

Abstract
Duchenne muscular dystrophy (DMD) is an X-linked genetic disorder characterized by progressive muscular weakness because of the loss of dystrophin. Extracellular Ca2+ flows into the cytoplasm through membrane tears in dystrophin-deficient myofibers, which leads to muscle contracture and necrosis. Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) takes up cytosolic Ca2+ into the sarcoplasmic reticulum, but its activity is decreased in dystrophic muscle. Here, we show that an allosteric SERCA activator, CDN1163, ameliorates dystrophic phenotypes in dystrophin-deficient mdx mice. The administration of CDN1163 prevented exercise-induced muscular damage and restored mitochondrial function. In addition, treatment with CDN1163 for 7 weeks enhanced muscular strength and reduced muscular degeneration and fibrosis in mdx mice. Our findings provide preclinical proof-of-concept evidence that pharmacological activation of SERCA could be a promising therapeutic strategy for DMD. Moreover, CDN1163 improved muscular strength surprisingly in wild-type mice, which may pave the new way for the treatment of muscular dysfunction.
Introduction
Duchenne muscular dystrophy (DMD) is a severe, progressive, X-linked muscle-wasting disease caused by mutations in the DMD gene (1). Recent studies estimated the incidence of DMD to be 1:3500–1:10 000 newborn males (2,3). Clinical symptoms of DMD appear at 2–3 years of age, and the loss of independent ambulation occurs by age 11–13 years. The mean age at death due to respiratory and cardiac complications without ventilator support is ~19 years (4).
The DMD gene encodes the dystrophin protein, which localizes under the sarcolemma and forms a complex with glycoproteins (the dystrophin-glycoprotein complex, DGC) at the sarcolemma, linking the extracellular matrix and cytoskeleton (5,6). In the absence of DGC, the sarcolemma is disrupted during muscular contraction/relaxation and extracellular Ca2+ flows into the cytoplasm through membrane tears (7). In addition, Iwata et al. reported that a stretch-activated channel, TRPV2, is translocated to the plasma membrane in dystrophic muscle fibers and allows abnormal Ca2+ entry (8). More recent studies further showed that stretch- and store-operated Ca2+ channels are activated in dystrophic muscle and elevate cytoplasmic Ca2+ levels (9,10).
Abnormally elevated cytosolic Ca2+ causes muscle contracture, mitochondrial dysfunction, activation of proteases, and finally muscular degeneration and necrosis (11). In addition, calcium regulation in the sarcoplasmic reticulum (SR) in the dystrophic muscle of mdx mice, an animal model of DMD, was shown to be chronically impaired (12,13). Ryanodine receptor 1 (RyR1), a Ca2+-release channel of the SR, was shown to be leaky due to S-nitrosylation by aberrantly activated nitric oxide synthase. FKBP12 (calstabin1) binds and stabilizes RyR1 but was reported to be reduced from the RyR1 complex when RyR1 is S-nitrosylated (12). Furthermore, the activity of Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) was shown to be decreased by upregulation of sarcolipin (SLN), an inhibitor of SERCA (13), in dystrophic muscle. SERCA removes >70% of Ca2+ from the cytosol (14); therefore, the dysfunction of this Ca2+ pump of the SR further increases Ca2+ levels in the cytoplasm (11). These reports suggest that the restoration of Ca2+ regulation of the SR to reduce the cytosolic Ca2+ level might be a good therapeutic strategy for DMD.
Previous reports showed that SERCA overexpression by a transgene (15–18) or enhancement through Hsp72 upregulation (19) rescued dystrophic phenotypes in mdx mice, sgcd−/− mice, and dystrophin- and utrophin-deficient dko mice; however, whether pharmacological activation of SERCA is beneficial to DMD phenotypes is still unknown. CDN1163 is an allosteric SERCA activator that was identified in high-throughput screening assays and increases the activity at saturating (Ca2+) (Vmax) (20,21). The therapeutic effects of CDN1163 have been shown in various animal models of oxidative stress-related diseases, such as 6-OHDA-lesioned rats as a model of Parkinson’s disease (22), APP/PS1 mice as a model of Alzheimer’s disease (23), ob/ob mice as a model of diabetes (24), SOD1-deficient mice (25) and aging mice (26).
SERCA1a is the major isoform in fast-twitch skeletal muscle, and SERCA2a is expressed in cardiac, smooth, and slow-twitch skeletal muscles. Importantly, CDN1163 is a pan-activator for SERCA and is not isoform-specific (22). Recently, Lindsay et al. (27) reported that the incubation of isolated mdx muscle with CDN1163 for 30 min attenuated the loss of eccentric contraction-induced force ex vivo, suggesting that CDN1163 may be applicable for DMD treatment.
As a next step, we treated dystrophin-deficient mdx mice by the administration of CDN1163. In this study, we demonstrated the reduction of cytosolic Ca2+ level in mdx myotubes and whole tibialis anterior (TA) muscles isolated from mdx mice by pharmacological activation of SERCA. We found that the administration of CDN1163 for 1 week restored mitochondrial function and prevented exercise-induced muscular damage in mdx mice. We further revealed that treatment with CDN1163 for 7 weeks mitigated DMD-associated pathology and improved muscular strength in mdx mice. Our findings provide preclinical proof-of-concept evidence that pharmacological activation of SERCA ameliorates the dystrophic phenotypes of DMD model mice and could be a promising therapeutic strategy for DMD.
Results
Administration of CDN1163 reduced cytosolic Ca2+ levels in vitro and ex vivo
CDN1163 increases SERCA activity in vitro and in vivo in an allosteric manner (20,21,24). Importantly, a previous report showed that treatment of isolated mdx muscle with CDN1163 modestly attenuated loss of eccentric contraction-induced force ex vivo (27). Therefore, we first determined whether CDN1163 would reduce Ca2+ levels in the H2K-mdx cell line. The fluorescence intensity of Fluo-4 AM in H2K-mdx myotubes was significantly reduced after 30 min of incubation with CDN1163 (Fig. 1A and B). In whole TA muscle dissected from mdx mice, the signal intensity was also significantly reduced after 30 min of incubation with CDN1163 to the same level as that in wild-type C57BL/6J (BL6) mice (Fig. 1C and D). This result confirmed that CDN1163 reduced cytosolic Ca2+ levels in dystrophin-deficient mdx myofibers.
Figure 1 .
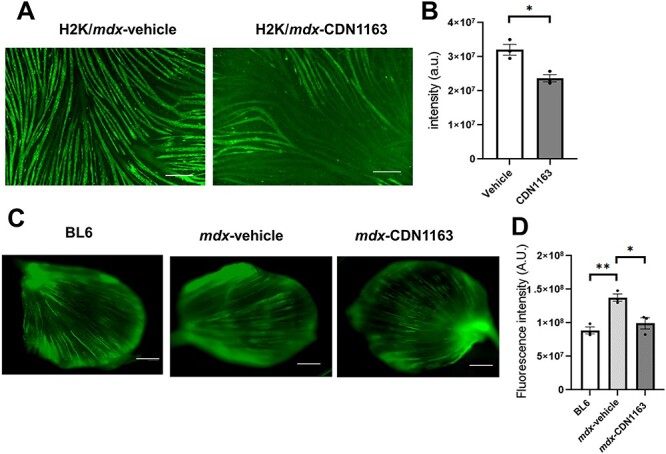
CDN1163 decreased cytosolic Ca2+ level in vitro and ex vivo. (A) Representative images showing cytosolic Ca2+ (Fluo-4 AM) of H2K-mdx myotubes established from C57BL10/mdx mice after 30 min of treatment with vehicle or CDN1163. Scale bar, 200 μm. (B) Quantitative analysis of Fluo-4 AM signal intensity in (A). n = 3. (C) Representative images showing cytosolic Ca2+ (Fluo-4 AM) in whole TA muscles dissected from wild-type C57BL/6J mice without treatment (BL6) or mdx mice after 30 min of treatment with vehicle (mdx-Vehicle) or CDN1163 (mdx-CDN1163). Muscles were incubated with Fluo-4 AM for 30 min. Scale bar, 1 mm. (D) Quantitative analysis of Fluo-4 AM signal intensity (n = 3 mice per group) in (C). *P < 0.01 by Student’s t-test (B) or ANOVA with the Tukey–Kramer test (D). Data are presented as the means ± SEMs.
