

Abstract
Vismodegib, a Smoothened antagonist, is clinically approved for treatment of human basal cell carcinoma (BCC), in the clinical trials of medulloblastoma (MB) and other cancers. However, a significant proportion of these tumors fail to respond to Vismodegib after a period of treatment. Here, we find that AMPK agonists, A769662, and Metformin, can inhibit GLI1 activity and synergize with Vismodegib to suppress MB cell growth in vitro and in vivo. Furthermore, combination of AMPK agonists with Vismodegib is effective in overcoming Vismodegib‐resistant MB. This is the first report demonstrating that combining AMPK agonist (Metformin) and SHH pathway inhibitor (Vismodegib) confers synergy for MB treatment and provides an effective chemotherapeutic regimen that can be used to overcome resistance to Vismodegib in SHH‐driven cancers.
Keywords: AMPK, Hedgehog pathway, Medulloblastoma, Metformin, Vismodegib
Abbreviations
- AMPK
adenosine 5′ monophosphate‐activated protein kinase
- FDA
food and drug administration
- GLI1
glioma‐associated oncogene 1
- MB
medulloblasoma
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- PTCH1
protein patched homolog 1
- S6 K
ribosomal protein S6 kinase
- SHH
sonic hedgehog
- SMO
smoothened
- WNT
wingless and Int‐1
1. INTRODUCTION
Medulloblastoma (MB) is the most common pediatric brain cancer. This highly metastatic cancer arises in the cerebellum in the posterior fossa of the skull. Although conventional therapies, comprised of surgical resection, radiation, and chemotherapy, have improved survival rates, treatment for MB faces several challenges. 1 , 2 For example, patients experience devastating side effects from these treatments, including learning disabilities, growth hormone imbalance, and secondary cancers after treatment. In addition, one of the major hurdles is tumor recurrence, indicating the likely existence of resistance to current chemotherapeutic approaches. 2 To better understand this disease and target these challenges, genomic sequencing to understand the molecular pathways enriched in different MB cases is needed. Of the four molecular subgroups (WNT, sonic hedgehog group, Group 3, and Group 4), the sonic hedgehog (SHH) subgroup comprises nearly a third of all MB cases. 1 , 3 In SHH‐MB, the aberrant activation of transcriptional activator GLI1 typically arises from activating mutations in Smoothened (SMO), a major upstream effector, or inactivating mutations in Ptch1, the 12‐pass transmembrane receptor at the beginning of the pathway. 4
The only FDA‐approved SMO inhibitors available to target SHH‐driven cancers are Vismodegib and Sonidegib, which were approved in 2012 and 2015, respectively, for the treatment of basal cell carcinoma (BCC). Anti‐SMO drug‐based chemotherapy is the latest advancement for MB and basal cell carcinoma (BCC). Unfortunately, cases where tumors acquire resistance to SMO inhibitors frequently happen in the clinic. Furthermore, Vismodegib resistance leads to terminal progression of SHH cancer which cannot be overcome with Sonidegib in the BCC patients. 5 Patients who have developed treatment resistance may not benefit from SMO inhibitors and it is mostly due to SMO mutations (such as D473G and W535L, the most common mutation sites in BCC and MB) and tumors will continue to progress even under treatment with other SMO inhibitors. 6
It has been shown that permanent defects in bone growth occur in younger mice treated with SMO inhibitor treatment, but not adult mice. 78 In a human clinical phase II trial (NCT01878617), three young patients with SHH‐MB treated with Vismodegib developed growth plate fusion. 9 These findings suggest that the use of SMO inhibitors in skeletally immature patients should be with discretion, and reducing the concentration of SMO inhibitors by combination with other therapies may diminish side‐effects in pediatric patients.
Emerging evidence shows that the AMPK‐mTOR‐S6 K signaling cascade controls the SHH/GLI1 pathway. As an energy and metabolism sensor, AMPK activation inhibits SHH/GLI1 signaling. 10 , 11 AMPK directly phosphorylates GLI1 at three sites, inducing GLI1 degradation. 10 , 12 Mutating GLI1 AMPK‐mediated phosphorylation sites extends GLI1 stability and activity under activation of AMPK. It is known that AMPK inhibits mTOR/S6 K signaling. 13 In addition, S6 K (downstream of mTOR) promotes GLI1 activity through non‐canonical SHH signaling in esophageal cancer. 14 Inhibition of GLI1 may be through AMPK‐mediated S6 K inhibition. Thus, we speculate that a similar relationship may also exist in medulloblastoma. Recently, genetic deletion of mTORC1 was found to inhibit MB development in a SmoM2 transgenic MB mouse model. 15 Thus, targeting AMPK‐mTOR signaling may shed light on the development of novel therapies in SHH‐driven cancers.
Here we find that AMPK activators, A769662 and Metformin, sensitize MB cells to the anti‐tumor activity of Vismodegib. Combination of AMPK activators and Vismodegib significantly suppresses MB in vitro cell growth and colony formation. Furthermore, in the Vismodegib‐resistant MB cell lines, reduced AMPK activity accompanies persistent HH activation. Notably, combining AMPK activators and Vismodegib can overcome Vismodegib resistance and inhibit growth of SMOD473G MB cells. In both mouse subcutaneous and intracranial models, Metformin and Vismodegib combination treatment exhibits synergistic suppression of MB tumor growth. Altogether, AMPK activator likely provides a new strategy for treating Vismodegib‐resistant MB.
2. METHODS
2.1. Reagents and Cell lines
AMPK activator Metformin was purchased from Sigma (St. Louis, MO, USA), A‐769662 and Vismodegib were purchased from LC Laboratories (Woburn, MA, USA). D‐Luciferin was purchased from Fisher Scientific (Hampton, NH, USA). DAOY (RRID: CVCL_1167) and Med1 (RRID: CVCL_7988) were from Dr. Matthew P. Scott (Stanford University, CA, USA). The Vandy‐MB11 cell line was a gift from Dr. Xi Huang (The Hospital for Sick Children, Toronto, Canada). ONS‐76 (RRID: CVCL_1624) and MED8A (RRID: CVCL_M137) cell lines were from Dr. Marc Remke (University Hospital Düsseldorf, Germany). Lentiviral expression plasmid DNA of Smoothened WT, D473G and W535L, and BCC SMOD473G and SMOW535L stable cell lines were from Dr. Scott X. Atwood (University of California Irvine, CA, USA).
