes for GEP-NET
Thirty-three somatic driver mutations were detected in a total of 30 genes. Two genes (PTEN and NF1) are associated with the mTOR pathway and defined as actionable genes in the current study. Both were included in the OncoKB database as level 4 actionable genes for multiple tumors (22). Three additional genes in which pathogenic variants were detected in the current analysis were either FDA-approved or used in clinical practice as actionable genes: EGFR was approved by the FDA as an actionable gene for non-small cell lung cancer (NSCLC; level 1), FGFR1 is currently being studied as a potential actionable gene for squamous cell carcinoma of the lung (level 3), and MET is used in clinical practice as an actionable gene in NSCLC and renal cell carcinoma (level 2).
DISCUSSION
In the current analysis we report a very low rate of actionable mutations in locally advanced and metastatic GEP-NET for targeted therapy. In addition, we detected driver somatic variants in nontarget genes using mutation analysis for 500 oncogenes and tumor-suppressor genes. We also found a higher rate of CN alterations among patients with no driver mutation detected, compared to patients in which a mutation was found.
CN alterations in patients in which no driver mutation was detected included Chr 18 CN loss and Chr 5 CN gain. Interestingly, Chr 18 CN loss was previously reported in patients with SINET (25), was associated with CDKN1B mutations together with CN gain of Chr 4, 5, 14, and 20 (26), and was associated with poor clinical outcome (8). Our data is novel for the following 2 reasons: (1), we detect-ed these alterations in patients harboring PNETs whereas previous reports were only on patients with SINETs, and (2), the CN alterations were more frequent among patients with no driver somatic mutations detected.
The low rate of actionable mutations, namely those associated with the mTOR pathway, was found in GEP-NET of various anatomic locations in the current analysis. Our prospective data supports a previous report, that 14% of somatic mutations in PNETs are associated with the mTOR pathway (10). Importantly, our findings extend our knowledge on the rates of actionable mutations to other nonpancreatic gastro-intestinal NETs.
The detection of driver somatic mutations in the DCC gene is also interesting, as it is a tumor suppressor gene that is located on Chr 18. This locus, which was found to have a high rate of CN loss in SINETs, encodes other genes such as SMAD2, SMAD4, and CABLES (27). Previous studies have shown loss of heterozygosity in DCC in gastric NET (28) and in poorly differentiated GEP-NET (27), and also a low expression level in SINET with Chr 18 CN loss (29). However, here we report DCC pathogenic variants in patients with well-differentiated gastric NET and SINET, strongly suggesting that this gene has a causative role in NET tumorigenesis.
In this study, we had an opportunity to analyze tumor genomic heterogeneity in a small number of patients with multiple lesions. We found different somatic variants in tumors from the same patient, and in another patient heterogeneity in terms of CN alterations. The heterogeneity that we found in a patient with MEN-1 may also stem from multiple primary tumors, as supported by the clonality that was found (Fig. 3). Tumor genomic heterogeneity in lung cancer was suggested as a fuel for tumor evolution and was associated with an adverse clinical outcome (30). This finding is important considering the limited data on tumor heterogeneity in NET (31), and due to its possible clinical importance. Tumor genomic heterogeneity may explain an unpredicted response to treatment of different tumors in the same patient. In addition, our findings emphasize the limited clinical utility of single lesion sampling, both in terms of histopathologic evaluation, and in terms of genetic analysis.
In our analysis, no driver genetic alterations were detected in 3 lesions (1 lesion of patient SE0017 and in 2/4 lesions of patient SE0029), neither pathogenic variant, nor CN alteration. Since the OncoVAR-NET assay consists only of oncogenes and tumor suppressor genes, we set high coverage and allele fraction thresholds for defining a variant as pathogenic, as described in Fig. 1. The desired increase in the specificity of variant detection was associated with decreased sensitivity, explaining the nondetection of somatic mutations in the PTEN and MEN1 genes in patients with a known germline mutation in those genes (Table 1).
Altered DNA methylation is a known driver for tumorigenesis in several cancer types, and leads to methylation-targeted therapies (32). Hence, since NET are relatively “genetically silent” tumors with a low mutation rate, epigenetic analysis is of special interest. Moreover, since epigenetic alterations may lead to mTOR pathway alterations, our findings may suggest using methylome alterations as actionable factors in treatment selection for patients with NET, in addition to mutation targeted therapy.
Our study has several limitations. First, it is based on a small sample size, but GEP-NET are relatively rare. Second, CN analysis based next-generation sequencing is not a direct analysis of CN alterations; however, several investigators have demonstrated that such an approach is accurate for large CN alterations (33,34). Third, a retest concordance analysis would have improved our results reliability, but was not possible due to technical issues. We reduced the risk for false-positive results by setting high thresholds for the variant calling. Finally, structural variant analysis is not reported due to its limited accuracy in a targeted gene sequencing assay.
CONCLUSION
In conclusion, genome-wide sequencing may identify novel actionable genes and lead to the identification of novel target genes. Tumor genomic heterogeneity in NET should be taken into consideration both in terms of sampling mistakes and tumor response to various treatment modalities. Future studies should thoroughly characterize GEP-NET, to further reduce the “dark matter” in GEP-NET genetic drivers.
