

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent for the COVID-19. The Sulfonamides groups have been widely introduced in several drugs, especially for their antibacterial activities and generally prescribed for respiratory infections. On the other hand, imidazole groups have the multipotency to act as drugs, including antiviral activity. We have used a structure-based drug design approach to design some imidazole derivatives of sulfonamide, which can efficiently bind to the active site of SARS-CoV-2 main protease and thus may have the potential to inhibit its proteases activity. We conducted molecular docking and molecular dynamics simulation to observe the stability and flexibility of inhibitor complexes. We have checked ADMET (absorption, distribution, metabolism, excretion and toxicity) and drug-likeness rules to scrutinize toxicity and then designed the most potent compound based on computational chemistry. Our small predicted molecule non-peptide protease inhibitors could provide a useful model in the further search for novel compounds since it has many advantages over peptidic drugs, like lower side effects, toxicity and less chance of drug resistance. Further, we confirmed the stability of our inhibitor-complex and interaction profile through the Molecular dynamics simulation study. Our small predicted molecule
Communicated by Ramaswamy H. Sarma
Keywords: SARS-CoV-2 main protease, structure-based drug design, molecular docking, ADMET, drug-likeness, MD simulation
1. Introduction
COVID19 has affected more than 15 million individuals and caused more than 640 thousand deaths within a few months. Coronavirus 2 [(SARS-CoV-2)] virus responsible for COVID1 (World Health Organization, 2020). It infects humans and other animals and causes a variety of highly prevalent and severe diseases like previously reported severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS-CoV) (Chan et al., 2015). One of the best-characterized drug targets among coronaviruses is the main protease: Mpro, also called 3CL Protease. Along with the papain-like proteases, this enzyme is essential for processing the polyproteins that are translated from the viral RNA. The replicase gene of SARS-CoV-2 encodes two overlapping polyproteins-pp1a and pp1ab that are required for viral replication and transcription (Zhang et al., 2020). Since no human proteases with a similar cleavage specificity are known, inhibitors to this target are much less likely to be toxic and cause side effects which makes Mpro as an attractive target for the design of antiviraldrugs (Liu et al., 2020). To deal with four separate types of coronaviruses, we have tried to design a broad-spectrum inhibitor targeted against the progenitor. For this purpose, we have searched for the potency of designed drugs with other same families of viruses like SARS-CoV (Severe acute respiratory syndrome- related coronavirus) Bat-CoV (Tylonycteris bat coronavirus) and MERS-CoV (Middle East respiratory syndrome-related coronavirus). We have conducted a virtual screening using a combinatorial library of FDA-approved drugs (Sulfonamides) combined with imidazole to see if some of these are predicted to bind to the protease. We have introduced nonpeptidic drug-like molecules because (i) It would efficiently mimic peptide binding in the active site of the Mpro and provide enhanced bioavailability to the inhibitor,(ii) non-peptide molecules are small in size and obey drug-likeness rules like Lipinski's rule of five and so there will be less undesirable (side) effects in patents body and less toxicity (iii) Since most of the drugs that target Mpro are peptide are in nature, virus will try to resist it, non-peptide drugs are rare so less chance of drug resistance will be there.
Sulfonamides have a variety of pharmacological properties such as respiratory diseases, antibacterial, antidiabetic, anti-inflammatory and anticancer activity (Bano et al., 2011; Pradhan & Sinha, 2017, 2018a, 2018b). Imidazole is another crucial chemical that has occupied a unique position in heterocyclic chemistry. Darunavir is a sulfonamide derivative being considered as a possible treatment for SARS-CoV-2, clinical trials are underway (Harrison, 2020). Other sulfonamide derivatives are also being used in in silico studies (Calligari et al., 2020; Mary et al., 2021). Imidazole is a nitrogen-containing heterocyclic ring that possesses biological and pharmaceutical importance. The imidazole derivatives possess an extensive spectrum of biological activities such as anticancer, antibacterial, antifungal and antiviral activities (Hebishy et al., 2020; Khabnadideh et al., 2003; Pradhan et al., 2016; Sharma et al., 2009). These attributes of sulfonamides and imidazole provoked us to explore the properties of these two molecules, when they and combined in a single drug-like molecule.
In this present work, we have tried to observe the antiviral (anticovid19) properties of these imidazole derivatives of smx, targeting the novel viral protein Mpro. Initially, we docked the molecules against Mpro, based on the docking results, we have screened out the most potent smx derivative,4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐(5‐methyl‐1,2‐oxazol‐3‐yl)benzene‐1‐sulfon (M10). Further, we have checked its drug-likeness and ADMET properties to achieve the most potent therapeutic. We have also performed comparative interaction profiles of M10/Mpro of four different kinds of viruses (SARS-CoV-2, SARS-CoV, Bat-CoV and MERS-CoV). We measured molecular orbital energies of M10 to assess the chemical reactivity, intermolecular interactions and kinetic stability of the compound. Finally, we have conducted MD simulation of M10-Mpro complex to observe the stability and interaction profiles of the complex. Our detailed systemic analysis portrays that our newly designed novel smx derivative has the high possibility of acting as a potent anti-COVID 19 agents by specifically inhibiting the viral protein Mpro.
2. Material and methods
2.1. Sequence alignment
In the process of developing broad-spectrum antiviral drugs, we have searched for the common amino acid residues in the main proteases (Mpro) of SARS-CoV-2, SARS-CoV, BAT-CoV and MERS-CoV. We have done Multiple Sequence Alignment (MSA) to find homology and the evolutionary relationships between four types of virus sequences. We have also compared with dihydropteroate synthase (DHPS) of Escherichia coli (E.coli), for which the drug sulfamethoxazole (Sulfonamide) is made for(Pradhan & Sinha, 2018b). We have used Clustal Omega. Sequence alignment results are obtained from clustal omega webserver after feeding four types of virus Mpro FASTAsequences in the Clustal Omega web server (Sievers & Higgins, 2014).
2.2. Virtual screening
Structures of four viruses Mpro are available in protein databank, so we have applied a structure-based drug discovery method (Anderson, 2003). Structure-based drug design is usually used to observe the binding interactions of drug-like molecules and then to identify modifications that result in better interactions and higher potency. Moreover, by measuring the distances between the atoms of the drug-like molecules and neighboring amino acid residues in the binding site, it is possible to identify critical binding interactions between the drug-like molecules and the protein/enzyme binding site. Virtual Screening (VS) is a method to facilitated drug design for structure-based drug discovery (Lounnas et al., 2013). It is a procedure of screening a large number of drugs-like molecules against selected and specific drug targets. VS is used for the screening of various types of libraries, including combinatorial chemistry, genomics, protein, and peptide libraries. VS has some steps; they are described below. We have used VS to screen the chemical agents (smx-derivatives) to find the best-fitted molecule in the active site of four types of Mpro.