Exercise-induced muscular damage was prevented by 1-week administration of CDN1163
Next, we daily injected CDN1163 (40 mg/kg) to mdx mice for 1 week (Fig. 2A). We selected this dose based on a previous report that it effectively restored muscle mass and force in Sod−/− mice without harmful side effects (25). A higher dose of CDN1163 was not applicable to mice, because CDN1163 is insoluble in an aqueous solution. Before histological analysis, we forced vehicle- and CDN1163-treated mdx mice to run for 60 min on a treadmill to induce muscular damage and then intraperitoneally injected Evans blue dye (EBD) as previously described (28). Rupture of the sarcolemma often causes uptake of this dye in dystrophin-deficient myofibers (29). As expected, 1 h of running on the treadmill caused a significant increase in EBD-positive fibers in the TA muscle of mdx mice (Fig. 2B and C). Importantly, the administration of CDN1163 significantly reduced EBD uptake into myofibers after treadmill exercise, indicating that CDN1163 protected dystrophic muscle against exercise-induced damage.
Figure 2 .
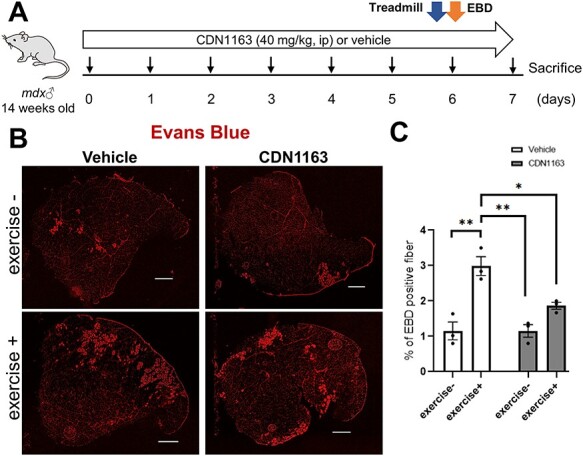
One-week administration of CDN1163 decreased the uptake of EBD by damaged myofibers after treadmill exercise load. (A) Experimental design of EBD injection and exercise load in 14-week-old male mdx mice administered vehicle or CDN1163. n = 3 mice/group. Just after treadmill exercise on day 6, EBD was injected into the peritoneal cavity. (B) Representative images of EBD uptake (red fluorescence) in the TA muscles of mdx mice treated with vehicle or CDN1163 with or without treadmill running (exercise + or −). Scale bar, 500 μm. (C) Percentages of EBD-positive fibers in the TA muscles in (B). Data are presented as the means ± SEMs. *P < 0.05, **P < 0.01 by ANOVA with the Tukey–Kramer test.
Mitochondrial respiratory function was restored by pharmacological SERCA activation
Sustained elevation of cytosolic Ca2+ levels was reported to cause mitochondrial dysfunction by the formation of mitochondrial permeability transition pores (mPTPs) (30,31). Previous reports showed that mitochondria isolated from dystrophic mice were swollen because of mPTPs, leading to myofiber necrosis (15,30). To test whether CDN1163 prevents mPTP formation, we daily administrated CDN1163 (40 mg/kg) to mdx mice. After 1 week, freshly isolated mitochondria from each group were assessed. The baseline absorbance of mitochondria isolated from vehicle-treated mdx mice tended to be lower than that of CDN1163-treated mdx mice or BL6 mice. This suggests that mdx mitochondria are swollen at the baseline, though the difference was not statistically significant (Fig. 3A). The maximal absorbance change with a high-Ca2+ solution (200 μm) for mitochondria isolated from the vehicle-treated mdx mouse muscles was significantly smaller than that from the BL6 mouse muscle. These results confirmed that mitochondria were swollen in mdx mouse muscles, as previously described (15,30). On the other hand, mitochondria isolated from mdx mice treated with CDN1163 showed a significant increase in the maximal absorbance with a high-Ca2+ solution (Fig. 3B), indicating that the administration of CDN1163 reversed the mitochondrial swelling in mdx mice.
Figure 3 .
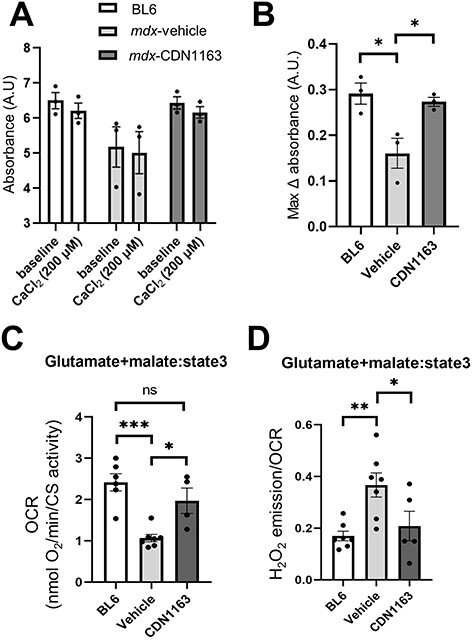
Pharmacological activation of SERCA restored mitochondrial swelling, OCR and ROS production in mdx mice. (A) Absorbance at 540 nm by mitochondria in response to an exogenous high-Ca2+ solution (200 μm CaCl2). After daily administration of CDN1163 to 14-week-old male mdx mice for 1 week, mitochondria were isolated from the TA and GC muscles. Absorbance was measured before and 10 min after Ca2+ addition. A decrease in absorbance indicated swelling of the mitochondria. (B) The maximal change in absorbance at 540 nm by mitochondria in response to high external Ca2+ solution. (C and D) The OCR (OCR) and ROS production in mitochondria isolated from BL6 and mdx mice treated with vehicle or CDN1163. Data are presented as the means ± SEMs. *P < 0.05, **P < 0.01, ***P < 0.001 by ANOVA with the Tukey–Kramer test.
Next, we measured the glutamate malate-supported oxygen consumption rate (OCR) and reactive oxygen species (ROS) production from mitochondria. Previous reports showed that cytosolic Ca2+ overload opens the mPTPs, leading to mitochondrial depolarization, oxidative stress, inhibition of mitochondrial ATP synthesis, and finally death of the myofibers in dystrophic mice (11,30–34). Mitochondria isolated from the gastrocnemius (GC) muscles of vehicle-treated mdx mice showed significantly lower OCR and higher production of ROS than those of BL6 mice (Fig. 3C and D). The administration of CDN1163 significantly increased OCR and reduced ROS production in mdx mice.
Administration of CDN1163 for 7 weeks decreased muscular degeneration and fibrosis in mdx mice
To know the effects of CDN1163 on dystrophic phenotypes, we injected the compound into mdx mice for 7 weeks (Fig. 4A). Six-week-old male mdx mice were randomly divided into two experimental groups (n = 7/per group): the vehicle-injected group and the CDN1163-treated group. CDN1163 was injected intraperitoneally at 40 mg/kg into mdx mice three times per week for 7 weeks. The administration of CDN1163 did not change the body weight or organ weight (brain, liver, kidneys and spleen) of mdx mice (Supplementary Material, Fig. S1A and B).
Figure 4 .
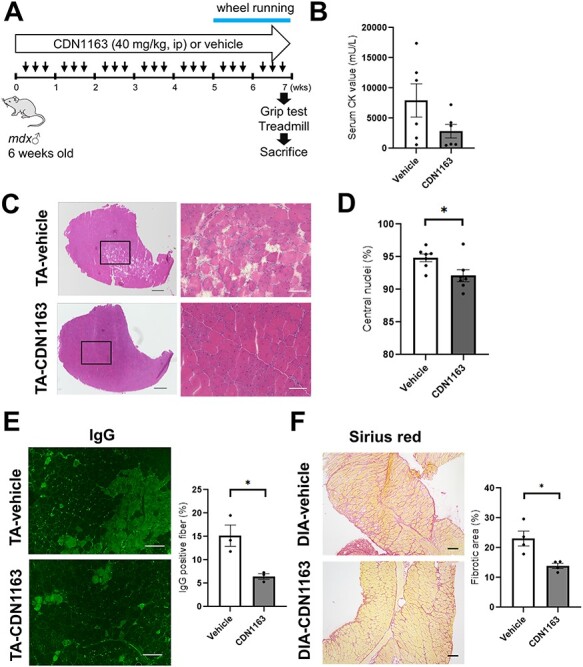
Pharmacological activation of SERCA decreased muscular degeneration and fibrosis in mdx mice. (A) Experimental design for CDN1163 administration. Male mdx mice (6-week-old, n = 7 per group) were randomly selected and treated with vehicle or CDN1163 (40 mg/kg BW) three times per week for 7 weeks. ip: intraperitoneal injection. (B) Quantitation of serum CK levels (n = 6 per group). (C) Representative H&E staining of TA muscles from mdx mice treated with vehicle (Vehicle-TA) or CDN1163 (CDN1163-TA) for 7 weeks. Scale bars, 500 μm (left) and 100 μm (right). (D) Percentage of myofibers with central nuclei (n = 6 mice per group). (E) Representative images of endogenous IgG detected with Alexa 488-labeled anti-mouse IgG antibody in TA muscles of vehicle-treated (Vehicle-TA) or CDN1163- treated mice (CDN1163-TA). Scale bar, 100 μm. Quantitative analysis of IgG positive fibers in TA muscle (right panel, n = 3 mice per group). (F) Representative images of Sirius red staining of the DIA muscles of vehicle-treated (Vehicle-DIA) or CDN1163-treated mice (CDN1163-DIA). Scale bar, 100 μm. Quantitative analysis of the fibrotic area in the DIA muscle (n = 4 mice per group). We injected DMSO and Tween-80 at a final concentration of 10% v/v each into all experimental mice in vehicle group. Data are presented as the means ± SEMs. *P < 0.05 by Student’s t-test.
