

Abstract
The COVID-19 pandemic infection has claimed many lives and added to the social, economic, and psychological distress. The contagious disease has quickly spread to almost 218 countries and territories following the regional outbreak in China. As the number of infected populations increases exponentially, there is a pressing demand for anti-COVID drugs and vaccines. Virtual screening provides possible leads while extensively cutting down the time and resources required for ab-initio drug design. We report structure-based virtual screening of a hundred plus library of quinoline drugs with established antiviral, antimalarial, antibiotic or kinase inhibitor activity. In this study, targets having a role in viral entry, viral assembly, and viral replication have been selected. The targets include: 1) RBD of receptor-binding domain spike protein S 2) Mpro Chymotrypsin main protease 3) Ppro Papain protease 4) RNA binding domain of Nucleocapsid Protein, and 5) RNA Dependent RNA polymerase from SARS-COV-2. An in-depth analysis of the interactions and G-score compared to the controls like hydroxyquinoline and remdesivir has been presented. The salient results are (1) higher scoring of antivirals as potential drugs (2) potential of afatinib by scoring as better inhibitor, and (3) biological explanation of the potency of afatinib. Further MD simulations and MM-PBSA calculations showed that afatinib works best to interfere with the the activity of RNA dependent RNA polymerase of SARS-COV-2, thereby inhibiting replication process of single stranded RNA virus.
Communicated by Ramaswamy H. Sarma
Keywords: SARS-COV-2, RNA dependent RNA polymerase, Bruton Tyrosine kinase inhibitors, quinoline based FDA approved Drugs

1. Introduction
The pandemic outbreak of novel severe acute respiratory syndrome 2 or COVID-19 has claimed many lives and added to the social, economic, and psychological distress (Huang et al., 2020). Initially, the outbreak was local in Wuhan, China. With time the virus spread exponentially across borders through human contact. Considering the grave gravity, the World Health Organization (WHO) declared COVID-19 pandemic, a public health emergency of international concern (Law, 2020).
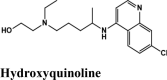
The continuously growing numbers of infections and mortality worldwide have called for a prompt therapeutic solution against COVID-19. Currently, no drugs or vaccines can specifically target the proteins in the corona virus to prevent diseases; hence the discovery of drugs or vaccines may be a milestone for all researchers. Based on clinical experiences while treating moderate to severe cases, three drugs-hydroxyquinoline, (Rothan & Byrareddy, 2020) remdesivir (Ko et al., 2020) and, lopinavir/ritonavir (Chu et al., 2004) have emerged with varied and contentious potential. Vaccine development is under progress. However, the chances of a breakthrough are bleak in the immediate future.
The pressing and expeditious demand for an effective therapeutic clubbed with limited biochemical knowledge, and complex-tedious-resource intensive drug designing have compelled researchers to switch to virtual screening for drug molecules. Drug repurposing through virtual screening is an innovative approach in the current time to quickly arrive at the promising scaffold (Kiplin Guy et al., 2020; Shah et al., 2020).
Taking leads from the limited and not-so successful clinical experiences, we hypothesize that virtual screening of drugs similar tohydroxyquinoline (HQ), remdesivir, and lopinavir/ritonavir might provide potential scaffolds. The three drugs target different pathways in effective scenarios: hydroxyquinoline acts as inhibitors during the entry of viral particles (Liu et al., 2020), remdesivir interfere with RNA replication (Yin et al., 2020), lopinavir/ritonavir (Cao et al., 2020) inhibits the activity of the virus by interfering with essential protein necessary for their life cycle. Among them, our interest focuses on hydroxyquinoline derived molecules because: (1) It is a proven antimalarial drug and antiviral, primarily acting as entry inhibitor and in some cases as endosomal pH modulator interfering with viral release, (2) It is an attractive pharmacophore for many protease inhibitors like the inhibitors for Fibroblast activated protein (FAP: Ramser et al., 2009), Bacillus thuringiensis serotype Kurstaki(BTK) proteases: (Barnard et al., 2014), Platelet-Derived Growth Factor (PDGFR), and as ALK5 inhibitors for TGF-β RI Kinase, and (3) It also acts as an immunomodulator. Thus, the heterocycle compound quinoline and it’s derivatives have found applications as an anticancer, anti(myco)bacterial, antiviral, anticonvulsant, anti-inflammatory, and cardiovascular activity regulator (Marella et al., 2013).
A detailed insight into quinoline's mechanism as an anti-COVID reflects three potential targetclasses: Class 1. As an inhibitor during viral entry, Class 2. As an inhibitor for transmembrane proteases, and Class 3. As a modulator of the immune response (Alexpandi et al., 2020). The first two target classes are primarily related to coronavirus, whereas the third class refers to the host.
The coronavirus entry into the host cell relies on the interaction of its spike glycoprotein with the Angiotensin receptor (ACE-2) of the host (human) (Shang et al., 2020). This entry mechanism is nearly universal for other members of the betacoronavirus of the coronaviridae family. The attachment to the host cells occurs through the S1 subunit of the betacoronavirus spike proteins, marking the viral fusion (H. Chakraborty et al., 2020). Quinoline derivatives have been reported to be an antagonist for ACE2 receptors. Figure 1 summarizes some potent antagonists for the ACE2 receptor.
Figure 1.