2.2.1. Drug-like smx-derivatives molecule preparation
We have collected sulfa drugs from the PubChem database (Kim et al., 2016). We have drawn the smx-derivatives by compiling sulfa drugs with imidazole attached to the amino group of sulfa drugs by using Accelrys Draw v4 (Draw). The 3 D coordinates and changes of ionization are done by Autodock software's ligand preparation wizard. We have acquired the 3 D optimized structure for docking by adding Gasteiger charge, detecting routes and selecting torsion from the torsion tree of the Autodock Tools panel (Gasteiger & Marsili, 1980; Huey & Morris, 2008).
2.2.2. Docking preparation of Mpro
We retrieved the crystal structure of Mpro of four viruses in complex with inhibitors from the RCSB PDB database (PDB id: 5R80, PDB id: 2VJ1, PDB id: 4YOI and PDB id: 5WKL for SARS-CoV-2 SARS-CoV Bat-CoV, MERS-CoV, respectively). We have used a monomeric unit (A chain) from the PDB files from the dimer of two chains in the docking studies. We are removed the bounded inhibitors and waters from the PDB structures before docking. We added hydrogens to the protein structure to make PDB structures compatible for docking with the help of Autodock Tools. We executed the docking process by using the Lamarckian genetical algorithm and default parameters (Morris et al., 1998).
2.2.3. Molecular docking
We have carried out molecular docking of selected Mpro inhibitors (smx-derivatives) to the receptor enzyme in the present study by using Autodock vina (Trott & Olson, 2010). For the assurance of potential relationships between Mpro and smx-derivatives, predicted Autodock vina score of the best- docked conformations of small-molecule inhibitors (smx-derivatives) are selected as preliminary binding conformations and saved for observing interactions between Mpro and smx-derivatives.
2.3. Quantum chemistry calculation
We have observed surfaces (molecular orbital, density, potential) and potential electrostatics charges (EPS) to calculate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). HOMO and LUMO (the frontier molecular orbitals) are the most significant orbitals in a molecule (Loukova, 2002). The interaction pattern of the molecule is dependent on these orbitals with other molecules: chemical reactivity, intermolecular interactions and kinetic stability of a molecule are characterized by the energy difference of HOMO-LUMO functions (Aihara, 2000; Karunakaran & Balachandran, 2012). We have used Argus lab to calculate HOMO and LUMO and their differences (Chikhi & Bensegueni, 2008).
2.4. Druglikeness
As a rule of thumb, orally absorbed drugs tend to obey Lipinski's rule of five (Lipinski et al., 1997). The rule of five was derived from an analysis of compounds from the World Drugs Index database aimed at identifying features that were important in making a drug orally active. It was found that the factors concerned involved numbers that are multiples of five: a molecular weight less than 500; no more than 5 hydrogen bond do nor groups; no more than 10 hydrogen bond accept or groups; a calculated log P value less than +5 (Lipinski et al., 1997). In an attempt to improve the predictions of drug-likeness, the rules have laid many extensions, like the Ghose's rule which states that the partition coefficient log P should be in −0.4 to +5.6 range; molar refractivity should be from 40 to 130; molecular weight should be from 180 to 480; Number of atoms should be from 20 to 70 (Ghose et al., 1999). All the 15 molecules are passed through Lipinski's rule of five and Ghose's rule to check their drug-likeness. Besides, we have pass M10 by Veber and Muegge drug-likeness filter (Muegge et al., 2001; Veber et al., 2002). We have used multiple drug-like filters to observe M10 pharmacological potency. According to Muegge (Bayer) filter, molecular weight should be in the range of 200 to 600 daltons. X LOG P should be in −2 to 5 range drug-like molecules should have 10 or fewer rotatable bonds and the total polar surface area should be equal to or less than 150 Å2. The number of rings, Number of carbons, Number of heteroatoms and Number of rotatable bonds should be in 7, 4 and 1 and 15, respectively. Hydrogen bond acceptor should be in 10 Hydrogen bond donors should be in 5.ADMET
The acronym ‘ADMET’ refers to ‘absorption, distribution, metabolism, excretion, and toxicity’. These parameters, in addition to efficacy, are critical in determining whether a drug-like molecule will become a clinical candidate and, subsequently, a commercially viable production of a series of factors (Hodgson, 2001). Optimization of the compounds' pharmacokinetic, metabolic, and toxic properties are not anymore postponed to later stages but along the discovery process rather than at the final stages. In silico ADMET screening models and software approaches are often used to guide medicinal chemistry efforts to design molecules with desired properties. ADMET has been done by evaluating water solubility, human intestinal absorption, oral bioavailability, blood-brain barrier penetration, transporter, plasma protein binding, the volume of distribution, CYP 450, toxicity, etc. by support vector machine (SVM) algorithm (Cheng et al., 2012).
2.5. Molecular dynamics simulation
All molecular dynamics (MD) simulations for the docked complex were performed using NA78 with the CHARMM36m force field parameter for protein and ions (Brooks et al., 1983; Brooks & Karplus, 1989; MacKerell et al., 1998). Swiss Param server was used to generate parameter and topology files for the docked ligands. All system was solvated in a periodic truncated water box using the TIP3P water model. Autoionize tool in VMD (Humphrey et al., 1996) was used to neutralize the system and to maintain the salt concentration at 0.15 M by replacing water molecules with Na+ and Cl- ions. Systems were subjected to energy minimization for 5000 steps using the steepest descent method, followed by NVT and NPT equilibration for 1 and 2 ns, respectively. During the equilibration, positional restraint was applied to both protein and ligand. The temperature of the system was maintained at 310 K using the damping coefficient (γ) of 1ps−1 by Langevin dynamics. To calculate long-range electrostatic interactions in protein, Particle Mesh Ewald (PME) method was used (Darden et al., 1993). The direct non-bonded potential cut-off was set to 12 with 2 Å pair-list distance cut-off, while the scaling factor in the range 1–4 was used during the simulation. The Langevin piston Nosé–Hoover method was applied to maintain 1 atm constant pressure (Feller et al., 1995; Martyna et al., 1994). After equilibration, a 100 ns production run was performed for the system in NPT condition without any restraint with a 2 fs time step interval.
RMSD of protein and ligand were calculated to check the equilibration of the system. RMSF of protein from M10 and smx complex were calculated to measure the residue-wise fluctuation of protein. H-bonds between protein and ligand were defined as formed when the donor-acceptor distance cut-off was less than 3.5 Å and the donor hydrogen acceptor angle was greater than 135º.