Mdx mouse muscle originally shows hypertrophy, which is a compensatory effect because of muscular degeneration and subsequent activation of satellite cells to regenerate myofibers (18); however, neither muscular atrophy nor hypertrophy was observed in mdx muscle treated with CDN1163 compared with the vehicle-injected group (Supplementary Material, Fig. S1C). The serum creatine kinase (CK) level, which is an indirect marker of muscular damage, was reduced in treated mdx mice, although this difference was not statistically significant (Fig. 4B). A significant reduction in CK level was observed in 14-week-old mdx mice treated daily with CDN1163 for 16 days (Supplementary Material, Fig. S2A and B). On the other hand, a 16-day treatment of 3-week-old mdx mice tended to increase the CK level (Supplementary Material, Fig. S3A and B). Next, we examined whether CDN1163 administration ameliorated the histopathology in mdx mouse muscle. Hematoxylin and eosin (H&E) staining of the TA muscle showed a reduction in the number of necrotic fibers (Fig. 4C). The presence of centrally located nuclei in the mouse muscle indicates that the myofibers had been regenerated after damage. Importantly, the percentage of myofibers with centrally nucleated fibers was significantly decreased by CDN1163 treatment (Fig. 4D). The administration of CDN1163 also significantly reduced endogenous IgG accumulation in necrotic fibers (Fig. 4E). In addition, Sirius red staining showed that CDN1163 significantly reduced collagen deposits in diaphragm (DIA) muscles, which is one of the most severely compromised muscles in mdx mice (Fig. 4F). Amelioration of the dystrophic histopathology of mdx mice was also obtained by 16 days of treatment in 14-week-old mdx mice (Supplementary Material, Fig. S2C), whereas that of 3-week-old mdx mice was unchanged (Supplementary Material, Fig. S3C).
SERCA Ca2+-ATPase activity was enhanced by CDN1163 without a change in protein expression of SERCA
The expression level of SERCA1 (ATP2A1) in the GC muscle of mdx mice was the same as that of non-treated BL6 mice, and not changed by the administration of CDN1163 (Fig. 5A). SERCA2 (ATP2A2) expression was elevated in mdx mice compared with non-treated BL6 mice, but not affected by CDN1163 treatment (Fig. 5B). Importantly, the SERCA activity of mdx mice treated with CDN1163 for 7 weeks was increased by 50% compared with that of vehicle-treated mdx mice (Fig. 5C).
Figure 5 .
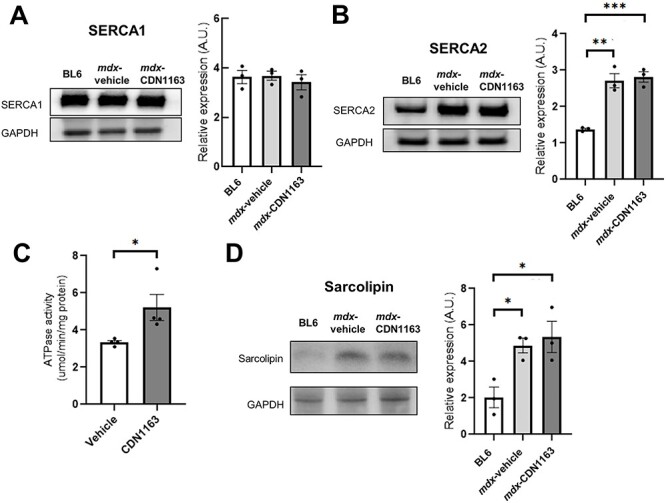
SERCA Ca2+-ATPase activity was enhanced by the administration of CDN1163. (A and B) Representative western blot analyses of SERCA1 (A) and SERCA2 (B) in GC muscles of non-treated BL6 and mdx mice treated with vehicle or CDN1163 for 7 weeks. Quantitation of the signal intensities of SERCA1 (A) and SERCA2 (B) are also shown. n = 3 mice per group. GAPDH was used as a loading control. (C) Ca2+-ATPase activity in enriched SR fractions isolated from the GC muscles of mdx mice treated with vehicle or CDN1163 for 7 weeks. n = 4 mice per group. (D) Western blot analysis and quantification of the signal intensities of SLN in the DIA muscles of non-treated BL6 and mdx mice treated with vehicle or CDN1163 for 7 weeks. n = 3 mice per group. GAPDH was used as a loading control. Data are presented as the means ± SEMs. *P < 0.05, **P < 0.01, ***P < 0.001 by ANOVA with the Tukey–Kramer test (A, B and D) or Student’s t-test (C).
We also examined the expression of SLN, which binds SERCA and inhibits its function. The protein level of SLN in the DIA muscle of mdx mice was upregulated, as previously described (13), but unchanged in mdx mice treated with CDN1163 (Fig. 5D), suggesting that CDN1163 directly activates SERCA function without regulating the protein expression of SERCA nor SLN.
A Ca2+ overload in dystrophic skeletal muscle is thought to activate Ca2+-dependent proteases, such as calpain and caspase-3, and contribute to muscular degeneration in DMD muscles (11). Therefore, we assessed calpain activity in mdx muscles after CDN1163 administration for 7 weeks. Calpain activity in GC muscles tended to be lower in CDN1163-treated mdx muscle than in vehicle-treated mdx muscle, but the effects were not large enough to explain the therapeutic effects of CDN1163 on the mdx dystrophic phenotype (Supplementary Material, Fig. S4A). Caspase-3 is also a key protease that leads to apoptosis. The protein level of caspase-3 was slightly but significantly upregulated in mdx muscle; however, it was not decreased by the administration of CDN1163 (Supplementary Material, Fig. S4B). Technically, however, it was difficult to accurately measure calpain activity in homogenized tissue samples, because artificial activation of calpains might occur. Therefore, we cannot exclude the possibility that CDN1163 changed calpain activity.
Pharmacological activation of SERCA improved in vivo exercise capacity and contraction force of isolated muscle ex vivo
We further assessed in vivo muscle function in mdx mice after 7 weeks of CDN1163 administration. CDN1163 significantly increased grip strength in mdx mice (Fig. 6A). Treadmill running time until exhaustion tended to be increased by the administration of CDN1163 (Fig. 6B). In the voluntary wheel running test, there was no difference in total activity between the vehicle-injected and CDN1163-treated groups (Fig. 6C). In addition, CDN1163 administration increased both the specific twitch and tetanic force of TA and GC muscles isolated from mdx mice (Fig. 6D). Furthermore, the improvement of grip strength in 14-week-old mdx mice and 3-week-old mdx mice was observed even after 16 days of daily treatment with CDN1163 (Supplementary Material, Figs S2D and S3D). Treadmill running time significantly increased in 3-week-old mdx mice, though there was no difference in 14-week-old mdx mice (Supplementary Material, Figs S2E and S3E).
Figure 6 .
