

Abstract
Neurodegenerative and neuropsychiatric disorders are pervasive and debilitating conditions characterized by diverse clinical syndromes and comorbidities, whose origins are as complex and heterogeneous as their associated phenotypes. Risk for these disorders involves substantial genetic liability, which has fueled large-scale genetic studies that have led to a flood of discoveries. In turn, these discoveries have exposed substantial gaps in our knowledge with regards to the complicated genetic architecture of each disorder and the substantial amount of genetic overlap among disorders, which implies some degree of shared pathophysiology underlying these clinically distinct, multifactorial disorders. Understanding the role of specific genetic variants will involve resolving the connections between molecular pathways, heterogeneous cell types, specific circuits and disease pathogenesis at the tissue and patient level. We consider the current known genetic basis of these disorders and highlight the utility of molecular systems approaches that establish the function of genetic variation in the context of specific neurobiological networks, cell-types, and life stages. Beyond expanding our knowledge of disease mechanisms, understanding these relationships provides promise for early detection and potential therapeutic interventions.
Introduction
Neurological and psychiatric disorders are the leading cause of disability and death globally [1]. The root of neurodegenerative and psychiatric diseases is thought to be a combination of genetic variation and environmental exposures during a person’s lifetime. The genetic causes involve virtually all forms of DNA sequence variation and transmission, from rare de novo mutations (autosomal dominant or X-linked, imprinted etc.) and rare recessive conditions, to inherited common and rare variation across the genome [2–4]. Currently, we lack how this disease-causing variation influences specific cell types, biological processes and pathological trajectory, all of which help explain disease mechanisms and provide better substrate for putative effective therapies. For rare, protein disrupting mutations, connecting genetic mechanisms to cell types and molecular pathways has been facilitated by the ability to focus on a single protein and its major effects in model systems, and yet still this has been a challenging path [5]. For common genetic variation, this process is further complicated by the large number of contributing variants, the small effect size of individual variants, and the difficulty in identifying the actual causal variants and genes that they impact [6,7].
Here, we will first discuss what we have learned from the genetic variation associated with a number of prominent neurodegenerative and neuropsychiatric diseases. While many genetic risk loci have been identified, they explain only a small portion of heritability, far more remain to be found, and only a small percentage of patient cases can be directly explained by a single specific major mutation [7]. We concentrate on disorders where substantial functional genomics investigations have been performed to try to link loci to pathophysiology. For neurodegenerative disorders, we focus particular attention on Alzheimer’s (AD) and related tauopathies and dementia (frontal-temporal dementia, FTD; progressive supranuclear palsy, PSP; and Parkinson’s disease, PD). With respect to neuropsychiatric with neurodevelopmental origins, we focus primarily on schizophrenia (SCZ) and autism spectrum disorder (ASD), in comparison with Bipolar disorder (BD) and ADHD which each have both overlapping and distinct genetic architectures. We explore new findings that connect common genetic risk variants to specific genes and altered transcriptional networks. It is vital to find better means of filtering and exploring these data to help both identify and focus on causative genes, as well as to explore new multi-loci or polygenic paradigms which is further complicated by the apparent shared genetic features overlap among many conditions [8,9]. Therefore, we also touch on new avenues of research that aim to incorporate multi-omic data from patients across cell-types, brain regions, and timeframes. By establishing functional relationships between a given causal variant, or a set of contributory genetic variants, and neurobiological phenotypes, these approaches hold promise to begin to fill gaps in our mechanistic understanding of the origins of disease traits and facilitate development of more effective approaches for prevention and treatment.
Genetic Architecture (shared, polygenic risk)
The increasing feasibility of interrogating the genome through high-throughput genotyping and sequencing technologies has allowed dissection of the genetic variation that underlie many disorders. Modern application of genotyping arrays, whole exome sequencing (WES), and whole-genome sequencing (WGS) to case control studies have permitted identifying hundreds of loci, or specific mutations, increasing disease susceptibility [10]. In a general sense, all of these disorders have rare and common forms, although the relative contributions from these different frequency pools differ. For example, disorders with clinical onset in infancy, such as ASD have a relatively higher proportion of cases harboring major gene forms, either dominant de novo or Mendelian (15-20% [11,12]) than SCZ or BD (<5%, [13–15]). By GWAS, where similar samples sizes in SCZ and BD have uncovered more genome wide significant common loci than in ASD, consistent with more heterogeneity and a higher contribution from rare genetic variation in the latter [16–18]. In neurodegenerative disorders, the contribution of Mendelian gene forms also varies widely, contributing at least 20-30% to FTD [19,20] but substantially less to AD (~5%;[21], [22]), with PD in between (5-10%; [23]). As a general rule, although these cases share substantial overlap in clinical phenotypes with familial forms, they also can show some atypical aspects in their presentations, such as earlier age of onset in dementias (FTD, PD, AD) [21,24,25], or a higher frequency of motor or intellectual disability (ID) in the case of ASD or SCZ [26–29]. In all cases, common variants do not explain all of the heritability suggesting a role for rare inherited variants, which has been supported by early, relatively small studies in dementia and ASD [30,31].
Neurodegenerative dementias.
