

Abstract
Remdesivir and hydroxychloroquine derivatives form two important classes of heterocyclic compounds. They are known for their anti-malarial biological activity. This research aims to analyze the physicochemical properties of remdesivir and hydroxychloroquine compounds by the computational approach. DFT, docking, and POM analyses also identify antiviral pharmacophore sites of both compounds. The antiviral activity of hydroxychloroquine compound's in the presence of zinc sulfate and azithromycin is evaluated through its capacity to coordinate transition metals (M = Cu, Ni, Zn, Co, Ru, Pt). The obtained bioinformatic results showed the potent antiviral/antibacterial activity of the prepared mixture (Hydroxychloroquine/Azithromycin/Zinc sulfate) for all the opportunistic Gram-positive, Gram-negative in the presence of coronavirus compared with the complexes Polypyridine-Ruthenium-di-aquo. The postulated zinc(II) complex of hydroxychloroquine derivatives are indeed an effective antibacterial and antiviral agent against coronavirus and should be extended to other pathogens. The combination of a pharmacophore site with a redox [Metal(OH2)2] moiety is of crucial role to fight against viruses and bacteria strains.

Communicated by Ramaswamy H. Sarma
Keywords: Remdesivir, hydroxychloroquine, azithromycin, anti-COVID-19 drugs, DFT, docking, POM (Petra/Osiris/Molinspiration) theory
1. Introduction
Coronavirus is one of the most recent leading causes of mortality worldwide. The limited success of currently used antiviral drugs is a driving force for searching of new compounds with antiviral potential (Fang & Lerner, 2020). A limited number of such compounds are being tested against coronavirus in laboratories worldwide (Hoft, 2020; Hunt, 2020; Subramanian, 2020). The main challenge is to find a relatively simple, efficient, and generally synthetic method that enables the preparation of libraries of anti-corona drugs containing one or more pharmacophoric groups (hybrid molecules) with hope for new, effective anti-corona agents. In addition, great attention of world chemists is given to the new treatments of COVID-19, which have been proposed by French, Chinese, and American Governments.
On January 27th/2020, science published a news release suggesting that the ideal treatment for 2019-nCoV is likely combine a drug called Remdesivir and monoclonal antibodies. Remdesivir is thought to be effective for both MERS and new coronaviruses. In a mouse study led by Ralph Baric of the University of North Carolina, published in the journal Nature-Communications, the study tested interferon beta-1b with Remdesivir, an experimental drug manufactured by Gilead, USA.
On the other hand, the study published online Friday night, by the controversial Professor Didier Raoult and the Marseille University Hospital Institute (IHU), involving 80 peoples, was immediately criticized by the scientific community. Carried out without a control group, it still does not confirm the beneficial effect of hydroxychloroquine on the symptoms and the evolution of COVID-19. The positive development and the drop in viral load observed could be natural or not. The first results of the most framed European clinical trial Discovery, which includes hydroxychloroquine, could be known by the weekend, said Monday, March 30, 2020, Frédérique Vidal, the Minister of French Research (Richard & Gambert, 2020).
Lack of time, and to avoid more stress resulting of innocent Americans death, Sir President Donald Trump and his Government-authorized without hesitation this synergetic combination to fight with highly pathogenic coronavirus for five days:1-Hydroxychloroquine (200 mg), 2 Azithromycin (500 mg), 3-Zinc sulfate (220 mg) (Figure 1).
Figure 1.

Proposed known antipaludism drugs to fight against COVID-19.
Molecular modeling has become a very practical and powerful tool. This approach is an application of theoretical calculation methods to explain the molecular structure problems and chemical reactivity (Liotta, 1988). In the domain of the study of the reactivity of organic and pharmaceutical molecules, the researchers have encountered two main problems. The first problem concerns the explanation of the reactivity of some compounds in comparison to others. In addition, the second difficulty is to explain why some sites of the molecule are more reactive than other sites. To solve these problems, several theories have been established for the study of the chemical reactivity. Such as the transition state theory (Eyring,1931), Frontier Molecular Orbital Theory (Fukui et al., 1952), Hard and Soft Acids and Bases Theory (Pearson, 1963), and the density functional theory (DFT) (Klopman 1968).
In fact, the organic/organometallic synthesis constitutes an important field in the synthesis of novel bioactive molecules, particularly heterocyclic ones bearing anti-coronavirus bioactivity (Richard & Gambert, 2020). The hydroxychloroquine is an important nucleus, thanks to its structure (Figure 2), which contains various nucleophilic and electrophilic reactive sites, which may yield to the synthesis of transition metal complexes.
Figure 2.