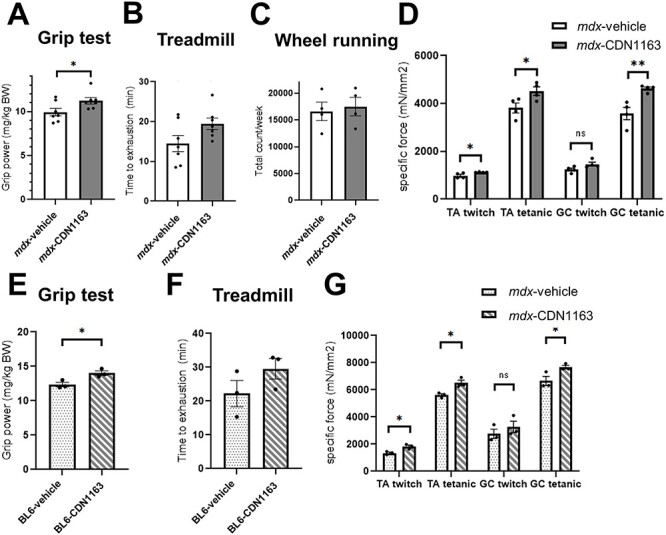
Pharmacological activation of SERCA increased muscular strength in mdx mice and C57BL/6J mice. (A and B) Grip strength measurement (A) and time to exhaustion in the treadmill test (B) in mdx mice treated with vehicle or CDN1163 for 7 weeks (n = 7 mice per group). (C) Voluntary wheel running test (n = 4 mice per group). The number of wheel revolutions was recorded over a week, and the total wheel count per week is shown. (D) Specific twitch and tetanic force of TA and GC muscles (n = 4 mice per group). (E and F) Grip strength measurement (E) and time to exhaustion in the treadmill test (F) in BL6 mice treated with vehicle or CDN1163 for 16 days (n = 3 mice per group). (G) Specific twitch and tetanic force of isolated TA and GC muscles of BL6 mice (n = 3 mice per group). We injected DMSO and Tween-80 at a final concentration of 10% v/v each into all experimental mice in vehicle group. Data are presented as the means ± SEMs. *P < 0.05, **P < 0.01, by Student’s t-test.
We also performed the same set of experiments using BL6 mice to investigate the effects of pharmacological activation of SERCA on muscular function in wild-type mice. Interestingly, the administration of CDN1163 (40 mg/kg) for 16 days significantly increased the grip strength and the specific muscle force even in BL6 mice, although treadmill running time did not change significantly (Fig. 6E–G). The performance of mdx mice treated with CDN1163 was lower than that of vehicle-injected BL6 mice (Fig. 6B, F, D and G).
Genes associated with inflammation were downregulated by CDN1163 administration
To further clarify the mechanisms by which CDN1163 ameliorated the dystrophic phenotypes of mdx mice, we examined the gene expression profiles using the TA muscles of 14-week-old mdx mice treated with vehicle or CDN1163 (10 mg/kg) daily for 16 days by RNA sequencing (RNA-seq). Many genes were differentially expressed in mdx mouse muscle compared with non-treated wild-type muscle (Fig. 7A). In contrast, only 40 genes were significantly altered of which 15 were upregulated and 25 were downregulated by CDN1163 treatment in mdx mice (FDR < 0.05 and log2-fold change > 0.5, Fig. 7B). We found that several genes associated with inflammation (Matrix metalloproteinase 9 (Mmp-9), Il-1β, Ptgs2, and CXC chemokine family) were downregulated by CDN1163. There was a significant difference in the expression of Mmp-9 (Fig. 7B and C). Mmp-9 is involved in tissue remodeling, inflammation, and interstitial fibrosis in many diseases (35,36). Importantly, Mmp-9 is upregulated in mdx mice, and the inhibition of Mmp-9 improves myopathy and myofiber regeneration in mdx mice (37).
Figure 7 .
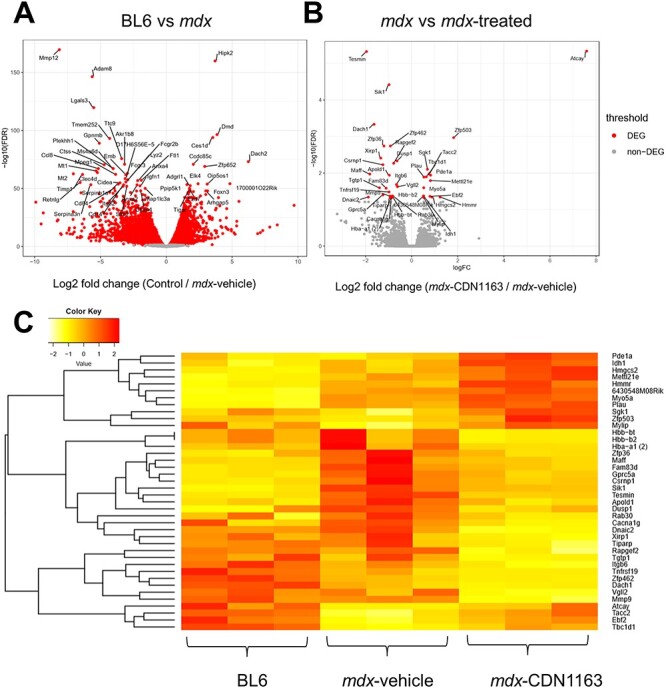
RNA-seq gene expression analysis of mdx mouse muscle after the administration of CDN1163 for 16 days. (A) Volcano plot showing differential expression of all genes between BL6 (non-treated) and vehicle-treated mdx mice. (B) Volcano plot showing differential expression of all genes between mdx mice treated with vehicle and CDN1163 (1.0 mg/ml in 10% DMSO/PBS, 10 mg/kg BW) for 16 days (14 weeks old, n = 3 per group). Top 50 DEGs with FDR < 0.05 and log2 fold change (log2FC) > 0.5 are indicated. (C) Heat map illustrates DEGs in non-treated BL6 mice, mdx mice treated with vehicle, and treated with CDN1163. The heat map is based on normalized expression levels of significantly changed genes with the Benjamini–Hochberg adjusted P-value < 0.05 between vehicle and CDN1163-treated mdx mice. Z-scores for the DEGs were transformed from the TMM value by using the gene filter package and visualized as a heat map by using the gplots package.
Increased cytosolic Ca2+ levels were reported to induce fast-to-slow fiber transformation (38–40). Dusp1 and Vgll2, which promote fast-to-slow fiber-type switching (41–43), were significantly downregulated; however, the expression of myosin heavy chain genes (Myh1, Myh2, Myh4, Myh7) was not significantly altered (data not shown). Furthermore, the percentage of type I and type IIa fibers in the GC/plantaris muscle of mdx mice treated with CDN1163 for 7 weeks were not different from those in vehicle-treated mdx mice (Supplementary Material, Fig. S5).
Discussion
In this study, we revealed that pharmacological activation of SERCA attenuated dystrophic phenotypes in mdx mice. We demonstrated that the administration of CDN1163 indeed decreased the cytosolic Ca2+ level in vitro and ex vivo. Notably, 1-week administration of CDN1163 reduced EBD uptake by damaged myofibers after exercise load. OCR and ROS production in isolated mitochondria were also restored after the treatment. Our findings suggest that the therapeutic effects of CDN1163 on mdx pathophysiology can partly be explained by the restoration of mitochondrial function, because previous reports showed that the mitochondrial dysfunction due to cytosolic Ca2+ overload in dystrophic mice is related to dystrophic phenotypes (11,30–34). In particular, recovered mitochondrial function might have positive effects on EBD uptake after exercise load because mitochondria are indispensable for cell membrane repair (44).
The expression of SLN, which binds SERCA and inhibits its function, did not change in mdx mice treated with CDN1163, suggesting that CDN1163 directly activated SERCA function without regulating the protein expression of SLN. A previous report showed that allosteric SERCA activators directly perturbed the structure of SERCA and altered SERCA-phospholamban FRET as a result (20). In this study, we did not examine the effects of CDN1163 on the interaction between SERCA and SLN, but it is possible that CDN1163 changed the binding of SLN to SERCA.
Treatment with CDN1163 for 7 weeks successfully decreased muscular degeneration. We also found that CDN1163 significantly reduced fibrosis in the DIA muscle, which represents one of the most severely compromised muscles in mdx mice. These findings indicate that pharmacological activation of SERCA may be a promising therapeutic strategy for DMD. In terms of elevation of the serum CK level in 3-week-old mdx mice treated with CDN1163, there is a possibility that increased muscular strength due to CDN1163 treatment rather leads to muscular damage in 3-week-old mdx mice because they are at the severe early necrosis-degeneration stage of mdx mice (45).
Grip strength and contraction force of isolated muscles were increased by CDN1163 treatment. Interestingly, the administration of CDN1163 significantly improved the muscular function in wild-type mice also. This suggests that SERCA activation has the potential for enhancing the muscular function, even in wild-type muscle, by increasing the speed of the return to baseline of a Ca2+ transient. In this study, we dissolved CDN1163 in DMSO, which itself has anti-inflammatory effects (46). Nevertheless, the therapeutic effects of CDN1163 were observed in each experiment even with the anti-inflammatory effects.