3. Results and discussion
3.1. Sequence alignment analysis
MSA results showed that there is a small similarity between (E. coli DHPS) bacteria and 4 types of viruses. Percentage identity matrix results have shown that SARS-CoV-2 has a 95.4 percent similarity with SARS-CoV, whereas 50.2 and 50.8 percent similarity with Bat-CoV, MERS-CoV, respectively (Table 1), but has only 16.99 percent similarity with DHPS.
Table 1.
Percent identity matrix, created by Clustal omega.
| Species | Percentage (SARS-CoV) |
Percentage (SARS-CoV-2) |
Percentage (Bat-CoV) |
Percentage (MERS-COV) |
Percentage (DHPS) |
|---|---|---|---|---|---|
| SARS-CoV | 100 | 95.4 | 50.8 | 51.2 | 16.99 |
| SARS-CoV-2 | 95.4 | 100 | 50.2 | 50.8 | 16.99 |
| Bat-CoV | 50.8 | 50.2 | 100 | 81.1 | 17.90 |
| MERS-COV | 51.2 | 50.8 | 81.1 | 100 | 16.73 |
| DHPS | 16.99 | 16.99 | 17.90 | 16.73 | 100 |
The sequence alignment result has shown that four types of viruses have configurable similarities, whereas E. coli DHPS has some differences. Four types of viruses have some common amino acid residues like Histidine 41 and Cysteine 145, while E. coli DHPS has valine 41 instead of histidine 41 (Figure 1(a)). There are many fully conserved regions among four types of viruses, but E. coli DHPS is denoted by asterisk marks in the MSA results. At the same time, colon designates conservation between groups of strongly similar properties and period indicates conservation between groups of weakly similar properties. The Mpro structure alignment result of the four types of viruses is well fitted with each other (Figure 1(b)) and common amino acids are found in the catalytic domain of the four Mpros.
Figure 1.

(a) Multiple sequence alignment results showing the similarity between four types of viruses and E.coli. (b) Superimposed Mpro structures of four viruses after multiple sequence alignment, the catalytic domain is highlighted by a box. SARS-CoV-2, SARS-CoV, Bat-CoV MERS-CoV are represented by light orange, green, red and pink, respectively.
3.2. Docking and bond analysis
To identify the inhibitor of Mpro we have conducted virtual screening and docked 15 smx- derivatives; out of these M10 found to possess best docking score (binding affinity). We have docked smx-derivatives with SARS-CoV-2 by autodock vina standard docking protocol.
Structure, docking score and IUPAC names of the other smx-derivatives are given in Table 2. All of the smx-derivatives has shown better docking score than smx interestingly we have found all the smx-derivatives has significantly better docking score than the smx itself.
Table 2.
Chemical structures, IUPAC names and their corresponding docking score (autodock vina) of smx and its derivative with conformational diversities (qualified Lipinski’s rule).
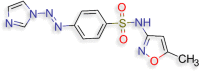 (4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐(5‐ methyl‐1,2‐oxazol‐3‐yl)benzene‐1‐sulfonamide (M10) (D.S −9.2 kcal/mol) |
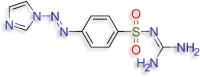 N''‐{4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐ N''‐{4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]benzenesulfonyl}guanidine (D.S-7.2 kcal/mol ) |
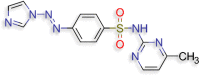 4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐(4‐ methylpyrimidin‐2‐yl)benzene‐1‐sulfonamide (D.S −8.0 kcal/mol ) |
 N‐{4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐ yl]benzenesulfonyl}acetamide (D.S −7.3 kcal/mol ) |
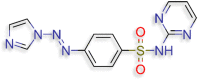 4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐ (pyrimidin‐2‐yl)benzene‐1‐sulfonamide (D.S −8.2 kcal/mol ) |
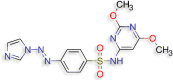 N‐(2,6‐dimethoxypyrimidin‐4‐yl)‐4‐[(E)‐2‐(1H‐ N‐(2,6‐dimethoxypyrimidin‐4‐yl)‐4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]benzene‐1‐sulfonamide (D.S −8.4 kcal/mol ) |
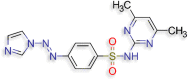 N‐(4,6‐dimethylpyrimidin‐2‐yl)‐4‐[(E)‐2‐(1H‐ N‐(4,6‐dimethylpyrimidin‐2‐yl)‐4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]benzene‐1‐sulfonamide (D.S −8.8 kcal/mol ) |
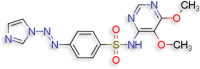 N‐(5,6‐dimethoxypyrimidin‐4‐yl)‐4‐[(E)‐2‐(1H‐ imidazol‐1‐yl)diazen‐1‐yl]benzene‐1‐sulfonamide (D.S −8.5 kcal/mol ) |
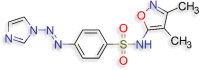 N‐(3,4‐dimethyl‐1,2‐oxazol‐5‐yl)‐4‐[(E)‐2‐(1H‐ imidazol‐1‐yl)diazen‐1‐yl]benzene‐1‐sulfonamide (D.S −8.6 kcal/mol ) |
 4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐(3‐ methoxypyrazin‐2‐yl)benzene‐1‐sulfonamide (D.S −8.3 kcal/mol ) |
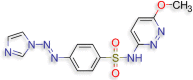 4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐(6‐ methoxypyridazin‐3‐yl)benzene‐1‐sulfonamide (D.S −8.1 kcal/mol ) |
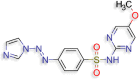 4‐[(E)‐2‐(1H‐imidazol‐1‐yl)diazen‐1‐yl]‐N‐(5‐ methoxypyrimidin‐2‐yl)benzene‐1‐sulfonamide (D.S −8.2 kcal/mol ) |
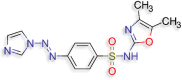 N‐(4,5‐dimethyl‐1,3‐oxazol‐2‐yl)‐4‐[(E)‐2‐(1H‐ imidazol‐1‐yl)diazen‐1‐yl]benzene‐1‐sulfonamide (D.S −8.3 kcal/mol ) |
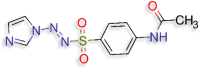 N‐{4‐[(E)‐[(1H‐imidazol‐1‐ yl)imino]sulfamoyl]phenyl}acetamide (D.S −8.5 kcal/mol ) |
 N‐(2,6‐dimethylpyrimidin‐4‐yl)‐4‐[(E)‐2‐(1H‐ imidazol‐1‐yl)diazen‐1‐yl]benzene‐1‐sulfonamide (D.S −8.2 kcal/mol ) |
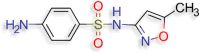 4-Amino-N-(5-methylisoxazol-3-yl)-benzenesulfonamide (smx) (D.S −6.1 kcal/mol ) |
3.2.1. Interaction analysis M10 with different Mpro
Each monomer of the SARS Mpro consists of three domains. The catalytic dyad is composed of Histidine 41 and Cysteine 145, which are found in the active site domains. From the pH dependency of enzymatic activity, the pKa's have been determined as 6.4 for the histidine and 8.3 for the cysteine (Huang et al., 2004). The cysteine acts as the nucleophile in the proteolytic cleavage reaction with His 41 acting as a general base (Solowiej et al., 2008; Świderek & Moliner, 2020). There is also a close correlation between dimer formation and the enzyme catalytic activity. A flip-flop mechanism is proposed for Mpro in which its two subunits are alternately used in acylation and deacylation steps (Mary et al., 2021). Details of M10 and SARS- CoV-2 Mpro is given below. Based on the docking score table, we have chosen M10 and observed the interaction of M10 and Mpro (Table 4).