Recent GWAS have identified over 90 PD-associated loci [32–35], 30 for AD [36–38], 29 loci for FTD [39,40], and 5 for PSP [41,42] (Figure 1A). Given that these disorders share certain general features such as protein aggregation, neuronal degeneration and neuro-inflammation, one might expect to see a high amount of genetic overlap among these disorders. Genetic correlations based on common variants from each GWAS reveal the highest relationship was found between PSP and FTD (0.6, [41]), both of which can involve pure tauopathy [43] (Figure 1A). AD common variation was most correlated with FTD (0.29), whereas PD was more correlated with PSP (0.4) and FTD (0.31), all of which share Parkinsonian features or pathology (Figure 1A; [9,41]). Interestingly, neurodegenerative disorders exhibit striking enrichment of risk-variants associated with immune-mediated diseases, such as Crohn disease (CD), ulcerative colitis (UC), rheumatoid arthritis (RA), type 1 diabetes (T1D), celiac disease (CED), and psoriasis (PSOR). After correcting for the MHC-associated SNPs, AD is found to be moderately enriched with CD, UC, T1D, and RA (3-8 fold enrichment, listed in decreasing order [44]) whereas FTD was mostly enriched in T1D, CED, CD and RA (5-35 fold enrichment, listed in decreasing order [45]). Thus, there does seem to be a relationship between genetic sharing and common pathology across neurodegenerative disorders including risk-overlap with several common immunological disorders, suggesting some common pathophysiological mechanisms, consistent with models based on cell biological and biochemical studies [46–49].
Figure 1. Correlation of Common Variation underlying Neurodegenerative and Neuropsychiatric disorders.
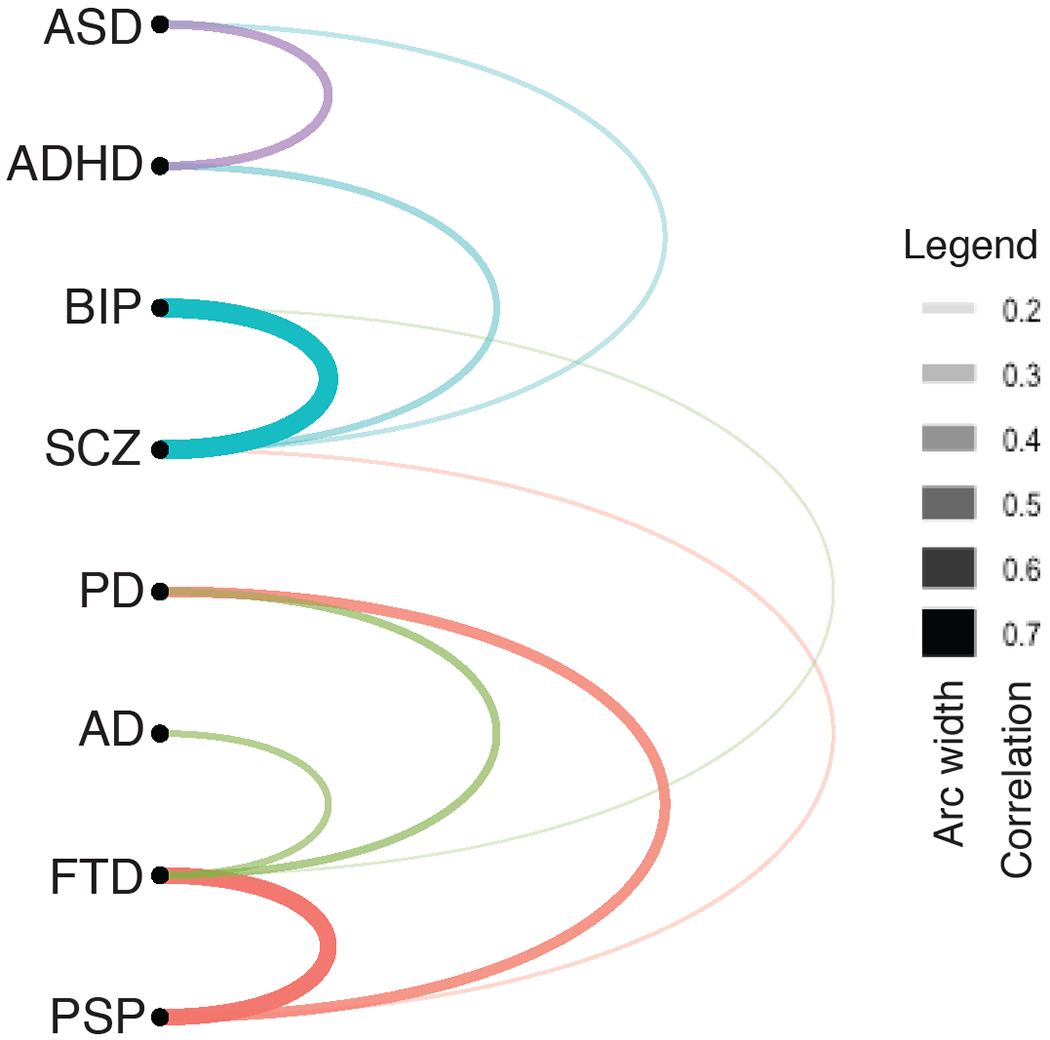
Arc network of variant correlations (> 0.15) exhibiting shared risk between common neurodevelopmental, ASD and ADHD colored by purple, neuropsychiatric, BIP and SCZ colored by blue, and neurodegenerative disorders, PD, AD, FTD, PSP colored by red and green. The width of the colored connecting arc depicts the size of correlation. Correlations taken from {Chen:2018ks, AutismSpectrumDisorderWorkingGroupofthePsychiatricGenomicsConsortium:2019ex, Demontis:2018do}. ASD; Autism spectrum disorder, ADHD; Attention-deficient hyperactivity disorder, BIP;Bipolar, SCZ; Schizophrenia, PD; Parkinson’s disorder, AD; Alzheimer’s disorder, FTD; Frontal-temporal lobe Dementia, PSP; Progressive supranuclear palsy.