Previous studies showed that transgene-mediated SERCA overexpression or indirect activation through Hsp72 induction ameliorated dystrophic phenotypes in mdx and Sgcd−/− mice (15–19). Mázala et al. (18) showed that transgenic SERCA overexpression reduced the weights of the TA, soleus, plantaris, and extensor digitorum longus muscles in mdx mice. Muscular hypertrophy is commonly observed in mdx mice, and the authors considered that upregulated SERCA1 rescued pathological hypertrophy in mdx mice. In the current study, however, the administration of CDN1163 did not reduce muscular weight, even with a 50% increase in SERCA Ca2+-ATPase activity. Similarly, the therapeutic effects of CDN1163 were milder than those in SERCA1-transgenic mice. Previous reports showed a 2–3-fold increase in SR Ca2+-ATPase activity and SR Ca2+ uptake in SERCA1-overexpressing mice (15,18). It is likely that the high levels of transgenic SERCA1 expression from the neonatal period had much stronger effects on mdx mice than pharmacological activation of SERCA.
To further clarify the mechanisms by which CDN1163 decreased muscular damage in mdx mice, we assessed the gene expression profiles by RNA-seq. Unexpectedly, a very small percentage of the genes were differentially expressed between vehicle- and CDN1163-treated mdx muscles; however, several genes associated with inflammation were downregulated by CDN1163, indicating that CDN1163 treatment prevented muscular degeneration. Because CDN1163 activates SERCA2b which is expressed in macrophages, improvement of the function and viability of macrophages may also contribute to the downregulation of the genes associated with inflammation. The expression of genes related to fast-to-slow fiber-type conversion was also significantly downregulated by the administration of CDN1163, but the conversion of fiber type was not evident in our study. Recently, Qaisar et al (26). showed that CDN1163 treatment of 16-month-old male mice for 10 months mitigated age-related muscular atrophy and weakness. Interestingly, ingenuity upstream regulator analysis suggested that CDN1163 activated PGC1-α, UCP1, HSF1, and APMK signaling and suppressed MEF2C and p38 MAPK signaling. Based on the findings, the authors speculate that CDN1163 mitigated age-related muscular atrophy and weakness, at least in part by restoring energy production and muscular regeneration activity. In dystrophic mdx mice, however, RNA-seq analysis did not suggest that CDN1163 altered these signaling pathways. This could be because we treated young mdx mice, which actively regenerate injured skeletal muscle. Unexpectedly, the expression level of many genes in mdx muscle treated with CDN1163 did not show intermediate values between vehicle-treated mdx mice and wild-type mice. This might be because CDN1163 did not completely suppress muscular degeneration. Many myofibers were still in active degeneration/regeneration cycles, and regenerating myofibers might show different gene expression patterns from mature intact myofibers. Therefore, it may look like an unfavorable gene expression pattern.
Another therapeutic strategy focuses on RyR1, a Ca2+-release channel on the SR. Compounds that stabilize the binding of RyR1 to calstabin are called rycals, and a previous report showed that rycals reduced RyR1 Ca2+ leakage and improved dystrophic phenotypes in mdx mice (12). The combination of CDN1163 and rycals might have synergistic therapeutic effects on dystrophic phenotypes.
CDN1163 would have effects on cardiac muscle which express SERCA2a. Further investigations, including assessments of the safety of CDN1163 and its therapeutic effects on the heart, ideally using other dystrophic murine models with severe dystrophic phenotypes, such as the double knock-out mdx mouse or the dystrophic rat, remain to be performed.
Materials and Methods
Animals
C57BL/6J (BL6) mice were purchased from Nihon Crea (Tokyo, Japan). mdx mice with a BL6 background were a generous gift from Dr Sasaoka (Niigata University). The experimental mice were 6–14 weeks old. Only male mice were used in the study. The mice were bred and kept in plastic cages in a 12:12-h light–dark cycle at the specific pathogen-free animal facility at the National Institute of Neuroscience, NCNP. The mice were allowed free access to food and drinking water. Body weight was measured weekly during the experiment. Our experimental design and procedures were based on the three R principle (replacement, reduction and refinement) and approved by the Experimental Animal Care and Use Committee of the National Institute of Neuroscience of the NCNP (approval ID: 2018041). The mice were randomly assigned to experimental groups.
CDN1163
CDN1163, an allosteric SERCA activator, was purchased from Sigma-Aldrich (St. Louis, MO). CDN1163 was reported to be selective against > 160 off-targets (23). CDN1163 showed an acceptable pharmacokinetic profile in mice (20,21), and membranes showed high permeability to CDN1163. Vehicle (10% DMSO, 10% Tween-80 in PBS) or CDN1163 (1.0 mg/ml stock solution, final dose of 40 mg/kg) was intraperitoneally injected into mdx mice.
Cell culture and intracellular Ca2+ measurements
Murine H2K-mdx myoblasts (47) were seeded in plates coated with matrigel (Corning, Corning, NY, USA) at 8 × 104 cells/well in growth medium. After 2 days, they were differentiated using 5% horse serum-containing differentiation medium. Intracellular Ca2+ was monitored using Fluo-4 AM as previously described (48). Briefly, whole fresh TA muscles and H2K-mdx myotubes were incubated in buffer containing 4 μm Fluo-4 AM (Dojindo, Kumamoto, Japan), 140 mm NaCl, 5 mm KCl, 2.5 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 10 mm glucose (pH 7.4) combined with vehicle or CDN1163 (100 μm) for 30 min at room temperature. The fluorescence intensity was evaluated using a BZ-X810 fluorescence microscope (Keyence, Osaka, Japan).
Serum CK level
Mice were anesthetized with isoflurane, and blood was taken via the inferior vena cava and kept at room temperature overnight. The serum was then collected from the blood by centrifugation at 3000 × g for 10 min. The serum level of CK, an indicator of muscular damage, was assayed using the Fuji Drychem system (Fuji Film Medical Co. Ltd, Tokyo, Japan) according to the manufacturer’s instructions.
Measurement of muscle contraction force ex vivo
The twitch and tetanic force of TA and GC muscles were measured using an in vitro muscle test system (Aurora Scientific Inc., Aurora, ON, Canada) as previously described (49). Briefly, the entire TA and GC muscles were dissected and kept in a buffer containing 137 mm NaCl, 24 mm NaHCO3, 5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 11 mm glucose, 1 mm NaH2PO4 (pH 7.4), and 0.025 mm d-tubocurarine at 25°C with continuous perfusion with 95% O2–5% CO2 gas (50,51). After a muscle was placed in the bath, a single 0.4-msec stimulation pulse was used to generate a twitch force, and the length of the muscle was carefully adjusted to determine the optimal muscular length (L0) to obtain the maximal twitch force. Tetanic force was obtained by stimulation of the muscle at L0 for a period of 300 msec (TA) or 600 msec (GC) with a series of 3-msec pulses. The force developed during trains of stimulation pulses (83 Hz) was recorded. Specific twitch and tetanic forces were calculated by normalizing to the physiological cross-sectional area, which was determined by the ratio of muscular weight to L0 and the density of mammalian skeletal muscle.
Grip strength measurements
The grip strength of the combined forelimbs and hind limbs was recorded using a grip strength meter (Muromachi Kikai Co., Ltd; model MK-380M). The grip strength meter was positioned horizontally, and the mouse was held by its tail and allowed to securely grip the metal mesh. After the mouse obtained a solid grasp of the metal mesh, the mouse was pulled backward parallel to the device. The force that was applied to the mesh at the time of release was recorded as the peak grip strength. The measurement was repeated three times, and the average force was determined for each mouse. Grip strength values were normalized to the body weight (kg) of each mouse (mg/kg BW).
Treadmill running test
The mice were placed in individual lanes of an electrically driven 10-lane treadmill (MK-680, Muromachi, Tokyo, Japan) and acclimated to the treadmill at a speed of 5 m/min for 30 min twice a week before the experiment. The test began at a speed of 5 m/min for 5 min and gradually increased by 1 m/min every minute until exhaustion. Exhaustion was defined as the time when the mouse could no longer run despite repeated gentle nudges (52).
Voluntary exercise test
Voluntary exercise in an individual cage for 1 week was recorded using a running wheel (SW-15, Melquest) as described previously (53). The mice were kept in plastic cages with a running wheel for 2 weeks. The first week was used to acclimate the mice to the wheel. Voluntary exercise over the second week was quantified as the average of the total number of wheel revolutions per day.