Showing antagonist for inhibiting the activity of SARS-COV-2 at Different stages and mechanism of SARS-COV-2 from entry into the host cell to generation of new viral species.
The ACE2 receptor facilitates the entry of the viral particles through endocytosis and allows the transfer of a single stranded RNA strand into the host cell. Proteases also mediate the entire process at different steps. Main Protease is a cysteine protease that processes itself and then cleaves into several non-structural viral proteins having roles in viral replication. Thus, the protease has been suggested as one of the most facile and pragmatic target for drug repurposing owing to its role in the viral cycle and the ease of its biochemical assays (Dai et al., 2020).
Besides the above two targets that focus on viral particles, the host immune response can help prevent the replication and infection of the virus. However, an overactive immune system can cause a cytokine storm (C. Chakraborty & Bhattacharjya, 2020) leading to life-threatening conditions. An anti-COVID agent that can avoid the overactivation of human cells and modulates the immune response can be of therapeutic utility.
Hypothesizing that quinoline derivatives can emerge as a potent anti-COVID agent, targeting either of the above targets individually or in combination, we have screened an extensive library of hundred plus FDA approved quinoline based drugs using structure-based methods. Our focus has been to target the coronavirus, and hence the first two classes of targets have been considered. Among the class 1, we selected Receptor binding Domain of Spike protein of SARS-COV-2 (PDB ID 6M0J: Target 1), and among class 2 targets we have chosen: (a) Replicase polyprotein through Main Protease Mpro of SARS-COV-2 (PDB ID 5R80: Target 2) and Papain like protease (PDB ID 6W9C: Target 3) (b) Viral assembly through N-terminal RNA binding domain of Nucleocapsid protein of SARS-COV-2 (PDB ID 6M3M: Target 4), and (c) Viral RNA synthesis by targeting RNA Dependent RNA Polymerase (PDB ID 7BTF: Target 5)
2. Methods
2.1. Drugs Screened for analysing repurposing potential
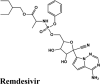
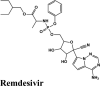
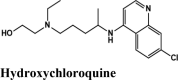
One hundredthirty-onequinoline based different category of drugs that are FDA approved as antimalarial, antiviral, inhibitors of BTK and PDGFR, antibiotics and respiratory specificdrugs were selected for structure-based screening. Appropriate controls viz., hydroxychloroquine and the non-quinoline drugs- remdesivir and galidesivir were chosen to compare the interactions. Molecular modelling Schrödinger Software (v 2020) and Maestro 11.1 platform have been used for computational studies.
2.2. Targets selected to identify anti-COVID drug
Five targets were chosen for the study: (1) Receptor binding domain of SARS-COV-2 interacting with human ACE2 receptor (PDB ID: 6M0J) (2) chymotrypsin-like main protease of the virus MPro or 3CLPro, (MPro, PDB ID: 5R80) (3) Papain-like Protease from SARS-COV-2PLPro (PDB ID: 6W9C), (4) N-terminal RNA binding domain of Nucleocapsid protein of SARS-COV-2 (PDB ID: 6M3M), and (5) RNA dependent RNA Polymerase from SARS-COV-2 (PDB ID: 7BTF). Also, two additional PDBs (6LU7, 6WTT) for the target Main Protease were chosen that are co-crystallized structures with different inhibitors. The coordinates and detailed sequence information was obtained from RCBS Protein Data Bank (www.rcsb.org). The drugs were drawn using the Marwin Sketch tool as Mol2 format and imported in the software. After the ligand preparation using LigPrep v2.9 (OPLS3 force field, pH 7.0 ± 2.0) and protein preparation using Protein Preparation Wizard followed by binding site identification using SiteMap. The grid was generated with the box-dimensions (a) 120*120*120 for 6M0J against amino acids residue within the active site having Tyr495, Tyr505, Gly496, Asn487, and Gly502 (b) 80*80*80 for 6W9C within the active site having Asp103, Gly164, and Gly270, (c) 88*88*88 for 7BTF within the active site having Asp760 and Asp761, and (d) 112*112*112 for 6M3M including residues Ala51, Tyr112, and Tyr124. For the main of protease of SARS-COV-2, grid was generated against bound co-crystallized ligand N3 inhibitor with box dimension 72*72*72. After generation of grid docking of energy minimized ligands was performed using Extra Precision mode in Glide module.
For identification of possible receptor-ligand interaction analysis, more than five poses per ligand were selected, and docking parameters were computed using XP-visualizer. The drug interactions with the target, GScores, docking scores, and Glide EModel were thoroughly analysed to get the best interaction pose of ligand (drug) with the receptor. For all targets, appropriate controls were selected. Hydroxychloroquine serves as a control for Target S protein and MPro. Since galidesivir is screened as a potent drug for targeting RdRp polymerase, we used it as its control (Elfiky, 2020).
2.3. In silico ADME analysis
The pharmacokinetic (PK) properties of quinoline-based library viz., absorption, distribution, metabolism, excretion, and toxicity (ADMET) were calculated using the BioLuminate module of the SchrödingerMolecular Modelling Software (M/s Schrödinger, LLC, New York, NY, v. 2020).
2.4. Enrichment studies
Enrichment studies have been performed to assess the enrichment of active compounds in a screening process that includes a set of actives and a set of decoys (1000 decoys). The screening can be done with any program: Glide, Shape Screening, Phase. We used Glide program (docking tool) for the screening process. The active ligands input for the panel was taken from the output from the screening program having highest GScore with each therapeutic targets of SARS-COV-2 and, set of decoys were used from Schrodinger Maestro 11.0.
2.5. Molecular Dynamics simulation
Molecular dynamics simulations have been used extensively to explore the biological processes and ligand interactions in recent years (Dror et al., 2012; Duan et al., 2019; Hollingsworth & Dror, 2018; Santhanam et al., 2019). As through docking we have screened that afatinib is the best drug among all quinoline based drugs to target proteases of SARS-COV-2. Therefore, MD simulations have been performed with Afatinib drugs with all five therapeutics targets of SARS-COV-2. We performed all-atom explicit solvent MD simulations on the docked protein-afatinib (6M0J-afatinib, 5R80-afatinib, 6W9C-afatinib, 7BTF-RdRp, 6M3M-Nprotein) complexesto evaluate the binding of the afatinib at the active site of the protein with respect to the simulations run length using AMBER software (Yang et al., 2016). The geometry of the ligands was optimized, and the bond, angle, dihedral, and partial charges [RESP] were generated using HF/6-31G* in Gaussian09 (Vanquelef et al., 2011) All the ligand parameters were saved in an AMBER compatible library file for each ligand considered for the study. All the complexes were immersed in a cubic water box (TIP3P water molecules) with counterions to ensure the overall electroneutrality of the systems. The protein counterparts were simulated with modified ff99SB force field (Maier et al., 2015), and the parameters for the counterions were taken from the literature (Joung & Cheatham, 2008).
We used Particle Mesh Ewald treatment (Cheatham et al., 1995) (for long-range electrostatics) with periodic boundary conditions for performing the simulations. All the systems underwent minimization to remove close contact in the systems, if any, followed by heating (50 ps, NVT) and equilibrating (5 ns). Finally, 100 ns long MD simulations were performed. For analysis, the Cpptraj code (Roe & Cheatham, 2013) was used for computing RMSD fluctuations, structure clustering, and the number of hydrogen bonds between ligand and protein molecules.
2.6. MM-PBSA calculation
LigPlot + software (Laskowski & Swindells, 2011) was used to sketch the interactions of the afatinib with protein. Molecular Mechanic/Poison Boltzmann Surface Area (MM-PBSA) (Onufriev et al., 2000) calculations were performed to evaluate the binding proclivity of the ligand to the protein. The binding free energy gives information about different kind of interactions (potential energy and polar and non-polar solvation energy) and computed by using the following equation: (Bhardwaj et al., 2020)
where ΔG binding refers to change in energy after the formation of afatinib- ligand complex and G receptor is energy of free receptor without afatinib and G ligand is the energy of afatinib + in unbound form.
3. Results and discussion
3.1. Docking and analysis
Quinoline pharmacophore is an important moiety according to the biological point of view. Its derivatives have been used in many fields for the progression of Alzheimer's diseases (Sureshkumar et al., 2020) as an antimalarial drugand target serine protease as an anticancer agent, and as an antimicrobial and antifungal agent (Marella et al., 2013; Desai et al., 2017). Hence in present work, we have reported in silico studies of quinoline-based, FDA-approved drugs for docking studies with crystal structures of SARS-COV-2. We have screened a total of hundred plus FDA approved quinoline based drugs. They are categorised based on their approved clinical application. (their detailed properties and mode of action are displayed in supplementary).
Five targets are used for this study:
Class 1: Targeting viral entry
Target 1: RBD S protein that provides a viral surface for the attachment to host cell receptor ACE2.
Class 2: Targeting viral replication
Class 2a: Replicase polyprotein
Target 2: MPro and PLPro both are responsible for proteolysis of viral polyprotein into functional unit.
Target 3: Papain-like proteases
Class 2b:Viral assembly
Target 4: Nucleocapsid proteins
Class 2c:Viral RNA synthesis by targeting RNA Dependent RNA Polymerase
Target 5: RdRp is responsible for replicating viral genome
Tables 1 and 2 summarizes the top-ranking compounds with their respective targets and their 2 D LigandInteraction Diagram (LID) are displayed in Figures 2–6.
Table 1.
GScore of top-ranking drugs for different category of targets 1) 6M0J 2)5R80 3) 6W9C 4) 6M3M 5) 7BTF.
| Target 1 6M0J | Target 2 5R80 | Target 3 6W9C | Target 4 6M3M | Target 5 7BTF |
|---|---|---|---|---|
| −10.8 | −9.04 | −8.57 | −8.51 | −8.97 |
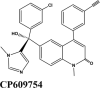
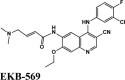
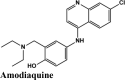
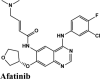
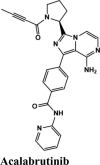
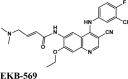
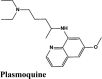
| CP609754 | Afatinib | Amodiaquine | Primaquine | EKB-569 |
| −9.72 | −8.77 | −8.53 | −8.50 | −7.81 |
| Afatinib | Tezacaftor | Afatinib | Amodiaquine | Campothecin |
| −9.6 | −8.48 | −8.4 | −7.61 | −7.53 |
| Saquinavir | EKB-569 | Saquinavir | Saquinavir | Amodiaquine |
| −9.52 | −8.00 | −8.15 | −7.57 | −7.10 |
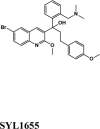
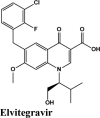
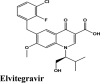
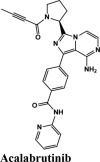
| Acalabrutinib | Saquinavir | SYL1655 | Elvitegravir | Primaquine |
| −9.41 | −7.75 | −8.09 | −7.29 | −7.04 |
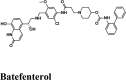
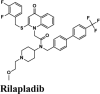
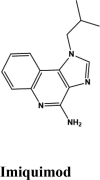
| Rilapladib | Batefenterol | Batefenterol | Imiquimod | Dequalinium |
| −9.02 | −7.48 | −7.57 | −7.26 | −7.04 |
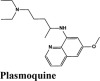
| Plasmoquine | Alatrofloxacin | Quarfloxin | Afatinib | Elvitegravir |
| −7.4 | −7.4 | −7.68 | −7.23 | −6.7 |
| Elvitegravir | Elvitegravir | Campothecin | Pamaquine | Imiquimod |
| −6.91 | −6.3 | −6.67 | −5.15 | −6.4 |
| Amodiaquine | Amodiaquine | Elvitegravir | Acalabrutinib | Saquinavir |
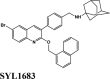
| −6.93 | ||||
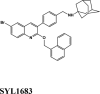
| SYL1683 | ||||
| −6.75 | −8.02 | −8.42 | −6.85 | −8.4 |
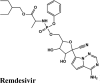
| Remdesivir | Remdesivir | Remdesivir | Remdesivir | Remdesivir |
| −5.37 | ||||
| Galidesivir | ||||
| −6.05 | −5.3 | −5.11 | −4.99 | −6.1 |
| HQ | HQ | HQ | HQ | HQ |
| −4.9 | −4.32 | −5.77 | −3.58 | −5.9 |
| EKB-569 | Acalabrutinib | Plasmoquine | EKB-569 | Afatinib |
| −4.01 | −4.84 | −2.63 | −5.4 | |
| Plasmoquine | Acalabrutinib | Plasmoquine | Acalabrutinib |
Table 2.
Illustrations of top-ranking antiviral, antimalarial and, antibiotic, kinase inhibitor and anti-asthmatic Drugs.
 |
 |
 |
 |
 |
 |
 |
 |
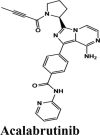 |
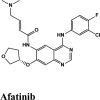 |
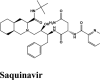 |
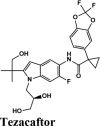 |
 |
 |
 |
 |
 |
 |
 |
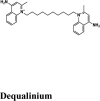 |
Figure 2.

2D Ligand Interaction Diagram for Top scores drugs to target RBD of SARS-COV-2.
Figure 3.

2D Ligand Interaction Diagram for Top score drugs to target Main Protease of SARS-COV-2.
Figure 4.

2D Ligand Interaction Diagram for Top score drugs to target Papain Protease of SARS-COV-2.
Figure 5.

2D Ligand Interaction Diagram for Top score drugs to target Nucleocapsid Protein of SARS-COV-2.
Figure 6.

2D Ligand Interaction Diagram for Top score drugs to target RNA dependent RNA polymerase of SARS-COV-2.
3.1.1. Docking results for class 1, target 1: viral entry
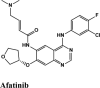
The antivirals were the top scorers when averaged over the docking scores and energy (Figure 7(a)). Even though kinase inhibitors average values were lower, the top rankers included inhibitors with better potential like Afatinib, Acalabrutinib and Rilapladib.
Figure 7.