Neuropsychiatric disorders.
GWAS on common neuropsychiatric disorders has identified a similar magnitude of disease-risk variants: 145 for SCZ [16,50], 30 for BD [17], 12 for ADHD [51], and 12 for ASD [52] (Figure 1A). The genetic correlations between SCZ and BIP are relatively high (0.68, [53]; 0.70, [17]}), whereas SCZ is correlated with both ADHD and ASD, albeit at lower, but still significant rates (0.21 ASD and 0.36 ADHD, [9,52]}). Smaller orthogonal studies utilizing family history find high risk for ASD in offspring with relatives diagnosed with SCZ, BIP, ASD, and/or ADHD [54–56] and vice versa for each disorder (ADHD/BIP [57], ADHD/ASD [58]). Recent studies actually approach the concept of cross-disorder genetic liability directly, by pooling major psychiatric diagnosis (including anorexia nervosa, major depression, obsessive-compulsive disorder, and Tourette syndrome in addition to ADHD, ASD, BIP, and SCZ) into one “affected” category [59,60], together finding 4-6 loci with shared risk for all disorders, 23 shared by more than four disorders and over 100 loci with shared-risk by two disorders. Additionally, hierarchal analysis and exploratory factor analysis (EFA) of the common risk variants organized disorders into major phenotypical categories such as compulsivity (anorexia nervosa, obsessive-compulsive disorder, and Tourette syndrome), mood/psychosis (Major depression, BIP and SCZ) and early onset neurodevelopmental disorders (ASD, ADHD, and Tourette syndrome)[61]. Thus, despite having major differences in diagnostic criteria and behavioral phenotypic onset, there is considerable evidence that SCZ and BIP, SCZ and ASD, ADHD and ASD share some similarities in origins, but the extent of sharing varies across disorders seemingly dependent on the extent of co-morbidity of specific traits. Thus, one major challenge in connecting genetic variants to specific clinical phenotypes is understanding the intermediate phenotypes or biological pathways modulated by shared and disorder-specific risk variants [7]. Although this is a challenge, it also provides an opportunity, in that we have the tools in hand to begin to dissect this complexity [4,6,62].
Connecting risk loci to genes
One feature that unites all common disorders, brain or otherwise, is that the majority of common genetic risk-variants are found in non-coding, regulatory regions of genome [63,64]. In AD, only 2% of common risk variants are found in protein-coding regions and 50% of risk variants are found in intergenic loci, making their association with a specific gene more difficult [38]. Similarly, BIP-, ADHD-, and ASD-SNPs are found significantly enriched within non-coding regions (~15-20 fold enrichment) whereas the enrichment in coding regions are both low (~7 fold) and not significant [17,18,51]. In SCZ virtually all risk-SNPs are found to lie in intergenic and intronic loci (<1% of variants are located in exonic loci; [16,50]).
Altogether, these findings have illuminated a complex connection between genotype and disorder. That is, these disorders are polygenic, with substantial shared risk despite distinct clinically defined phenotypes, and whose risk is imparted by variants that reside primarily in regulatory regions, challenging the proper mapping of variant interactions to specific genes [3,65,66]. Moreover, in neuropsychiatric disorders, this architecture is further complicated by the presence of rare de novo or inherited larger copy-number variations, whose clinical presentation in individuals may be modified by polygenic risk [67–69]. To properly leverage these genetic findings, so as to fuel a deeper understanding of the biological origins and mechanisms, one needs to combine these population genetic findings with functional genomic maps [4], and integrate them using a systems biology approach [6,62,70]. Such integrative genomic studies attempt to connect loci to specific genes that they regulate, and understand what biological pathways, developmental periods, cell types, and circuits these genes control, and further, how specific variation they might impact them.
Causal variant to gene.
A first step in connecting variation to pathways is to understand which specific genes are regulated by disease-associated variants. This process initially involves identifying which variant(s) within a loci are functional. Often the most significant SNP from GWAS is not the causal variant due to linkage disequilibrium (LD; [71]), thus it is necessary to utilize bio-informatic methods that incorporate LD (i.e. Caviar [72], Paintor [73]) in conjunction to annotation within human genomic functional maps and validation through high throughput assays, such as multiple parallel reporter assays [66,74]. This process is further complicated by the fact that such regulation likely mechanistically occurs through long-range chromatin interactions, often does not affect the closest gene [75,76] and is not influenced by patterns of linkage disequilibrium that are used in mapping the loci [77]. Given these precautions researchers have made advances by mapping variants onto core functional maps derived from the integration of multi-omic investigations (Figure 2).
Figure 2. Functional Genomics Links Genetic origins to Disease Mechanisms.