Molecular structure of proposed new anti-Covid-19 drugs.
According to the World health organization, 42,512,186 confirmed cases with 1,147,301 deaths and 215 countries affected by coronavirus. By reports, most confirmed cases reported by the Americas (1,147,301) and the death cases (222,507), which is presently spreading by community transmission. The first wave of COVID-19 is still going on, but in some countries, the 1st wave was over, and the second wave was expected to increase. Although coronavirus is a global pandemic, no particular drugs or vaccines for this viral disease are still available. The treatment is suggestive, and oxygen therapy symbolizes the initial phase for the respiratory deficiency (Wang et al., 2020; Xu et al., 2020). At present, the immunization against coronavirus infection is not scientifically confirmed. In the research, various compounds from other viral infections have been repurposed to treat COVID-19, but the treatment advantage has, in many cases, been minimal or non-existent (Rakib et al., 2020).
We report the docking, DFT, and POM analyses of newly proposed treatments in the current training. The three products are subjected to comparative study. In addition, the influence of the substituents carried by the aromatic nucleus on the antibacterial and antiviral activities is emphasized. The observed anti-COVID-19 and antibacterial activities are interpreted through the analysis of DFT/Docking/POM calculations. The presence of Zinc sulfate in the American treatment presents a crucial role because its presence with hydroxychloroquine as N-ligand monodentate lead us to the formation of the complex with a potential antiviral activity, carrying two optional therapeutic functions
2. Materials and methods
2.1. DFT calculations
The optimization of the molecular structure was accomplished by integrating the functional three-parameter exchange of Becke (1993), with the functional Lee-Yang-Peer association (Lee et al., 1988). The method (B3LYP) with a basis set of 6-31G(d) is applied with the software package Gaussian 9.0. HOMO-LUMO energy differences have been obtained to understand the stability and reactivity of compounds. Mulliken charges were calculated for molecular polarization and electronic structure.
2.2. Molecular docking
2.2.1. Selection of protein structure from protein data bank
The three-dimensional coordinates of COVID-19 Mpro in complex with peptidomimetic inhibitor, N3, (PDB ID: 6LU7) were downloaded from the Protein Data Bank (PDB, https://www.rcsb.org/). Water molecules were removed from protein structure using BIOVIA Discovery Studio 4.5 (Dassault Systems, USA).
2.2.2. Generation of ligand data
The 3D structure of remdesivir was optimized using the molecular mechanics force field (MM+) (Hocquet & Langgard, 1998) and subsequently by the semiempirical PM3 method (Stewart, 1989) using the Avogadro 1.2.0 (University of Pittsburgh, Pittsburgh, PA, USA). Water molecules were removed using BIOVIA Discovery Studio 4.5 (Dassault Systems, USA).
2.2.3. Molecular docking using discovery studio
Genetic parameters for molecular docking were set on: population size 300; generations 100; the number of solution or poses: 10. Remdesivir was docked into the binding site, and after the docking procedure, protein-compound interaction profiles of electrostatic (Elec), hydrogen-bonding (H-bond), and van der Waals (vdW) interactions were generated. Docking poses were ranked by combining the pharmacological interactions and energy-based scoring function (E) is: E = vdW + H-bond + Elec. Results were viewed and analyzed with BIOVIA Discovery Studio (Ver 4.5).
2.3. Mechanism of docking
Docking was accomplished by iGEMDOCK molecular docking software (Hsu et al., 2011). During docking, at first, the molecules were prepared, and bonds, bond orders, explicit hydrogen's, charges, flexible torsions were assigned to both the protein and ligands. From the docking, wizard ligands were selected, and the scoring function used was iGEMDOCK score. If hydrogen bonding is likely, the hydrogen bond energy contribution to the docking score is assigned a penalty based on the deviations from the ideal bonding angle. This prospect can significantly reduce the number of unlikely hydrogen bonds and also internal electrostatic interaction; internal hydrogen bond sp2-sp2 torsions are calculated from the pose by enabling the ligand evaluation terms.
The search algorithm is taken as iGEMDOCK and numbers of runs taken are 10, and max interactions were 2000 with population size 300 and with an energy threshold of 100 also at each step least 'min' torsions/translations/rotations are tested and the one giving lowest energy is chosen. If the energy is positive (i.e. because of a clash or an unfavourable electrostatic interaction), then additional 'max' positions will be tested. If the pose being docked is closer to one of the ligands in the list than specified by the Root Mean Square Deviation (RMSD) threshold, an additional penalty term (the energy penalty) is added to the scoring function. This ensures greater diversity of the returned solutions since the docking engine will focus its search on poses different from earlier poses found. The energy penalty was set to 100, the RMSD threshold was 2.00, and RMSD calculation by atom ID (fast) was set. Docking was conducted between protein and drugs, which results in binding affinities in kcal/mol and docking run time. The compound which gives lowest binding energy is chosen as the best inhibitor. iGEMDOCK showed better overall performance in docking simulations when compared with other software.
2.4. Petra/Osiris/Molinspiration (POM) analyses
Assessments of Petra/Osiris/Molinspiration (POM) were used to determine the pharmacophore site type and to evaluate the impact of the position and physical/chemical properties of substituents affecting biological activity. The presence of several side effects is a major concern associated with synthetic drugs. In addition to an excellent biological action, a drug molecule must have an excellent pharmacokinetic profile in the biological system. In the in silico method, Petra/Osiris/Molinspiration (POM), authenticated by almost 7,000 drug molecules that are available on the market, we have used well-known pharmacokinetic profiles in comparison with the five rules of Lipinski that found bio-availability predictions imperfectly (Ben Hadda et al., 2020).
3. Results and discussion
3.1. DFT calculation of remdesivir
The geometrical, electronic, and energy parameters were extracted from GuassView 5.0 program based on the optimized structures (Francl et al., 1982; Frisch, 2009). The molecular geometry of remdesivir and their substituents' nature is often correlated with their stability and reactivity. To specify the relationship between the experimental results of the activities with the structure of the molecules and to evaluate this relationship, theoretical studies were carried out by molecular modeling. Thus, modeling gives some important and necessary information on the structure and reactivity of remdesivir.
3.2. Conformational determination
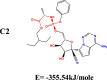
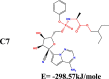
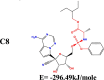
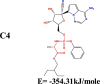
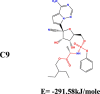
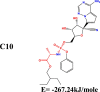
The conformation of a molecule influences its physical and chemical properties. Therefore, reliable conformational analysis plays a key role in the understanding of structure. Initial conformational searching of the title compound C1 was performed by the Spartan 8.0 program (Deppmeier et al., 2002) with MMFF (Brintzinger et al., 1999; Huang et al., 2006) molecular mechanics force field (Table 1).
Table 1.
Energy (kcal/mole) of different conformers of Remdesivir.
| Entry | Structure | Entry | Structure |
|---|---|---|---|
| C1 | 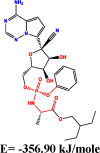 |
C6 |  |
 |
 |
||
 |
 |
||
 |
 |
||
 |
 |
In this study, molecular modeling was used to study the studied compounds' reactivity to determine the most stable structure (Figures 3). The results show that the molecular structure of DFT optimization is consistent with the crystal structure determined by X-ray single crystal diffraction (Siegel et al., 2017).
Figure 3.