Average GScore variation for different category of Drugs with all therapeutics targets of SARS-COV-2 (Antimalarial, Antiviral, kinase inhibitor, Antiviral, Respiratory specific).
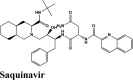
RBD S protein is responsible for the entry of SARS-COV-2 into a host cell, which simultaneously binds with ACE2 and TMPRSS into the host cell. Therefore, targeting RBD Spike protein of SARS-COV-2 is the most prior step (Lan et al., 2020). Among all the screened drugs CP-609754 had the highest G-Score of −10.8 kcal/mol. The binding was approximately 78.51% higher than hydroxyquinoline (having a G-score of −6.05 kcal/mol). An insight into its binding highlight the additional hydrophobic energy due to the terminal propargyl group. However, not all the amino acids lining the binding pocket adds to the interaction, and the significant contributors are given in Table 3. A good ligand should have a combination of best fit and docking parameters. The next ranking molecules were Saquinavir and Afatinib, showing a G-score of −9.6 kcal/mol and −9.72 kcal/mol, respectively. Docking pose reveals a higher contribution from polar forces like the H bonding. Saquinavir and Afatinib have the best combination of both G-score and amino acid participation resulting in a better fit. Saquinavir has more fitting in the active site of binding pocket as there are five hydrogen bonds between heteroatoms of saquinavir and within the active site of RBD. Three oxygen forms hydrogen bonds with Gln96, Arg403, Gln406, Gln409 and Lys417. Also, the amine group of the alkyl chain forms a hydrogen bond with Glu406. Aromatic ring in saquinavir forms п-п stacking with Arg403. The residue in the active site with which saquinavir binds are Arg408, Lys417, Tyr505, Gly416, Ile418, which are crucial for binding with RBD of SARS-COV-2 (Sachdeva et al., 2020).
Table 3.
Interacting amino acids for Drugs with RBD of SARS-COV-2 (6M0J) with G-Score above Hydroxyquinoline and active side residue are marked bold.
| Compound (6M0J) | Mode of Action | Hydrophobic | Polar | Hydrogen bonding | п-п stacking | Charged |
|---|---|---|---|---|---|---|
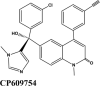 |
Farnesyl transferase inhibitor | Val93, Leu29, Ala387, Pro389, Phe390 | His33, Gln96, Thr92, Gln388 | Thr92 | Lys26, Asp30, Glu37, Arg393 | |
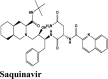 |
Anti-HIV Protease inhibitor | Chain A: Phe390, Pro389, Ala387, Ala386, Val93, Leu29 Chain E: Tyr505, Ile418, Gly416 |
Chain A: Gln388, Asn33, Gln96, Thr92, Gln E: 409 | Chain A: Gln96, Chain E: Lys417, Gln409, Gln406, Arg403 | Chain E Arg403, | Chain A: Glu37, Arg393, Asp30, Lys26 Chain E: Glu406, Asp405, Arg403, Arg408, Lys417 |
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Chain A: Ala387, Ala386, Leu29, Val93, Pro389 Chain E: Tyr495, Gln496, Tyr453 |
Chain A: His33, Gln388, Gln96 Chain E: Gln 493, Ser494 | Chain E: Tyr 453, Arg403, | Chain A: Glu37, Asp38, Asp30, Lys353, Lys26, Arg393 Chain E: Arg403, |
|
 |
Bruton Tyrosine Kinase Inhibitor | Chain A: Pro389, Ala 387, Ala386, Met383, Gly354, Phe356 Chain E: Tyr505, Gly504, Val503, Gly502 |
Chain A: Asn33, Gln388, Thr324 | Chain E: Gly504, Arg403, Asp405 | Chain E: Tyr505 | Chain A: Glu37, Arg393 Chain E: Asp405, Arg408, Arg403, |
 |
Lipoprotein associated phospholipase (A2) Lp-plA2 Inhibitor | Chain A: Pro389, Ala387, Phe390, Ala386, Leu29, Val93 Chain E: Tyr505, Leu455, Gly504 |
Asn33, Gln388, Thr92, Gln96 | Chain A: Asp30 | Chain E: Arg403 | Chain A: Asp30, Lys26, Arg393 Chain E: Lys417, Arg408, Arg403 |
 |
Synthetic Antimalarial Drug | Chain A: Pro389, Ala387, Ala386, Val93, Leu 29 Chain E: Tyr505 |
Chain A: Asn33, Gln388, Thr92, Gln96 | Chain A: Asn33, Gln96, Arg393, Glu37 Chain E: Tyr505, Arg403 |
Chain A: Arg393, Asn33 | Chain A: Lys26, Asp30, Glu37, Arg393, Chain E: Arg403, Asp405 |
 |
Antimalarial Drug Anti-arthritis |
Chain E: Tyr453, Tyr495, Gln496, Phe497, Tyr505 Chain A: Phe390, Pro389, Ala387, Ala386 |
Chain E: Ser494, Gln493 | Chain A: Lys353, Chain E: Asp405 | Chain E: Arg403 | Chain A: Asp38, Glu37, Arg393, Lys353, Chain E: Arg403, Asp405, Glu406 |
 |
Antimalarial Drug | Chain A: Leu29, Val93, Phe390, Pro389, Ala387, Ala386 Chain E: Gly504, Tyr505 |
Chain A: Asn33, Gln96 | Chain E: Asp405 | Chain A: Lys26, Arg393, Asp30, Glu37 Chain E: Arg403 |
|
 |
Anti-HIV Inhibitor | Chain A: Pro389, Ala387, Ala386, Met383, Phe356, Gly354 Chain E: Tyr505, Gly504, Val503, Gly502, Ile418, Gly416 |
Chain A: Asn33, Gln388, Thr324 Chain E: Gln409 |
Chain E: Asp405, Arg403, Lys417, Gln409 | Chain E: Asp405, | Chain E: Lys417, Arg408, Arg403 Chain A: Arg393 |
 |
Potent Irreversible EFGR receptor | Chain A: Leu29, Val93, Phe390, Pro389, Ala387, Ala386 Chain E: Tyr505, Tyr495, Gly496, Tyr453 |
Chain A: Asn33, Gln96, Gln388 Chain E: Ser494, Gln493 | Chain E: Arg403, Tyr453 Chain A: Asn33 |
Chain A: Asp30, Lys26, Arg393, Lys353, Glu37, Asp38 Chain E: Arg403 |
|
 |
Anti-HIV Inhibitor | Chain A: Pro389, Ala387, Phe390 Chain E: Tyr505 |
Chain A: Asn33, Gln388, Gln96 Chain E: Gln409 |
Chain A: Asp30, Glu37 Chain E: Arg403 |
Chain E: Arg408 | Chain A: Asp30, Glu37, Arg393 Chain E: Lys417, Arg408, Arg403, Asp405, Glu406 |
 |
Antiviral Drug |
Chain A: Pro389, Phe390 Chain E: Tyr505, Tyr495, Gly496, Tyr453, Ile418, Gly416 |
Chain A: Asn33, Chain E: Ser494, Gln493, Gln409, Tyr415 | Chain A: Asp30 Chain E: Glu406, Gln409, Tyr505, Arg403 |
Chain A: Asp30, Glu37, Glu35, Asp38 Arg393, Lys353 Chain E: Lys417, Arg403, Asp405, Glu406 |
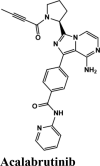
Acalabrutinib exhibited the G-score of −9.52 kcal/mol with key interactions between oxygen atoms and Arg403 and Gly505 as H-bonding and п-п stacking of aromatic ring with Tyr505. The protonated nitrogen forms a salt bridge with Asp405.
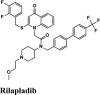
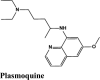
Next in series were rilapladib and plasmoquine with G-score −9.41 kcal/mol and −9.02 kcal/mol (Docking Parameters Table 4). Rest of drugs details have provided in Table S7*SI.
Table 4.
Docking Parameters for Highest scoring drugs with receptor binding domain of SARS-COV-2 (PDB ID 6M0J).
| Drugs | GScore | DScore | Lipophilic EVDW | Hbond | EModel |
|---|---|---|---|---|---|
| CP609754 | −10.8 | −8.26 | −6.23 | −2.34 | −90.242 |
| Saquinavir | −9.6 | −8.32 | −4.46 | −1.96 | −83.616 |
| Afatinib | −9.72 | −7.24 | −4.39 | −1.45 | −82.436 |
| Acalabrutinib | −9.52 | −8.73 | −3.3 | −1.1 | −80.606 |
| Rilapladib | −9.41 | −8.24 | −4.89 | −0.29 | −79.456 |
| Plasmoquine | −9.02 | −8.13 | −6.23 | −2.6 | −75.299 |
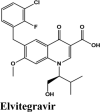
| Elvitegravir | −7.4 | −7.2 | −5.23 | −1.3 | −78.818 |
| Amodiaquine | −6.91 | −5.64 | −3.15 | −1.08 | −49.763 |
| SYL1683 | −6.93 | −5.92 | −4.25 | 0 | −70.42 |
| Remdesivir | −6.75 | −6.75 | −5.22 | −2.73 | −80.717 |
| HQ | −6.05 | −5.64 | −3.15 | −1.08 | −43.363 |
| EKB-569 | −4.9 | −4.4 | −4.4 | −1.1 | −64.352 |
The screened drugs having a G-Score greater than 8.00 kcal/mol showed in general, the binding interactions with the following residues: H-bonding with Gln496, Lys417, and Arg408, pi-pi stacking with Tyr505, Tyr453, Tyr449, Glu37, Asp38, Lys68 are considered as potent drugs for blocking the Spike-ACE2 interactions.
3.1.2. Docking results for targets of Class 2: Replication Target 1: Interaction analysis within active site of SARS-COV-2 main protease MPro (three PDB IDs: 5R80- complexed withZ18197050, 6WTT-complexed with inhibitor GC376, 6LU7-with inhibitor N3)
When the quinoline library was docked on MPro PDB (5R80), and the average docking scores and binding energies compared, respiratory specificand antivirals emerged as most promising (Figure 7(b)).
MPro or the chymotrypsin like protease (3CLpro)/C30 Endopeptidase produces non-structural proteins that later play a role in mediating the replication of the virus (Elzupir, 2020). Therefore, inhibiting the activity of this enzyme can block viral replication. Once inside the host cell, the proteases of the virus cleave the mRNA into structural and non-structural proteins. The protease belongs to cysteine protease family with cysteine-histidine catalytic dyad. 3CLpro monomer has three domains, domain I (residues 8-101), domain II (residues 102-184) and domain III (residues 201-303), and a long loop (residues 185-200) connects domains II and III. The active site of 3CLpro is located in the gap between domains I and II, and has a Cys-His catalytic dyad (Cys145 and His41 (Vatansever et al., 2020). Recently, aminoquinolines have been reported as inhibitors of certain cysteine proteases (Braga et al., 2017). However, a greater number of antivirals and inhibitors scored above hydroxyquinoline. Only tezacaftor, that is a cystic fibrosis transmembrane conductance regulator (CFTR) was able to score a higher G-Score.
Molecules with docking scores more than that of hydroxyquinoline (G-score −5.4) are summarised in the Table 1. Ligand Interaction Diagram for top scorers with 5R80 are shown in Figure 3 and their interaction information are provided in (Table 5). Afatinib has the best G-score of −9.04 kcal/mol, followed by tezacaftor G-score of −8.77 kcal/mol (Docking Parameters) (Table 6). Table G-score of rest of drugs have represented in Table S8*SI.
Table 5.
Ligand Interaction information for top scoring drugs to inhibit the activity of main protease of SARS-COV-2 (5R80).
| Compound | Mode of Action | Hydrophobic | Polar | п-п stacking | H-Bond | Charged |
|---|---|---|---|---|---|---|
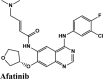 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Phe140, Leu141, GLY143, Cys145, Leu27, Cys44, Tyr54, Pro52, Met49, Met165, Leu167, Pro168 | Asn142, Ser144, Thr26, Thr25, His163, Hie164, Gln189, Thr190, Gln192 | Glu166 | Hip41 Asp187 Arg188 Glu166 |
|
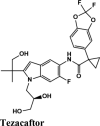 |
Cystic Fibrosis Transmembrane Conductance regulator |
Leu141, Gly143 Cys145, Met49 Met165 |
Gln189, Hie164, His163, Gln189, Hie164, His163, Ser46 | Gly143, Thr25, Thr24, Ser46 | Hip41, Arg188, Glu166 | |
 |
An irreversible Epidermal Receptor Growth receptor Tyrosine kinase |
Phe140, Leu141, Gly143, Cys145, Met165, Cys44, Met49, Pro52, Tyr54 | Hie172, Asn142, Ser144, Thr190, Gln189, Gln192, Thr25, Hie164, His163 | Glu166, Thr190 | Hip41 | Hip41 Asp187 Arg188 Glu166 |
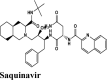 |
Anti-HIV Protease inhibitor |
Tyr54, Cys44, Met49, Phe140, Leu141, Cys145, Met165, Leu167, Pro168 | Thr45, Ser46, Hie172, Asn142, Ser144, Thr25, His163, Hie164 | Glu166 | Hip41 | Hip41 Asp187 Arg188 Glu166 |
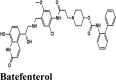 |
ꞵ2 adrenoceptors agonist, muscarinic receptor antagonist | Leu167, Met165, Pro168, Phe140, Leu141, Met49, Gly170, Gly138, Val171 | Gln189, Hie164, Thr190, His163, Gln192, Thr169, Hie172, Ser139, Asn142 | Gly138, Glu166, Thr169 | Hip41 | Hip41, Arg188, Asp187, Glu166 |
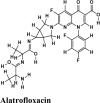 |
Antibacterial Antineoplastic DNA topoisomerase inhibitor |
Phe140, Leu141, Cys145, Gly143, Met49, Tyr54 | Gln189, Thr190, Hie164, Asn142, Hie172 | Glu166, Asn142, Phe140, Hie164, Glu166 | Hip41 | Hip41, Arg188, Asp187, Glu166 |
 |
Pro168, Leu167, Met165, Met49, Cys44, Tyr54 | Gln189, Thr190, Gln192 | Hip41, Arg188, Asp187, Glu166 | |||
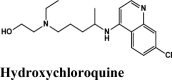 |
Antimalarial Drug Anti-arthritis |
Cys145, Met165, Gly143, Leu141, Phe140, Tyr54, Pro52, Met49, Cys44 | Hie164, His163, Ser144, Asn142 | Glu166 | Hip41 | Hip41, Arg188, Asp187, Glu166 |
 |
Anti-HIV Inhibitor | Pro168, Leu167, Met165, Val186, Tyr54, Pro52, Met49, Cys44, Cys145 | Gln189, Thr190, Gln192, Hie164, Asn142 | Hie164 | Hip41, Arg188, Asp187, Glu166 | |
 |
Antimalarial Drug | Met49, Leu27, Pro168, Leu167, Met165, Gly170, Leu141, Gly143 | Gln189, Thr190, Gln192, Hie164, His163, Asn142, Hie172, Ser144, Thr25, Thr26 | Glu166 | Hip41, Arg188, Asp187, Glu166 | |
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Leu167, Met165, Pro168, Met49, Tyr54, Cys44, Gly143, Cys145 | Gln192, Gln189, Thr190, Hie164, Ser46, Thr45, Thr25 | Gln189, Arg188 | Hip41, Arg188, Asp187, Glu166 | |
 |
Antimalarial Drug | Met165, Pro168, Met49, Phe140, Leu141, Gly143, Cys145 | Gln189, Thr190, Hie164, His163, Ser144, Asn142, Hie172 | Asn142 | Hip41, Arg188, Glu166 | |
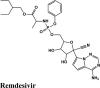 |
Antiviral Drug |
Ala191, Pro168, Phe140, Leu141, Cys145, Met49, Met165, Gly143 | Gln189, Thr190, Hie164, His163, Ser144, Asn142, Hie172 | Asn142 | Glu166, Arg188, Hip41 |
Table 6.
Docking Parameters for top scoring drugs with main protease of SARS-COV-2 (PDB ID 5R80).
| Drugs | GScore | DScore | Lipophilic EVDW | Hbond | EModel |
|---|---|---|---|---|---|
| Afatinib | −9.04 | −7.86 | −5.44 | −2.03 | −82.179 |
| Tezacaftor | −8.77 | −8.77 | −3.44 | −3.75 | −73.611 |
| EKB-569 | −8.48 | −7.29 | −4.91 | −0.6 | −80.502 |
| Saquinavir | −8.0 | −8.32 | −4.46 | −1.96 | −113.245 |
| Batefenterol | −7.75 | −6.95 | −3.46 | −0.56 | −77.398 |
| Alatrofloxacin | −7.48 | −6.23 | −3.73 | −2.9 | −80.502 |
| Elvitegravir | −7.4 | −7.2 | −5.23 | −1.3 | −83.219 |
| Amodiaquine | −6.3 | −6.3 | −4.52 | −1.57 | −63.984 |
| Remdesivir | −8.02 | −8.018 | −5.22 | −2.73 | −82.640 |
| HQ | −5.3 | −5.2 | −1.2 | −0.6 | −82.284 |
| Acalabrutinib | −4.32 | −4.31 | −3.85 | −0.82 | −72.269 |
| Plasmoquine | −4.01 | −3.78 | −3.85 | −0.5 | −40.857 |
Among all the drugs, afatinib with GScore −9.04 was well fitted into the binding pocket of MPro and the binding was 67.4% higher than that of HCQ. A similar trend was observed when the molecules were docked on other PDBs of MPro (6WTT, 6LU7). Afatinib was the top scorer with GScore of −9.3 kcal/mol with 6WTT and −9.943 kcal/mol with 6LU7 and also showed binding with catalytic dyad forming п-п stacking with Hip41 and interaction with Cys145. The binding pocket is primarily marked by the catalytic dyad of amino acids Cys145 and His41 (Khan et al., 2020). All reported residues in the active site of binding pocket of Mpro and as evident in the co-crystallized PDB bind to afatinib. The quinoline ring in afatinib showed п-п stacking with Hip41 along with the H-bond between protonated nitrogen with Glu166 and covalent interaction of chlorine atom with Asn142 and Gly143. Also, afatinib bind with 12 Hydrophobic residues and with ten polar residues. Therefore, afatinib can be considered the potent drug for targeting main protease of SARS-COV-2.
3.1.3. Target 3: Interaction characterization of quinoline based drugs with SARS-COV-2 papain like protease
When the quinoline library was docked on PLPro PDB (6W9C), averagedG-scores, docking scores and binding energies were compared, respiratory specific are served as most promising (Figure 7(c)).
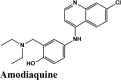
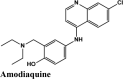
PLPro is responsible for the cleavages of N-terminus of the replicate poly-protein to release non-structured proteins (Nsp1-3), essential for correcting virus replication. PLPro was also confirmed to be significant in antagonizing the innate immunity of the host. As an indispensable enzyme in the process of coronavirus replication and infection of the host, PLPro has been a popular target for coronavirus inhibitors. It is very valuable for targeting PLPro to treat coronavirus infections, but no inhibitor has been approved by the FDA for marketing. All quinoline based drugs were docked with crystal structure of PLpro (PDB ID 6W9C). Docking Parameters for high scoring drugs are displayed in Table 7. Remdesivir was considered as control with GScore of −8.4 kcal/mol. Among all screened drugs again, amodiaquine, afatinib and saquinavir having G-scores −8.57, −8.53 and −8.4 respectively scored above remdesivir. The binding pocket is primarily marked by the amino acids Gly270, Asp103, Gly164 and their interaction information are provided in Table 8. Heteroatoms of amodiaquine viz., protonated nitrogen and nitrogen atom of quinoline ring form H-bond with Asn109, Asp108, Val159 and Glu161. Other residues in the binding pocket of PLPro with Amodiaquine forms covalent interaction are Cys270, Leu162, Trp106, Val159, Gly160.
Table 7.
Docking parameters of top scorer drugs to target papain protease of SARS-COV-2 (PDB ID 6W9C).
| Drugs | GScore | DScore | Lipophilic EVDW | HBond | EModel |
|---|---|---|---|---|---|
| Amodiaquine | −8.57 | −8.56 | −5.34 | −1.62 | −90.424 |
| Afatinib | −8.53 | −7.49 | −5.22 | −0.32 | −88.913 |
| Saquinavir | −8.4 | −8.38 | −5.78 | −1.99 | −105.105 |
| SYL1655 | −8.15 | −8.11 | −7.76 | −0.48 | −86.282 |
| Batefenterol | −8.09 | −8.03 | −5.23 | −1.49 | −116.512 |
| Quarfloxin | −7.57 | −7.57 | −6.87 | −0.7 | −90.9 |
| HCQ | −5.11 | −5.06 | −3.76 | −0.7 | −44.246 |
| Remdesivir | −8.42 | −8.42 | −5.57 | −2.29 | −89.549 |
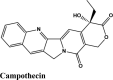
| Campothecin | −7.68 | −7.68 | −6.52 | −0.42 | −72.146 |
| Elvitegravir | −6.67 | −6.54 | −5.27 | −1.05 | −62.598 |
| Plasmoquine | −5.77 | −5.54 | −4.44 | −0.85 | −58.285 |
| Acalabrutinib | −4.84 | −4.82 | −4.72 | −0.04 | −73.443 |
Table 8.
Interacting amino acids for Drugs having G-Score above Hydroxyquinoline for Papain Protease of SARS-COV-2 (PDB ID: 6W9C).
| Compound | Mode of Action | Hydrophobic | Polar | п-п Stacking | H-Bond | Charged |
|---|---|---|---|---|---|---|
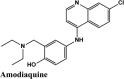 |
Antimalarial, Anti-inflammatory | Chain A: Gly160, Leu162 Chain B: Leu162, Val159, Cys270, Gly160 Chain C: Leu162, Val159 Gly160, Trp106, Cys270 |
Chain A: Asn109, Gln269 Chain B: Asn109, Gln269 Chain C: Asn109, His89, Gln269 |
Chain B: Glu161 Chain C: Asn109, Asp108 |
Chain A: Glu161 Chain B: Glu161, Asp108 Chain C: Asp108 |
|