Table 4.
Docking score of different Mpro.
| Species | Docking score kcal/mol |
|---|---|
| SARS-CoV-2 | –9.2 |
| SARS-CoV | –8.7 |
| Bat-CoV | –7.9 |
| MERS-COV | –6.8 |
Potential inhibition mechanism: M10 is a nonpeptidic inhibitor of SARS-CoV-2 Mpro that will lodge itself in the active site of Mpro through a number of hydrogen bonds. Strong interaction will come from increased hydrogen bonds between M10 and the backbone of the Mpro active site.M10 binding in the active site of Mpro will make unavailable for the viral polyproteins to cut them into pieces. Uncut viral polyproteins effect viral growth. In this way M10 will inhibit SARS-CoV-2 and thus covid-19.
3.2.1.1. M10 and SARS-CoV-2 Mpro
We have docked M10 in the active site of Mpro of SARS-CoV-2. It forms four hydrogen bonds with amino acids of the Mpro in the distances 2.566 to 3.080 Å (Table 3, Figure 2.1). It also forms five hydrophobic bonds with amino acids of the MRO (2.844 to 5.108 Å). The hydrogen bond-forming amino acids are GLY143, ASP187 GLN189, MET165, ARG188, PRO168, HIS41, CYS145, MET49. Details of smx and SARS-CoV2 Mpro are given in the supporting information (Table S1).
Table 3.
Non-covalent bond distances between M10, SARS-CoV2 Mpro and SARS-CoV.
| Non-covalent bond distances between M10 and SARS-CoV2 Mpro | Distance (Å) | Bonding category | Non-covalent bond distances between M10 and SARS-CoVMpro | Distance (Å) | Bonding category |
|---|---|---|---|---|---|
| A:GLY143:HN - :M10:N18 |
2.566 | Hydrogen bond |
A:HIS163:HE2 - :M10:O1 |
2.042 | Hydrogen bond |
| A:ASP187:HA - :M10:O8 |
2.812 | Hydrogen bond |
:M10:H1 - A:GLU166:OE2 |
2.262 | Hydrogen bond |
| A:GLN189:HA - :M10 |
2.844 | Hydrophobic | A:SER144:HG - :M10:O2 |
2.443 | Hydrogen bond |
| :M10:H28 - A:MET165:SD |
2.919 | Hydrogen bond | A:LEU141:HA - :M10:N3 |
2.721 | Hydrogen bond |
| A:ARG188:HA - :M10:O7 |
3.080 | Hydrogen bond | :M10:H12 - A:CYS44:O |
2.783 | Hydrogen bond |
| :M10:C1 - A:PRO168 | 4.435 | Hydrophobic | :M10:H11 - A:CYS44:O |
3.094 | Hydrogen bond |
| A:HIS41 - :M10 | 4.543 | Hydrophobic | A:CYS145:SG - :M10 | 3.571 | Other |
| :M10 - A:CYS145 | 4.705 | Hydrophobi c | A:LEU141:C,O;ASN142:N - :M10 | 3.725 | Hydrophobic |
| :M10 - A:MET49 | 5.108 | Hydrophobi c | A:MET49:SD - :M10 | 4.889 | Other |
| A:CYS145:SG - :M10 | 5.121 | Other | :M10:S1 -A:HIS163 | 5.054 | Other |
| A:MET165:SD - :M10 | 5.410 | Other | A:HIS41 - :M10 | 5.078 | Hydrophobic |
| :M10: S1 - A: HIS172 | 5.971 | Other |
Figure 2.
(2.1) Comprehensive perception of Mpro and M10 after docking. (a) The secondary structure of Mpro represented by cartoon and M10 represented is by ball and stick model and has been coloured according to elements. (b) The secondary structure of Mpro represented by hydrophobic surface and M10 represented is by stick model (c) Interactions of M10 with Mpro amino acids. Bonds are in dots. M10 surrounding amino acids are in three letters code represented in blue. (2.2) Comprehensive perception of SARS-CoV Mpro and M10 after docking. Figure legends are the same as Figure 2.1. (2.3) Comprehensive perception of BAT-CoVMpro and M10 after docking. Figure legends are the same as Fig. 2. (2.4) Comprehensive perception of MERS-COV Mpro and M10 after docking. Figure legends are the same as Figure 2.




3.2.1.2. M10 and SARS-CoV Mpro
We have docked M10 in the active site of Mpro of SARS-CoV. It forms six hydrogen bonds with amino acids of the Mpro in the distances 2.09 to 3.10 Å (Table 3, Figure 2.2). It also includes five hydrophobic bonds with amino acids of the Mpro (2.844 to 5.108 Å). The hydrogen bond-forming amino acids are HIS41, MET49, GLY143, CYS145, MET165, PRO168, ASP187, ARG188, GLN18.
3.2.1.3. M10 and BAT-CoV Mpro
We have docked M10 in the active site of Mpro of SARS-CoV. It forms six hydrogen bonds with amino acids of the Mpro in distances 2.042 to 3.094 Å (Table S2 in supporting information, Figure 2.2). It also includes two Hydrophobic bonds with amino acids of the Mpro (3.725 Å and 5.078 Å). The hydrogen bond-forming amino acids are HIS41, CYS44, SER144, CYS145, HIS163, GLU166, HIS172.
3.2.1.4. M10 and MERS-CoV Mpro
We have docked M10 in the active site of Mpro of MERS-CoV. It forms five hydrogen bonds with amino acids of the Mpro in distances 2.279 to 3.555 Å (Table S2 in supporting information, Figure 2.4). It also includes five hydrophobic bonds with amino acids of the Mpro (3.999 Å and 5.322 Å). The hydrogen bond-forming amino acids are HIS41, MET49, GLY143, CYS145, MET165, PRO168 ASP187, GLN189, ARG188.