Optimized structure of Remdesivir and Hydroxychloroquine obtained at B3LYP/6-31G (d) level in Gas phase.
In addition, the molecular electrostatic potential and the frontier molecular orbital of the title compound were further studied using DFT, indicating that the compound has certain nucleophilic reactivity and good chemical stability. The frontier molecule orbitals (FMOs), which refer to two frontier molecular orbitals called HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) play a very important role in the reactional mechanisms. Molecular electronic and physiochemical properties such as ionization potential (Table 2), electron affinity, chemical reactivity, kinetic stability, electronegativity, and electrophilicity (Parr & Pearson, 1983), are a chemical property that informs us about the stability of the molecule.
Table 2.
Calculated EHOMO, ELUMO, ΔEgap, and quantum molecular descriptors of Remdesivir and Hydroxychloroquine.
| Molecular descriptors | Remdesivir | Hydroxychloroquine |
|---|---|---|
| Log P | 3.2 | 2.87 |
| αTot (Bohr3) | 353.11 | 231.68 |
| Total Energy (a.u.) | −2321.63 | −1401.2388 |
| ELUMO (eV) | −1.2438 | −1.1632 |
| EHOMO (eV) | −6.0790 | −5.6711 |
| ΔEgap(eV)) EHOMO − ELUMO | 4.8352 | 4.5079 |
| Ionization Potential (I = −EHOMO) | 6.0790 | 5.6711 |
| Electron Affinity (A = −ELUMO) | 1.2438 | 1.1632 |
| Chemical hardness (η = (I − A)/2) | 2.4176 | 2.2539 |
| Chemical softness (s = 1/2 η) | 0.2068 | 0.2218 |
| Chemical Potential (μ = −(I + A)/2). | −3.6614 | 3.4171 |
| Electronegativity (χ = (1+ A)/2) | 3.6614 | −3.4171 |
| Electrophilicity index (ω = μ2/2 η) | 2.7725 | 2.5903 |
In the present investigations, the energy gap of hydroxychloroquine is 4.5079 eV. A small energy gap of HOMO-LUMO means more chemical active and low kinetic stability. The other parameters like Ionisation potential (I), electron affinity (A), chemical potential (μ), global hardness (η), global softness (S), electronegativity (χ) and electrophilicity index (ω) of title molecule are computed by using HOMO and LUMO energy values.
The electrophilicity index helps in describing the biological activity of the molecule, (ω = 2.5903) for hydroxychloroquine.
3.3. Frontier molecular orbitals (FMOs)
To investigate the chemical stability of conformer 8, the energies of the highest occupied molecular orbital (EHOMO), the lowest unoccupied molecular orbital (ELUMO), and their orbital energy gap (△E) were calculated using the B3LYP/6-31G (d) method. The pictorial illustration of the FMOs and their respective positive and negative regions represented by red and green colors are shown in Figure 4. The values of EHOMO and ELUMO are −6.0790 eV and −1.2438 eV respectively and the value of the energy separation between the HOMO and LUMO is 4.8352 eV for the most stable conformer. The large HOMO-LUMO energy gap means high excitation energy of the excited state, good chemical stability and large hardness for the calculated conformer.
Figure 4.

HOMO, LUMO orbitals and their energy gap (ΔEgap) for Remdesivir and Hydroxychloroquine.
3.4. Molecular electrostatic potential
Electrostatic potential maps give information on the molecular regions preferred by an electrophile or a nucleophile. Any chemical system creates an electrostatic potential around itself (Murray & Sen, 1996). The different electrostatic potentials at the surface of the molecule are represented by different colors. Electrostatic potentials increase in red < orange < yellow < green < blue, and red indicates the electron-rich region and blue indicate the electron deficient region. Figure 5 shows phosphonate groups and the N atoms of the conformational isomer and is surrounded by negative charges, indicating some possible nucleophilic attack sites. Besides, the positive charge regions are located on the H atoms on the rings.
Figure 5.

Molecular electrostatic potential MESP for Remdesivir and Hydroxychloroquine.
The Mulliken charge calculations (Table 3) indicate the presence of the antiviral O,O-pharmacophore site with Oδ−/Oδ−, as it was postulated based on fundamental concepts of POM Theory.
Table 3.
Mulliken charge of Remdesivir.
| N-P | −0.727 | P-O-CH2 | −0.520 | Antiviral pharmacophore site | |
|---|---|---|---|---|---|
| O = P | −0.562 | C = O | −0.491 | (P = Oδ− …… δ−O = C) | |
| P-O-cycle | −0.617 | O = C-O | −0.466 |
3.5. Docking study of remdesivir
Remdesivir exhibited high potential for the binding to the COVID-19 Mpro. The best pose (pose 9) of inhibitor achieved the total energy value (−146.197 kcal/mol) of which the van der Waals forces participate with 109.165 kcal/mol, while the hydrogen bonds with 38.032 kcal/mol. Energies of the main interactions between COVID-19 Mpro and remdesivir are presented in Table 4.
Table 4.
Energies of the main interactions COVID-19 Mpro residues and ligand Remdesivir (M = main chain; S = side chain).
| H bond | Energy/kcal/mol | van der Waals interaction | Energy/kcal/mol |
|---|---|---|---|
| S-THR 24 | −6.98 | M-THR 24 | 5.17 |
| S-THR 25 | −3.5 | S-THR 25 | 4.93 |
| S-THR 45 | −3.5 | S-MET 49 | 5.97 |
| S-SER 46 | −6 | M-LEU 141 | 5.43 |
| M-GLY 143 | −3.5 | M-ASN 142 | 11.57 |
| M-SER 144 | −3.5 | S-ASN 142 | 9.75 |
| M-CYS 145 | −3.5 | M-GLY 143 | 8.43 |
| S-CYS 145 | 4.28 | M-SER 144 | 4.09 |
| M-CYS 145 | 4.27 | ||
| M-HIS 164 | 4.27 | ||
| M-LEU 4 | 7.55 |
Figure 6 shows the docking and interactions of remdesivir at the N3-binding site of COVID-19 Mpro.
Figure 6.

(a) 3D representation and atomic charge surface of the binding site. (b) Interactions of Remdesivir with residues binding site of COVID-19 Mpro.
The binding site of COVID-19 Mpro was defined according to the binding mode of synthetic glutamine analogs N3 as an inhibitor (PDB ID: 6LU7). The substrate-binding pocket is located in a cleft between domain I (residues 8–100), and domain II (residues 101–183) and it is composed of Cys144 and His41 (Figures 7 and 8). Remdesivir forms two strong hydrogen bonds: one between the nitrogen atom from triazine ring and Thr24, and second with hydroxyl group from dihydroxyoxolan group and Ser46. Other hydrogen bonds formed amino nitrogen atom and propanoate oxygen atom with Thr25, Thr45, Gly143, Ser144, Cys145. The strongest van der Waals interactions are formed with Asn142. Aminopyrrole ring forms amide-π interactions with Met 49. Inhibitor N3 in complex with human coronavirus HCoV-NL63 Mpro forms standards covalent bonds with Cys144, similar to hydrogen bonds with Cys 145 in complex COVID-19 Mpro and remdesivir (Wang et al., 2016). The hydrogen bond between the lactam oxygen of N3 and His163 could be compared with van der Waals interaction with His164. The docking study indicated that remdesivir exhibited high potential for binding to the active site of COVID-19 Mpro and could be potential therapeutic agents in COVID-19 infections.
Figure 7.

The binding site of COVID-19 Mpro with docked inhibitor Remdesivir.
Figure 8.

Potential surface representation of COVID-19 Mpro binding site with docked inhibitor Remdesivir. (Range of potential: from min. 1.77 mV (blue) to max. 0.541 mV (red)).
3.6. POM analyses and identification of pharmacophore sites
POM physicochemical analysis or ADME/T is important to qualify drugs and their efficacy as leading candidates against various biotargets, as like antibacterial (Grib et al., 2020; Hatzade et al., 2015; Jamalis et al., 2020; Jarrahpour et al.,2019; Mabkhot et al., 2015, 2016; Messali et al., 2014, 2015; Nasruddin et al., 2018; Rad et al., 2017; Rbaa et al., 2019; Sajid et al., 2016; Tatar et al., 2016), antifungal (Al-Maqtari et al., 2017; Khan et al., 2017; Rachedi et al., 2020; Radi et al., 2015; Tighadouni et al., 2016; Titi et al., 2019), antiviral (Chander et al., 2017; Lahsasni et al., 2015), antitumoral (Bechlem et al., 2020; Kamal et al., 2019; Piaz et al., 2018; Rachedi et al., 2019, 2020; Youssoufi et al., 2015), antiparasitic drugs and various enzymes inhibitors (Amirkhanov et al., 2019; Ben Hadda et al., 2018, 2020; Mabkhot et al., 2014; Rauf et al., 2017).
The POM physicochemical calculations included a partition coefficient (cLogP), aqueous solubility, donor hydrogen bond, and drug-likeness, evaluated in terms of Lipinski’s rule-of-five. To qualify oral bioavailability, the topological polar surface (TPSA) should be <140 Å2. The results of POM physicochemical analyses of compounds are shown in (Figure 9), and these compounds showed good oral bioavailability (cLogP range = 0.31–3.08). Drug Score (DS) and Drug Likeness (DL) analyses were also within the required limits. The comparison data is shown in (Tables 5 and 6).
Figure 9.

Osiris calculations of Remdesivir, Hydroxychloroquin and Azithromycin, respectively.
Table 5.
Osiris calculations of toxicity risks of compounds.
| Compd. | MW | Toxicity risks[a] |
Osiris calculations[b] |
||||||
|---|---|---|---|---|---|---|---|---|---|
| MUT | TUM | IRRI | REP | cLogP | cLogS | DL | DS | ||
| RMDS | 602 | +++ | −−− | −−− | −−− | 0.31 | −4.99 | −30.36 | 0.05 |
| HCQS | 335 | −−− | +++ | +++ | +++ | 3.08 | −3.55 | 6.54 | 0.48 |
| AZTM | 748 | +++ | +++ | +++ | +++ | 1.66 | −3.09 | 13.85 | 0.48 |
RMDS: Remdesivir, HCQS: Hydroxychloroquin, AZTM: Azithromycin.
Highly toxic: (−−), slightly toxic: (+), Not toxic (+++). [a] MUT: Mutagenic, TUM: Tumorigenic, IRRIT: Irritant, RE: Reproductive effective. [b] cLogS: Solubility, DL: Druglikeness, DS: Drug-score.
Table 6.
Molinspiration calculations of compounds.
| Compd. |
Mol inspiration calculationsa |
Drug-likenessb |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TPSA | nOHNH | NV | VOL | GPCRL | ICM | KI | NRL | PI | EI | |
| RMDS | 204 | 5 | 2 | 523 | 0.27 | −0.35 | 0.20 | −0.48 | 0.49 | 0.38 |
| HCQS | 48 | 2 | 0 | 321 | 0.35 | 0.30 | 0.44 | −0.12 | 0.12 | 0.15 |
| AZTM | 180 | 5 | 2 | 736 | −0.60 | −1.50 | −1.35 | −1.40 | −0.28 | −0.82 |
RMDS: Remdesivir, HCQS: Hydroxychloroquin, AZTM: Azithromycin.
TPSA: Total molecular polar surface area; nNHOH: number of OH and NH interaction, NV: number of violation of five Lipinsky rules; VOL: volume.
GPCRL: GPCR ligand; ICM: Ion channel modulator; KI: Kinase inhibitor; NRL: Nuclear receptor ligand; PI: Protease inhibitor; EI: Enzyme inhibitor.
3.7. Mechanistic study: How each drug works?
The total atom charge calculations of remdesivir (Figure 10) indicate clearly the antiviral O,O-pharmacophore site with cis-Oδ−/Oδ−, as it was exactly confirmed above by Mulliken charge calculations of optimized conformer. On the other hand, when we inspect the molecular structure of hydroxychloroquine, we do not found any one of the antiviral cis-O,O or cis-N,O or cis-N,N-pharmacophore sites. The N,OH is classified as an antibacterial pharmacophore site as it was always postulated by POM Theory. So its mechanism of interaction with coronavirus should be different. In fact, Nsp2 of heterocyclic ring constitutes an excellent center of ligation of transition metals (Figure 11). The presence of ZnSO4 in the American treatment is of crucial role because its presence with hydroxychloroquine as potential monodentate N-ligand should lead us to the formation of the complex bellow, bearing two optional therapeutic functions (Figure 12).
Figure 10.

Atomic charges of anti-COVID-19 agents.
Figure 11.

Hypothetic molecular structure of resulting Zinc complex of Hydroxychloroquine bearing antibacterial N,OH-pharmacophore site antiviral redox [(Zn(OH2)2] moiety.
Figure 12.

Organometallic approach of Prof Taibi BEN HADDA team as proposal to fight against COVID-19 by using Ruthenium (II) complexes of Polypyridines in presence of SATIVEX spray as Supplementary synergetic drug in goal to obtain immediate relaxation and more antiviral therapeutic effect.
All complexes bearing [M-(OH2)2] moiety are of great importance in modern medicinal chemistry. Many such selected metal-based compounds contain [M-(OH2)n], [M-(OH)n], [M = O], which have shown interesting and potential anti-tumor or antibacterial activity (Ben Hadda et al., 2009, 2015; Chatterjee et al., 2006; Chohan et al., 2006; Chohan & Supuranm, 2006) (Figure 13). Bipyridyl-derived ruthenium(II) complexes with antimycobacterial in vitro have been reported with our group. The ruthenium(Ii) complex of Bpy* also showed important activity (%Inhibition = 100%; MIC < 6.5 µg/mL). In another report, a ruthenium(II) complex displayed a fantastic anti-tumor activity with MIC 18.000-fold lower than that of Staurosporine derivative (https://grantome.com/grant/NIH/R01-GM071695-04).
Figure 13.

Molecular structure of some bioactive complexes bearing [Metal(OH2)2] moiety and pharmaceutical SATIVEX drug (https://sweetseeds.es/fr/sativex-medicamento-con-extractos-de-marihuana-ya-en-espana/#).
For the reason cited above, we can propose some ruthenium(II) complexes as a potential perspective to fight with COVID-19 with success (Figure 13).
4. Conclusion
In this study, three proposed anti-COVID-19 supportive drugs/treatments (Remdesivir, Hydroxychloroquine, Azithromycin) have been analysed, and their pharmacophore sites are characterized via bioinformatic DFT, docking and POM analysis. Osiris calculations of analysed compounds reveal that hydroxychloroquine and azithromycin are the safest ones. The most important antiviral activities of hydroxychloroquine and azithromycin agree with the POM results obtained and the tested hydroxychloroquine compound via a redox mechanism.
The addition of zinc sulfate to hydroxychloroquine in the presence of azithromycin is far away from being innocent. The addition of ZnSO4 can play various roles in the coordination of ligands and template effect. It should generate ‘in situ’ important Zn(II) complexes of hydroxychloroquine bearing [Zn(H2O)2] moiety, source of redox properties, and an antibacterial N,OH-pharmacophore site. For these reasons, the hypothetic resulting complex cis-diaquo-Zn-hydroxychloroquine is efficient against coronavirus and should be tested against opportunistic bacteria and other viruses.
Now, it is clear that the Zn(II) is well involved in antiviral activity via the in situ formation of [(Zn(Hydroxychloroquine)2(H2O)2] complex. So this is encouraging to prepare and test similar complexes of ruthenium(II) against coronavirus. The ruthenium(II) complexes are more stable and safe.
5. Perspectives
Identification of a miRNA-peptide with theoretical Exosome Affinity according to COVID-19 structure using Exosomes as potential biomarkers of vaccination (Figure 14) (Cruz-Rodriguez et al., 2020).
Synthesis and validation of an effective vaccine, allowing prevention against COVID (Cruz-Rodriguez et al., 2020).
Clinical trials of the candidate vaccine, using the Food and Drug Administration (FDA) protocols.
Figure 14.

Exosomes as potential biomarkers of vaccination.
Acknowledgements
T. Ben Hadda acknowledges the offer of the local facilities by Prof Yassine Zarhloule, the President of Mohammed Premier University. The authors would like to thank the deanship of scientific research at Umm Al-Qura University for supporting this work by Grant Code: (20UQU0044DSR).
Funding Statement
This work was financially supported by The General Directorate for Scientific Research and Technological Development (DG-RSDT), Algerian Ministry of Scientific Research, Applied Organic Laboratory (FNR 2000).
Disclosure statement
All authors declare no conflict of interest.
References
- Al-Maqtari, H. M., Jamalis, J., Ben Hadda, T., Sankaranarayanan, M., Chander, S., Ahmad, N. A., Mohd Sirat, H., Althagafi, I. I., & Mabkhot, Y. N. (2017). Synthesis, characterization, POM analysis and antifungal activity of novel heterocyclic chalcone derivatives containing acylated pyrazole. Research on Chemical Intermediates, 43(3), 1893–1907. 10.1007/s11164-016-2737-y [DOI] [Google Scholar]
- Amirkhanov, V., Rauf, A., Ben Hadda, T., Ovchynnikov, V., Trush, V., Saleem, M., Raza, M., Rehman, T., Zgou, H., Shaheen, U., & Farghaly, T. A. (2019). Pharmacophores modeling in terms of prediction of theoretical physicochemical properties and verification by experimental correlations of carbacylamidophosphates (CAPh) and sulfanylamidophosphates (SAPh) tested as new carbonic anhydrase inhibitors. Mini-Reviews in Medicinal Chemistry, 19(12), 1015–1027. 10.2174/1389557519666190222172757 [DOI] [PubMed] [Google Scholar]
- Bechlem, K., Aissaoui, M., Belhani, B., Rachedi, K. O., Bouacida, S., Bahadi, R., Djouad, S.-E., Ben Mansour, R., Bouaziz, M., Almalki, F., Ben Hadda, T., & Berredjem, M. (2020). Synthesis, X-ray crystallographic study and molecular docking of new α-sulfamidophosphonates: POM analyses of their cytotoxic activity. Journal of Molecular Structure, 1210, 127990–127998. 10.1016/j.molstruc.2020.127990 [DOI] [Google Scholar]
- Becke, A. D. (1993). Density‐functional thermochemistry III. The role of exact exchange. Journal of Chemical Physics, 98(7), 5648–5652. 10.1063/1.464913 [DOI] [Google Scholar]
- Ben Hadda, T., Deniz, F. S. S., Orhan, I. E., Zgou, H., Rauf, A., Mabkhot, Y. N., & Maalik, A. (2020). Spiro heterocyclic compounds as potential anti-alzheimer agents (Part 2): Their metal chelation capacity, POM analyses and DFT studies. Medicinal Chemistry, 16, 1. 10.2174/1573406416666200610185654 [DOI] [PubMed] [Google Scholar]
- Ben Hadda, T., Genc, Z. K., Masand, V. H., Nebbache, N., Warad, I., & Jodeh, S. (2015). Computational POM and DFT evaluation of experimental in-vitro cancer inhibition of Staurosporine-Ruthenium(II) complexes: The power force of organometallics in drug design. Acta Chimica Slovenica, 62, 679–688. 10.17344/acsi.2015.1357 [DOI] [PubMed] [Google Scholar]
- Ben Hadda, T., Mehmet, A., Baba, M. F., Daoudi, M., Bennani, B., & Kerbal, A. (2009). Anti-tubercular activity of ruthenium (II) complexes with polypyridines. Journal of Enzyme Inhibition and Medicinal Chemistry, 24, 457–463. 10.1080/14756360802188628 [DOI] [PubMed] [Google Scholar]
- Ben Hadda, T., Talhi, O., Silva, A. S. M., Senol, F. S., Orhan, I. E., & Rauf, A. (2018). Cholinesterase inhibitory activity of some semi-rigid spiro heterocycles: POM analyses and crystalline structure of pharmacophore site. Mini - Reviews in Medicinal Chemistry, 18, 711–716. 10.2174/1389557517666170713114039 [DOI] [PubMed] [Google Scholar]
- Brintzinger, H. H., Prosenc, M. H., Schaper, F., Weeber, A., & Wieser, U. (1999). Alternative force field models for ansa-zirconocene complexes-vibrational and structural studies on Me2Si-bridged and tert-butyl-substituted representatives. Journal of Molecular Structure, 485–486, 409–419. 10.1016/S0022-2860(99)00184-2 [DOI] [Google Scholar]
- Chander, S., Tang, C. R., Al-Maqtari, H. M., Jamalis, J., Penta, A., & Ben Hadda, T. (2017). synthesis and study of anti-hiv-1 rt activity of 5-benzoyl-4-methyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-2-one derivatives. Bioorganic Chemistry., 72, 74–79. 10.1016/j.bioorg.2017.03.013 [DOI] [PubMed] [Google Scholar]
- Chatterjee, B. D., Mitra, A., & De, G. S. (2006). Ruthenium polyaminocarboxylate complexes. Platinum Metals Review, 50(1), 2–12. 10.1595/147106705X82874 [DOI] [Google Scholar]
- Chohan, Z. H., Arif, M., Akhtar, M. A., & Supuran, C. T. (2006). Metal-based antibacterial and antifungal agents: Synthesis, characterization, and in vitro biological evaluation of Co(II), Cu(II), Ni(II), and Zn(II) complexes with amino acid-derived compounds. Bioinorganic Chemistry and Applications, 2006, 1–13. 10.1155/BCA/2006/83131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chohan, Z. H., & Supuranm, C. T. (2006). Metalloantibiotics: Synthesis, characterization and in-vitro antibacterial studies on cobalt (II), copper (II), nickel (II) and zinc (II) complexes with cloxacillin. Journal of Enzyme Inhibition and Medicinal Chemistry, 21, 441–448. 10.1080/14756360500397307 [DOI] [PubMed] [Google Scholar]
- Cruz-Rodriguez, L., Dilsiz, N., Ziarati, P., Lambert Brown, D., Hochwimmer, B., & Zayas Tamayo, A. M. (2020). A mirna-peptide fusion as a vaccine candidate against the novel coronavirus (COVID-19). Exosomes as potential biomarkers of sars-cov-2 in lung: After and before vaccination LCR 2020 B008-13. Journal of Bioscience and Bioengineering, 1, 1–11. [Google Scholar]
- Deppmeier, B. J., Driessen, A. J., Hehre, T. S., Hehre, W. J., Johnson, J. A., & Klunzinger, P. E. (2002). SPARTAN 02. Wavefunction Inc. [Google Scholar]
- Eyring, H. (1931). The energy of activation for bimolecular reactions involving hydrogen and the halogens, according to the quantum mechanics. Journal of the American Chemical Society, 53(7), 2537–2549. 10.1021/ja01358a014 [DOI] [Google Scholar]
- Fang, L., Lerner, S. (2020). Coronavirus treatment developed by Gilead sciences granted “rare disease” status, potentially limiting affordability. The Intercept. March 24, 3:03 a.m. https://theintercept.com/2020/03/23/gilead-sciences-coronavirus-treatment-orphan-drug-status/
- Francl, M. M., Pietro, W. J., Hehre, W. J., Binkley, J. S., Gordon, M. S., DeFrees, D. J., & Pople, J. A. (1982). Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. Journal of Chemical Physics., 77(7), 3654–3665. 10.1063/1.444267 [DOI] [Google Scholar]
- Frisch, M. J. (2009). Gaussian-09 revision A.02. Gaussian Inc. [Google Scholar]
- Fukui, K., Yonezawa, T., & Shingu, H. (1952). A molecular orbital theory of reactivity in aromatic hydrocarbons. Journal of Chemical Physics., 20(4), 722–725. 10.1063/1.1700523 [DOI] [Google Scholar]
- Grib, I., Berredjem, M., Rachedi, O. K., Djouad, S. E., Bouacida, S., Bahadi, R., Ouk, T. S., Kadri, M., Ben Hadda, T., & Belhani, B. (2020). Novel N-sulfonylphthalimides: Efficient synthesis, X-ray characterization, spectral investigations, POM analyses, DFT computations and antibacterial activity. Journal of Molecular Structure, 1217, 128423–128423. 10.1016/j.molstruc.2020.128423 [DOI] [Google Scholar]
- Hatzade, K., Sheikh, J., Taile, V., Ghatole, A., Ingle, V., Genc, M., Lahsasni, S., & Ben Hadda, T. (2015). Antimicrobial/antioxidant activity and POM analyses of novel 7-o-β-d-glucopyranosyloxy-3-(4, 5-disubstituted imidazol-2-yl)-4H-chromen-4-ones. Medicinal Chemistry Research, 24(6), 2679–2693. 10.1007/s00044-015-1326-8 [DOI] [Google Scholar]
- Hocquet, A., & Langgard, M. (1998). An evaluation of the MM + force field. Journal of Molecular Modeling, 4(3), 94–112. 10.1007/s008940050128 [DOI] [Google Scholar]
- Hoft, J. (2020). Video: Coronavirus Treatment: New York Doctor Vladimir Zelenko finds 100% success rate in 350 patients using hydroxychloroquine with zinc. Region: USA. Theme: Science and Medicine. Global Research, March 24. Gateway Pundit 23 March 2020. https://www.globalresearch.ca/video-ny-doctor-vladimir-zelenko-finds-100-success-rate-350-patients-using-hydroxychloroquine-zinc/5707381.
- Hsu, K.-C., Chen, Y.-F., Lin, S.-R., & Yang, J.-M. (2011). iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics, 12(Suppl 1), S33. 10.1186/1471-2105-12-S1-S33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, N., Kalyanaraman, C., Bernacki, K., & Jacobson, M. P. (2006). Molecular mechanics methods for predicting protein ligand binding. Physical Chemistry Chemical Physics, 8, 5166–5177. 10.1039/b608269f [DOI] [PubMed] [Google Scholar]
- Hunt, J. (2020). Japan is racing to test a drug to treat COVID-19.
- Jamalis, J., Yusof, F. S. M., Chander, S., Wahab, R. A., P. Bhagwat, D., Sankaranarayanan, M., Almalki, F., & Ben Hadda, T. (2020). Psoralen derivatives: Recent advances of synthetic strategy and pharmacological properties. Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry, 19(3), 222–239. 10.2174/1871523018666190625170802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahpour, A., Heiran, R., Sinou, V., Latour, C., Bouktab, L. D., & Brunel, J. M. (2019). Synthesis of new β-lactams bearing the biologically important morpholine ring and POM analyses of their antimicrobial and antimalarial activities. Iranian Journal of Pharmaceutical Research, 18, 34–48. PMID: 31089342. [PMC free article] [PubMed] [Google Scholar]
- Kamal, A. M., Abdelhady, M. I. S., & Ben Hadda, T. (2019). Two novel flavone C-glycosides isolated from Podocarpus gracilior: POM analyses and in-vitro anticancer activity against hepatocellular carcinoma. International Journal of Pharmacy and Pharmaceutical Sciences, 11, 57–62. 10.22159/ijpps.2019v11i7.33163 [DOI] [Google Scholar]
- Khan, H., Khan, Z., Amin, S., Mabkhot, Y. N., Mubarak, M. S., & Ben Hadda, T. (2017). Plant bioactive molecules bearing glycosides as lead compounds for the treatment of fungal infection: A review. Biomedicine & Pharmacotherapy, 93, 498–509. 10.1016/j.biopha.2017.06.077 [DOI] [PubMed] [Google Scholar]
- Klopman, G. (1968). Chemical reactivity and the concept of charge-and frontier controlled reactions. Journal of the American Chemical Society, 90(2), 223–234. 10.1021/ja01004a002 [DOI] [Google Scholar]
- Lahsasni, S., Ben Hadda, T., Masand, V., Pathan, N. B., Parvez, A., Warad, I., Shaheen, U., Bader, A., & Aljofan, M. (2015). POM analyses of raltegravir derivatives: A new reflection enlighting the mechanism of HIV-integrase inhibition. Research on Chemical Intermediates, 41(8), 5121–5136. 10.1007/s11164-014-1616-7 [DOI] [Google Scholar]
- Lee, C., Yang, W., & Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B, 37, 785–789. 10.1103/PhysRevB.37.785 [DOI] [PubMed] [Google Scholar]
- Liotta, D. (1988). Advances in molecular modeling. JAI Press.
- Mabkhot, Y., Alatibi, F., El-Sayed, N., Al-Showiman, S., Kheder, N., Wadood, A., Rauf, A., Bawazeer, S., & Ben Hadda, T. (2016). Antimicrobial activity of some novel armed thiophene derivatives and petra/osiris/molinspiration (POM) analyses. Molecules, 21(2), 222–237. 10.3390/molecules21020222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabkhot, Y., Aldawsari, F., Al-Showiman, S., Barakat, A., Ben Hadda, T., Mubarak, M., Naz, S., Ul-Haq, Z., & Rauf, A. (2015). Synthesis, bioactivity, molecular docking and POM analysis of novel substituted thieno[2,3-b]thiophenes and related congeners. Molecules, 20(2), 1824–1841. 10.3390/molecules20021824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabkhot, Y. N., Barakat, A., Yousuf, S., Choudhary, M. I., Frey, W., & Ben Hadda, T. (2014). Substituted thieno[2,3-b]thiophenes and related congeners: Synthesis, β-glucuronidase inhibition activity, crystal structure, and POM analyses. Bioorganic and Medicinal Chemistry, 22, 6715–6725. 10.1016/j.bmc.2014.08.014 [DOI] [PubMed] [Google Scholar]
- Messali, M., Aouad, M. R., Ali, A. A. S., Rezki, N., Ben Hadda, T., & Hammouti, B. (2015). Synthesis, characterization and POM analysis of novel bioactive imidazolium-based ionic liquids. Medicinal Chemistry Research, 24(4), 1387–1395. 10.1007/s00044-014-1211-x [DOI] [Google Scholar]
- Messali, M., Aouad, M. R., El-Sayed, W. S., Ali, A. A., Ben Hadda, T., & Hammouti, B. (2014). New eco-friendly 1-alkyl-3-(4-phenoxybutyl) imidazolium-based ionic liquids derivatives: A green ultrasound-assisted synthesis, characterization, antibacterial activity and POM analyses. Molecules, 19, 11741–11759. 10.3390/molecules190811741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, J. S., & Sen, K. (1996). Molecular electrostatic potentials: Concepts and applications (1st ed.). Elsevier. [Google Scholar]
- Nasruddin, G. U., Rauf, A., Khan, H., Mamadalieva, N. Z., Khan, A., & Farooq, U. (2018). Phytochemical analysis, urease inhibitory effect and antimicrobial potential of Allium humile. Zeitschrif für Arznei- und Gewürzpflanzen. Journal of Medicinal & Spice Plants, 22, 173–175. [Google Scholar]
- Parr, R. G., & Pearson, R. G. (1983). Absolute hardness: Companion parameter to absolute electronegativity. Journal of the American Chemical Society, 105(26), 7512–7516. 10.1021/ja00364a005 [DOI] [Google Scholar]
- Pearson, R. G. (1963). Hard and soft acids and bases. Journal of the American Chemical Society, 85(22), 3533–3539. 10.1021/ja00905a001 [DOI] [Google Scholar]
- Piaz, F. D., Bader, A., Malafronte, N., D’Ambola, M., Petrone, A. M., & Porta, A. (2018). Phytochemistry of compounds isolated from the leaf-surface extract of Psiadia punctulata (DC.) Vatke growing in Saudi Arabia. Phytochem, 155, 191–202. 10.1016/j.phytochem.2018.08.003 [DOI] [PubMed] [Google Scholar]
- Rachedi, K. O., Bahadi, R., Aissaoui, M., Ben Hadda, T., Belhani, B., Bouzina, A., & Berredjem, M. (2020). DFT Study, POM analyses and molecular docking of novel oxazaphosphinanes: Identification of antifungal pharmacophore site. Indonesian Journal of Chemistry, 20(2), 440–450. 10.22146/ijc.46375 [DOI] [Google Scholar]
- Rachedi, K. O., Ouk, T.-S., Bahadi, R., Bouzina, A., Djouad, S.-E., Bechlem, K., Zerrouki, R., Ben Hadda, T., Almalki, F., & Berredjem, M. (2019). Synthesis, DFT and POM analyses of cytotoxicity activity of α-amidophosphonates derivatives: Identification of potential antiviral O,O-pharmacophore site. Journal of Molecular Structure, 1197, 196–203. 10.1016/j.molstruc.2019.07.053 [DOI] [Google Scholar]
- Rad, J. A., Jarrahpour, A., Latour, C., Sinou, V., Brunel, J. M., Zgou, H., Mabkhot, Y., Ben Hadda, T., & Turos, E. (2017). Synthesis and antimicrobial/antimalarial activities of novel naphthalimido trans-β-lactam derivatives. Medicinal Chemistry Research, 26(10), 2235–2242. 10.1007/s00044-017-1920-z [DOI] [Google Scholar]
- Radi, S., Tighadouini, S., Feron, O., Riant, O., Mabkhot, Y. N., & Al-Showiman, S. S. (2015). One pot synthesis, antitumor, antibacterial and antifungal activities of some Sciff base heterocycles. International Journal of Pharmaceutics, 5(1), 39–45. [Google Scholar]
- Rakib, A., Sami, S. A., Mimi, N. J., Chowdhury, M. M., Eva, T. A., Nainu, F., Paul, A., Shahriar, A., Tareq, A. M., Emon, N. U., Chakraborty, S., Shil, S., Mily, S. J., Ben Hadda, T., Almalki, F. A., & Emran, T. B. (2020). Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Computers in Biology and Medicine, 124, 103967. 10.1016/j.compbiomed.2020.103967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauf, A., Orhan, I. E., Ertas, A., Temel, H., Ben Hadda, T., & Saleem, M. (2017). Elucidation of phosphodiesterase-1 inhibitory effect of some selected natural polyphenolics using in vitro and in silico methods. Current Topics in Medicinal Chemistry, 17, 412–441. 10.2174/1568026616666160824103615 [DOI] [PubMed] [Google Scholar]
- Rbaa, M., Oubihi, A., Anouar, E. H., Ouhssine, M., Almalki, F., Ben Hadda, T., Zarrouk, A., & Lakhrissi, B. (2019). Synthesis of new heterocyclic systems oxazino derivatives of 8-hydroxyquinoline: Drug design and POM analyses of substituent effects on their potential Antibacterial properties. Chemical Data Collections., 24, 100306. 10.1016/j.cdc.2019.100306 [DOI] [Google Scholar]
- Richard, P., & Gambert, P. (2020). Coronavirus. L’hydroxychloroquine: ce qu’on en sait aujourd’hui. Ouest-France Magazine.
- Sajid, Z., Ahmad, M., Aslam, S., Ashfaq, U. A., Zahoor, A. F., Saddique, F. A., Parvez, M., Hameed, A., Sultan, S., Zgou, H., & Ben Hadda, T. (2016). Novel armed pyrazolobenzothiazine derivatives: Synthesis, X-ray crystal structure and POM analyses of biological activity against drug resistant clinical isolate of staphylococus aureus. Pharmaceutical Chemistry Journal, 50(3), 172–180. 10.1007/s11094-016-1417-y [DOI] [Google Scholar]
- Siegel, D., Hui, H. C., Doerffler, E., Clarke, M. O., Chun, K., Zhang, L., Neville, S., Carra, E., Lew, W., Ross, B., Wang, Q., Wolfe, L., Jordan, R., Soloveva, V., Knox, J., Perry, J., Perron, M., Stray, K. M., Barauskas, O., … Mackman, R. L. (2017). Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of ebola and emerging viruses. Journal of Medicinal Chemistry, 60(5), 1648–1661. 10.1021/acs.jmedchem.6b01594 [DOI] [PubMed] [Google Scholar]
- Stewart, J. J. P. (1989). Optimization of parameters for semiempirical methods I. Journal of Computational Chemistry, 10(2), 209–220. 10.1007/s00894-007-0233-4 [DOI] [Google Scholar]
- Subramanian, S. (2020). How Wuhan virologists pegged chloroquine as a potential COVID-19 cure. The Diplomate, April 01. https://thediplomat.com/2020/04/how-wuhan-virologists-pegged-chloroquine-as-a-potential-covid-19-cure.
- Tatar, E., Şenkardeş, S., Sellitepe, H. E., Küçükgüzel, Ş. G., Karaoğlu, Ş. A., Bozdeveci, A., DE Clercq, E., Pannecouque, C., Ben Hadda, T., & Küçükgüzel, İ. (2016). Synthesis, and prediction of molecular properties and antimicrobial activity of some acylhydrazones derived from N-(arylsulfonyl)methionine. Turkish Journal of Chemistry, 40, 510–534. 10.3906/kim-1509-21 [DOI] [Google Scholar]
- Tighadouni, S., Radi, S., Sirajuddin, M., Akkurt, M., Ozdemir, N., & Ahmad, M. (2016). In vitro antifungal, anticancer activities and POM analyses of a novel bioactive schiff base 4-{[(E)-furan-2-ylmethylidene]amino}p-henol: Synthesis, characterization and crystal structure. Journal of the Chemical Society of Pakistan, 38, 157–165. [Google Scholar]
- Titi, A., Messali, M., Alqurashy, B. A., Touzani, R., Shiga, T., & Oshio, H. (2019). Synthesis, characterization, X-ray crystal study and bioactivities of pyrazole derivatives: Identification of antitumor, antifungal and antibacterial pharmacophore sites. Journal of Molecular Structure, 1205, 127625. https://doi.10.1016/j.molstruc.2019.127625. [Google Scholar]
- Wang, F., Chen, C., Tan, W., Yang, K., & Yang, H. (2016). Structure of main protease from human coronavirus NL63, insights for wide spectrum anti-coronavirus drug design. Scientific Reports 6, 22677. 10.1038/srep22677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Chen, X., Lu, Y., Chen, F., & Zhang, W. (2020). Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. BioScience Trends, 14, 64–68. 10.5582/bst.2020.01030 [DOI] [PubMed] [Google Scholar]
- Xu, Z., Peng, C., Shi, Y., Zhu, Z., Mu, K., Wang, X., & Zhu, W. J. B. (2020). Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv Preprint. 10.1101/2020.01.27.921627 [DOI] [Google Scholar]
- Youssoufi, M. H., Ben Hadda, T., Warad, I., Naseer, M. M., Mabkhot, Y. N., & Bader, A. (2015). POM analyses of anti-kinase activity of thirteen peptide alkaloids extracted from Zizyphus species. Medicinal Chemistry Research, 24(1), 267–274. 10.1007/s00044-014-1117-7 [DOI] [Google Scholar]