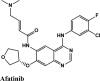 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Chain A: Val159, Gly160, Leu162 Chain B: Val159, Gly160, Trp106, Cys270 Chain C: Val159, Leu162, Gly160, Cys270 |
Chain A: Asn109, Gln269, Thr158 Chain B: Asn109, Gln269, His89 Chain C: Thr158, Gln 269, Asn109 |
Chain C: Asn109 | Chain A: Glu161, Chain B: Asp108, Chain C: Glu161 | |
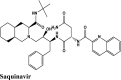 |
Anti-HIV Protease inhibitor |
Chain A: Leu162, Val159, Gly160 Chain B: Leu162, Val159, Cys270 Chain C: Leu162, Val159, Cys270, Gly160 |
Chain A: Thr158, Asn109, Gln269 Chain B: Thr158, Asn109, Gln269, His89, Ser85 Chain C: Thr158, Asn109, Gln269, |
Chain B: Leu162, Val159, Gly160 Chain C: Asp108, Asn109 |
Chain A: Glu161 Chain B: 161 Chain C: Glu161, Asp108 |
|
 |
Anti-HIV Protease inhibitor |
Chain A: Val159, Gly160, Leu162 Chain B: Leu162, Cys270, Gly160 Chain C: Cys270, Leu162, Val159 |
Chain A: Asn109, Thr158, Gln269 Chain B: Asn109, Gln269 Chain C: His89, Gln269 |
Chain A: Asp108, Glu161 Chain B: Glu161 Chain C: Glu161, Asp108 |
||
 |
ꞵ2 adrenoceptors agonist, muscarinic receptor antagonist | Chain A: Val159, Ala86 Chain B: Leu162, Tyr171, Gly160 Chain C: Trp93, Ala107, Val159 |
Chain A: Thr58, Ser85, His89 Chain B: Asn156, His89 |
Chain C: Asp108, Chain B: Glu161 | Chain A: Glu161, Arg82, Lys157 Chain B: Glu167, Glu161 Chain C: Lys92, Asp108 |
|
 |
Antineoplastic Inhibits RNA Polymerase activity |
Chain A: Val159, Leu162, Ala86, Gly160 Chain B: Val159, Cys270 Chain C: Leu162, Cys270, Gly160 |
Chain A: Thr158, Ser85, His89, Gln269 Chain B: Gln269, His89, Asn109 Chain C: Asn109, Gln269, Thr158 |
Chain A: Val159 | Chain A: Arg82, Glu161 Chain B: Asp108 Chain C: Glu161 |
|
 |
Antimalarial Drug Anti-arthritis |
Chain A: Leu162, Gly160 Chain B: Leu162, Val159, Cys270, Gly160 |
Chain A: Asn109, Gln269, Thr158, Chain B: Thr158, Gln269, Asn109 Chain C: Asn109, Gln269 |
Chain A: Gly160 Chain B: Val159, |
Chain A: Gly160 Chain B: Val159 |
Chain A: Glu161 Chain B: Glu161 |
 |
Topoisomerase Inhibitor | Chain A: Leu162, Val159 Chain B: Val159, Gly160, Leu162, Cys270 Chain C: Val159, Leu162, Gly160, Cys270 |
Chain B: Thr158, Gln269, Asn109 Chain A: Gln269, Thr158, Asn109 Chain C: Asn109, Gln269 |
Chain B: Gly160 | Chain B: Glu161, Asp108 Chain A: Arg108 Glu161 Chain C: Glu161, Asp108 |
|
 |
Anti-HIV Inhibitor | Chain A: Leu162 Chain B: Leu162, Gly160, Cys270, Val159 Chain C: Leu162, Gly160, Val159 |
Chain A: Thr158, Asn109, Gln269 Chain B: Thr158, Gln269, Asn109 Chain C: Asn109, Gln269 |
Chain B: Leu162, Val159 | Chain B: Glu161 Chain A: Glu161 Chain C: Asp108 |
|
 |
Antimalarial Drug | Chain A: Leu162, Gly160 Chain B: Leu162, Gly160, Cys270, Val159 Chain C: Leu162, Gly160 |
Chain A: Thr158, Gln269 Chain B: Thr158, Gln269, Asn109, His89 Chain C: Asn109, Gln269 |
Chain B: Asn109 | Chain A: Glu161 Chain B: Asp108 Chain C: Glu161 |
|
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Chain A: Leu162, Gly160 Chain B: Leu162, Gly160, Cys270, Chain C: Leu162, Gly160, Ala86, Val159, Cys270 |
Chain A: Asn109, Gln269 Chain B: Asn109, Gln269 Chain C: Asn109, Gln269, His89, Thr158, Ser85 |
Chain C: Val159 | Chain A: Glu161 Chain B: Glu161 Chain C: Glu161, Asp108 | |
 |
Antiviral Drug |
Chain A: Leu162, Gly160, Val159 Chain B: Leu162, Gly160, Cys270, Val159 Chain C: Leu162, Gly160, Val159, Cys270 |
Chain A: Asn109, Thr158, Gln269 Chain B: Gln269, Asn109, His89, Thr158 Chain C: Gln269, Asn109, Thr158 |
Chain C: Asn109 Chain B: Asn109, Gly160 |
Chain A: Glu161 Chain B: Asp108, Glu161 Chain C: Glu161, Asp108 |
Ligand Interaction Diagram for top scorers with 6W9C are displayed in Figure 4. Docking parameters for rest of drugs with 6W9C are provided in Table 9*SI.
Table 9.
Ligand Interaction information for top ranking drug with Nucleocapsid Protein of SARS-COV-2 (PDB ID 6M3M).
| Compound | Mode of Action | Hydrophobic | Polar | H-Bond | п-п stacking | Charged |
|---|---|---|---|---|---|---|
 |
Antimalarial | Chain A: Ala91, Ala51, Tyr110, Tyr112, Pro118 Chain B: Trp133, Val134, Tyr124, Pro68, Ile132, Phe67, Gly125 |
Chain A: Asn49, Thr50 | Chain B: Trp133 | Chain A: Tyr110, Chain B: Lys66 | Chain A: Arg108, Arg90, Arg90, Arg93 Chain B: Arg89, Arg69, Lys66 |
 |
Anti-HIV Protease inhibitor |
Chain A: Tyr112, Pro118, Ala51, Tyr110 Chain C: Val159, Tyr173, Trp133, Val134, Tyr124, Ala135, Pro68, Ile132, Phe67, Gly70, Gly125 |
Chain A: Asn49, Thr50, Ser52 Chain C: Thr55 |
Chain A: Thr50, Lys66, Chain B: Phe67 | Chain A: Arg108, Arg150 Chain B: Arg69, Lys66 | |
 |
Anti-HIV inhibitor |
Chain A: Tyr112, Tyr110, Ala51, Ala91, Pro152 Chain B: Phe67, Pro68, Trp133 Chain C: Val159 |
Chain A: Ser52, Thr50, Asn49 | Chain A: Tyr110, Tyr112, Ser52 Chain B: Arg69 |
Chain A: Tyr110, Lys66 | Chain A: Arg108, Arg93 Chain B: Lys66, Arg69 |
 |
Antibacterial | Chain A: Pro118, Ala91, Ala51, Tyr110, Tyr112 Chain B: Pro68, Phe67, Pro169 |
Chain A: Ser52 Chain B: Thr92 Chain C: Thr167 |
Chain A: Tyr112 Chain B: Lys66 |
Chain B: Lys66, Arg89 | Chain A: Arg150, Arg108, Arg93 Chain B: Arg89, Lys66 |
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Chain A: Tyr110, Ala51, Pro152, Ala157 Chain B: Gly70, Pro68 Chain C: Tyr173, Val159, Leu162, Leu168, Leu160, Leu57 |
Chain A: Asn49 Chain B: Thr167, Gln71 Chain C: Gln161 |
Chain B: Arg69 | Chain A: Arg150, Arg93, Arg108 Chain B: Arg69, Lys66 |
|
 |
Antimalarial | Chain C: Leu160, Leu162, Leu168, Pro163, Gly165, Tyr173, Val159, Leu57 Chain B:Pro163, Gly70, Pro81, Pro68 |
Chain C: Gln161, Thr166, Gln164, Thr167 Chain B: Thr136, Gln71, Gln164, Gln84, Thr167 |
Chain C: Gly165, Chain B: Glu137 | Chain B: Glu137, Arg69, Chain A: Arg108 | |
 |
Antimalarial Drug Anti-arthritis |
Chain B: Pro81, Ile75, Pro74, Val73, Pro163, Gly70 Chain C: Leu168, Pro163, Leu162, Gly165 |
Chain B: Gln71, Gln164, Gln84, Ser79, Ser80 Chain C: Gln161, Thr166, Gln164, Thr167 |
Chain C: Leu162 | Chain B: Glu137 | |
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Chain B: Trp133, Tyr124, Pro68, Phe67, Gly125 Leu168, Pro169 Chain A: Tyr112, Ala91, Ala51, Tyr110, Pro152 Chain C: Tyr173 |
Chain A: Asn49, Thr50, Ser52, Thr92 Chain B: Thr167, |
Chain A: Thr50 | Chain B Lys66 |
Chain A: Arg93, Arg108, Arg94 Chain B: Arg69, Lys66 |
 |
An irreversible Epidermal Receptor Growth receptor Tyrosine kinase |
Chain C: Val159, Tyr173, Pro118 Chain A: Tyr112, Ala51 Chain B: Trp133, Val134, Tyr124, Ala135, Pro68, Ile132, Phe67, Gly70 |
Chain C: Gln161 Chain B: Gln71 Chain A: Asn49, Thr50, Ser52 |
Chain B: Arg69, Trp133, Phe67 | Chain B: Lys66, Gly70, Ala135 | Chain B: Arg69, Lys66 |
 |
Antimalarial Drug | Chain C: Leu162, Pro163, Leu168, Gly165, Chain B: Pro163, Gly70, Pro81, Ile 75 | Chain C: Thr167, Thr166, Gln164, Gln161 Chain B: Gln84, Ser79, Asn76, Thr136, Thr77, Gln164, Gln71, Thr166 |
Chain B: Gly70 | Chain B: Glu137 | |
 |
Antiviral Drug |
Chain B: Ala126, Tyr124, Ile131, Ile 132, Trp133, Val134, Ala135, Gly125, Gly70, Chain A: Tyr110, Pro152, Ala51 Chain C: Val159 |
Chain B: Asn127, Gln71, Chain A: Thr50, Asn49 | Chain A: Arg150, Thr50, Chain B: Trp133, Lys66 | Chain A: Arg150, Lys66 Chain B: Lys128, Arg69 |
3.1.4. Target 4: Interaction characterization of quinoline based drugs with RNA binding domain of nucleocapsid protein of SARS-COV-2
When the quinoline library was docked on PDB (6M3M), average GScoresand the average docking scores and binding energies compared, respiratory specific drugs emerged as most promising (Figure 7(d)).
The SARS-COV-2 nucleocapsid RNA binding protein plays a vital role in viral RNA transcription and replication. As the name is suggestive of its function, the primary function of the N-protein is binding to the viral RNA genome and packing into a long helical nucleocapsid structure or ribonucleoprotein (RNP) complex (Dutta et al., 2020). Experimental studies revealed that N-protein maintains highly ordered RNA conformation suitable for replicating and transcribing the viral genome. The protein is speculated to regulate host-pathogen interactions, such as actin reorganization, host cell cycle progression, and apoptosis. The N protein itself is highly immunogenic and abundantly expressed protein during infection, capable of inducing protective immune responses against SARS-CoV-2 (Kang et al., 2020).
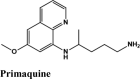
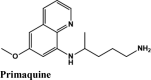
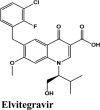
The docking Parameters and Ligand Interaction amino acids are provided in Tables 9 and 8 respectively. The drugs that have GScore greater than or equal to −7 are considered best candidates and bind with residues Ala51, Tyr112, Tyr124 within the active site are considered potent drugs for targeting N protein. Docking with Nucleocapsid proteins of SARS-COV-2 suggest that primaquine and amodiaquine antimalarial drugs serve as the best inhibitor with G-score −8.5, followed by saquinavir with GScore −7.61 kcal/mol and and elvitegravir with −7.57 kcal/mol respectively (Table 1). Key interactions are the H-bond between protonated nitrogen atom in primaquine and Trp133, п-п stacking with Tyr110 and Lys66, and covalent interactions with Ala51, Thr50, Asn49, Ty124 and Tyr110, which are crucial for binding with RNA binding domain of N Protein, and makes it the best drug to target Nucleocapsid protein of SARS-COV-2. The GScore of primaquine is 70.2% higher than HCQ. Ligand Interaction Diagram for top scorers are displayed in Figure 5 with 6M3M. Docking parameters for rest of drugs with 6M3M are provided in Table S10*SI.
Table 10.
Docking parameters of top scorer drugs for nucleocapsid protein of SARS-COV-2 (PDB ID 6M3M).
| Drugs | GScore | DScore | Lipophilic EVDW | HBond | EModel |
|---|---|---|---|---|---|
| Primaquine | −8.51 | −7.58 | −2.56 | −0.86 | −60.692 |
| Amodiaquine | −8.5 | −5 | −2.6 | −0.9 | −56.71 |
| Saquinavir | −7.61 | −6.65 | −4.64 | −1 | −78.879 |
| Elvitegravir | −7.57 | −7.44 | −2.76 | −1.4 | −67.765 |
| Imiquimod | −7.29 | −7.03 | −2.14 | −2.98 | −56.262 |
| Afatinib | −7.26 | −6.03 | −3.29 | −0.97 | −58.428 |
| Pamaquine | −7.23 | −7.23 | −5.19 | −1.33 | −81.96 |
| HCQ | −4.99 | −4.94 | −3.38 | −1.28 | −40.568 |
| Remdesivir | −6.85 | −6.85 | −4.64 | −1.75 | −69.954 |
| Acalabrutinib | −5.15 | −5.14 | −4.05 | −0.92 | −72.062 |
| EKB-569 | −3.58 | −1.76 | −3.91 | −1.1 | −55.435 |
| Plasmoquine | −2.63 | −2.4 | −4.43 | −0.96 | −56.61 |
3.1.5. Target 5: Interaction characterization of quinoline based drugs with SARS-COV-2 RNA dependent RNA polymerase (PDB ID 7BTF)
The quinoline library was docked on PDB (7BTF), and the average docking scores and binding energies compared kinase inhibitors emerged as most promising (Figure 7(e)).
RDRP is a vital enzyme for the life cycle of the single-stranded RNA coronavirus (Elfiky, 2020). The function of RdRp is to convert a single-stranded RNA virus into many single-stranded RNA viruses. RdRp active site is conserved among different organisms, while two successive, surface-exposed aspartate residues are protruding from a beta-turn motif (Yin et al., 2020).
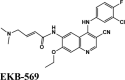
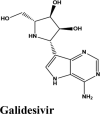
Ligand Interaction Diagram for top scorers with 7BTF are displayed in Figure 6. The binding pocket is primarily marked by the amino acids Asp760 and Asp761. As galidesivir is considered as the best ligand for RdRp, it was used as a control with a GScore of −5.375 kcal/mol. Docking with RdRp suggests that EKB-569 has the highest binding with GScore value −8.97 kcal/mol followed by campothecin with GScore −7.81 kcal/mol. The docking parameters and interaction amino acids informations are provided in Tables 11 and 12 respectively. The binding of EKB-569 with RdRp was approximately 66.8% higher than that of galidesivir. The protonated nitrogen and oxygen atoms of EKB-569 form hydrogen bonding with Asp760 and Asp761, which is crucial for binding within the active site of RNA dependent RNA polymerase from SARS-COV-2. The interactions also include п-пstacking between the aromatic and quinoline rings with Lys621, Arg553, and a hydrogen bond with Arg553.
Table 11.
Docking Parameters of Top scoring Drugs to target RNA dependent RNA Polymerase from SARS-COV-2 (PDB ID 7BTF).
| Drugs | GScore | DScore | Lipophilic EVDW | HBond | EModel |
|---|---|---|---|---|---|
| EKB-569 | −8.97 | −6.45 | −4.4 | −2.1 | −87.789 |
| Campothecin | −7.81 | −7.1 | −6.5 | −0.4 | −72.146 |
| Amodiaquine | −7.53 | −7.53 | −5.3 | −1.6 | −78.285 |
| Primaquine | −7.1 | −7 | −6.2 | −2.5 | −71.433 |
| Dequalinium | −7.04 | −6.4 | −5.2 | −0.7 | −78.145 |
| Elvitegravir | −7.04 | −4.19 | −1.92 | −1.78 | −80.599 |
| Imiquimod | −6.7 | −6.6 | −4 | −1.9 | −59.016 |
| Saquinavir | −6.4 | −6.4 | −5.8 | −2 | −87.479 |
| Afatinib | −5.9 | −4 | −5.2 | −0.5 | −73.441 |
| Acalabrutinib | −5.4 | −5 | −4.3 | −0.7 | −73.387 |
| Hydroxychloroquine | −6.1 | −5.11 | −4.4 | −1.2 | −44.626 |
| Remdesivir | −8.4 | −8.4 | −5.6 | −2.3 | −82.944 |
| Galidesivir | −5.4 | −5.3 | −0.7 | −3.3 | −42.332 |
Table 12.
Interacting amino acids for compounds having G-Score above Hydroxyquinoline and Galidesivir for RNA dependent RNA Polymerase of SARS-COV-2 (PDB ID 7BTF).
| Compound | Mode of Action | Hydrophobic | Polar | H-Bond | п-п stacking | Charged |
|---|---|---|---|---|---|---|
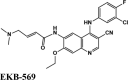 |
Potent Irreversible EFGR receptor | Tyr455, Ala554, Val557, Tyr619, Pro620, Cys622 | Thr556, Ser682, Ser759 | Asp760, Asp760, Arg553, Arg553, Asp623, Asp623, Asp623 | Lys621, Arg553, Arg553 | Lys621, Arg553, Arg624, Arg555, Arg545, Asp452, Asp623, Asp760, Asp761 |
 |
DNA Topoisomerase inhibitor | Tyr455, Tyr619, Pro620, Cys622, Ala550 | Ser549 | Asp760, Asp760, Asp623, Lys621, Tyr619 | Lys621, Arg553, Arg553 | Lys798, Arg621, Arg624, Arg555, Arg553, Lys551, Asp623, Asp618, Asp761, Asp760 |
 |
Antimalarial, Anti-inflammatory | Tyr455, Cys622, Pro620, Tyr619, Trp617, Ala762 | Asp761, Trp617, Asp760, Lys621, Asp761 | Arg553, Lys621 | Lys551, Arg553, Arg555, Arg624, Lys621, Lys798, Asp623, Asp618, Asp761, Asp760 | |
 |
Antimalarial | Tyr455, Cys622, Pro620, Tyr619, Trp617, Ala762 | Ser759 | Asp760, Asp760, Asp761 | Lys621, Arg553, Arg553, Asp452 | Lys551, Arg553, Asp452, Arg624, Asp623, Lys621, Asp618, Asp760, Asp761, Lys798 |
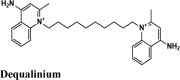 |
Antibiotic | Tyr455, Ala554, Cys622, Pro620, Tyr619, Trp617, Ala762, Trp800, Phe812, Cys814 | Thr556, Ser682, Ser814 | Asp452, Asp623, Asp761, Trp617 | Arg553, Arg553 | Arg553, Arg555, Arg624, Lys621, Lys798, Asp452, Asp623, Asp618, Asp761, Asp760, Glu811 |
 |
Anti-HIV inhibitor | Tyr619, Pro620, Cys622, Tyr455, | Asn691, Thr680, Ser682, Thr556 | Arg553, Asn691, Arg555 | Lys621, Arg553 | Asp618, Asp760, Asp623, Lys621, Arg624, Arg553, Arg555, |
 |
Antiviral activity against positive and negative sense RNA viruses for example: Ebola, Marburg, Yellow fever, Zika virus | Tyr619, Pro620, Cys622, Tyr455 | Thr556 | Asp623, Asp618, Tyr619 | Asp618, Asp760, Asp623, Asp452, Lys798, Lys621, Arg624, Arg553, Lys551 | |
 |
Antimalarial Drug Anti-arthritis |
Cys622, Pro620, Tyr619, Tro617, Trp800, Ala762, Phe812, Cys813 | Ser814 | Asp760, Asp761, Asp761, Asp761, Asp623 | Lys621 | Asp623, Lys621, Asp618, Asp760, Asp761, Glu811, Lys798 |
 |
Toll-like receptor 7 Agonist | Tyr619, Pro620, Cys622, Tyr455 | Asp623, Arg553, Arg555 | Lys621, Arg553 | Lys621, Asp623, Asp618, Lys798, Arg624, Asp760, Asp452 | |
 |
β2 adrenoceptors agonist, muscarinic receptor antagonist | Cys622, Pro620, Tyr619, Trp617, Tyr455, Ala554 | Ser759, Asn691, Thr556, Ser814 | Lys621, Asp618, Asp760, Ala554 | Lys621, Tyr455, Arg553 | Lys621, Arg624, Asp623, Asp618, Lys551, Arg553, Arg555, Asp760, Asp761 |
 |
Anti-HIV Protease Inhibitor | Tyr619, Trp617, Pro620, Cys622, Ala762, Ala688 | Ser682, Thr687, Asn691, Thr556, Ser759 | Asp760, Asp623, Arg553, Arg555, Thr556 | Asp618, Glu811, Lys621, Arg624, Asp623, Lys545, Asp684, Arg555, Arg553, Asp760, Asp761 | |
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Chain A: Tyr455, Ala554, Ala448 | Chain A: Asn552, Ser451, Asn447, Gln444 Chain C: Gln34 |
Chain A: Ala554, Asp445 | Chain A: Arg553, Ala554 | Chain A: Lys621, Arg624, Lys551, Arg553, Asp623, Asp445 |
 |
Tyrosine Kinase inhibitor; Epidermal Growth Factor Receptor (EGFR) inhibitor | Pro620, Cys622, Val166, Tyr455, Ala688 | Ser759, Asn691, Thr680, Ser681, Ser682 | Asp623, Lys621 | Arg553 | Lys798, Glu167, Asp760, Asp623, Arg624, Lys621, Arg553, Lys551 |
 |
Antiviral Drug | Tyr619, Val166, Pro620, Cys622, Met626, Tyr455 | Ser759, Thr680, Asn691, Asn552 | Lys798, Lys621, Cys622 | Arg553, Lys621 | Asp618, Lys798, Lys621, Arg624, Asp623, Lys551, Arg553, Asp760 |
The drugs having GScore greater than or equal to −7.00 kcal/mol and bind with active site residue Asp760 and Asp761 within the active site are considered as potent drugs to target RdRp from SARS-COV-2
Ligand Interaction Diagram for top scorers with 7BTF are displayed in Figure 6. Docking parameters rest of drugs with 7BTF are provided in Table S11*SI.
A preliminary analysis based on higher score than the control hydroxyquinoline (Table 1) reflects the following. Overall, among all the drugs amodiaquine serves as best for all the targets. Afatinib and saquinavir were above the HQ in four of the targets. This analysis brings out the contenders that may target multiple targets. Elvitegravir and EKB-569 reserved their roles as inhibitors of proteases and RdRp polymerase. Rilapladib emerged as a potential candidate for inhibition of viral entry with binding potential with ACE2 (Tables 11 and 12).
3.3. In silico ADME properties
The compilation of bioactivity parameters is presented in Table S6*SI. Results of in silico ADME analysis indicate the following results: (Figure 8)
Figure 8.