3.3. Density functional theory analysis
The difference in HOMO and LUMO energy indicates the electronic excitation energy (Figure 3) that is necessary to compute the molecular reactivity and stability of the compounds. The smaller HOMO-LUMO gap of M10 (–0.19662) than smx (–0.19781) proves quick adaptation in the protein environment to start with biochemical activity.
Figure 3.

(a, b) The plots of HOMO and LUMO of M10. The positive electron density in pink and negative electron density in blue. (c, d) The plots of HOMO and LUMO of smx. The positive electron density in yellow and negative electron density in violet.
3.4. Druglikness
The physicochemical properties of M10 showed that it had passed all (Lipinski's, Ghose's, Veber's, Muegge's) the drug-likeness filters. A Bioavailability Score, ABS, is formulated as the probability that a compound will have >10% bioavailability in rat or measurable Caco-2 permeability. ABS is 0.11 for anions for which PSA is >150 Å2, 0.56 if PSA is between 75 and 150 Å2, and 0.85 if PSA is <75 Å2. For the remaining compounds, ABS is 0.55 if it passes the rule-of-five and 0.17 if it fails. ABS also identify poorly and well-absorbed compounds tested in humans. Bioavailability Score of M10 is 0.55 that supports the fact that it has passed the rule-of-five (Table 5). In comparison with approved drug smx (Table 5), M10 results have shown that its quite similar pharmacokinetic properties to smx, which is another positive sign because if smx has shown efficacy and potency for almost sixty years.
Table 5.
Physicochemical and pharmacokinetics properties of M10 and smx.
| Physicochemical properties M10 |
Pharmacokinetics | Physicochemical properties smx |
Pharmacokinetics | |||
|---|---|---|---|---|---|---|
| Formula | C13H12N6O3S | Lipinski's | C10H11N3O3S | Lipinski's | Yes | |
| Molecular | 332.34 g/mol | Ghose’s | 253.28 g/mol | Ghose’s | Yes | |
| weight | ||||||
| Number of heavy atoms | 23 | Veber's | 17 | Veber's | Yes | |
| Number of aromatic heavy atoms | 16 | Muegge's | 11 | Muegge's | Yes | |
| Fraction Csp3 | 0.08 | Bioavailability score | 0.10 | Bioavailability score | 0.55 | |
| Number of rotatable bonds | 5 | 3 | ||||
| Number of H-bond acceptors | 7 | Leadlikeness | 4 | Leadlikeness | Yes | |
| Number of H-bond donors | 2 | Synthetic accessibility | 2 | Synthetic accessibility | 2.73 | |
| Molar refractivity | 81.35 | 62.99 | ||||
| TPSA | 133.98 Ų | 106.60 Ų | ||||
| Consensus Log P | 1.88 | 0.90 | ||||
| Log S (ESOL) | –3.23 | –2.25 | ||||
| Solubility | 1.95e-01mg/ml; 5.86e-04mol/l | 4.34e-02 mg/ml; 1.71e-04 mol/l | ||||
| Class | Soluble | Soluble | ||||
3.5. ADMET property analysis
There are total of 26 parameters in ADMET data, which are extracted from PubMed and Google Scholar searches within 2002 to 2011 publications (Cheng et al., 2012). Pharmacokinetic properties and toxicities are predicted by ADMET, which can predict permeability for, BBB (blood-brain barrier), HIA (human intestinal absorption), P-glycoprotein Substrate/Inhibitor, renal organic cation transporter, etc. ADMET results suggest M10 has positive (+) attributes in Blood-Brain Barrier Permeability, human intestinal absorption, which suggests that (Table 6) the molecule can be well absorbed in the human body (Ajay et al., 1999; De Vrieze et al., 2015). Inhibition and initiation of P-glycoprotein have been reported as the cause of drug-drug interactions (Chen et al., 2011). The data in Table 6, show best-fitted M10 is in the permissible limit (Cheng et al., 2012). Organic cation transporters are responsible for drug absorption and disposition in the kidney, liver, and intestine (Zhang et al., 1998). ADMET result shows that M10 is non-inhibitor to renal organic cation transporter. The human cytochromes P450 (CYPs), particularly is a forms of 1A2, 2C9, 2D6, and 3A4, are responsible for about 90% oxidative metabolic reactions. Inhibition of CYP enzymes will lead to inductive or inhibitory failure of drug metabolism (Uttamsingh et al., 2005). Low CYP inhibitory promiscuity (Table 6) results showed that M10 has also passed the metabolism filter. The Ames test is a widely employed method that uses bacteria to test whether a given chemical can cause cancer. More formally, it is a biological assay to assess the mutagenic potential of chemical compounds (Forman, 1991; Mortelmans & Zeiger, 2000). ADMET results suggest that M10 qualifies the Ames test; thus, it is non- Ames toxic and non-carcinogenic. Human Ether-à-go-go-Related Gene (hERG) is a gene, sensitive for drug binding (Sanguinetti & Tristani-Firouzi, 2006). The result shows M10 is a weak inhibitor and non-inhibitor of hERG week inhibition (predictor I and II). That means M10 will bind well with the receptor (SARS-CoV-2). Based on the ADMET results, we can conclude that M10 will not harm the patient's body and will not cause any adverse effects.
Table 6.
ADMET properties of M10 and smx.