ACKNOWLEDGMENT
The study was funded by the intramural research program of the National Cancer Institute.
Abbreviations:
- Chr
chromosome
- CN
copy number
- DNA
deoxyribonucleic acid
- FDA
Food and Drug Administration
- GEP
gastro-enteropancreatic
- MEN-1
multiple endocrine neoplasia syndrome type 1
- mTOR
mammalian target of rapamycin
- NET
neuroendocrine tumor
- PFS
progression-free survival
- PNET
pancreatic neuroendocrine tumors
- SINET
small-intestine neuroendocrine tumor
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
REFERENCES
- 1.Dasari A, Shen C, Halperin D, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017; 3:1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. [DOI] [PubMed] [Google Scholar]
- 3.Caplin ME, Pavel M, Ćwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–233. [DOI] [PubMed] [Google Scholar]
- 4.Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebocontrolled, phase 3 study. Lancet. 2016;387:968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. [DOI] [PubMed] [Google Scholar]
- 7.Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karpathakis A, Dibra H, Pipinikas C, et al. Prognostic impact of novel molecular subtypes of small intestinal neuroendocrine tumor. Clin Cancer Res. 2016;22:250–258. [DOI] [PubMed] [Google Scholar]
- 9.Scarpa A, Chang DK, Nones K, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. [DOI] [PubMed] [Google Scholar]
- 10.Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF V600 -mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464–472. [DOI] [PubMed] [Google Scholar]
- 12.Cabel L, Fuerea A, Lacroix L, et al. Efficacy of histology-agnostic and molecularly-driven HER2 inhibitors for refractory cancers. Oncotarget. 2018;9:9741–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross JS, Fakih M, Ali SM, et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer. 2018;124:1358–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neychev V, Steinberg SM, Cottle-Delisle C, et al. Mutation-targeted therapy with sunitinib or everolimus in patients with advanced low-grade or intermediate-grade neuroendocrine tumours of the gastrointestinal tract and pancreas with or without cytoreductive surgery: protocol for a phase II clinical trial. BMJ Open. 2015;5:e008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg L, Gough SM, Lee F, et al. Somatic mutations in murine models of leukemia and lymphoma: disease specificity and clinical relevance. Genes Chromosomes Cancer. 2017;56:472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ Data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picard Tools. Available at: http://picard.sourceforge.net. Accessed January 1, 2019.
- 19.Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayakonda A, Koeffler HP. Maftools: efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. BioRxiv. Available at: https://www.biorxiv.org/content/10.1101/052662v1.article-info. Accessed January 1, 2019. [Google Scholar]
- 21.Douville C, Carter H, Kim R, et al. CRAVAT: cancer-related analysis of variants toolkit. Bioinformatics. 2013;29:647–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shell J, Patel D, Powers A, et al. Somatic VHL mutation in a patient with MEN1-associated metastatic pancreatic neuroen-docrine tumor responding to sunitinib treatment: a case report. J Endocr Soc. 2017;1:1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murtaza M, Jolly LA, Gecz J, Wood SA. La FAM fatale: USP9X in development and disease. Cell Mol Life Sci. 2015;72: 2075–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banck MS, Kanwar R, Kulkarni AA, et al. The genomic land-scape of small intestine neuroendocrine tumors. J Clin Invest. 2013;123:2502–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francis JM, Kiezun A, Ramos AH, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013;45:1483–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pizzi S, Azzoni C, Bassi D, Bottarelli L, Milione M, Bordi C. Genetic alterations in poorly differentiated endocrine carcinomas of the gastrointestinal tract. Cancer. 2003;98:1273–1282. [DOI] [PubMed] [Google Scholar]
- 28.Rindi G, Villanacci V, Ubiali A. Biological and molecular aspects of gastroenteropancreatic neuroendocrine tumors. Digestion. 2000;62(suppl 1):19–26. [DOI] [PubMed] [Google Scholar]
- 29.Nieser M, Henopp T, Brix J, et al. Loss of chromosome 18 in neuroendocrine tumors of the small intestine: the enigma remains. Neuroendocrinology. 2017;104:302–312. [DOI] [PubMed] [Google Scholar]
- 30.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376:2109–2121. [DOI] [PubMed] [Google Scholar]
- 31.Walter D, Harter PN, Battke F, et al. Genetic heterogeneity of primary lesion and metastasis in small intestine neuroendocrine tumors. Sci Rep. 2018;8:3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanwal R, Gupta K, Gupta S. Cancer Epigenetics: An Introduction. In: Verma M, ed. Methods in Molecular Biology (Methods and Protocols). New York, NY: Humana Press; 2015: 3–25. [DOI] [PubMed] [Google Scholar]
- 33.Tattini L, D’Aurizio R, Magi A. Detection of genomic structural variants from next-generation sequencing data. Front Bioeng Biotechnol. 2015;3:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bansal V, Dorn C, Grunert M, et al. Outlier-based identification of copy number variations using targeted resequencing in a small cohort of patients with Tetralogy of Fallot. PLoS One. 2014;9:e85375. [DOI] [PMC free article] [PubMed] [Google Scholar]