ADME Properties of top-ranking drugs (*red for toxicity and inhibitor, *green for safety and non-inhibitor and *orange for less toxic).
Absorption: Primaquine, amodiaquine, and HCQ have high Caco-2 (heterogeneous human epithelial colorectal adenocarcinoma cells) permeability.
Human Intestinal Absorption: All drugs are majorly absorbed by human intestine. (For Ideal drug, HIA percentage should be higher than 30%) and primaquine and amodiaquine are highly absorbed by the intestine.
P-Glycoprotein substrate and Inhibitor: P-Glycoprotein is mainly known as multidrug resistance protein; ATP binding cassette subfamily B member is an integral part of cell membrane which flush out foreign substances out of the cell. The results indicate that only primaquine and HCQ are non-inhibitors substrates for P-Glycoprotein.
Distribution: The Distribution results indicate the following observation.
BBB Permeation: For ideal drug Log BBB value, much be greater than 0.3. All drugs are able to cross the Blood-Brain Barrier.
Metabolism:
CY2D6, CY34A: Cytochrome 450 is an enzyme that is encoded by both CY2D6 and CY34A gene, which are primarily expressed and metabolized in the liver. The results indicate that all drugs are except amodiaquine and primaquine are non-inhibitors to CY2D6 substrate and inhibitor and afatinib, acalabrutinib, and primaquine are non-inhibitors to CY34A substrate.
OATP1B1, OATP1B3: OATP1B1 and OAT1B3 are uptake transporters that are expressed on the sinusoidal site of hepatocytes, and these are responsible for drug uptake and endogenous compounds from the blood. The results indicate that all quinoline drugs except afatinib is non-inhibitor for OATP1B1. All drugs are inhibitors of OATP1B1 and OATP1B3, which led to the conclusion that drugs may be metabolized by the liver.
Excretion:
OCT2, MATE-1 Inhibitor:
All quinoline based drugs except primaquine are non-inhibitor to Organic cation Transporter 2 (OCT2) and Multidrug and toxic extrusion (MATE-1) inhibitor which concluded that all drugs are eliminated from urine.
Toxicity:
The toxic analysis indicates that all quinoline drugs are non-carcinogenic and non-toxic.
Hence all ADME results indicate that best scorer quinolines drugs have ideal properties to work as anti-SARS-COV-2 therapeutic drugs.
3.4. Enrichment studies
Enrichment studies help to assess the active set of compounds statically among large set of databases through virtual screening. The parameters that are calculated through enrichment studies are Receiver Operator Characteristics area under the curve (ROC), Boltzmann-enhanced DiscriminationReceiver Operator Characteristic area under the curve and Enrichment Factor (BERDOC), and Enrichment factor calculated with respect to the number of total ligands. EF = (a/n)/(A/N), where a is the number of actives found in sample size n, A is the total number of actives, and N is the total number of ligands (decoys and actives). All these parameters are summarized in the Table 13.
Table 13.
Enrichment Parameters for all therapeutics targets of SARS-COV-2.
| Parameters | 6M0J | 5R80 | 6W9C | 7BTF | 6M3M |
|---|---|---|---|---|---|
| BERDOC | 0.234 | 0.363 | 0.335 | 0.216 | 0.320 |
| ROC | 0.76 | 0.78 | 0.76 | 0.62 | 0.55 |
| EF | 20% | 40% | 40% | 30% | 30% |
The ideal value for BERDOC and ROC parameters should be between 0 and 1. The active ligands among whole databases and decoys with all five targets main protease, spike proteins, RdRp enzymes, papain protease as well as for N protein of SARS-CO-2 have value below 1 which indicates that the active ligands are ideal to work again therapeutics targets of SARS-COV-2. The ROC curves for active ligands with each target are shown in Figure 9.
Figure 9.