Tissue preparation
Mice were sacrificed by cervical dislocation, and the body was weighed. The TA, GC, EDL, SOL, DIA, and H muscles and organs (brain, liver, kidneys and spleen) were collected using standard dissection methods, and their weights were measured. Muscles and organs were then quickly frozen in isopentane cooled by liquid nitrogen for histological analysis and RNA and protein isolation. Frozen samples were stored at −80°C.
Histology and IgG staining
The TA and DIA muscles were sectioned at 8 μm using a CryoStar NX70 cryostat (Thermo Fisher) and stained with H&E or Sirius red. IgG staining was performed to detect degenerating fibers as previously described (54). In brief, the cryosections were preincubated with 5% BSA in PBS for 1 h, followed by incubation with Alexa 488-conjugated goat anti-mouse IgG antibody (A-11029, Invitrogen, Carlsbad, CA). Fluorescence images were obtained using a BZ-X810 fluorescence microscope (Keyence).
Acute exercise load and EBD uptake
After the daily administration of vehicle or CDN1163 (40 mg/kg) for 1 week, muscular damage was induced by a modified treadmill protocol as previously described (26). Briefly, the exercise load began at a speed of 5 m/min for 5 min and gradually increased by 1 m/min every minute until the speed reached 10 m/min. The mice were kept on the treadmill for 30 min under repeated gentle nudges. After a 10-min interval, the session was repeated. All mice were able to carry out the test until the end of the experiment. After treadmill exercise, the mice were injected intraperitoneally with EBD (10 mg/ml, 0.1 ml/10 g BW). The next day, the mice were sacrificed, and their GC muscles were cut into cryosections to observe EBD uptake (red fluorescence) by degenerating fibers. Images were taken with a BZ-X810 fluorescence microscope (Keyence, Osaka, Japan).
Western blot analysis
The protein was extracted with a sample buffer containing 2% SDS, 15% glycerol, 125 mm Tris–HCl, 1 mm dithiothreitol, and cOmplete protease inhibitor cocktail (11 873 580 001; Roche, Indianapolis, IN, USA). The protein lysate was then denatured at 100°C for 5 min and centrifuged at 15 000 rpm for 5 min. After the supernatant was collected, the protein concentration was determined using a protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with BSA as a standard. The protein lysate was separated on an SDS-polyacrylamide gel and electrically transferred to polyvinylidene difluoride membranes (Millipore, Darmstadt, Germany). The blots were then incubated with primary antibodies at 4°C overnight. The next day, the membranes were incubated with secondary antibodies. Signals were detected using ECL Prime Western Blotting Detection Reagent (GE Healthcare, Buckinghamshire, UK; #RPN2232) and a ChemiDoc MP imaging system (Bio-Rad, Hercules, CA, USA). Data were analyzed using Image Lab 6.0 (Bio-Rad).
Antibodies
Antibodies against SERCA1 (#12293) and SERCA2a (#4388) were purchased from Cell Signaling Technologies (Beverly, MA, USA). Anti-caspase-3 antibody (#67341A) was obtained from BD Pharmingen (San Diego, CA, USA). BA-F8 (type I MHC), SC-71 (type IIa MHC), and BF-F3 (type IIb MHC) were obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA, USA). An antibody against SLN was a kind gift from Dr Muthu Periasamy (University of Central Florida). HRP-conjugated secondary antibodies (anti-rabbit IgG, horseradish peroxidase-linked F(ab′)2 fragment; #NA9340 V) were purchased from GE Healthcare. HRP-conjugated rabbit anti-goat IgG (H + L) antibody (#611620) was obtained from Invitrogen (Carlsbad, CA, USA).
Mitochondrial swelling assay
After the daily administration of vehicle or CDN1163 (40 mg/kg) for 1 week, mitochondria were isolated from the GC muscles of mdx mice. Light scattering from the isolated mitochondria was measured using 200 μg of mitochondria suspended in 400 μL of isolation buffer, and 200 μm CaCl2 was used to induce mitochondrial swelling and shrinking as previously described (15). The change in absorbance at 540 nm for 10 min after the addition of CaCl2 was measured by a plate reader (BioTek, Winooski, VT, USA).
Isolation of mitochondria and measurement of OCR
After daily administration of the vehicle or CDN1163 (40 mg/kg) for 1 week, GC muscles were collected. The mitochondrial fraction was obtained by differential centrifugation as previously described (55). Immediately after tissue collection, GC muscles were placed in buffer [PBS, 10 mm EDTA (349–01885, Dojindo Molecular Technologies, Kumamoto, Japan), pH 7.4]. Tissues were well-minced using scissors, supplemented with 0.025% trypsin (209–16 941, Fujifilm Wako Pure Chemical Corporation), and then incubated for 5 min on ice. To remove the trypsin, tissue suspensions were centrifuged at 200 g for 5 min at 4°C, and then supernatants were discarded. The tissue pellets were resuspended in buffer [50 mm MOPS (194 837, BM Bio Japan, Tokyo, Japan), 100 mm KCl (163–03545, Fujifilm Wako Pure Chemical Corp.), 1 mm EGTA (342–01314, Dojindo Molecular Technologies), 5 mm MgSO4 (83585–0401, Junsei Chemical, Tokyo, Japan) and 2.0 g/L BSA (015–21 274, Fujifilm Wako Pure Chemical), pH 7.1] and were homogenized by 20 strokes using a Potter glass homogenizer. The tissue homogenates were centrifuged at 500 g for 10 min at 4°C, and the supernatants derived were collected. The supernatants were centrifuged at 10000 g for 10 min at 4°C to obtain mitochondrial pellets. The mitochondrial pellets were washed in buffer (50 mm MOPS, 100 mm KCl, 1 mm EGTA and 5 mm MgSO4) and resuspended in buffer (10 mm Tris, 30 mm KCl, 10 mm KH2PO4, 5 mm MgCl2, 1 mm EGTA and 2.5 g/L BSA, pH 7.2). The protein concentration of each was determined using the BCA method and then adjusted to 2.0 mg/ml.
The mitochondrial OCR was measured as previously described with minor modifications (55). Briefly, freshly isolated mitochondria (20 μg) were incubated in a reaction buffer (105 mm potassium-MES, 10 mm Tris, 30 mm KCl, 10 mm KH2PO4, 5 mm MgCl2, 1 mm EGTA, 2.5 g/L BSA, pH 7.2). Complex I-driven state III (ATP synthesis coupled) respirations were stimulated by adding 10 mm glutamate (070–00502, Fujifilm Wako Pure Chemical Corp.), 2 mm malate (138–07512, Fujifilm Wako Pure Chemical Corp.) and 2.5 mm ADP (306–50 501, Fujifilm Wako Pure Chemical Corp.). Mitochondrial oxygen consumption was measured using a Tecan Spark multimode plate reader (Spark 20 M, Tecan, Männedorf, Switzerland) with an oxygen-monitoring 96-well microplate (OP96C, PreScan Precision Sensing, Regensburg, Germany) (excitation (Ex): 560 (20) nm; emission (Em): 670 (25) nm). The relative change in fluorescence per minute was measured, and this was converted to the OCR according to the manufacturer’s instructions. To normalize the efficiency of mitochondrial isolation and purification, citrate synthase (CS) activity in isolated mitochondria was measured as follows: 1 μg of mitochondrial protein was mixed with reaction buffer [100 μm DTNB (346–08551, Dojindo Molecular Technologies), 300 μm acetyl-CoA (00546–54; Nacalai Tesque, Kyoto, Japan), 50 μm oxaloacetate (25804–81, Nacalai Tesque)] in a 96-well plate. Absorbance changes at 412 nm/min were calculated. The OCR was normalized to CS activity.
Mitochondrial ROS emission kinetics
Mitochondrial ROS emission was measured as previously described with minor modifications (56). Briefly, mitochondria were incubated in a black 96-well plate with mitochondrial respiration buffer and 5 μm Amplex Red (Thermo Fisher Scientific, Waltham, MA, USA), 1 U/ml Horseradish peroxidase (169-10791, Fujifilm Wako Pure Chemical Corp.), and 5 U/ml superoxide dismutase (192-11281, Fujifilm Wako Pure Chemical Corp.). The kinetics of ROS emission were assessed under state 3 respiratory conditions through the addition of 2.5 mm ADP, 10 mm glutamate and 2 mm malate. The relative changes in fluorescence per minute were measured using a fluorescence plate reader (Spark 20 M, Tecan) (Ex: 560 [20] nm; Em: 620 [20] nm). The relative change in fluorescence per minute was measured and normalized to OCR.