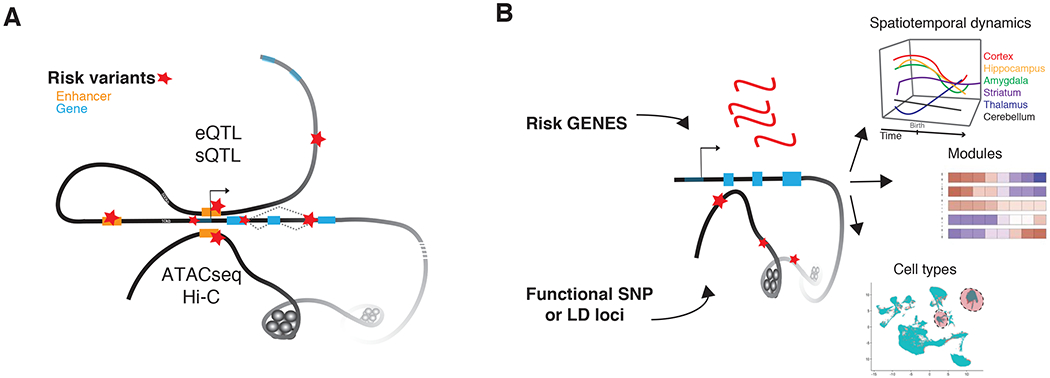
A) Depiction of annotation of risk variation to specific genes. DNA is depicted as a black line with a simple gene model: right hand arrow depicts a transcriptional start site, blue rectangle represents exons, and grey diagonal, connections between exons as isoform options. Enhancers are depicted as orange rectangles and risk variants are marked by red stars. The top extension of DNA from the gene body illustrates mapping by either eQTL or sQTL of a proximal risk variant to a gene. The bottom extension of DNA illustrates long-range DNA contacts (cis or trans), through the combination of ATACseq and Hi-C datasets, to map risk variants to specific genes. B) Cartoon of the two basic integrative approaches used to collapse polygenic disease variation to specific temporal epochs, regions, cell types and network modules. These methods are often thought of interchangeably, but should be distinguished: 1) top, risk genes (red) identified through the annotation with core functional maps in (A) are used in comparison to, 2) bottom, SNPs or multi-SNP loci (LD, red stars) are mapped directly to regulatory regions (open chromatin) to look for enrichment in regulatory regions that are active in specific spatiotemporal datasets across time and brain regions, modules of specific pathways and processes, cell types from single-cell technological approaches from normal human reference and/or postmortem case datasets.
Functional maps for variant annotation are typically derived from normal healthy tissue and often are based on specific quantitative-trait loci that map common genetic variants onto their expression (eQTL) or splicing effects (sQTL) [78–81]; Figure 2A). Small sample size, and the restricted representation of cis-interactions only, can limit the number of genes covered in a given QTL functional dataset (for comparison see: [80,82] and [78]). Thus, genomic datasets that leverage biochemical methods, such as correlation of open chromatin regions by ATAC-seq, or presumed enhancer – promotor regulator loops identified by chromatin confirmation methods, such as Hi-C, are useful adjuncts to QTL maps ([75,83,84]; Figure 2A). Moreover, although about half of gene regulatory variants are shared across cells [76], a substantial amount of regulatory variation is tissue and stage specific [80,85]. Thus, we need functional regulatory maps of brain tissue at all stages and regions, and across major psychiatric disorders to understand variant effects on gene expression and splicing, the core goal of the psychENCODE consortium [78,86,87] which is complemented by broader framework endeavors such as ENCODE [88], GTEX [89] and the 4D genome [90]. Similar efforts to map gene expression and proteomics are being conducted by the AMP-AD consortium in dementias [91–93]. Validation of regulatory predictions from these resources using in vitro models and CRISPR engineering of enhancers or splicing regulatory regions has provided additional confidence that these predictions are biologically relevant [75,83,94,95].
Genes to pathways and cells.
Most studies to date have used standard ontological databases to map risk genes onto pathways [96]. Although these approaches are a valuable step, they are limited by current knowledge and incomplete annotation of most pathways with respect to CNS specific processes [62], leading to specific efforts to expertly curate nervous system specific knowledge such as SynGO [97]. Moreover, solely relying on gene ontology or KEGG pathways for testing enrichment of common or rare risk genes is more biased than analyses based on functional genomic data sets [62,98,99]. More advanced methods look for gene or protein networks, which are built from the bottom up using expression or protein interaction data and are increasingly becoming widely applied [99–101]. A major advantage of network analysis is that it is able to organize multiple levels of the hierarchical molecular structure of the brain into significant modules, or highly connected set of genes, that specifically represent brain region, maturation state, cell type, organelle function, and molecular pathways [102–104].
Further annotation of GWAS signals to specific biological pathways, brain regions and cell types is achieved through the mapping of risk-variants onto gene or protein networks from brain regional and single cell transcriptomes or epigenomic data (e.g. ATAC-seq) (Figure 2B). For example, network analysis utilizing the BrainSpan database [105] illustrated a striking enrichment of ASD-risk genes in modules representing transcriptional regulation and chromatin modification as well as specific excitatory neuronal cell types during neurogenesis, linking ASD risk to midgestational development [102], findings that have been replicated and extended since [31,106–109]. Altogether, exemplified by recent studies in ASD, this approach has yielded substantial evidence of pathways impacting transcriptional regulation and chromatin modification, microtubule function and synaptic function, primarily affecting projection neurons and neurogenesis and less so inhibitory neurons, during fetal brain development [31,109].
In contrast to major gene mutations, polygenicity poses a challenge to the paradigm of single gene to function, how does one understand or summarize the biological mechanisms of hundreds or thousands of common variants, each with variable but minute effect sizes on disease risk? Perhaps paradoxically, polygenic risk has actually proven quite powerful in elucidating pathways. Analogous to expression data, one can partition the polygenic signal measured by disease GWAS-risk across the genome utilizing functional chromatin datasets, asking if disease-risk is in specific pathways defined by co-expression or PPI modules, brain regions, or cell types (Figure 2B bottom). In this way, a complex association signal can be broken down into the biological elements that it affects. These approaches have made substantial contribution to our understanding of disease exemplified below.