Receiver Operator Characteristics Curve for all active drugs with therapeutics targets of SARS-COV-2.
3.5. Molecular dynamics simulation
The protein-Afatinib complexes: afatinib-5R80, afatinib-6M0J, afatinib-6W9C, and afatinib-7BTF showed very stable RMSD fluctuations (< 3.6 Å) throughout the simulation’s trajectories. Among the four stable complexes, 7BTF and 5R80 had the least variation in the backbone. Afatinib-6M3M complex displayed dramatic RMSD fluctuations (2-8.25 Å with spikes upto 11 Å) during the course of simulations. On visualizing the MD simulation trajectories, it was observed that the high RMSD fluctuations in 6M3M are due to the movement of one of the protein subunits in the protein, though the ligand remained in the active site of the protein. The RMSD fluctuations are shown in Figure 10.
Figure 10.

RMSD Plot for all targets of SARS-COV-2 with afatinib drug.
The RMSF fluctuations with respect to the residue number of the protein-ligand (Afatinib) complexes considered for the study is shown in Figure 11. Except for the afatinib-6M0J complex, it was observed that the active site residues and the ligand displayed RMSF fluctuations. Due to random coil structural elements present in the complexes, which are known to display huge structural fluctuations, we noticed high RMSF fluctuations in these regions. Overall, the stable RMSF profile suggests that the protein-ligand complexes are stable during the course of simulations.
Figure 11.