2.2. Cell culture, MTT cell growth, and anchorage‐independent growth and colony formation assay
The Vandy‐MB11(MB11), DAOY, ONS‐76, and MED8A human MB cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Corning, Manassas, VA, USA) supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. MB11‐ and DAOY‐ Vismodegib‐resistant cell lines were obtained in vitro by prolonged culture of parental WT cells with sublethal Vismodegib concentrations as showed in Figure 3A and cultured in DMEM with 200 μM Vismodegib. The cell growth rate was determined using MTT assays. 16 Briefly, cells (3 × 103 per well) were plated in 96‐well culture plates. After cells adhered, at the required time points, MTT was added (20 μM final) to each well, incubated for 2 h, and the purple formazan crystals were dissolved in 200‐µL DMSO. After the 30‐min incubation, absorbance was read at 595 nm using an LB960 microplate reader (Bio‐Tek Instruments, Winooski, VT, USA). For anchorage‐independent growth (soft agar colony formation) assay, 5 × 104 cells were suspended in complete medium containing 0.3% low melting agarose and overlaid onto wells containing 3‐ml DMEM with 10% FBS and 0.6% agarose in a six‐well plate. Culture medium containing either DMSO, Vismodegib, Metformin or both Vismodegib and Metformin as indicated was applied, and the medium was replenished every 3 days. After 2–3 weeks, colonies larger than 1.0 mm in diameter were counted.
FIGURE 3.
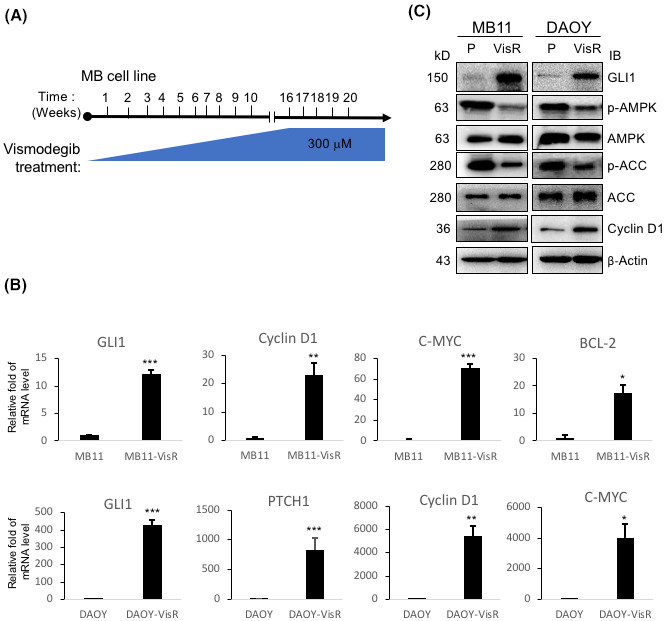
HH/GLI1 activation and suppression of AMPK in Vismodegib‐resistant medulloblastoma cell lines. (A) To generate Vismodegib‐resistant MB cells, MB11 and DAOY cells were treated with Vismodegib daily with a gradual increase in concentration from 10 to 300 μM in 16 weeks. (B) From two pairs of MB parental and Vismodegib‐resistant cell lines (VisR), MB11 and DAOY, mRNA was collected, and the amount of HH/GLI1 downstream targets, GLI1, PTCH1, Cyclin D1, C‐MYC, and BCL‐2 mRNA was analyzed using qRT‐PCR with GAPDH mRNA as the internal control for normalization. The bars indicate mRNA level relative to that of parental cells. The experimental points were in triplicate and independently repeated three times (*p < 0.05, **p < 0.01, *** p < 0.001). (C) Lysates from MB11 and DAOY (P: parental; VisR: Vismodegib‐resistant) cells were analyzed by immunoblot with the indicated antibodies
For clonogenic assay, 1000 cells/well were seeded in 12‐well plates and treated with DMSO, Vismodegib, Metformin, or both Vismodegib and Metformin as indicated. The medium was refreshed every 3 days with the drug until the end of assay. After 10 days, cells were fixed with acetic acid/methanol 1:7 (vol/vol) at room temperature (RT) for 10 min and then stained with 0.5% crystal violet solution at RT for 2 hours. The colonies were counted and compared with DMSO controls. All experiments were done in triplicates.
2.3. Immunoblotting
Immunoblotting was performed as previously described 16 with the following antibodies: β‐actin (1:5,000; Sigma (#A5441)), AMPK (1:1000; Cell Signaling Technology (#2532)), GLI1 (1:1,000; Cell Signaling Technology (#3538 s)), p‐AMPK (1:1000; Cell Signaling Technology (#2535)), Cleaved Caspase‐3 (1:1000; Cell Signaling Technology (#9661 s)), and cleaved PARP (1:1000; Cell Signaling Technology (#5625p)).
2.4. Immunohistochemistry staining
The immunoperoxidase staining method was modified from the avidin–biotin complex technique as described previously. 17 In brief, slides (5 μm) were deparaffinized. After antigen retrieval, the slides were digested in 0.05% trypsin. The endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide, and the slides were then treated with 10% normal goat for 30 min at RT. After overnight incubation with primary antibodies (a) Cleaved Caspase‐3 (1:100 dilution; Cell Signaling Technology); (b) Ki67 (1:100 dilution; Cell Signaling Technology), the slides were incubated with biotinylated secondary antibodies and subsequently with avidin–biotin–horseradish peroxidase complex (Vector Laboratories, Burlingame, CA, USA). Antibody detection was performed with the 0.125% aminoethyl carbazole chromogen substrate solution (AEC substrate) from Sigma (St. Louis, MO, USA). After counterstaining with Mayer's hematoxylin (Sigma (St. Louis, MO, USA)), the sides were mounted and imaged.