| ADMET Properties |
M10 |
smx |
||
|---|---|---|---|---|
| Value | Probability | Value | Probability | |
| Absorption | ||||
| Blood-brain barrier | BBB+ | 0.9328 | BBB+ | 0.9382 |
| Human intestinal absorption | HIA+ | 0.9971 | HIA+ | 1.0000 |
| Caco-2 permeability | Caco2- | 0.5714 | Caco2- | 0.5346 |
| P-glycoprotein substrate | Non-substrate | 0.8746 | Non-substrate | 0.8884 |
| P-glycoprotein inhibitor | Non-inhibitor | 0.8631 | Non-inhibitor | 0.9362 |
| Non-inhibitor | 0.8064 | Non-inhibitor | 0.8754 | |
| Renal organic cation transporter | Non-inhibitor | 0.8804 | Non-inhibitor | 0.9195 |
| Distribution | ||||
| Subcellular localization | Plasma membrane | 0.4290 | Mitochondria | 0.4068 |
| Metabolism | ||||
| CYP450 2C9 substrate | Non-substrate | 0.6336 | Non-substrate | 0.7652 |
| CYP450 2d6 substrate | Non-substrate | 0.8388 | Non-substrate | 0.9115 |
| CYP450 3A4 substrate | Non-substrate | 0.6644 | Non-substrate | 0.7632 |
| CYP450 1A2 inhibitor | Non-inhibitor | 0.8589 | Non-inhibitor | 0.9045 |
| CYP450 2C9 inhibitor | Inhibitor | 0.5135 | Non-inhibitor | 0.9071 |
| CYP450 2D6 inhibitor | Non-inhibitor | 0.8387 | Non-inhibitor | 0.9231 |
| CYP450 2C19 inhibitor | Non-inhibitor | 0.5000 | Non-inhibitor | 0.9025 |
| CYP450 3A4 inhibitor | Non-inhibitor | 0.8371 | Non-inhibitor | 0.9744 |
| CYP inhibitory promiscuity | Low CYP inhibitory promiscuity | 0.7490 | Low CYP inhibitory promiscuity | 0.7151 |
| Toxicity | ||||
| Human ether-a-go-go-related gene inhibition | Weak inhibitor | 0.8231 | Weak inhibitor | 0.9143 |
| Non-inhibitor | 0.8905 | Non-inhibitor | 0.8956 | |
| AMES toxicity | Non AMES toxic | 0.6540 | Non AMES toxic | 0.9132 |
| Carcinogens | Non-carcinogens | 0.7516 | Non-carcinogens | 0.8193 |
| Fish toxicity | High FHMT | 0.6105 | Low FHMT | 0.9103 |
| Tetrahymena pyriformis toxicity | High TPT | 0.6844 | Low TPT | 0.5751 |
| Honey bee toxicity | Low HBT | 0.7783 | Low HBT | 0.7986 |
| Biodegradation | Ready biodegradable | 0.5407 | Not ready biodegradable | 0.9882 |
| Acute oral toxicity | III | 0.5716 | IV | 0.6184 |
| Carcinogenicity (three-class) | Non-required | 0.4830 | Non-required | |
3.6. MD Simulation
MD simulations study of docked complexes of SARS-CoV-2 Mpro with smx and M10 compounds. To validate the docked result of SARS-CoV-2 Mpro with smx and M10, we have done the comparative MD simulation study between smx and M10 complex. Initially, we calculated the backbone RMSD of SARS-CoV-2 Mpro from the docked complex (smx and M10) (Figure 4(a,b)). The data reveal that SARS-CoV-2 Mpro from M10 complex has less RMSD over smx complex. In the case of smx complex, there is a certain increase in the fluctuation of RMSD after 50 ns and the value goes to around 4 Å, suggesting that the complex from smx is less stable. However, there is not much significant difference for ligand RMSD from smx and M10 and the values remained within 2.5 Å. To explore the interaction between protein and ligand, we further examined the number of H-bonds from docked complexes of smx and M10. H-bonds was measured between donor and acceptor atom of protein and ligand within 3.5 Å distance cut-off and angle less than 135 degree. We found that M10 interacts well in the pocket of SARS-CoV-2 Mpro with the average number of H-bonds ∼3.45 as compared to smx (average ∼1.90). The most important H-bond forming residues from SARS-CoV-2 Mpro with M10 are His41, Glu166, Asp187, Arg188, Gln192, Gln189, Thr190. We noticed that compounds (smx and M10) also alters the residue-wise fluctuation of SARS-CoV-2 Mpro (Figure 5b). Although, RMSF indicates that residues 100–250 of SARS-CoV-2 Mpro become the least stable in the presence of smx over M10. Overall, we conclude that M10 compound is more effective in binding with SARS-CoV-2 Mpro as compared to the smx.
Figure 4.

Comparative MD simulation study of the docked complex of SARS-CoV-2 Mpro with smx and M10 compounds. RMSD of protein and ligand from the complex of (a) SARS-CoV-2 Mpro with smx and (b) SARS-CoV-2Mpro with M10 compound. (c) Number of H-bonds between SARS-CoV-2 Mpro with smx (red) and M10 (blue). (d) RMSF of SARS-CoV-2 Mproin the presence of smx (red) and M10 (blue).
Figure 5.

Effectiveness of M10 compound against other viruses SARS-CoV (PDB ID: 2VJ1), BAT-CoV (PDB ID: 4YOI) and MERS (PDB ID: 5WKL). (a) RMSD of protein and ligand from docked complex of SARS-CoV (left), BAT-CoV (middle) and MERS (right) with M10 compound. (b) Number of H-bonds between SARS-CoV (left), BAT-CoV (middle) and MERS (right) with M10.
3.6.1. Effectiveness of M10 compound against other viruses SARS-CoV, BAT-CoV and MERS
Earlier, we have seen that M10 compound is most prominent in binding with SARS-CoV-2 Mpro. So, it will be very interesting to see whether same M10 compound is effective against other SARS family viruses. Therefore, we have done comparative MD simulation study of docked complexes of SARS-CoV, BAT-CoV and MERS protein with M10 compound. Both RMSD of protein and ligand show some extent of stability (Figure 5(a,b)). To further analyze the effectiveness of binding of M10 in complex, we calculated the number of H-bonds formed for the individual protein SARS-CoV (PDB ID: 2VJ1), BAT-CoV (PDBID:4YOI) and MERS (PDBID:5WKL) with the same ligand (M10) (Figure 5b). We found that M10 is the most stable inhibitor in forming H- bonds in the pocket of MERS having average H-bonds (5.71) as compared to BAT-CoV (3.52) and SARS-CoV (2.29). Overall, we propose that our designed compound (M10) is also effective against other viruses too.
4. Conclusion
Through this study, we have adopted structure-based drug design and predict a series of imidazole derivatives of sulfonamide group of drugs which can interact with the COVID 19 Mpro. Through molecular docking, we have screened out the best-docked molecule M10. Further, we docked the same molecule with other reported coronavirus Mpro proteins and found that this M10 molecule is able to bind with almost all the Mpro protein active site with high to moderate affinity. Moreover interaction profiles of imidazole derivatives with Mpro are completely different with its known target DHPS. We have checked the molecular orbital, pharmacophore, drug-likeness and ADMET property of the compound and interestingly the compound passed all the tests to act as an effective drug molecule. To nail the specificity and the binding of the molecule with Mpro of all the coronaviruses we performed MD stimulation of M10 docked Mpro and in agreement with our docking result from our MD simulation data also supports the fact that M10 has the very specific and strong binding with the active protease site of Mpro. These results undoubtedly depict the fact that the molecule M10 has the high potency to act as an anti-COVID 19 drugs. As this drug has the potential to be an inhibitor against the other Mpro of the coronaviruses, this drug can have the potency to act as broad-spectrum anti-coronavirus drug. This is noteworthy that sulfonamide group of drugs usually prescribed for respiratory bacterial infection, whereas the imidazole group has the proven antiviral activity. On the other-hand, coronavirus-induced disease including COVID19 causes an acute respiratory problem, and has the high possibility to induce secondary lung infection. In this scenario, this sulfonamide group may provide additional protection by ceasing the secondary lung infection. Although our present theoretical study has provided a concrete base to predict the above-mentioned compound as a potential Anti-COVID 19 drug, further study is required to synthesize the compound and experimentally establish it's inhibitory effect against coronavirus Mpro.