Calpain activity assay
Calpain activity in GC muscles was assessed using a calpain activity assay kit (ab65308, Abcam, Cambridge, UK) according to the manufacturer’s protocol. In brief, muscles were lysed in an extraction buffer and centrifuged at 20 000 × g. The supernatant was collected, and the protein concentration was determined using a protein assay (Bio-Rad Laboratories, Inc.). Protein (200 μg) was incubated in a 96-well plate with calpain as a substrate for 1 h at 37°C. The fluorescence intensity (Ex/Em = 400/505 nm) of the samples was measured using a Synergy plate reader (BioTek). Active calpain 1 and calpain inhibitor were used as positive and negative controls, respectively.
ATPase activity assay
Microsomes were prepared as previously described (24). In brief, fresh GC muscles were homogenized in lysis buffer containing 0.25 M sucrose, 2 mm Tris–HCl (pH 7.4), 1 mm DTT and cOmplete protease inhibitor cocktail (Roche). The microsome fraction was obtained after a series of centrifugation steps (800 × g for 15 min, 6000 × g for 15 min, 100 000 × g for 1 h) and resuspended in 250 mm sucrose. Ca2+-dependent ATP hydrolysis by the microsomal preparations was measured using a colorimetric ATPase Assay Kit (Novus Biologicals, Littleton, CO, USA) following the manufacturer’s instructions. One microgram of the microsome fraction was incubated in reaction buffer containing 50 mm Tris (pH 7.5), 2.5 mm MgCl2 and 0.5 mm ATP at room temperature for 1 h. PiColorLock Gold reagent and accelerator were added to the solution to stop the reaction, followed by mixing with a stabilizer. Absorbance at 650 nm was measured and normalized to the total protein concentration. ATPase activity (μmol/min/mg protein) was calculated from the concentration of free phosphate (μm) determined by the standard curve.
RNA isolation and RNA-seq
Total RNA was extracted from the TA muscles of BL6 mice and mdx mice with vehicle and CDN1163 (1.0 mg/ml in 10% DMSO/PBS, 10 mg/kg BW) for 16 days (14-week-old, n = 3 per group) using an RNeasy Mini Kit (Qiagen, Hilden, Germany), and RNA-seq was performed by Takara Bio (lot ID: PR1433). The library was prepared by using the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed using an Illumina NovaSeq 6000 (150 bp, paired-end). Read alignment was performed by STAR (ver. 2.5.2b). Data analysis was performed by using Genedata Profiler Genome (ver. 10.1.15a). The gene expression level was normalized by fragments per kilobase of exon per million reads mapped. Differential expression analysis was performed using edgeR (ver. 3.28.1) (57, 58). Genes with the Benjamini–Hochberg adjusted P-values < 0.05 were identified and defined as differentially expressed genes (DEGs). The Z-scores of the DEGs were transformed from the trimmed mean of the M (TMM) value by using the gene filter (ver. 1.68.0) package and visualized as a heat map by using the gplots (ver. 3.0.3) package. All the codes used in this study are available from the corresponding author.
Statistics
Data were analyzed and plotted using GraphPad Prism 8 software. All values are expressed as the means ± standard error of mean (SEMs). Statistical differences were assessed by unpaired Student’s t-test or one-way analysis of variance (ANOVA) with Tukey–Kramer post hoc analysis. Differences with probabilities <5% (*P < 0.05), 1% (**P < 0.01), or 0.1% (***P < 0.001) were considered to be statistically significant.
Supplementary Material
Acknowledgements
We are grateful to Dr Muthu Periasamy for providing SLN-specific antibody. We also thank Dr Terence Partridge, Dr Jennifer Morgan and Dr James Novak for providing the H2K-mdx cell line. We gratefully acknowledge the work of past and present members of our laboratory and Dr Katsuya Miyake.
Conflict of Interest statement. None declared.
Funding
This study was supported by (1) the Japan Agency for Medical AQ7 Research and Development (AMED, grant number 19bm0804005-h0103 to Y.M.-S. and 20lm0203086h0002 to Y.A.); (2) Grant-in-aid for Scientific Research (C) (19K075190001) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan; and (3) Neurological and Psychiatric Disorders of the National Center of Neurology and Psychiatry intramural research grants 28-6 to S.T. (28-6) and Y.M.-S. (30-9).
References
- 1. Hoffman, E.P., Brown, R.H. and Kunkel, L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 2. Birnkrant, D.J., Bushby, K., Bann, C.M., Apkon, S.D., Blackwell, A., Brumbaugh, D., Case, L.E., Clemens, P.R., Hadjiyannakis, S., Pandya, S. et al. (2018) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol., 17, 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mah, J.K., Korngut, L., Dykeman, J., Day, L., Pringsheim, T. and Jette, N. (2014) A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord., 24, 482–491. [DOI] [PubMed] [Google Scholar]
- 4. Passamano, L., Taglia, A., Palladino, A., Viggiano, E., D'Ambrosio, P., Scutifero, M., Rosaria Cecio, M., Torre, V., D.E.L., Picillo, E. et al. (2012) Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta. Myol., 31, 121–125. [PMC free article] [PubMed] [Google Scholar]
- 5. Arahata, K., Ishiura, S., Ishiguro, T., Tsukahara, T., Suhara, Y., Eguchi, C., Ishihara, T., Nonaka, I., Ozawa, E. and Sugita, H. (1988) Immunostaining of skeletal and cardiac muscle surface membrane with antibody against Duchenne muscular dystrophy peptide. Nature, 333, 861–863. [DOI] [PubMed] [Google Scholar]
- 6. Ozawa, E. (2010) Our trails and trials in the subsarcolemmal cytoskeleton network and muscular dystrophy researches in the dystrophin era. Proc. Jpn. Acad. Ser B. Phys. Biol. Sci., 86, 798–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsumura, K. and Campbell, K.P. (1994) Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle Nerve, 17, 2–15. [DOI] [PubMed] [Google Scholar]
- 8. Iwata, Y., Katanosaka, Y., Arai, Y., Komamura, K., Miyatake, K. and Shigekawa, M. (2003) A novel mechanism of myocyte degeneration involving the Ca2+-permeable growth factor-regulated channel. J. Cell Biol., 161, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruegg, U.T. (2013) Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr. Opin. Neurol., 26, 577–584. [DOI] [PubMed] [Google Scholar]
- 10. Zhao, X., Moloughney, J.G., Zhang, S., Komazaki, S. and Weisleder, N. (2012) Orai1 mediates exacerbated Ca2+ entry in dystrophic skeletal muscle. PLoS One, 7, e49862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen, D.G., Whitehead, N.P. and Froehner, S.C. (2016) Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev., 96, 253–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bellinger, A.M., Reiken, S., Carlson, C., Mongillo, M., Liu, X., Rothman, L., Matecki, S., Lacampagne, A. and Marks, A.R. (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med., 15, 5–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneider, J.S., Shanmugam, M., Gonzalez, J.P., Lopez, H., Gordan, R., Fraidenraich, D. and Babu, G.J. (2013) Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J. Muscle Res. Cell Motil., 34, 349–356. [DOI] [PubMed] [Google Scholar]
- 14. Periasamy, M. and Kalyanasundaram, A. (2007) SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve, 35, 430–442. [DOI] [PubMed] [Google Scholar]
- 15. Goonasekera, S.A., Lam, C.K., Millay, D.P., Sargent, M.A., Hajjar, R.J., Kranias, E.G. and Molkentin, J.D. (2011) Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest., 121, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morine, K.J., Sleeper, M.M., Barton, E.R. and Sweeney, H.L. (2010) Overexpression of SERCA1a in the mdx diaphragm reduces susceptibility to contraction-induced damage. Hum. Gene Ther., 21, 1735–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shin, J.H., Bostick, B., Yue, Y., Hajjar, R. and Duan, D. (2011) SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J. Transl. Med., 9, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mázala, D.A., Pratt, S., Chen, D., Molkentin, J.D., Lovering, R.M. and Chin, E.R. (2015) SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell. Physiol., 308, C699–C709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gehrig, S.M., van der Poel, C., Sayer, T.A., Schertzer, J.D., Henstridge, D.C., Church, J.E., Lamon, S., Russell, A.P., Davies, K.E., Febbraio, M.A. et al. (2012) Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature, 484, 394–398. [DOI] [PubMed] [Google Scholar]