Two recent studies combining AD GWAS with either bulk or single-cell transcriptional analysis of post mortem brain from AD donors found that multiple GWAS risk loci were enriched in half of their top 20 significant modules disrupted in AD via bulk analysis [110], whereas only three modules from single-cell analysis exhibited significant enrichment (most likely a technical limitation) [111]. While the majority of these AD-modules seem to represent multiple functional changes in astrocytes and microglia (inflammatory and metabolic pathways; [110], the single cell data points to clear enrichment of AD risk-signal specifically within microglia [111]. These findings are further supported by large-scale proteomic AD brain donor cohort that found strikingly analogous and complimentary AD-PPI networks to the prior AD-expression networks and displayed strong enrichment for AD-risk signal within microglia modules [112].
In comparison, a recent study focused on chromatin confirmation data in human midgestational fetal cortical tissue utilized partitioned heritability (LD score regression) with a number of GWAS disease-associated datasets to determine if and when these variants can impact development ([83], Figure 2B). They found no overlap between common AD-variants and any of these developmental regulatory regions. In contrast, they found both SCZ and ADHD were significantly enriched in regulatory chromatin regions related to neurogenesis within the dividing germinal zone in comparison to the maturing cortical plate, a region comprised mainly of neurons. Analogously, another study focused on chromatin confirmation and function in adult human cortex found significant enrichment of risk-variants in isolated populations of neurons, oligodendrocytes, microglia, and astrocytes [113]. The authors found AD-associated risk variants specifically and only enriched within microglia enhancers. On the other hand, their analysis showed a strong enrichment of SCZ-, ASD-, BD-, and ADHD-risk variants in both the enhancers and promoters of genes expressed in neurons. BD and SCZ variants were also found in oligodendrocyte and astrocyte gene promoters. These findings are supported by single-cell transcriptomic analysis obtained earlier in mid-fetal development that find an enrichment of risk variants in transcriptome profiles of radial glia responsible for producing excitatory neurons, astrocytes and oligodendrocytes and select maturing excitatory neurons [108].
Conclusions
Despite underlying causal heterogeneity, polygenicity and phenotypic complexity contributing to common neurological and neuropsychiatric disorders there is early emergence of biologic themes that have been uncovered via integrative functional genomic analyses. Here we highlighted research utilizing integrative frameworks to better resolve disease mechanisms. As discussed above, although being highly heritable, AD-risk is expressed during adulthood, that most disease-variants function within glial cells and these findings are further supported by transcriptomics. The missing enrichment in fetal data of AD-risk variants within glial cells is most likely technical as these cell types have yet to be produced in large enough number for analysis or are masked within the bulk datasets as proof of principle it would be of interest to follow up investigation across postnatal development. SCZ and ASD genetic risk can be found as early as mid-gestation stage in fetal development particularly enriched in enhancers specific to radial glia progenitor cells and, later in adulthood, this enrichment can be extended to specifically to select excitatory neurons and glial cells supported by both bulk and single-cell transcriptomic analysis.
These approaches are in their infancy and there still remains much to discovered in terms of true mechanistic understanding. However the integrative analyses we have described show great promise by not only connecting genotype to phenotype but increasing the resolution of disease mechanisms and intermediate phenotypes.
Footnotes
Declaration of interest: none
References
- 1.James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, Abbastabar H, Abd-Allah F, Abdela J, Abdelalim A, et al. : Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet 2018, 392:1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.la Torre Ubieta de L, Won H, Stein JL, Geschwind DH: Advancing the understanding of autism disease mechanisms through genetics. Nature Publishing Group 2016, 22:345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan PF, Daly MJ, O’Donovan M: Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nature Publishing Group 2012, 13:537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan PF, Geschwind DH: Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 2019, 177:162–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders SJ, Sahin M, Hostyk J, Thurm A, Jacquemont S, Avillach P, Douard E, Martin CL, Modi ME, Moreno-De-Luca A, et al. : A framework for the investigation of rare genetic disorders in neuropsychiatry. Nature Medicine 2019, doi: 10.1038/s41591-019-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gandal MJ, Leppa V, Won H, Parikshak NN, Geschwind DH: The road to precision psychiatry: translating genetics into disease mechanisms. Nature Neuroscience 2016, 19:1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geschwind DH, Flint J: Genetics and genomics of psychiatric disease. Science 2015, 349:1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wray NR, Wijmenga C, Sullivan PF, Yang J, Visscher PM: Common Disease Is More Complex Than Implied by the Core Gene Omnigenic Model. Cell 2018, 173:1573–1580. [DOI] [PubMed] [Google Scholar]
- 9.Brainstorm Consortium, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, Escott-Price V, Falcone GJ, Gormley P, et al. : Analysis of shared heritability in common disorders of the brain. Science 2018, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lappalainen T, Scott AJ, Brandt M, Hall IM: Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. : Most genetic risk for autism resides with common variation. Nat Genet 2014, 46:881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.lossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. : The contribution of de novo coding mutations to autism spectrum disorder. Nature Publishing Group 2014, 515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Schizophrenia Psychiatric Genome-Wide Association Study Consortium (PGC-SCZ), The International Schizophrenia Consortium (ISC), The Molecular Genetics of Schizophrenia Collaboration (MGS), Lee SH, DeCandia TR, Ripke S, Yang J, Sullivan PF, Goddard ME, Keller MC, et al. : Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet 2012, 44:247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howrigan DP, Rose