2.5. Tumor xenograft
Six‐week‐old female BALB/c‐nu mice were purchased from the Jackson Laboratory. (Bar Harbor, ME, USA). MB11 cells (3x106) were injected subcutaneously into the abdomen of the mice. Mice will be anesthetized with 5% isoflurane in an induction box and then placed on a nose cone receiving 2–3% isoflurane for maintenance. The depth of anesthesia will be monitored by toe pinch and depth of respiration, and the percentage of anesthetic agent will be adjusted accordingly. When tumor size reached a volume of approximately 50 mm3, mice were randomized into four treatment groups (10 mice per group, 5 mice per cage): DMSO (control group), Vismodegib (10 mg/kg), Metformin (150 mg/kg), and Vismodegib (10 mg/kg) and Metformin (150 mg/kg). The reagents were diluted in DMSO and were delivered daily by intraperitoneal injections in the animal facility. Tumor volume was measured using a caliper and calculated with the formula 1/2 × L × W2 as described previously. 16 Animals were euthanized by CO2 asphyxiation and followed by cervical dislocation to ensure death. All procedures have been reviewed and approved by the Institutional Animal Use and Care Committee at Purdue University.
2.6. Intracranial brain tumor xenograft
The mouse intracranial brain tumor xenograft injection was followed as previously described. 18 Briefly, mice were first anesthetized using standard anesthetic isoflurane (described in the previous paragraph) and Stereotaxic (RWD). A small amount of ophthalmic ointment was placed on the eyes of the mouse during surgery. A hair clipper was used to shave the hair on the posterior skull above the position of the midbrain and cerebellum. Betadine solution was applied on the exposed scalp, followed by scrubbing with an alcohol pad, and this sterilization step was repeated two times. A quarter‐inch incision was made through the skin on the posterior scalp. A 0.5‐mm burr hole was drilled 2 mm to the right and 2 mm posterior to the lambda using a sterile dental drill. The mouse was then positioned in a stereotactic frame by hooking its incisors onto the frame hold. A bevel‐tipped 10‐μL syringe loaded with 4 μl of tumor cell (1.5 × 105) solution was inserted into the burr hole. Once the bevel of the syringe needle was below the surface of the skull, the syringe was inserted an additional 3 mm and then elevated by 0.5 mm. The tumor cells were slowly injected, with a steady force in a 30 s time frame, into the cerebellum. The syringe was held in place after completion of injection for an additional 2 min, then removed, and the incision was closed with surgical staples. Each mouse was placed on a warming blanket during and following surgery to help maintain its body temperature. Mobility and respiratory patterns were observed continuously during recovery. Four treatments (10 mice per group): DMSO, Vismodegib 5 mg/kg (3 mg/ml), Metformin 200 mg/kg (120 mg/ml) or Vismodegib and Metformin were then injected once daily by intraperitoneal injection. Tumor size was measured weekly by placing each mouse in an in vivo bioluminescence imaging system (AMI), 10 min following an intraperitoneal injection with 150 mg/kg D‐luciferin (Goldbio, St. Louis, MO, USA) in PBS. The peak luminescence signal was recorded using the AMI. The Living Image software was used to measure photon flux within a region of interest to quantify the bioluminescent signals emanating from the tumors. Randomization was done just before treatment, that is before week 2. Treatment started on the same day of the week and continued on the same day of 2nd to 5th week. Animals were euthanized by CO2 asphyxiation and followed by cervical dislocation to ensure death. All procedures have been reviewed and approved by the Institutional Animal Use and Care Committee at Purdue University.
2.7. Quantitative real‐time PCR
The RNA extraction kit (Zymo Research, Irvine, CA, USA) was used to extract total RNA, which was reverse transcribed into cDNA using the cDNA Synthesis Kit (ABI, Foster City, CA, USA). The cDNA was amplified with SYBR Premix (Roche, Basel, Switzerland) using a Light Cycler 96 Real‐Time PCR System (Roche, Basel, Switzerland). The PCR cycling parameters were 95°C for 10 min, then 95°C for 10 s, 60°C for 10 s, and 72°C for 20 s repeated 40 times. A single primer for each sample was carried out in triplicate. Relative mRNA levels were calculated by comparative Ct method (Applied Biosystem instruction manual) and presented by their ratio to their biological controls. The fold change in expression of each target mRNA relative to GAPDH was calculated as 2Δ(ΔCt), where ΔCt = ΔCtGAPDH‐Ctgene. GAPDH transcript levels were confirmed to correlate well with total RNA amounts and therefore were used for normalization throughout. The primers used for real‐time PCR were designed by primer bank (http://pga.mgh.harvard.edu/primerbank/).
2.8. Lentivirus infection
pCDH‐CMV‐MCS‐EF1‐Puro SMO‐lentivirus packaging and infection was done according to the manual from SBI System Bioscience (Cat. #sCD500–CD700). Infected MB11 and DAOY cells were treated with 1.5 μg/ml puromycin for 2 weeks to eliminate non‐infected cells.
2.9. Statistical analysis
All the experiments were performed at least three times, independently. The data obtained were expressed as “Mean ±Standard Error.” Significance was calculated as per student's t‐test using Graph Pad Prism Version 7 (GraphPad Software, San Diego, CA). A univariate analysis was used to determine the variable distributions. Categorical variables among the groups were compared with the Pearson's chi‐square test. A p‐value of <0.05 was considered statistically significant.
3. RESULTS
3.1. AMPK agonist and Vismodegib synergistically suppresses MB cell growth
Our previous work has shown that activation of AMPK inhibits SHH signaling through suppression of GLI1 activity and expression by inducing GLI1 protein degradation. 10 , 12 Thus, targeting AMPK may be employed for treatment in SHH‐driven cancers.