Supplementary Material
Funding Statement
This work was funded by IACS intramural Project.
Disclosure statement
The authors declare that they have no conflict of interest.
References
- Aihara, J.-I. (2000). Correlation found between the HOMO–LUMO energy separation and the chemical reactivity at the most reactive site for isolated-pentagon isomers of fullerenes. Physical Chemistry Chemical Physics, 2(14), 3121–3125. 10.1039/b002601h [DOI] [Google Scholar]
- Ajay Bemis, G. W., & Murcko, M. A. (1999). Designing libraries with CNS activity. Journal of Medicinal Chemistry, 42(24), 4942–4951. [DOI] [PubMed] [Google Scholar]
- Anderson, A. C. (2003). The process of structure-based drug design. Chemistry & Biology, 10(9), 787–797. 10.1016/j.chembiol.2003.09.002 [DOI] [PubMed] [Google Scholar]
- Bano, S., Javed, K., Ahmad, S., Rathish, I., Singh, S., & Alam, M. (2011). Synthesis and biological evaluation of some new 2-pyrazolines bearing benzene sulfonamide moiety as potential anti-inflammatory and anti-cancer agents. European Journal of Medicinal Chemistry, 46(12), 5763–5768. 10.1016/j.ejmech.2011.08.015 [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. A., & Karplus, M. (1983). CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry, 4(2), 187–217. 10.1002/jcc.540040211 [DOI] [Google Scholar]
- Brooks, C. L., III, & Karplus, M. (1989). Solvent effects on protein motion and protein effects on solvent motion. Dynamics of the active site region of lysozyme. Journal of Molecular Biology, 208(1), 159–181. 10.1016/0022-2836(89)90093-4 [DOI] [PubMed] [Google Scholar]
- Calligari, P., Bobone, S., Ricci, G., & Bocedi, A. (2020). Molecular investigation of SARS–CoV-2 proteins and their interactions with antiviral drugs. Viruses, 12(4), 445. 10.3390/v12040445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J. F., Lau, S. K., To, K. K., Cheng, V. C., Woo, P. C., & Yuen, K.-Y. (2015). Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clinical Microbiology Reviews, 28(2), 465–522. 10.1128/CMR.00102-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L., Li, Y., Zhao, Q., Peng, H., & Hou, T. (2011). ADME evaluation in drug discovery. 10. Predictions of P-glycoprotein inhibitors using recursive partitioning and naive Bayesian classification techniques. Molecular Pharmaceutics, 8(3), 889–900. 10.1021/mp100465q [DOI] [PubMed] [Google Scholar]
- Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., Lee, P. W., & Tang, Y. (2012). admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. ACS Publications. [DOI] [PubMed] [Google Scholar]
- Chikhi, A., & Bensegueni, A. (2008). Docking efficiency comparison of Surflex, a commercial package and Arguslab, a licensable freeware. Journal of Computer Science & Systems Biology, 01 (01), 081–086. 10.4172/jcsb.1000007 [DOI] [Google Scholar]
- Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98(12), 10089–10092. 10.1063/1.464397 [DOI] [Google Scholar]
- De Vrieze, M., Janssens, P., Szucs, R., Van der Eycken, J., & Lynen, F. (2015). In vitro prediction of human intestinal absorption and blood-brain barrier partitioning: Development of a lipid analog for micellar liquid chromatography. Analytical and Bioanalytical Chemistry, 407(24), 7453–7466. 10.1007/s00216-015-8911-z [DOI] [PubMed] [Google Scholar]
- Draw, A. Version 4.0. Accelrys Inc. [Google Scholar]
- Feller, S. E., Zhang, Y., Pastor, R. W., & Brooks, B. R. (1995). Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics, 103(11), 4613–4621. 10.1063/1.470648 [DOI] [Google Scholar]
- Forman, D. (1991). Ames, the Ames test, and the causes of cancer. BMJ (Clinical Research Ed.), 303(6800), 428–429. 10.1136/bmj.303.6800.428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger, J., & Marsili, M. (1980). Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron, 36(22), 3219–3228. 10.1016/0040-4020(80)80168-2 [DOI] [Google Scholar]
- Ghose, A. K., Viswanadhan, V. N., & Wendoloski, J. J. (1999). A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. Journal of Combinatorial Chemistry, 1(1), 55–68. 10.1021/cc9800071 [DOI] [PubMed] [Google Scholar]
- Harrison, C. (2020). Coronavirus puts drug repurposing on the fast track. Nature Biotechnology, 38(4), 379–381. 10.1038/d41587-020-00003-1 [DOI] [PubMed] [Google Scholar]
- Hebishy, A. M., Abdelfattah, M. S., Elmorsy, A., & Elwahy, A. H. (2020). ZnO nanoparticles catalyzed synthesis of bis-and poly (imidazoles) as potential anticancer agents. Synthetic Communications, 50(7), 980–996. 10.1080/00397911.2020.1726396 [DOI] [Google Scholar]
- Hodgson, J. (2001). ADMET-turning chemicals into drugs. Nature Biotechnology, 19(8), 722–726. 10.1038/90761 [DOI] [PubMed] [Google Scholar]
- Huang, C., Wei, P., Fan, K., Liu, Y., & Lai, L. (2004). 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry, 43(15), 4568–4574. 10.1021/bi036022q [DOI] [PubMed] [Google Scholar]
- Huey, R., & Morris, G. M. (2008). Using AutoDock 4 with AutoDocktools: A tutorial. The Scripps Research Institute, USA, 54–56. [Google Scholar]
- Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics, 14(1), 33–38. 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Karunakaran, V., & Balachandran, V. (2012). FT-IR, FT-Raman spectra, NBO, HOMO–LUMO and thermodynamic functions of 4-chloro-3-nitrobenzaldehyde based on ab initio HF and DFT calculations. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 98, 229–239. 10.1016/j.saa.2012.08.003 [DOI] [PubMed] [Google Scholar]
- Khabnadideh, S., Rezaei, Z., Khalafi-Nezhad, A., Bahrinajafi, R., Mohamadi, R., & Farrokhroz, A. (2003). Synthesis of N-alkylated derivatives of imidazole as antibacterial agents. Bioorganic & Medicinal Chemistry Letters, 13(17), 2863–2865. 10.1016/s0960-894x(03)00591-2 [DOI] [PubMed] [Google Scholar]
- Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., Han, L., He, J., He, S., Shoemaker, B. A., Wang, J., Yu, B., Zhang, J., & Bryant, S. H. (2016). PubChem substance and compound databases. Nucleic Acids Research, 44(D1), D1202–D1213. 10.1093/nar/gkv951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23(1–3), 3–25. 10.1016/S0169-409X(96)00423-1 [DOI] [PubMed] [Google Scholar]