SA, Samocha KE, Fromer M, Cerrato F, Chen WJ, Churchhouse C, Chambert K, Chandler SD, Daly MJ, et al. : Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nature Neuroscience 2020, doi: 10.1038/s41593-019-0564-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rees E, Han J, Morgan J, Carrera N, Escott-Price V, Pocklington AJ, Duffield M, Hall LS, Legge SE, Pardinas AF, et al. : De novo mutations identified by exome sequencing implicate rare missense variants in SLC6A1 in schizophrenia. Nature Neuroscience 2020, doi: 10.1038/s41593-019-0565-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.as AFPX, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, Legge SE, Bishop S, Cameron D, Hamshere ML, et al. : Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 2018, doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, Mattheisen M, Wang Y, Coleman JRI, Gaspar HA, et al. : Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 2019, doi: 10.1038/s41588-019-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grove J, Ripke S, Als TD, Mattheisen M, Walters R, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, et al. : Common risk variants identified in autism spectrum disorder. 2017, doi: 10.1101/224774. [DOI] [Google Scholar]
- 19.Chow TW, Miller BL, Hayashi VN, Geschwind DH: Inheritance of frontotemporal dementia. Arch. Neurol 1999, 56:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, et al. : The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009, 73:1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, et al. : Early-Onset Autosomal Dominant Alzheimer Disease: Prevalence, Genetic Heterogeneity, and Mutation Spectrum. The American Journal of Human Genetics 1999, 65:664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernandez DG, Reed X, Singleton AB: Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem 2016, 139:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lesage S, Brice A: Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet 2009, 18:R48–59. [DOI] [PubMed] [Google Scholar]
- 24.Li Y-J, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, et al. : Age at Onset in Two Common Neurodegenerative Diseases Is Genetically Controlled. The American Journal of Human Genetics 2002, 70:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Duijn CM, Farrer LA, Cupples LA, Hofman A: Genetic transmission of Alzheimer’s disease among families in a Dutch population based study. Journal of Medical Genetics 1993, 30:640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, Bijlsma EK, Oortveld MAW, Ekici AB, Reis A, et al. : CNTNAP2 and NRXN1 Are Mutated in Autosomal-Recessive Pitt-Hopkins-like Mental Retardation and Determine the Level of a Common Synaptic Protein in Drosophila. The American Journal of Human Genetics 2009, 85:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H-G, Kishikawa S, Higgins AW, Seong I-S, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, et al. : Disruption of Neurexin 1 Associated with Autism Spectrum Disorder. The American Journal of Human Genetics 2008, 82:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E, et al. : De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Molecular Psychiatry 2019, doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N: Autism Spectrum Disorders and Childhood-Onset Schizophrenia: Clinical and Biological Contributions to a Relation Revisited. Journal of the American Academy of Child & Adolescent Psychiatry 2009, 48:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen JA, Wang Q, Davis-Turak J, Li Y, Karydas AM, Hsu SC, Sears RL, Chatzopoulou D, Huang AY, Wojta KJ, et al. : A multiancestral genome-wide exome array study of Alzheimer disease, frontotemporal dementia, and progressive supranuclear palsy. JAMA Neurol 2015, 72:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruzzo EK, Pérez-Cano L, Jung J-Y, Wang L-K, Kashef-Haghighi D, Hartl C, Singh C, Xu J, Hoekstra JN, Leventhal O, et al. : Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178:850–866.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.International Parkinson’s Disease Genomics Consortium, 23andMe Research Team, Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, et al. : A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017, 49:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. : Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet Neurology 2019, 18:1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, et al. : Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 2009, 41:1303–1307. [DOI] [PubMed] [Google Scholar]
- 35.Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. : Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009, 41:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.European Alzheimer’s Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer’s Disease (GERAD), Alzheimer’s Disease Genetic Consortium (ADGC), Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE), Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. : Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013, 45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bis JC, Jian X, Kunkle BW, Chen Y, Hamilton-Nelson KL, Bush WS, Salerno WJ, Lancour D, Ma Y, Renton AE, et al. : Whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Molecular Psychiatry 2019, doi: 10.1038/s41380-018-0112-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. : Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019, doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JBJ, Dobson-Stone C, Brooks WS, Schofield PR, Halliday GM, et al. : Frontotemporal dementia and its subtypes: a genome-wide association study. The Lancet Neurology 2014, 13:686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang L-S, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, et al. : Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010, 42:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JA, Chen Z, Won H, Huang AY, Lowe JK, Wojta K, Yokoyama JS, Bensimon G, Leigh PN, Payan C, et al. : Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. 2018, doi: 10.1186/s13024-018-0270-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang L-S, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, et al. : Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 2011, 43:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamb R, Rohrer JD, Lees AJ, Morris HR: Progressive Supranuclear Palsy and Corticobasal Degeneration: Pathophysiology and Treatment Options. Current Treatment Options in Neurology 2016, doi: 10.1007/s11940-016-0422-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yokoyama JS, Wang Y, Schork AJ, Thompson WK, Karch CM, Cruchaga C, McEvoy LK, Witoelar A, Chen C-H, Holland D, et al. : Association Between Genetic Traits for Immune-Mediated Diseases and Alzheimer Disease. JAMA Neurol 2016, 73:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Broce I, Karch CM, Wen N, Fan CC, Wang Y, Tan CH, Kouri N, Ross OA, Höglinger GU, Muller U, et al. : Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15:e1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hetz C, Saxena S: ER stress and the unfolded protein response in neurodegeneration. Nature Publishing Group 2017, 13:477–491. [DOI] [PubMed] [Google Scholar]