To test this hypothesis, we first examined the ability of AMPK‐specific activator A769662 to sensitize SHH‐MB cells to Vismodegib. Both human and mouse SHH‐MB cell lines DAOY and Med1 (From Ptch +/−; p53 −/− MB tumor) were treated with Vismodegib (GDC‐0449) alone, A769662 or Metformin alone, or a combination of both and then subjected to MTT assay to assess viability and to colony formation assay (Figure S1A,B,C). Combination treatment with A769662 and Vismodegib showed higher efficacy against cell viability compared with single‐drug treatment, suggesting that AMPK activation in combination with Vismodegib may be applied for MB treatment. Next, we selected Metformin as an AMPK activator to test in combination with Vismodegib in three human SHH‐MB cell lines and group 3, MED8A cell line for MTT assay (Figure 1A‐D). Metformin is an FDA‐approved therapeutic agent demonstrated to exhibit low toxicity in patients and has been shown to cross the blood–brain barrier and activate AMPK. 19 Consistent with what we observed with A769662, Metformin (used at supraphysiological dose) treatment in combination with Vismodegib significantly reduced viability in all four MB cell lines (Figure 1A‐D). Then, we examined whether AMPK activators and Vismodegib synergistically suppress MB cell growth. Indeed, both A769662 and Metformin worked synergistically with Vismodegib in inhibition of MB cell growth (Figure 1E‐H). Furthermore, based on Chou‐Talalay quantitative method, an algorithm for simulation of drug synergism and antagonism at any effect and dose level, 20 combination of Metformin and Vismodegib showed combination index (CI) in the range of 0.59 to 0.98 (lower CI shows greater killing effect) in both MB cell lines, indicating the synergism (CI<1) of both drugs in MB cells (Figure S2A, B).
FIGURE 1.
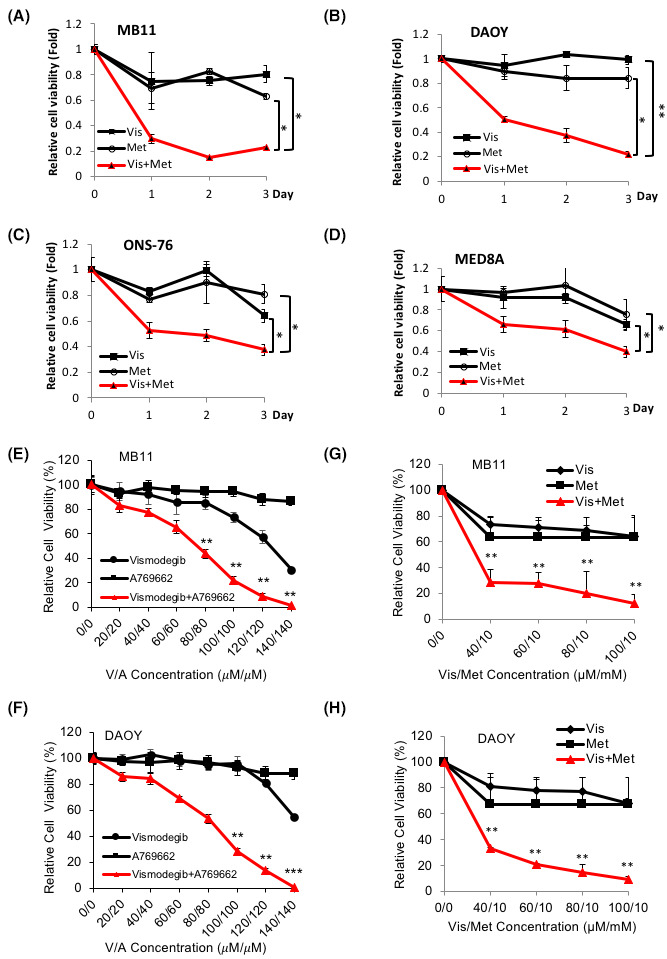
AMPK agonist synergizes with Vismodegib and suppresses medulloblastoma cell growth. Human MB cell lines, (A) MB11, (B) DAOY, (C) ONS‐76, and (D) MED8A were treated with DMSO, Vis (Vismodegib, 10 μM), Met (Metformin, 5 mM), Vis and Met (Vis+Met, 10 μM+5 mM) and subjected to cell growth assay for three consecutive days. (E) MB11 and (F) DAOY cells were treated with Vismodegib (from 20 to 140 μM), A769662 (from 20 to 140 μM), Vismodegib and A769662 for 48 hours and subjected for cell growth assay. (G) MB11 and (H) DAOY cells were treated with Vismodegib (from 40 to 100 μM), Metformin (10 mM), Vismodegib and Metformin for 48 hours and subjected to cell growth assay. All the experimental points were in triplicate and repeated three times independently (*p < 0.05, **p < 0.01, ***p < 0.001)
In addition, we performed soft agar colony formation and clonogenicity assays in two SHH‐MB cell lines 21 , 22 : DAOY and Vandy‐MB‐11 (MB11) for 2–3 weeks. We found that compared with single drug treatment, Vismodegib (Vis) and Metformin (Met) combination treatment remarkably reduced the number of colonies formed on soft agar (Figure 2A,B, red bar); similar result was observed with the clonogenicity assay (Figure 2C). The reduction in cell viability and growth suggested an activation of the cell apoptotic process with decreased cell proliferation. To test this possibility, we treated MB11 and DAOY cells with Vismodegib and Metformin as single drugs or in combination and analyzed expression levels of cleaved PARP (as an apoptotic marker) and cyclin D1 (as a marker of cellular growth) (Figure 2D). As expected, cleaved‐PARP was increased whereas cyclin D1 was reduced in the cells with combination drug treatment. Furthermore, the GLI1 protein level was significantly repressed and p‐AMPK was increased in the combination treatment group. These results were consistent with our previous finding showing that activation of AMPK inhibits GLI1, which leads to the subsequent cell growth inhibition.
FIGURE 2.