- Liu, S., Zheng, Q., & Wang, Z. (2020). Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics (Oxford, England), 36(11), 3295–3298. 10.1093/bioinformatics/btaa224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukova, G. V. (2002). The first experimental approach to probing frontier orbitals and HOMO–LUMO gap in bent metallocenes. Chemical Physics Letters, 353(3–4), 244–252. 10.1016/S0009-2614(02)00031-3 [DOI] [Google Scholar]
- Lounnas, V., Ritschel, T., Kelder, J., McGuire, R., Bywater, R. P., & Foloppe, N. (2013). Current progress in structure-based rational drug design marks a new mindset in drug discovery. Computational and Structural Biotechnology Journal, 5(6), e201302011. 10.5936/csbj.201302011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A. D., Bashford, D., Bellott, M., Dunbrack, R. L., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., Joseph-McCarthy, D., Kuchnir, L., Kuczera, K., Lau, F. T., Mattos, C., Michnick, S., Ngo, T., Nguyen, D. T., Prodhom, B., … Karplus, M. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B, 102(18), 3586–3616. 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- Martyna, G. J., Tobias, D. J., & Klein, M. L. (1994). Constant pressure molecular dynamics algorithms. The Journal of Chemical Physics, 101(5), 4177–4189. 10.1063/1.467468 [DOI] [Google Scholar]
- Mary, S. J., Pradhan, S., & James, C. (2021). Molecular structure, NBO analysis of the hydrogen-bonded interactions, spectroscopic (FT–IR, FT–Raman), drug likeness and molecular docking of the novel anti COVID-2 molecule (2E)-N-methyl-2-[(4-oxo-4H-chromen-3-yl) methylidene]-hydrazinecarbothioamide (Dimer)-quantum chemical approach. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 251, 119388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary, S. J., Siddique, M. U. M., Pradhan, S., Jayaprakash, V., & James, C. (2021). Quantum chemical insight into molecular structure, NBO analysis of the hydrogen-bonded interactions, spectroscopic (FT–IR, FT–Raman), drug likeness and molecular docking of the novel anti COVID-19 molecule 2-[(4, 6-diaminopyrimidin-2-yl) sulfanyl]-N-(4-fluorophenyl) acetamide-dimer. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 244, 118825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., & Olson, A. J. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry, 19(14), 1639–1662. [DOI] [Google Scholar]
- Mortelmans, K., & Zeiger, E. (2000). The Ames Salmonella/microsome mutagenicity assay. Mutation Research, 455(1–2), 29–60. 10.1016/s0027-5107(00)00064-6 [DOI] [PubMed] [Google Scholar]
- Muegge, I., Heald, S. L., & Brittelli, D. (2001). Simple selection criteria for drug-like chemical matter. Journal of Medicinal Chemistry, 44(12), 1841–1846. 10.1021/jm015507e [DOI] [PubMed] [Google Scholar]
- Pradhan, S., Mondal, S., & Sinha, C. (2016). In search of Tuberculosis drug design: An in silico approach to azoimidazolyl derivatives as antagonist for cytochrome P450. Journal of the Indian Chemical Society, 93(9), 1067–1084. [Google Scholar]
- Pradhan, S., & Sinha, C. (2017). Combating prostate cancer by sulfonamide compounds: Theoretical prediction. Journal of the Indian Chemical Society, 94(10), 1113–1122. [Google Scholar]
- Pradhan, S., & Sinha, C. (2018a). High throughput screening based highly potent sulfonyl-benzamide anti-diabetic drug. Current Drug Therapy, 13(2), 162–173. 10.2174/1574885512666171023154352 [DOI] [Google Scholar]
- Pradhan, S., & Sinha, C. (2018b). Sulfonamide derivatives as Mycobacterium tuberculosis inhibitors: In silico approach. In Silico Pharmacology, 6(1), 4–17. 10.1007/s40203-018-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti, M. C., & Tristani-Firouzi, M. (2006). hERG potassium channels and cardiac arrhythmia. Nature, 440(7083), 463–469. 10.1038/nature04710 [DOI] [PubMed] [Google Scholar]
- Sharma, D., Narasimhan, B., Kumar, P., Judge, V., Narang, R., De Clercq, E., & Balzarini, J. (2009). Synthesis, antimicrobial and antiviral evaluation of substituted imidazole derivatives. European Journal of Medicinal Chemistry, 44(6), 2347–2353. 10.1016/j.ejmech.2008.08.010 [DOI] [PubMed] [Google Scholar]
- Sievers, F., & Higgins, D. G. (2014). Clustal Omega, accurate alignment of very large numbers of sequences (Multiple sequence alignment methods (pp. 105–116). Springer. [DOI] [PubMed] [Google Scholar]
- Solowiej, J., Thomson, J. A., Ryan, K., Luo, C., He, M., Lou, J., & Murray, B. W. (2008). Steady-state and pre-steady-state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro cysteine protease: Development of an ion-pair model for catalysis. Biochemistry, 47(8), 2617–2630. 10.1021/bi702107v [DOI] [PubMed] [Google Scholar]
- Świderek, K., & Moliner, V. (2020). Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 M pro by QM/MM computational methods. Chemical Science, 11(39), 10626–10630. 10.1039/D0SC02823A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamsingh, V., Lu, C., Miwa, G., & Gan, L.-S. (2005). Relative contributions of the five major human cytochromes P450, 1A2, 2C9, 2C19, 2D6, and 3A4, to the hepatic metabolism of the proteasome inhibitor bortezomib. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 33(11), 1723–1728. 10.1124/dmd.105.005710 [DOI] [PubMed] [Google Scholar]
- Veber, D. F., Johnson, S. R., Cheng, H.-Y., Smith, B. R., Ward, K. W., & Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. Journal of Medicinal Chemistry, 45(12), 2615–2623. 10.1021/jm020017n [DOI] [PubMed] [Google Scholar]
- World Health Organization. (2020). Coronavirus Disease 2019 (COVID-19): Situation Report, 82.
- Zhang, L., Brett, C. M., & Giacomini, K. M. (1998). Role of organic cation transporters in drug absorption and elimination. Annual Review of Pharmacology and Toxicology, 38(1), 431–460. 10.1146/annurev.pharmtox.38.1.431 [DOI] [PubMed] [Google Scholar]
- Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., Becker, S., Rox, K., & Hilgenfeld, R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, 368(6489), 409–412. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.