The RMSF fluctuations is shown for the whole complex (blue) and for the active site residues (orange). The RMSF fluctuations with respect to the residue number of the protein-ligand complexes considered for the study is shown in Figure Y. Except 58RO complex, we observed that the active site residues and the ligand displayed stable RMSF fluctuations. Due to random coil structural elements present in the complexes, which is known to display huge structural fluctuations, we noticed high RMSF fluctuations in these regions. Overall, the stable RMSF profile suggests that the protein-ligand complexes are stable during the course of simulations.
The average number of hydrogen bonds in protein-ligand (Afatinib) complexes 5R80, 6M0J, 6M3M, 6W9C, and 7BTF were 1, 1, 2, 1, and 3, respectively. Thus, the maximum number of hydrogen bond interactions were shown by ligand in complexation with (RdRp) RNA dependent RNA polymerase enzyme of SARS-COV-2 while maintaining several van-der Waals contacts. The number of H-bonds formed in the protein-ligand complexes considered for the study as a function of run-length is shown in Figure 12.
Figure 12.

Number of H-bonds vs. run-length for all the complexes considered for the study.
3.6. MM-PBSA calculation
MM-PBSA calculation provide overview about the molecular interaction and free binding energy of Afatinib-protein complex. We computed the Binding free energies of the ligand to the protein via MM-PBSA calculations. We also utilized the last 20 ns of the simulations trajectories and generated 80 frames for processing the data for binding energy calculations. The observed binding free energies were: 5R80 (-5.44 ± 0.69 kcal/mol), 6M0J (-4.29 ± 0.38 kcal/mol), 6M3M (-7.66 ± 0.54 kcal/mol), 6W9C (-4.16 ± 0.44 kcal/mol), and 7BTF (-11.19 ± 0.72 kcal/mol). The high binding free energy of the complex with RNA dependent RNA polymerase (7BTF) from SARS-COV-2 suggested (Table 14) that the afatinib could be helpful most in inhibiting the replication process of single stranded RNA virus.
Table 14.
MM-PBSA Energy calculation.
| S. No | PDB | Binding Free Energy |
|---|---|---|
| 1 | 5R80 | −5.44 ± 0.69 kcal/mol |
| 2 | 6M0J | −4.29 ± 0.38 kcal/mol |
| 3 | 6M3M | −7.66 ± 0.54 kcal/mol |
| 4 | 6W9C | −4.16 ± 0.44 kcal/mol |
| 5 | 7BTF | −11.19 ± 0.72 kcal/mol |
3.6.1. Biological Mechanism by which Afatinib inhibit the activity of SARS-COV-2 (Figure 13)
Figure 13.

SSRNA of SARS-COV-2 binds with toll like receptors (in macrophages) which activates the Bruton Tyrosine Kinase (BTK), triggering the production of various inflammatory cytokines (cytokine storm). Afatinib act as BTK Inhibitor and inhibit the activation of cytokine storm.
Afatinib belongs to the tyrosine kinase inhibitor family of medication. It is also used to treatnon-small lung cell carcinoma, which maintains mutation in the Epidermal Growth factor receptor in a gene (Roskoski, 2016). SARS-Cov-2 virus, when entered into the body simultaneously, binds with ACE-2 and TMPRSS-2 receptor through which it penetrates the lungs where it releases its single-stranded RNA virus to start multiplication to form multiple copies of a single-stranded virus.During this multiplication process, RNA of COV-2 binds with Toll-like receptors present inthe macrophages, further activating the Bruton Tyrosine Kinase. Bruton Kinase Inhibitorplays an essential role in patients suffering from the coronavirus due to macrophageactivation (Roschewski et al., 2020). BTK deals with macrophages signalling and activation, which leads to the hyperinflammatory immune response in corona patients. After activation, BTK sends signals to NF-KB, which triggers various inflammatory cytokines (IL-6, IL-12, IL-8, CCL2, TNF-ά). BTK also activates the NLPR3 inflammasomal to secrete the IL1B. A virus-induced hyperinflammatory response or “cytokine storm” may be an important pathogenic mechanism of ARDS in these patients by altering pulmonarymacrophages and neutrophils, which can lead to the death of patients (Figure 13).
Hence BTK plays a vital role in the activation of these inflammatory cytokines (Conti et al., 2020). BTK inhibitors can inhibit the activity of BTK signalling from macrophage to other inflammatory Cytokines. Afatinib is a potent Bruton tyrosine kinase inhibitor drug (de Bruin et al., 2020). Afatinib breaks the chain of signalling from macrophages activation to auto-immune cells (IL-6, IL-12, IL-8, CCL2, TNF-ά) (de Bruin et al., 2020). Therefore, it inhibits the process of activation and cytokine storm. Afatinib also supports human innate immune system response, thereby helping in controlling in replication and infection of virus therefore expected to enhance the immune response (Roschewski et al., 2020).
4. Conclusion
This study showed that among tested drugs in the present in silico study, Afatinib has the highest binding potential to the main protease of SARS-CoV-2, which is higher than HCQ and remdesivir, respectively. (Figure 14) Likewise, other drugs amodiaquine, saquinavir showed efficient binding with active sites on the main protease, papain protease, and RdRp. Among all the screened drugs, Afatinib serves as the best candidate inhibitor for binding with (a) main protease MPro of SARS-COV-2 with a GScore of −9.04 kcal/mol. Docking with 7BTF RdRp suggests that EKB-569 has the highest binding with GScore value −8.97 kcal/mol. Docking with papain-like protease PLPro, (PDB ID 6W9C) amodiaquine and Afatinib are active binders with GScore −8.57 kcal/mol and −8.53 kcal/mol, respectively. Docking with Nucleocapsid proteins of SARS-COV-2 suggests that primaquineand amodiaquine serve as the best inhibitor with GScore −8.51 kcal/mol and remdesivir used as control have GScore −6.8 kcal/mol. From docking analysis, it is concluded that Afatinib, amodiaquine, saquinavir, and primaquine are the best drugs to inhibit the entry replication and transcription of viral genome of SARS-COV-2. Further, as we screened Afatinib could be best candidate to overall inhibit the process of SARS-COV-2. Molecular dynamics simulations of Afatinib drug with each therapeutics target of SARS-COV-2, followed by binding free energy estimations via MM-PBSA methods, suggested that the Afatinib-7BTF complex is the most stable complex with the highest ligand binding energetics.
Figure 14.

Average GScore of top-ranking Drugs for Protease (Main and Papain) to inhibit the activity of SARS-COV-2.
Supplementary Material
Acknowledgements
The authors would like to thank Department of Chemistry, University of Delhi, New Delhi, India and Institute of Nuclear Medicine and Allied Sciences, Défense Research Development Organization, New Delhi, India for providing Schrodinger Software and other facilities to accomplish this work. The authors also would like to thank UGC for providing fellowship. The authors one and two have equal contribution.
Glossary
Abbreviations
- COVID-19
Corona Virus Disease-2019
- SARS-COV-2
Severe Acute Respiratory Syndrome-2
- FDA
Food and DrugAdministration
- PK
Pharmokinetic Properties
- HCQ
Hydroxychloroquine
- RdRp
RNA Dependent RNA Polymerase
- BTK
Bruton Tyrosine Kinase
- 2D
Two Dimensional
Disclosure statement
The authors declare no conflict of interest.
References
- Alexpandi, R., De Mesquita, J. F., Pandian, S. K., & Ravi, A. V. (2020). Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in silico Analysis. Frontiers in Microbiology, 11(July), 1796–1715. 10.3389/fmicb.2020.01796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard, R. A., Wittenburg, L. A., Amaravadi, R. K., Gustafson, D. L., Thorburn, A., & Thamm, D. H. (2014). Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy, 10(8), 1415–1425. 10.4161/auto.29165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj, V. K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., & Kumar, S. (2020). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1766572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga, S. F. P., Martins, L. C., da Silva, E. B., Sales Júnior, P. A., Murta, S. M. F., Romanha, A. J., Soh, W. T., Brandstetter, H., Ferreira, R. S., & de Oliveira, R. B. (2017). Synthesis and biological evaluation of potential inhibitors of the cysteine proteases cruzain and rhodesain designed by molecular simplification. Bioorganic & Medicinal Chemistry, 25(6), 1889–1900. 10.1016/j.bmc.2017.02.009 [DOI] [PubMed] [Google Scholar]
- Cao, B., Wang, Y., Wen, D., Liu, W., Wang, J., Fan, G., Ruan, L., Song, B., Cai, Y., Wei, M., Li, X., Xia, J., Chen, N., Xiang, J., Yu, T., Bai, T., Xie, X., Zhang, L., Li, C., … Wang, C. (2020). A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. The New England Journal of Medicine, 382(19), 1787–1799. 10.1056/NEJMoa2001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, H., & Bhattacharjya, S. (2020). Mechanistic insights of host cell fusion of SARS-CoV-1 and SARS-CoV-2 from atomic resolution structure and membrane dynamics. Biophysical Chemistry, 265(June), 106438. 10.1016/j.bpc.2020.106438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, C., Sharma, A. R., Bhattacharya, M., Sharma, G., Lee, S. S., & Agoramoorthy, G. (2020). COVID-19: Consider IL-6 receptor antagonist for the therapy of cytokine storm syndrome in SARS-CoV-2 infected patients. Journal of Medical Virology, 92(11), 2260–2263. 10.1002/jmv.26078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham, T. E. I. I. I., Miller, J. L., Fox, T., Darden, T. A., & Kollman, P. A. (1995). Molecular dynamics simulations on solvated biomolecular systems: The particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. Journal of the American Chemical Society, 117(14), 4193–4194. 10.1021/ja00119a045 [DOI] [Google Scholar]
- Chu, C. M., Cheng, V. C. C., Hung, I. F. N., Wong, M. M. L., Chan, K. H., Chan, K. S., Kao, R. Y. T., Poon, L. L. M., Wong, C. L. P., Guan, Y., Peiris, J. S. M., & Yuen, K. Y. (2004). Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax, 59(3), 252–256. 10.1136/thorax.2003.012658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti, P., Ronconi, G., Caraffa, A., Gallenga, C. E., Ross, R., Frydas, I., & Kritas, S. K. (2020). Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): Anti-inflammatory strategies. Journal of Biological Regulators and Homeostatic Agents, 34(2), 327–331. 10.23812/CONTI-E [DOI] [PubMed] [Google Scholar]
- Dai, W., Zhang, B., Jiang, X.-M., Su, H., Li, J., Zhao, Y., Xie, X., Jin, Z., Peng, J., Liu, F., Li, C., Li, Y., Bai, F., Wang, H., Cheng, X., Cen, X., Hu, S., Yang, X., Wang, J., … Liu, H. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science (New York, N.Y.), 368(6497), 1331–1335. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin, G., Demont, D., de Zwart, E., Verkaik, S., Hoogenboom, N., van de Kar, B., van Lith, B., Emmelot- van Hoek, M., Gulrajani, M., Covey, T., Kaptein, A., & Barf, T. (2020). Discovery of quinoline-based irreversible BTK inhibitors. Bioorganic & Medicinal Chemistry Letters, 30(14), 127261. 10.1016/j.bmcl.2020.127261 [DOI] [PubMed] [Google Scholar]
- Desai, N. C., Patel, B. Y., Jadeja, K. A., Dave, B. P., & Desai, N. C. (2017). Nov appro drug des dev landscaping of quinoline based heterocycles as potential antimicrobial agents: A mini review. Mini Review, 1(4), 1–4. 10.19080/NAPDD.2017.01.555570 [DOI] [Google Scholar]
- Dror, R. O., Dirks, R. M., Grossman, J. P., Xu, H., & Shaw, D. E. (2012). Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annual Review of Biophysics, 41(1), 429–452. 10.1146/annurev-biophys-042910-155245 [DOI] [PubMed] [Google Scholar]
- Duan, L., Guo, X., Cong, Y., Feng, G., Li, Y., & Zhang, J. Z. H. (2019). Accelerated Molecular Dynamics Simulation for Helical Proteins Folding in Explicit Water. Frontiers in Chemistry, 7(August), 540–518. 10.3389/fchem.2019.00540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta, N. K., Mazumdar, K., & Gordy, J. T. (2020). The Nucleocapsid Protein of SARS–CoV-2: A Target for Vaccine Development. Journal of Virology, 94(13), 1–2. 10.1128/JVI.00647-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky, A. A. (2020). SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1761882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzupir, A. O. (2020). Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. Journal of Molecular Structure, 1222, 128878). 10.1016/j.molstruc.2020.128878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth, S. A., & Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron, 99(6), 1129–1143. 10.1016/j.neuron.2018.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., Zhang, L., Fan, G., Xu, J., Gu, X., Cheng, Z., Yu, T., Xia, J., Wei, Y., Wu, W., Xie, X., Yin, W., Li, H., Liu, M., … Cao, B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395(10223), 497–506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung, I. S., & Cheatham, T. E. (2008). Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. The Journal of Physical Chemistry B, 112(30), 9020–9041. 10.1021/jp8001614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, S., Yang, M., Hong, Z., Zhang, L., Huang, Z., Chen, X., He, S., Zhou, Z., Zhou, Z., Chen, Q., Yan, Y., Zhang, C., Shan, H., & Chen, S. (2020). Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharmaceutica Sinica. B, 10(7), 1228–1238. 10.1016/j.apsb.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, R. J., Jha, R. K., Amera, G. M., Jain, M., Singh, E., Pathak, A., Singh, R. P., Muthukumaran, J., & Singh, A. K. (2020). Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. Journal of Biomolecular Structure and Dynamics, 39(8), 2679–2692. 10.1080/07391102.2020.1753577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiplin Guy, R., DiPaola, R. S., Romanelli, F., & Dutch, R. E. (2020). Rapid repurposing of drugs for COVID-19. Science (New York, N.Y.), 368(6493), 829–830. 10.1126/science.abb9332 [DOI] [PubMed] [Google Scholar]
- Ko, W.-C., Rolain, J.-M., Lee, N.-Y., Chen, P.-L., Huang, C.-T., Lee, P.-I., & Hsueh, P.-R. (2020). Arguments in favour of remdesivir for treating SARS-CoV-2 infections. In International Journal of Antimicrobial Agents, 55 (4), 105933. 10.1016/j.ijantimicag.2020.105933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., Zhang, Q., Shi, X., Wang, Q., Zhang, L., & Wang, X. (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature, 581(7807), 215–220. 10.1038/s41586-020-2180-5 [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- Law, P. K. (2020). COVID-19 Pandemic: Its Origin, Implications and Treatments. Open Journal of Regenerative Medicine, 09(02), 43–64. 10.4236/ojrm.2020.92006 [DOI] [Google Scholar]
- Liu, J., Cao, R., Xu, M., Wang, X., Zhang, H., Hu, H., Li, Y., Hu, Z., Zhong, W., & Wang, M. (2020). Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. In Cell Discovery. 10.1038/s41421-020-0156-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella, A., Tanwar, O. P., Saha, R., Ali, M. R., Srivastava, S., Akhter, M., Shaquiquzzaman, M., & Alam, M. M. (2013). Quinoline: A versatile heterocyclic. Saudi Pharmaceutical Journal : SPJ : The Official Publication of the Saudi Pharmaceutical Society, 21(1), 1–12. 10.1016/j.jsps.2012.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev, A., Bashford, D., & Case, D. A. (2000). Modification of the generalized born model suitable for macromolecules. The Journal of Physical Chemistry B, 104(15), 3712–3720. 10.1021/jp994072s [DOI] [Google Scholar]
- Ramser, B., Kokot, A., Metze, D., Weiss, N., Luger, T. A., & Böhm, M. (2009). Hydroxychloroquine modulates metabolic activity and proliferation and induces autophagic cell death of human dermal fibroblasts. The Journal of Investigative Dermatology, 129(10), 2419–2426. 10.1038/jid.2009.80 [DOI] [PubMed] [Google Scholar]
- Roe, D. R., & Cheatham, T. E. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- Roschewski, M., Lionakis, M. S., Sharman, J. P., Roswarski, J., Goy, A., Monticelli, M. A., Roshon, M., Wrzesinski, S. H., Desai, J. V., Zarakas, M. A., Collen, J., Rose, K., Hamdy, A., Izumi, R., Wright, G. W., Chung, K. K., Baselga, J., Staudt, L. M., & Wilson, W. H. (2020). Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Science Immunology, 5(48), eabd0110. 10.1126/sciimmunol.abd0110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski, R. (2016). Ibrutinib inhibition of Bruton protein-tyrosine kinase (BTK) in the treatment of B cell neoplasms. Pharmacological Research, 113(Pt A), 395–408. 10.1016/j.phrs.2016.09.011 [DOI] [PubMed] [Google Scholar]
- Rothan, H. A., & Byrareddy, S. N. (2020). The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. Journal of Autoimmunity, 109, 102433. 10.1016/j.jaut.2020.102433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva, C., Wadhwa, A., Kumari, A., Hussain, F., Jha, P., & Kaushik, N. K. (2020). In silico Potential of Approved Antimalarial Drugs for Repurposing Against COVID-19. OMICS: A Journal of Integrative Biology, 24(10), 568–580. 10.1089/omi.2020.0071 [DOI] [PubMed] [Google Scholar]
- Santhanam, V., Pant, P., Jayaram, B., & Ramesh, N. G. (2019). Design, synthesis and glycosidase inhibition studies of novel triazole fused iminocyclitol-δ-lactams. Organic & Biomolecular Chemistry, 17(5), 1130–1140. 10.1039/c8ob03084g [DOI] [PubMed] [Google Scholar]
- Shah, B., Modi, P., & Sagar, S. R. (2020). In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sciences, 252(March), 117652. 10.1016/j.lfs.2020.117652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J., Wan, Y., Luo, C., Ye, G., Geng, Q., Auerbach, A., & Li, F. (2020). Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences, 117(21), 11727–11734. 10.1073/pnas.2003138117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureshkumar, B., Mary, Y. S., Panicker, C. Y., Suma, S., Armaković, S., Armaković, S. J., Van Alsenoy, C., & Narayana, B. (2020). Quinoline derivatives as possible lead compounds for anti-malarial drugs: Spectroscopic, DFT and MD study. Arabian Journal of Chemistry, 13(1), 632–648. 10.1016/j.arabjc.2017.07.006 [DOI] [Google Scholar]
- Vanquelef, E., Simon, S., Marquant, G., Garcia, E., Klimerak, G., Delepine, J. C., Cieplak, P., & Dupradeau, F.-Y. (2011). R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res, 39(Web Server issue), W511–W517. 10.1093/nar/gkr288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatansever, E. C., Yang, K., Kratch, K. C., Drelich, A., Cho, C.-C., Mellot, D. M., Xu, S., Tseng, C.-T K., & Liu, W. R. (2020). Targeting the SARS-CoV-2 main protease to repurpose drugs for COVID-19. bioRxiv: The preprint server for biology, 10.1101/2020.05.23.112235 [DOI]
- Yang, L., Skjevik, Å. A., Han Du, W. G., Noodleman, L., Walker, R. C., & Götz, A. W. (2016). Data for molecular dynamics simulations of B-type cytochrome c oxidase with the Amber force field. Data in Brief, 8, 1209–1214. 10.1016/j.dib.2016.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, W., Mao, C., Luan, X., Shen, D.-D., Shen, Q., Su, H., Wang, X., Zhou, F., Zhao, W., Gao, M., Chang, S., Xie, Y.-C., Tian, G., Jiang, H.-W., Tao, S.-C., Shen, J., Jiang, Y., Jiang, H., Xu, Y., … Xu, H. E. (2020). Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science (New York, N.Y.), 368(6498), 1499–1504. 10.1126/science.abc1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.