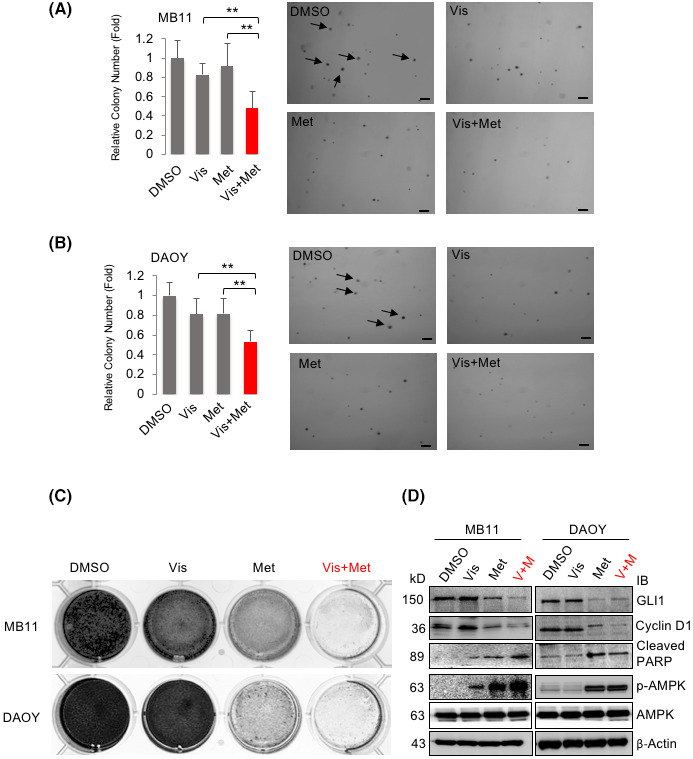
Combination of AMPK agonist and Vismodegib achieves better inhibitory effect on MB colony formation. (A) MB11 and (B) DAOY cells were seeded into six‐well plates and treated with DMSO, Vis, (Vismodegib, 10 μM), Met (Metformin, 5 mM), Vis and Met (Vis+Met, 10 μM+5 mM) for soft agar colony formation assay for 2 weeks. Colonies larger than 1.0 mm were counted (as indicated by arrows). The experiment was repeated three times (** p < 0.01,) Scale bar: 10 mm (C) MB11 and DAOY cells were seeded in 12‐wells plate and treated with DMSO, Vis, (Vismodegib, 10 μM), Met (Metformin, 5 mM), Vis and Met (10 μM+5 mM) for colony formation assay for 12 days. (D) MB11 and DAOY cells were treated with DMSO, Vis, (Vismodegib, 10 μM), Met (Metformin, 5 mM), Vis and Met (10 μM+5 mM) for 24 hours, and the cell lysates were analyzed by western blotting with the indicated antibodies
3.2. Activation of HH/ GLI1 signaling and reduction of AMPK activity in Vismodegib‐resistant MB cell lines
Drug resistance has limited the efficacy of FDA‐approved targeted therapies for SHH‐driven cancers. 23 Vismodegib resistance in BCC patients cannot be overcome with the other FDA‐approved SHH pathway inhibitor, Sonidegib, potentially due to SMO mutations that re‐activate HH signaling. 5 , 6 Based on our observations, AMPK activator may provide an option to overcome SMO inhibitor resistance. To test the hypothesis, we generated Vismodegib‐resistant cell (VisR) in two human MB cell lines, MB11 and DAOY (named as MB11‐VisR and DAOY‐VisR respectively). The VisR cell lines were generated in vitro by prolonged culture of the parental cells with Vismodegib at sublethal concentrations (Figure 3A). To verify the VisR MB cells, we treated the two VisR MB cell lines with Vismodegib ranging from 40 μM to 280 μM. The two VisR MB lines developed resistance to Vismodegib, even to higher concentration of Vismodegib compared with the parental MB cell lines (Figure S3A,B). We found that mRNA expression levels of GLI1, Cyclin D1, PTCH1, C‐MYC, and BCL‐2 (HH/GLI1 downstream targets) were highly elevated in the VisR cell lines (Figure 3B). Furthermore, both GLI1 and Cyclin D1 protein levels were increased whereas AMPK activity (p‐AMPK and p‐ACC) was suppressed in the VisR cells (Figure 3C). These data suggest that GLI1 activity is induced through inhibition of AMPK activity in the VisR MB cells.
3.3. Combination of AMPK activator and SMO inhibitor overcomes Vismodegib‐resistant MB
As VisR MB cells have gained GLI1 activity with repression of AMPK, we asked whether re‐activation of AMPK could overcome Vismodegib resistance through inhibition of GLI1 activity. To address this question, both MB11‐VisR and DAOY‐VisR cell lines were treated with Vismodegib alone, AMPK activator (A769662, Metformin) alone, or combination of Vismodegib and AMPK activator. The combination treatment resulted in significant synergism in reducing cell viability (Figure 4A‐D). Based on Chou‐Talalay quantitative method, 20 the CI of Metformin with Vismodegib was from 0.475 to 0.727 in MB11‐VisR and from 0.275 to 0.557 in DAOY‐VisR cell line, indicating the synergism (CI<1) of both drugs in VisR MB cell lines (Figure S4A,B).
FIGURE 4.