- 20. Cornea, R.L., Gruber, S.J., Lockamy, E.L., Muretta, J.M., Jin, D., Chen, J., Dahl, R., Bartfai, T., Zsebo, K.M., Gillispie, G.D. et al. (2013) High-throughput FRET assay yields allosteric SERCA activators. J. Biomol. Screen., 18, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gruber, S.J., Cornea, R.L., Li, J., Peterson, K.C., Schaaf, T.M., Gillispie, G.D., Dahl, R., Zsebo, K.M., Robia, S.L. and Thomas, D.D. (2014) Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J. Biomol. Screen., 19, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dahl, R. (2017) A new target for Parkinson’s disease: small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg. Med. Chem., 25, 53–57. [DOI] [PubMed] [Google Scholar]
- 23. Krajnak, K. and Dahl, R. (2018) A new target for Alzheimer's disease: A small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorg. Med. Chem., 28, 1591–1594. [DOI] [PubMed] [Google Scholar]
- 24. Kang, S., Dahl, R., Hsieh, W., Shin, A., Zsebo, K.M., Buettner, C., Hajjar, R.J. and Lebeche, D. (2016) Small Molecular Allosteric Activator of the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) Attenuates diabetes and metabolic disorders. J. Biol. Chem., 291, 5185–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qaisar, R., Bhaskaran, S., Ranjit, R., Sataranatarajan, K., Premkumar, P., Huseman, K. and Van Remmen, H. (2019) Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol., 20, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qaisar, R., Pharaoh, G., Bhaskaran, S., Xu, H., Ranjit, R., Bian, J., Ahn, B., Georgescu, C., Wren, J.D. and Van Remmen, H. (2020) Restoration of sarcoplasmic reticulum Ca2+ATPase (SERCA) activity prevents age-related muscle atrophy and weakness in mice. Int. J. Mol. Sci., 22, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindsay, A., Baumann, C.W., Rebbeck, R.T., Yuen, S.L., Southern, W.M., Hodges, J.S., Cornea, R.L., Thomas, D.D., Ervasti, J.M. and Lowe, D.A. (2020) Mechanical factors tune the sensitivity of mdx muscle to eccentric strength loss and its protection by antioxidant and calcium modulators. Skelet. Muscle, 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Terrill, J.R., Radley-Crabb, H.G., Grounds, M.D. and Arthur, P.G. (2012) N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord., 22, 427–434. [DOI] [PubMed] [Google Scholar]
- 29. Matsuda, R., Nishikawa, A. and Tanaka, H. (1995) Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J. Biochem., 118, 959–964. [DOI] [PubMed] [Google Scholar]
- 30. Millay, D.P., Sargent, M.A., Osinska, H., Baines, C.P., Barton, E.R., Vuagniaux, G., Sweeney, H.L., Robbins, J. and Molkentin, J.D. (2008) Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med., 14, 442–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reutenauer, J., Dorchies, O.M., Patthey-Vuadens, O., Vuagniaux, G. and Ruegg, U.T. (2008) Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol., 155, 574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hajnóczky, G., Davies, E. and Madesh, M. (2003) Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun., 304, 445–454. [DOI] [PubMed] [Google Scholar]
- 33. Ascah, A., Khairallah, M., Daussin, F., Bourcier-Lucas, C., Godin, R., Allen, B.G., Petrof, B.J., Des Rosiers, C. and Burelle, Y. (2011) Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol., 300, H144–H153. [DOI] [PubMed] [Google Scholar]
- 34. Percival, J.M., Siegel, M.P., Knowels, G. and Marcinek, D.J. (2013) Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet., 22, 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Page-McCaw, A., Ewald, A.J. and Werb, Z. (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell. Biol., 8, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vu, T.H. and Werb, Z. (2000) Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev., 14, 2123–2133. [DOI] [PubMed] [Google Scholar]
- 37. Li, H., Mittal, A., Makonchuk, D.Y., Bhatnagar, S. and Kumar, A. (2009) Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet., 18, 2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sreter, F.A., Lopez, J.R., Alamo, L., Mabuchi, K. and Gergely, J. (1987) Changes in intracellular ionized Ca concentration associated with muscle fiber type transformation. Am. J. Phys., 253, C296–C300. [DOI] [PubMed] [Google Scholar]
- 39. Chin, E.R., Olson, E.N., Richardson, J.A., Yang, Q., Humphries, C., Shelton, J.M., Wu, H., Zhu, W., Bassel-Duby, R. and Williams, R.S. (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev., 12, 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allen, D.L. and Leinwand, L.A. (2002) Intracellular calcium and myosin isoform transitions. Calcineurin and calcium-calmodulin kinase pathways regulate preferential activation of the IIa myosin heavy chain promoter. J. Biol. Chem., 77, 45323–45330. [DOI] [PubMed] [Google Scholar]
- 41. Shi, H., Scheffler, J.M., Pleitner, J.M., Zeng, C., Park, S., Hannon, K.M., Grant, A.L. and Gerrard, D.E. (2008) Modulation of skeletal muscle fiber type by mitogen-activated protein kinase signaling. FASEB J., 22, 2990–3000. [DOI] [PubMed] [Google Scholar]
- 42. Boyer, J.G., Prasad, V., Song, T., Lee, D., Fu, X., Grimes, K.M., Sargent, M.A., Sadayappan, S. and Molkentin, J.D. (2019) ERK1/2 signaling induces skeletal muscle slow fiber-type switching and reduces muscular dystrophy disease severity. JCI. Insight, 4, e127356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Honda, M., Hidaka, K., Fukada, S.I., Sugawa, R., Shirai, M., Ikawa, M. and Morisaki, T. (2017) Vestigial-like 2 contributes to normal muscle fiber type distribution in mice. Sci. Rep., 7, 7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vila, M.C., Rayavarapu, S., Hogarth, M.W., Van der Meulen, J.H., Horn, A., Defour, A., Takeda, S., Brown, K.J., Hathout, Y., Nagaraju, K. et al. (2017) Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ., 24, 330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pastoret, C. and Sebille, A. (1995) mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci., 129, 97–105. [DOI] [PubMed] [Google Scholar]
- 46. Morgan, J.E., Beauchamp, J.R., Pagel, C.N., Peckham, M., Ataliotis, P., Jat, P.S., Noble, M.D., Farmer, K. and Partridge, T.A. (1994) Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev. Biol., 162, 486–498. [DOI] [PubMed] [Google Scholar]
- 47. Ito, N., Ruegg, U.T., Kudo, A., Miyagoe-Suzuki, Y. and Takeda, S. (2013) Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat. Med., 19, 101–106. [DOI] [PubMed] [Google Scholar]
- 48. Moorwood, C., Liu, M., Tian, Z. and Barton, E.R. (2013) Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J. Vis. Exp., 71, e50036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lynch, G.S., Hinkle, R.T., Chamberlain, J.S., Brooks, S.V. and Faulkner, J.A. (2001) Force and power output of fast and slow skeletal muscles from mdx mice 6-28 months old. J. Physiol., 535, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Malicdan, M.C., Noguchi, S., Hayashi, Y.K. and Nishino, I. (2008) Muscle weakness correlates with muscle atrophy and precedes the development of inclusion body or rimmed vacuoles in the mouse model of DMRV/hIBM. Physiol. Genomics, 35, 106–115. [DOI] [PubMed] [Google Scholar]
- 51. Brunelli, S., Sciorati, C., D'Antona, G., Innocenzi, A., Covarello, D., Galvez, B.G., Perrotta, C., Monopoli, A., Sanvito, F., Bottinelli, R. et al. (2007) Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc. Natl. Acad. Sci. U. S. A., 104, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yonekawa, T., Malicdan, M.C., Cho, A., Hayashi, Y.K., Nonaka, I., Mine, T., Yamamoto, T., Nishino, I. and Noguchi, S. (2014) Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain, 137, 2670–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weller, B., Karpati, G. and Carpenter, S. (1990) Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci., 100, 9–13. [DOI] [PubMed] [Google Scholar]
- 54. Kitaoka, Y., Tamura, Y., Takahashi, K., Takeda, K., Takemasa, T. and Hatta, H. (2019) Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle. Physiol. Rep., 7, e13998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fisher-Wellman, K.H., Davidson, M.T., Narowski, T.M., Lin, C.T., Koves, T.R. and Muoio, D.M. (2018) Mitochondrial diagnostics: A multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Rep., 24, 3593–3606.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robinson, M.D., McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McCarthy, D.J., Chen, Y. and Smyth, G.K. (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res., 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.