- 47.Heneka MT: Microglia take centre stage in neurodegenerative disease. Nat. Rev. Immunol 2019, 19:79–80. [DOI] [PubMed] [Google Scholar]
- 48.Perry VH, Nicoll JAR, Holmes C: Microglia in neurodegenerative disease. Nature Publishing Group 2010, 6:193–201. [DOI] [PubMed] [Google Scholar]
- 49.Scrivo A, Bourdenx M, Pampliega O, Cuervo AM: Selective autophagy as a potential therapeutic target for neurodegenerative disorders. The Lancet Neurology 2018, 17:802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schizophrenia Working Group of the Psychiatric Genomics Consortium: Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, Baldursson G, Belliveau R, Bybjerg-Grauholm J, Bækvad-Hansen M, et al. : Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 2018, doi: 10.1038/s41588-018-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium, BUPGEN, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, 23andMe Research Team, Grove J, Ripke S, Als Td, Mattheisen M, Walters RK, Won H, et al. : Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019, 51:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH: The Emerging Picture of Autism Spectrum Disorder: Genetics and Pathology. Annu. Rev. Pathol. Mech. Dis 2015, 10:111–144. [DOI] [PubMed] [Google Scholar]
- 54.Cheng C-M, Chang W-H, Chen M-H, Tsai C-F, Su T-P, Li C-T, Tsai S-J, Hsu J-W, Huang K-L, Lin W-C, et al. : Co-aggregation of major psychiatric disorders in individuals with first-degree relatives with schizophrenia: a nationwide population-based study. Molecular Psychiatry 2017, 23:1756–1763. [DOI] [PubMed] [Google Scholar]
- 55.Sullivan PF, Magnusson C, Reichenberg A, Boman M, Dalman C, Davidson M, Fruchter E, Hultman CM, Lundberg M, Långström N, et al. : Family History of Schizophrenia and Bipolar Disorder as Risk Factors for Autism. Arch Gen Psychiatry 2012, 69:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larsson H, Rydén E, Boman M, Långström N, Lichtenstein P, Landén M: Risk of bipolar disorder and schizophrenia in relatives of people with attention-deficit hyperactivity disorder. Br J Psychiatry 2018, 203:103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faraone SV, Biederman J, Wozniak J: Examining the comorbidity between attention deficit hyperactivity disorder and bipolar I disorder: a meta-analysis of family genetic studies. Am J Psychiatry 2012, 169:1256–1266. [DOI] [PubMed] [Google Scholar]
- 58.Ghirardi L, Brikell I, Kuja-Halkola R, Freitag CM, Franke B, Asherson P, Lichtenstein P, Larsson H: The familial co-aggregation of ASD and ADHD: a register-based cohort study. Nature Publishing Group 2017, 23:257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Consortium C-DGOTPG: Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. The Lancet 2013, 381:1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pettersson E, Lichtenstein P, Larsson H, Song J, Attention Deficit/Hyperactivity Disorder Working Group of the iPSYCH-Broad-PGC Consortium, Autism Spectrum Disorder Working Group of the iPSYCH-Broad-PGC Consortium, Bipolar Disorder Working Group of the PGC, Eating Disorder Working Group of the PGC, Major Depressive Disorder Working Group of the PGC, Obsessive Compulsive Disorders and Tourette Syndrome Working Group of the PGC, Schizophrenia CLOZUK, Substance Use Disorder Working Group of the PgC, Agrawal A, Børglum AD, Bulik CM, Daly MJ, Davis LK, et al. : Genetic influences on eight psychiatric disorders based on family data of 4 408 646 full and half-siblings, and genetic data of 333 748 cases and controls. Psychol. Med 2018, 49:1166–1173.30221610 [Google Scholar]
- 61.Consortium C-DGOTPG, Lee PH, Anttila V, Won H, Feng Y-CA, Rosenthal J, Zhu Z, Tucker-Drob EM, Nivard MG, Grotzinger AD, et al. : Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 2019, 179:1469–1482.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parikshak NN, Gandal MJ, Geschwind DH: Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet 2015, 16:441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. : Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chatterjee S, Ahituv N: Gene Regulatory Elements, Major Drivers of Human Disease. Annu. Rev. Genom. Hum. Genet 2017, 18:45–63. [DOI] [PubMed] [Google Scholar]
- 65.Visscher PM: Challenges in understanding common disease. 2017, doi: 10.1186/s13073-017-0506-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edwards SL, Beesley J, French JD, Dunning AM: Beyond GWASs: Illuminating the Dark Road from Association to Function. The American Journal of Human Genetics 2013, 93:779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zarrei M, Burton CL, Engchuan W, Young EJ, Higginbotham EJ, MacDonald JR, Trost B, Chan AJS, Walker S, Lamoureux S, et al. : A large data resource of genomic copy number variation across neurodevelopmental disorders. npj Genomic Medicine 2019, doi: 10.1038/s41525-019-0098-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cardoso AR, Lopes-Marques M, Silva RM, Serrano C, Amorim A, Prata MJ, Azevedo L: Essential genetic findings in neurodevelopmental disorders. 2019, doi: 10.1186/s40246-019-0216-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malhotra D, McCarthy S, Michaelson JJ, Vacic V, Burdick KE, Yoon S, Cichon S, Corvin A, Gary S, Gershon ES, et al. : High Frequencies of De Novo CNVs in Bipolar Disorder and Schizophrenia. Neuron 2011, 72:951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geschwind DH, Konopka G: Neuroscience in the era of functional genomics and systems biology. Nature 2009, 461:908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, et al. : Linkage disequilibrium in the human genome. Nature 2001, 411:199–204. [DOI] [PubMed] [Google Scholar]
- 72.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, 2014: Identifying causal variants at loci with multiple signals of association. Genetics Soc America [no date], doi: 10.1534/genetics.114.167908/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kichaev G, Yang W-Y, Lindstrom S, Hormozdiari F, Eskin E, Price AL, Kraft P, Pasaniuc B: Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet 2014, 10:e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ernst J, Melnikov A, Zhang X, Wang L, Rogov P, Mikkelsen TS, Kellis M: Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat Biotechnol 2016, 34:1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Won H, la Torre Ubieta de L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. : Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.The GTEx Consortium, Ardlie KG, Deluca DS, Segre AV, Sullivan TJ, Young TR, Gelfand ET, Trowbridge CA, Maller JB, Tukiainen T, et al. : The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whalen S, Pollard KS: Most chromatin interactions are not in linkage disequilibrium. Genome Res. 2019, 29:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, et al. : Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362:eaat8464–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hauberg ME, Zhang W, Giambartolomei C, Franzén O, Morris DL, Vyse TJ, Ruusalepp A, Consortium C, Fromer M, Sieberts SK, et al. : Large-Scale Identification of Common Trait and Disease Variants Affecting Gene Expression. The American Journal of Human Genetics 2017, 100:885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walker RL, Ramaswami G, Hartl C, Mancuso N, Gandal MJ, la Torre Ubieta de L, Pasaniuc B, Stein JL, Geschwind DH: Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 2019, 179:750–771.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li YI, van de Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK: RNA splicing is a primary link between genetic variation and disease. Science 2016, 352:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O’Brien HE, Hannon E, Hill MJ, Toste CC, Robertson MJ, Morgan JE, McLaughlin G, Lewis CM, Schalkwyk LC, Hall LS, et al. : Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. 2018, doi: 10.1186/s13059-018-1567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.la Torre Ubieta de L, Stein JL, Won H, Opland CK, Liang D, Lu D, Geschwind DH: The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 2018, 172:289–295.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Short PJ, McRae JF, Gallone G, Sifrim A, Won H, Geschwind DH, Wright CF, Firth HV, FitzPatrick DR, Barrett JC, et al. : De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555:611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gamazon ER, Segrè AV, Bunt M, Wen X, Xi HS, Hormozdiari F, Ongen H, Konkashbaev A, Derks EM, Aguet F, et al. : Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 2018, doi: 10.1038/s41588-018-0154-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.The PsychENCODE Consortium*: Revealing the brain’s molecular architecture. Science 2018, 362:1262–1263. [DOI] [PubMed] [Google Scholar]
- 87.Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, Crawford GE, Jaffe AE, Pinto D, Dracheva S, Geschwind DH, et al. : The PsychENCODE project. Nature Neuroscience 2015, 18:1707–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ecker JR, Bickmore WA, Barroso I, Pritchard JK, Gilad Y, Segal E: ENCODE explained. Nature Publishing Group 2012, 489:52–54. [DOI] [PubMed] [Google Scholar]
- 89.Battle A, Brown CD, Engelhardt BE, Montgomery SB: Genetic effects on gene expression across human tissues. Nature Publishing Group 2017, 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, Lomvardas S, Mirny LA, O’Shea CC, Park PJ, Ren B, et al. : The 4D nucleome project. Nature Publishing Group 2017, 549:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hodes RJ, Buckholtz N: Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opinion on Therapeutic Targets 2016, 20:389–391. [DOI] [PubMed] [Google Scholar]
- 92.Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. : A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Systems 2017, 4:60–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Canchi S, Raao B, Masliah D, Rosenthal SB, Sasik R, Fisch KM, De Jager PL, Bennett DA, Rissman RA: Integrating Gene and Protein Expression Reveals Perturbed Functional Networks in Alzheimer’s Disease. CellReports 2019, 28:1103–1116.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matharu N, Rattanasopha S, Tamura S, Maliskova L, Wang Y, Bernard A, Hardin A, Eckalbar WL, Vaisse C, Ahituv N: CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363:eaau0629–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, et al. : A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019, 176:1516. [DOI] [PubMed] [Google Scholar]
- 96.Cirillo E, Parnell LD, Evelo CT: A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Front. Genet 2017, 8:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koopmans F, van Nierop P, Andres-Alonso M, Byrnes A, Cijsouw T, Coba MP, Cornelisse LN, Farrell RJ, Goldschmidt HL, Howrigan DP, et al. : SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 2019, 103:217–234.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schadt EE: Molecular networks as sensors and drivers of common human diseases. Nature 2009, 461:218–223. [DOI] [PubMed] [Google Scholar]
- 99.Carter H, Hofree M, Ideker T: Genotype to phenotype via network analysis. Current Opinion in Genetics & Development 2013, 23:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang B, Horvath S: A General Framework for Weighted Gene Co-Expression Network Analysis. Statistical Applications in Genetics and Molecular Biology 2011, 4:1–45. [DOI] [PubMed] [Google Scholar]
- 101.Mitra K, Carvunis A-R, Ramesh SK, Ideker T: Integrative approaches for finding modular structure in biological networks. Nat Rev Genet 2013, 14:719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH: Integrative Functional Genomic Analyses Implicate Specific Molecular Pathways and Circuits in Autism. Cell 2013, 155:1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, et al. : An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. : Transcriptional landscape of the prenatal human brain. Nature 2014, doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.BrainSpan Consortium†: BrainSpan: Atlas of the Developing Human Brain [Internet]. 2013. [Google Scholar]
- 106.Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, la Torre Ubieta de L, Huang J, et al. : Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, Kriegstein AR: Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Polioudakis D, la Torre Ubieta de L, Langerman J, Elkins AG, Shi X, Stein JL, Vuong CK, Nichterwitz S, Gevorgian M, Opland CK, et al. : A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 2019, 103:785–801.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, et al. : Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, De Luca GC, Crary JF, Casaccia P, Buxbaum JD, et al. : Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Medicine 2016, doi: 10.1186/s13073-016-0355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. : Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, et al. : Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine 2020, doi: 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. : Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 2019, 366:1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]