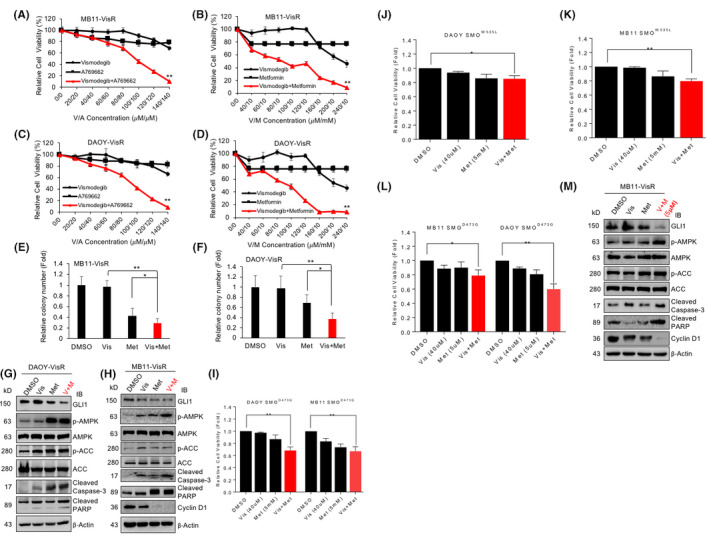
Combining AMPK activator and SMO inhibitor sensitizes Vismodegib‐resistant MB. (A) MB11‐VisR and (C) DAOY‐VisR cells were treated with DMSO, Vismodegib (From 20 to 140 μM), A769662 (From 20 to 140 μM), Vismodegib (From 20 to 140 μM), and A769662 (From 20 to 140 μM) for 48 hours and subjected to cell growth assay. (B) MB11‐VisR and (D) DAOY‐VisR cells were treated with DMSO, Vismodegib (From 40 to 240 μM), Metformin (10 mM), Vismodegib (From 40 to 240 μM), and Metformin (10 mM) for 48 hours and subjected to cell growth assay. (E) MB11‐VisR and (F) DAOY‐VisR cells were treated with DMSO, Vismodegib (20 μM), Metformin (10 mM), Vismodegib (20 μM), and Metformin (10 mM) for 2 weeks and subjected to soft agar colony formation assay. (G) DAOY‐VisR and (H) MB11‐VisR cells were treated with DMSO, Vismodegib (10 μM), Metformin (5 mM), Vismodegib (10 μM), and Metformin (5 mM) for 24 hours, and lysates were analyzed by immunoblot with the indicated antibodies. (I) MB11‐ and DAOY‐SMOD473G (Smoothened mutation D473G) stable cell lines were treated with DMSO, Vismodegib (40 μM), Metformin (5 mM), Vismodegib (40 μM) and Metformin (5 mM) for 48 hours and subjected to cell growth assay. Similarly, (J) DAOY‐SMOW535L and (K) MB11‐SMOW535L stable cell lines were treated with DMSO, Vismodegib (40 μM), Metformin (5 mM), Vismodegib (40 μM), and Metformin (5 mM) for 48 hours and subjected to cell growth assay. (L) MB11‐ and DAOY‐SMOD473G (Smoothened mutation D473G) stable cell lines were treated with DMSO, Vismodegib (40 μM), Metformin (5 μM), Vismodegib (40 μM) and Metformin (5 μM) for 48 hours and subjected to cell growth assay. (M) MB11‐VisR cells were treated with DMSO, Vismodegib (10 μM), Metformin (5 μM), Vismodegib (10 μM) and Metformin (5 μM) for 24 hours and lysates were analyzed by immunoblot with the indicated antibodies. All the cell growth assay points were in triplicate and each experiment repeated three times (*p < 0.05, ** p < 0.01)
To assess whether both pharmacologic activation of AMPK with Metformin and inhibition of Hh signaling with Vismodegib could reduce colony formation of VisR cells, we subjected the MB11‐VisR and DAOY‐VisR cells to Vismodegib and Metformin, alone or in combination, for soft agar colony formation assay. We found that similar to the results from the cell viability assay, combination treatment significantly reduced colony number in both of the VisR MB cell lines compared with the mock treated and single agent treated groups (Figure 4E,F). Combination treatment markedly inhibited GLI1 protein expression and enhanced AMPK activity, along with elevated cleaved caspase 3 and cleaved‐PARP levels (Figure 4G,H). These data suggest that combination treatment effectively increases apoptosis and suppresses growth of VisR cells.
Genomic mutation of SMO is known to cause Vismodegib‐resistance in MB and BCC, 6 and we asked whether combination of Metformin and Vismodegib was able to overcome SMO mutation‐related resistance in MB cells. First, we stably expressed wild‐type SMO protein (WT) and two mutant SMO proteins carrying known SMO mutations (D473G and W535L), 6 respectively, in MB11 and DAOY cells. We then tested whether these SMO mutant cells were resistant to Vismodegib by measuring mRNA expression levels of SHH/GLI1‐target genes, GLI1, PTCH1, Cyclin D1, and C‐MYC. We found that these SHH targets responded to Vismodegib treatment in the MB11‐vector and MB11‐SMOWT cells but not in the SMOD473G and SMOW535L cells (Figure S5A‐D). Furthermore, SMOD473G and SMOW535L cells had significantly higher endogenous GLI1 mRNA expression than the vector and SMOWT cells with or without Vismodegib treatment (Figure S5E,F); similar results were obtained from the DAOY‐SMO stable cell lines (Figure S6A‐F). Together, these data suggest that SMOD473G and SMOW535L mutations promote HH/GLI1 activity and resistance to Vismodegib.
Next, we tested whether combination of Vismodegib and Metformin suppresses SMO mutant MB cell lines. We treated MB11‐SMOD473G, MB11‐SMOW535L, and DAOY‐SMOD473G, DAOY‐SMOW535L with Vismodegib and Metformin, alone or in combination. The combination treatment significantly reduced the viability of SMOD473G and SMOW535L MB cells (Figure 4I‐K) as well as SMOD473G and SMOW535L BCC cells (Figure S7). Combined Vismodegib (40 µM) with Metformin at 1000‐fold lower concentration (5 µM) that is at therapeutic dose levels consistently suppressed viability of the SMOD473G cells. Additionally, in VisR cells, GLI1 and Cyclin D1 protein levels were reduced and phospho‐AMPK/ phospho‐ACC, cleaved caspase 3 along with cleaved‐PARP levels were increased (Figure 4L,M). Together, these results show that simultaneous AMPK activation and Vismodegib inhibition is effective for sensitizing resistant SHH‐MB to Vismodegib.
3.4. Combination of AMPK activator and SMO inhibitor effectively suppresses MB growth in vivo
To determine the therapeutic effect of the Metformin and Vismodegib combination treatment in a mouse xenograft model, we subcutaneously injected MB11 cells in nude mice that were then treated with (i) Metformin, (ii) Vismodegib, (iii) Metformin and Vismodegib, and the (iv) control vehicle. Drugs were injected intravenously for 5 consecutive days in a week and for 4 successive weeks. As show in Figure 5A, combination treatment significantly inhibited MB11 tumor growth in vivo. Tumors from the combination treatment group also showed significantly reduced expression of the proliferation marker ki67 and increased expression of the apoptosis marker cleaved caspase 3 staining compared with the mock‐treated or single agent‐treated group (Figure 5B,D).
FIGURE 5.
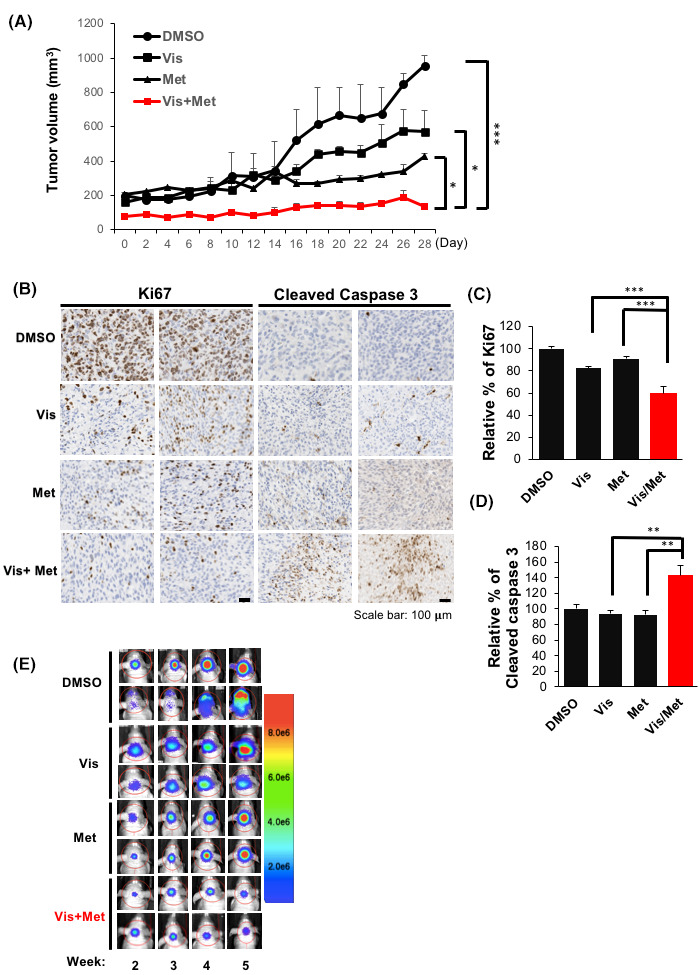
Combination of AMPK activator and SMO inhibitor effectively suppresses MB growth in vivo. (A) Tumor volume of the MB11 xenografts treated with DMSO, vismodegib, Metformin, and vismodegib plus Metformin was measured for 28 days. (B) The tumor sections of two individual samples from four different treatment groups were subjected to immunohistochemistry with a Ki67 and cleaved caspase 3 antibody. Scale bar: 100 µm. Relative percentages of Ki67 (C) and cleaved caspase 3 (D) expression by individual xenograft tumors from B were analyzed and the mean values of Ki67 and cleaved caspase 3 expression in DMSO, Vismodegib, Metformin and Vismodegib plus Metformin treated group were indicated as bars (**p < 0.01, ***p < 0.001). (E) Intracranial MB11 xenografts treated with DMSO, Vismodegib, Metformin and Vismodegib plus Metformin were measured by IVIS image system for 5 weeks. Color indicates the quantification of tumor growth from small (blue) to big size (red)
To further determine the therapeutic effect of the Metformin and Vismodegib combination treatment in an orthotopic mouse model, MB11 cells expressing firefly luciferase protein (Luc) were injected intracranially into the cerebellum of nude mice. Two weeks after injection, the treated mice were randomized into four groups for specific drug treatments as indicated in Figure 5A. Consistent with the data in Figure 5A, tumor growth was remarkedly inhibited in the combination treatment group (Figure 5E). Together, these results support the notion that combination of Metformin and Vismodegib has a synergistic effect in MB treatment.
4. DISCUSSION
Nearly a third of all MB cases are driven by aberrant SHH activity. However, limitations of current MB therapies include side effects, long‐term sequelae, and resistance to SMO inhibitors, the only FDA‐approved line of drugs used to target SHH‐cancers. Thus, it is urgent to find a better strategy to overcome the above limitations. Previously we showed that AMPK directly phosphorylates GLI1 on three residues, including Serine 408 which has also been confirmed by other independent research group. 11 AMPK phosphorylation on GLI1 results in increased binding of GLI1 with b‐TrCP which then targets the SHH transcriptional activator to the ubiquitin proteasome‐mediated degradation. 12 From these findings, AMPK activation that reduces GLI1 protein levels and transcriptional activity can potentially be employed for cancers driven by aberrant SHH activity, including MB, BCC, and glioblastoma.
Metformin initially came to the attention of cancer researchers as a potential therapeutic when it was observed that diabetes patients treated with Metformin exhibited a lower risk of tumor formation, and those already suffered from cancer showed reduced mortality. 24 It has been reported that a significantly improved overall and progression‐free survival is achieved in high‐grade glioma patients who take Metformin, but not in patients who take Sulfonylureas, Glitazones or Insulin. 25 Indeed, emerging evidence shows that Metformin alone or combined with other drugs can improve survival. Since Vismodegib is embryotoxic, and nearly all patients with Vismodegib treatment report side effects, such as fatigue, nausea, and diarrhea, 26 combination of Metformin and Vismodegib at a lowered dose will be necessary, especially for the pediatric patients.
Drug resistance is another major hurdle in cancer therapeutics. GLI1 activation has been reported in promoting drug resistance in acute myeloid leukemia (AML), 27 melanoma, 28 and colon cancer. 29 In AML drug‐resistant patients, GLI1 induces UDP glucuronosyltransferase (UGT1A), an enzyme that drives UGT1A‐dependent glucuronidation of Ribavirin and Ara‐C, two of the most commonly used AML treatments to modify the drug activities, thus leading to resistance to these treatments; inhibition of GLI1 is able to overcome resistance to chemotherapy in AML. 27 Consistently, we found GLI1 protein and activity were highly increased in the Vismodegib‐resistant MB cells (Figure 3B,C). Thus, targeting GLI1 presents a promising therapeutic strategy for cancer patients who have developed resistance to conventional cancer treatments.
Given the recognized compensatory alterations in HH‐dependent tumors, thus far, dual targeting the modulators of HH/GLI1 will likely provide new options for effective therapies. Indeed, our results suggest that AMPK activation works synergistically with SMO inhibitors to more efficiently downregulate SHH‐MB growth than the single drug treatment. With the advance of sequencing technology, the genome sequencing of SHH MB has revealed genetic alternation in SHH signaling components such as loss or mutation of SUFU, and amplification of GLI2 and other gene like MYCN was primarily found in the SMO inhibitors resistant MB. 30 , 31 Segal et al. showed that activation of RAS/MAPK pathway drives resistance to SMO inhibitor. 32 Other signaling or molecular pathway such as PI3 K/AKT, 33 atypical protein kinase C ι/λ (aPKC‐ι/λ) as GLI1 activator, 34 bromo, and extra C‐terminal (BET) bromodomain protein 4 (BRD4) 35 were all involved in rending MB resistant to SMO inhibitor. To gain more translational impact of MB treatment, following with current study the MB PDX or mouse genetic model will be applied. As both Vismodegib and Metformin are currently FDA‐approved human cancer treatments, the proposed combination therapy can be readily tested in future clinical trials.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
J.Y.Y designed, performed and coordinated research; S.G. performed experiments; J.Y.Y., G.Z., C.J.C. and S.G. analyzed the data and J.Y.Y. and S.G. wrote the paper; all of the authors contributed to discussion of results and interpretations.
Supporting information
ACKNOWLEDGMENTS
We thank Dr. Li‐Pin Kuo and Pei‐Chieh Tien for their early assistance on this project and Chu‐Hu Lai and Dr. Chi‐Pin Li for helping on the revised manuscript. We thank Dr. Russel Main (Purdue University) for commenting on this manuscript. This work was supported by Elsa U. Pardee Research award, China Medical University start up fund (10951L2*) and Ministry of Science and Technology (110‐2320‐B‐039‐007‐MY2) (J. ‐Y.Y.).
REFERENCES
- 1. Northcott PA, Robinson GW, Kratz CP, et al. Medulloblastoma. Nat Rev Dis Primers. 2019;5:11. [DOI] [PubMed] [Google Scholar]
- 2. Pollack IF, Agnihotri S, Broniscer A. Childhood brain tumors: current management, biological insights, and future directions. J Neurosurg Pediatr. 2019;23:261‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guerreiro Stucklin AS, Ramaswamy V, Daniels C, Taylor MD. Review of molecular classification and treatment implications of pediatric brain tumors. Curr Opin Pediatr. 2018;30:3‐9. [DOI] [PubMed] [Google Scholar]
- 4. Huang SY, Yang JY. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers (Basel). 2015;7:2110‐2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Danial C, Sarin KY, Oro AE, Chang AL. An Investigator‐Initiated Open‐Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin Cancer Res. 2016;22:1325‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/‐)p53(‐/‐) mice. Cancer Cell. 2004;6:229‐240. [DOI] [PubMed] [Google Scholar]
- 8. Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13:249‐260. [DOI] [PubMed] [Google Scholar]
- 9. Robinson GW, Kaste SC, Chemaitilly W, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. 2017;8:69295‐69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li YH, Luo J, Mosley YY, et al. AMP‐Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015;12:599‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Magno L, Basile A, Coni S, et al. The energy sensor AMPK regulates Hedgehog signaling in human cells through a unique Gli1 metabolic checkpoint. Oncotarget. 2016;7:9538‐9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang R, Huang SY, Ka‐Wai Li K, et al. Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through beta‐TrCP‐mediated proteasome degradation. Oncotarget. 2017;8:49869‐49881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91‐99. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Ding Q, Yen CJ, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21:374‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu CC, Hou S, Orr BA, et al. mTORC1‐Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling‐Driven Translation and Medulloblastoma. Dev Cell. 2017;43(673–688):e675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2‐mediated degradation. Nat Cell Biol. 2008;10:138‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia W, Chen JS, Zhou X, et al. Phosphorylation/cytoplasmic localization of p21Cip1/WAF1 is associated with HER2/neu overexpression and provides a novel combination predictor for poor prognosis in breast cancer patients. Clin Cancer Res. 2004;10:3815‐3824. [DOI] [PubMed] [Google Scholar]
- 18. Ozawa T, James CD. Establishing intracranial brain tumor xenografts with subsequent analysis of tumor growth and response to therapy using bioluminescence imaging. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Labuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacological Reports: PR. 2010;62:956‐965. [DOI] [PubMed] [Google Scholar]
- 20. Ashton JC. Drug combination studies and their synergy quantification using the Chou‐Talalay method–letter. Cancer Res. 2015;75:2400. [DOI] [PubMed] [Google Scholar]
- 21. Ivanov DP, Coyle B, Walker DA, Grabowska AM. In vitro models of medulloblastoma: Choosing the right tool for the job. J Biotechnol. 2016;236:10‐25. [DOI] [PubMed] [Google Scholar]
- 22. Huang X, Dubuc AM, Hashizume R, et al. Voltage‐gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev. 2012;26:1780‐1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang JY, Mackay‐Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with the basal‐cell nevus syndrome. N Engl J Med. 2012;366:2180‐2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC‐16. Diabetes Care. 2010;33:322‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seliger C, Luber C, Gerken M, et al. Use of metformin and survival of patients with high‐grade glioma. Int J Cancer. 2018. [DOI] [PubMed] [Google Scholar]
- 26. Meiss F, Andrlova H, Zeiser R. Vismodegib. Recent Results Cancer Res. 2018;211:125‐139. [DOI] [PubMed] [Google Scholar]
- 27. Zahreddine HA, Culjkovic‐Kraljacic B, Assouline S, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature. 2014;511:90‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faiao‐Flores F, Alves‐Fernandes DK, Pennacchi PC, et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib‐resistant human melanoma cells. Oncogene. 2017;36:1849‐1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang H, Ke F, Zheng J. Hedgehog‐glioma‐associated oncogene homolog‐1 signaling in colon cancer cells and its role in the celecoxib‐mediated anti‐cancer effect. Oncol Lett. 2014;8:2203‐2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kool M, Jones DT, Jager N. Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell. 2014;25:393‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sharpe HJ, Pau G, Dijkgraaf GJ, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27:327‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway‐Dependent Tumors. Cancer Res. 2015;75:3623‐3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang Y, Gholamin S, Schubert S, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20:732‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials