

Abstract
Microvesicles are a heterogeneous group of membrane‐enclosed vesicles that are released from cells into the extracellular space by the outward budding and pinching of the plasma membrane. These vesicles are loaded with multiple selectively sorted proteins and nucleic acids. Although interest in the clinical potential of microvesicles is increasing, there is only limited understanding of different types of microvesicles and the mechanisms involved in their formation. Here, we describe what is presently known about this expanding and complex field of research focusing on the mechanism of biogenesis, cargo loading, and release of microvesicles.
1. INTRODUCTION
Long thought of as little more than cell waste, continued investigation has placed extracellular vesicles (EVs) at the forefront of research into intercellular communication. The release of various types of membrane bound vesicles, broadly and generically referred to as extracellular vesicles, is an evolutionarily conserved process utilized by prokaryotic and eukaryotic cells of diverse origins from plants to bacteria to humans. 1 , 2 While the term extracellular vesicle is commonly used to classify the entire population of secreted membrane encapsulated vesicles, there is a growing list of classes and subpopulations highlighting the significant heterogeneity that exists and remains to be fully understood in the field. 3 , 4 , 5 The family of EVs has grown to include nanovesicles, 6 exomeres, 7 arrestin domain‐containing protein 1‐mediated microvesicles, 8 , 9 large oncosomes, 10 and apoptotic bodies. 11 The two most well‐characterized classes of EVs, however, remain exosomes and the class that is the primary focus of this review, microvesicles (MVs). Exosomes and microvesicles differ in many of the canonical characteristics used to define classes of MVs. These include distinct mechanisms of biogenesis, cargo composition, and size profiles, 1 , 12 , 13 as we describe in the following sections.
2. MICROVESICLE FORMATION AT THE CELL SURFACE
The mechanism of biogenesis is one of the predominant and defining differences between exosomes and microvesicles. Exosomes are formed initially as intraluminal vesicles (ILVs) following the invagination of the limiting membrane of an organelle of the endolysosomal pathway known as a multivesicular body (MVB) or multivesicular endosome. 12 This process can occur repeatedly leading to the accumulation of ILVs within each MVB. Mature MVBs can then fuse with lysosomes or autophagosomes for degradation or be trafficked to the cell periphery where they will fuse with the plasma membrane (PM) and release the ILV cargo into the extracellular space as exosomes. 13 For a more thorough review of exosome biogenesis, see references 12–14, and additional reviews in this series. Multiple pathways regulating the pinching and release of microvesicles have been uncovered thus far, as described further below. A common component of many microvesicle regulatory pathways is the influence of small GTPases, many of which have known roles regulating contractile machinery. 14 , 15 , 16 In contrast to the initial intracellular events during exosome biogenesis, microvesicle biogenesis begins with the direct outward budding of the plasma membrane. Mature microvesicles are then shed from the surface of cells, following a tightly regulated pinching and scission process that results in the release of MVs directly into the extracellular space.
Microvesicles span a significant and heterogeneous size range. Unlike exosomes, which are more regular in size, ranging from 50 to 100 nanometers (nm) in diameter, microvesicles span more than an order of magnitude in range. Originally thought of as occurring predominantly from 200 to 1000+ nm in diameter, several reports have highlighted the release of smaller particles via a similar outward budding and pinching. 4 These include the formation of ARMMs which when imaged by electron microscopy were found to have an average diameter of approximately 45 nm. 9 At the other end of the size spectrum, large oncosomes are released by tumor cells into the extracellular space following a similar biogenesis paradigm, and can range up to several microns in diameter. 10 , 17 The specifics of the aforementioned cell surface‐derived vesicles are further described below.
While increasing evidence suggests that normal cell types shed some quantity of MVs, the study of microvesicles has been, in large part, guided by the growing understanding of their roles during pathophysiological states and in particular cancers where MV production is a known means for tumor cell invasion and means to stimulate invasion in neighboring cells. 5 , 18 , 19 , 20 , 21 Malignant transformation of normal cells, has long been known to increase the quantity and the cargo content of shed MVs. 22 Comparative analysis, for example, between a normal melanocyte‐derived cell line (melan‐a) and a melanoma counterpart (Tm1) revealed a twofold increase in MV shedding by the malignant line. 23 This was further exemplified by experiments in which expression of the highly oncogenic variant of the EGF receptor (EGFRvIII) further increased the number of MVs detected at the cell surface. 24 More recent research has highlighted changes in bioactive cargo in MVs shed from transformed cells engineered to express an oncogenic form of diffuse B cell lymphoma (Dbl). Kreger and colleagues demonstrated that the oncogenic transformation of mouse embryonic fibroblasts led to the release of MVs carrying novel cargo including focal adhesion kinase. 25 Interestingly, the authors did not identify an increase in overall MV release with Dbl expression suggesting that there are likely driver and cell type specificities regulating MV release.
3. MECHANISMS AND REGULATION OF MICROVESICLE SHEDDING
As described previously, a fundamental property of microvesicles is their biogenesis pathway which stems from the outward budding and pinching of the plasma membrane (PM) releasing newly formed MVs directly into the extracellular environment. 26 The plasma membrane itself serves as a highly organized nexus, integrating intra‐ and extracellular signaling processes. In order to carry out myriad fundamental processes, the PM organizes into well‐defined domains, the dysregulation of which is increasingly linked to oncogenic signaling. 27 To date, much research has been aimed at understanding membrane curvature and shape regulation in the context of lipid components, phospholipid composition, and protein‐mediated bending. The biogenesis of MVs, for example, is documented to rely on a combination of plasma membrane phospholipid redistribution and coordination of actomyosin contractile machinery. 26 , 28 , 29 Further, formation of nascent MVs specifically requires cholesterol, as its depletion results in the loss of microvesicle release. 28 Though our understanding of PM dynamics and MV shedding has increased, little work has included analysis of the glycocalyx. This is despite long‐chain biopolymers within the glycocalyx frequently found anchored to the surfaces of cell surface organelles. 30 , 31 Recently, however, researchers concluded that microvesicles were released as a result of mucin‐induced membrane instabilities. 32 Exogenous expression of a construct containing 42 tandem repeats of Mucin 1 led to massive increases in particles ranging from 100 to 400 nm in conditioned cell culture media. Furthermore, as high mucin expression is a common feature of tumor cells, HeLa cells with high levels of endogenous MUC1 expression produced increased levels of microvesicles. 32
Extracellular factors have also been known to influence the shedding of MVs, including the formation of a hypoxic tumor environment which can increase MV release. 19 In solid tumors such as breast cancer, hypoxia is a hallmark microenvironmental stimulus that results in the expression of hypoxia‐inducible factors (HIFs) for cell survival. 33 In advanced breast cancer, HIF induction of RAB22A results in Rab22A‐associated MV release. 19 Under hypoxic conditions, RAB22A is a gene target of HIF‐1. When breast cancer cells were cultured in 1% of oxygen, HIF‐1 binds to a 5’‐untranslated sequence in the RAB22A gene to induce RAB22A expression. Rab22A colocalizes with budding vesicles and knockdown of RAB22A eliminates vesicle release suggesting a direct role for Rab22A in MV generation. 23 Interestingly, use of O2‐(3‐aminopropyl) diazeniumdiolate 3f (referred to as 3f) is able to significantly suppress growth and metastasis in vivo in triple negative breast cancer (TNBC) by attenuating MV formation. 19 Treatment with 3f compound results in epigenetic modification through an increase in levels of miR‐203, which acts to decrease RAB22A transcription. This decrease in RAB22A expression drastically reduces MV release, highlighting the potential for the therapeutic targeting of MV shedding. 34
In addition to alterations in membrane dynamics or signaling stemming from microenvironmental cues, tumor cells frequently exhibit perturbed calcium (Ca2+) homeostasis. 35 While Ca2+ has long been known as a secondary messenger with a regulatory role in EV shedding, particularly from cells of the vasculature where it was identified as a mediator of vesiculation, it has only recently been implicated in MV shedding from tumor cells. 36 , 37 In a series of experiments, Taylor and colleagues investigated the differential regulation of MV shedding from malignant and nonmalignant mammary cells. 38 These same researchers have previously also found that in malignant cells, the Ca2+‐mediated activation of the cysteine protease calpain, resulted in its redistribution to the cell periphery, where it can facilitate the focal remodeling of the cortical cytoskeleton. 39 While the complete signaling pathway leading from intracellular Ca2+ to MV release has yet to be elucidated, these results certainly warrant further investigation given the potential link to members of the S100 family of proteins, and TRPM7 which is known to mediate Ca2+ influx and can interact with calpain. 40 Additionally, tumor Ca2+ levels may also function as a double‐edged sword––working to increase MV shedding in an intra‐ and extracellular capacity. Elevated extracellular Ca2+ levels in line with serum concentrations of patients experiencing hypercalcemia, led to an increase in the production of microvesicles in vitro. 41
Coordination of actomyosin contraction for MV release is in part regulated by the ADP ribosylation factor 6 (ARF6) signaling axis. 26 , 42 ARF6, a member of the Ras GTPase family, plays significant roles in membrane trafficking, actin remodeling, and functions during cancer cell invasion. 18 , 43 , 44 Importantly, during MV shedding, ARF6 activity is required for subsequent phospholipase D activation and recruitment of extracellular signal‐regulated kinase (ERK) to the plasma membrane. 26 , 42 At the plasma membrane, ERK activity results in an activating phosphorylation on myosin light chain kinase (MLCK) which in turn phosphorylates the myosin light chain (MLC) to facilitate actomyosin‐based contractility. Phosphorylated MLC is enriched at the “necks” of vesicles where it can facilitate fission, releasing vesicles into the extracellular space. 26 Inhibition of myosin light chain phosphatase downstream of RhoA/Rho‐associated kinase (ROCK) signaling further promotes the release of MVs through a parallel pathway. 42 RhoA and ROCK signaling was also implicated in MV shedding through the activation of Lim kinase (LIMK). LIMK is necessary for an inhibitory phosphorylation on cofilin, which limits its actin severing capabilities and promotes MV biogenesis. 45 Additional research has demonstrated that the antagonistic action of Rab35 and ARF6 regulates MV biogenesis as well (Figure 1A). Here, Rab35 inactivation or depletion results in the redistribution of the actin bundling protein fascin, away from invadopodia, and to the cell cortex. 46 Peripheral fascin is able to form a ternary complex with ezrin and podocalyxin where it is then readily able to facilitate actin bundling and MV pinching in response to ARF6 activation. 46 Taken together, these results suggest that there are multiple layers of GTPase signaling responsible for regulating microvesicle release and the specific pathways utilized to form a given vesicle likely influence GTPase cargo.
FIGURE 1.
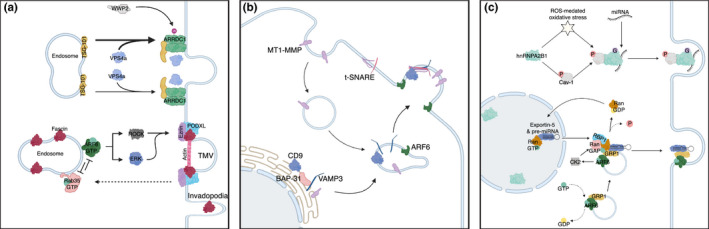
Mechanisms of microvesicle formation at the cell surface and cargo delivery. (A) ARF6 regulates actomyosin contraction at the necks of nascent microvesicles via activation of ERK and Rho signaling pathways. Mutually antagonistic Rab35 and ARF6 signaling govern the intracellular localization of the actin bundling protein, fascin, which in turn modulates microvesicle pinching and release. Similarly, ARRDC1‐mediated recruitment of TSG‐101 from endosomes to the plasma membrane facilitates microvesicle the shedding and release. (B) ARF6‐regulated endosomes serve as a nexus integrating newly synthesized and recycling the protease MT1‐MMP for delivery to nascent microvesicles in a VAMP3‐dependent manner. (C) The pre‐miRNA transporter Exportin‐5 serves as a chaperone to link the movement of TMV pre‐miRNA cargo to the ARF6‐endosomal network. Upon exit from the nucleus, Exportin‐5 together with pre‐miRNA cargo, is shuttled to a trafficking complex of ARF6 and cytohesin‐3 (GRP1) for incorporation into shedding microvesicles. Additionally, O‐GlcNAcylation of the RNA‐binding protein hnRNPA2B1 in response to ROS‐mediated oxidative stress and 14Y‐phosphorylated Caveolin‐1 regulates the binding of a distinct miRNA repertoire. The resulting complex including bound miRNA then traffics to the plasma membrane where it is included within shed vesicles. Figure created with BioRender
Large oncosomes derived from tumor cells similarly bud from the plasma membrane and are associated with the transition to an amoeboid morphology. 10 , 17 , 47 Formation of budding oncosomes is triggered by both stimulation with epidermal growth factor (EGF) in prostate cancer cells, and through overexpression of constitutively active Akt. 10 , 47 Overlapping roles of intrinsic cell signaling machinery have been identified between these large vesicles and other microvesicle populations. This includes RhoA and ROCK signaling which functions in concert with ARF6 to regulate tumor MV‐mediated cell invasion, and also acts to trigger the membrane blebbing of large oncosomes. 17 , 42 , 48 Additionally, ARF6 is localized to large oncosomes and may carry out similar functions for vesicle release. 47 Furthermore, the actin nucleating protein DRF3 acts as an inhibitor of large oncosomes formation suggesting that, like ARF6‐regulated MV shedding, there is a functional role for the actin cytoskeleton in regulating their formation. 10
Arrestin domain‐containing protein 1 (ARRDC1)‐mediated microvesicles (ARMMs) are a yet another subgroup of microvesicles that are produced directly from the plasma membrane. 9 ARMM formation is dependent upon ARRDC1 interaction with tumor susceptibility gene 101 (TSG101), a component of the endosomal sorting complexes required for transport (ESCRT)‐1 complex. Interaction with ARRDC1 results in the translocation of TSG101 from the endosome to the plasma membrane (Figure 1A). Abscission of the vesicle from the plasma membrane ultimately requires the ATPase activity of vacuolar protein sorting‐associated protein 4 (VPS4) and ubiquitination of ARRDC1 via E3 ligase family member, WWP2. 9 Evidence suggests that release of ARMMs also requires the ESCRT complex, classically associated with MVB formation and viral budding. 9 Critical to MV analysis, different subtypes have overlapping size profiles and physical characteristics which makes it difficult to fully separate these groups by size or density. Furthermore, a key regulator in the formation of microvesicles in tumor cells, ARF6 has been identified in oncosomes, and more recently, as being enriched 2.5 fold in ARMMs. 10 , 49 Given these results, additional research is needed to determine what, if any, similarities and differences exist between ARMMs, large oncosomes, and shed MVs, and to accurately attribute biological functions to distinct classes of EVs. Moreover, the overlap in molecular machinery utilized in the biogenesis of what appears to be distinct classes of extracellular vesicles highlights the need for continued research not only to delineate‐specific requirements for vesicle release, but also to more accurately determine the regulators of individual classes, and the relationships between them.
4. MICROVESICLE CARGO TRAFFICKING AND DELIVERY
Significant progress has been made in recent years toward a thorough and complete understanding of MV biology, including a more detailed understanding of the complex mixture of bioactive MV cargo content. Microvesicles are documented to contain a broad array of molecular cargo, which reflects not only the cell type from which the MV was released, but also the intracellular trafficking pathways delivering that cargo to the cell surface. Identified cargoes include multiple forms of proteins ranging from integrin receptors, active proteases, multiple small GTPases, to multidrug resistance proteins, and miRNA processing machinery. Though protein cargo is abundant, it is not the only bioactive cargo contained in shed MVs, which are known to also contain multiple forms of nucleic acids (both DNA and RNA), active lipids, ROS regulators, and mitochondria. 50 , 51 This list is by no means exhaustive, and the authors point readers to one of the many outstanding reviews on the subject for a more detailed analysis of MV cargo components. 1 , 14 , 52 As knowledge of MV cargo has increased, so too has our understanding of the pathways which traffic cargo for delivery to sites of MV biogenesis as we describe here.
4.1. Protein cargo trafficking
In mammalian cells, endocytic membrane trafficking has an essential role in the movement of many cellular components to distinct intracellular destinations. 53 Building on knowledge of the significant cross talk between regulators of endocytic membrane trafficking and MV biogenesis, researchers have begun to unravel the movement, sorting, and enrichment of MV cargo. In addition to the previously described regulatory functions governing MV pinching and release, the small GTPase ARF6 has increasingly been found to facilitate the selective enrichment of MV cargo. Activation of ARF6 led to the shedding of MVs containing MHC class I proteins, the active form of membrane type‐1 matrix metalloprotease (MT1‐MMP), β1‐integrin, together with an enrichment of active ARF6 itself. 26 This report also highlighted selective incorporation of vesicular SNARE (v‐SNARE) proteins, noting the inclusion of VAMP3 as MV cargo, but undetectable levels of VAMP7. 26
Subsequent work identified a VAMP3 specific interaction that was responsible for the delivery of MT1‐MMP to sites of MV biogenesis in tumor cells. 20 MT1‐MMP was found to associate with VAMP3 in a CD‐9‐dependent fashion, which serves to protect the protease from lysosomal degradation and allow for trafficking to the cell periphery where the VAMP3‐MT1‐MMP complex is incorporated into nascent MVs (Figure 1B). Furthermore, surface antibody labeling experiments demonstrated that in addition to newly synthesized protease, recycled MT1‐MMP is also included as MV cargo in a VAMP3‐dependent manner. The identification of VAMP3 positive endosomes which also contain both MT1‐MMP and ARF6 would suggest that ARF6‐regulated recycling endosomes are an integration and sorting point in which reserves of MT1‐MMP are held for rapid delivery to sites of active matrix degradation, and the multiple points at which ARF6 regulates MV biogenesis likely stem from its well‐documented roles regulating actin cytoskeletal remodeling and endocytic membrane trafficking. 20 , 44
The intricacies of the endocytic network extend deeply into the EV field through this regulated shuttling of cargo to sites of biogenesis. These elegantly choreographed movements result in the specific enrichment, inclusion, or exclusion of a host of transiting protein components. It has long been known that cargo which is preferentially recycled to the plasma membrane is less likely to be present on exosomes. 54 , 55 Interestingly, the same is potentially true for microvesicles, as the transferrin receptor, despite trafficking through ARF6‐regulated endosomal compartments in some cell types, appears to be excluded from tumor‐derived microvesicles. 26 Additionally, while currently only understood in the context of exosomes, the syndecan–syntenin–ALIX axis also functions to link the endosomal targeting of EV cargo. Syndecan interacts with syntenin, which also interacts with ALIX, through LYPX(n)L motifs, to support the biogenesis of ILVs and the recruitment of syndecan cargo to nascent exosomes. 56 Similarly, syntenin acts to recruit CHMP4 via ALIX and more recently, ALIX was identified as a binding partner for cSrc within exosomes, and an ALIX‐ESCRT‐III pathway promotes the sorting and delivery of tetraspanins to exosomes, highlighting the intersection of the EV biogenesis and the canonical ESCRT pathway. 56 , 57 , 58 Previous reports have also linked syndecan recycling to ARF6, and while little is known about syndecan content in microvesicles, both syndecan and ARF6 have been identified in a mixed population of EVs isolated from glioma patient plasma. 59 , 60 Additional research is needed to elucidate the role of ESCRT proteins in cargo recruitment to microvesicles, though emerging data from the ARMM subpopulation suggests this may be a common mechanism shared between exosomes, and small MVs.
ARMMs, which like ARF6‐mediated MVs, shed directly from the surface of the plasma membrane were initially identified based on their ARRDC1‐dependent mode of biogenesis. ARRDC1 localizes to the cytosolic side of the plasma membrane, where it recruits TSG‐101, a component of the ESCRT‐I complex frequently found on late endosomal membranes, to initiate budding. TSG‐101, together with ARRDC1, are both subsequently included as cargo in the shed ARMM. 9 Interestingly, fusion proteins containing ARRDC1 fused to the N‐terminal of p53, resulted in the efficient loading of p53 into ARMMs. Similar work has exploited ARRDC1 interacting domains to load CRISPR‐Cas9 for secretion as ARMM cargo. 8 Additional research, however, identified 177 proteins which were significantly enriched (>1.5 fold) in ARMMs. 49 These included members of the integrin family, proteasome components, additional components of the ESCRT complexes including CHMP6, proteases, NEDD4 E3 ligases, and plasma membrane proteins including NOTCH2. The incorporation of NOTCH2 is dependent upon the E3 ligase ITCH, and the metalloprotease ADAM10, both of which are also secreted as ARMM cargo. 49 It remains to be determined whether ESCRT‐III and ALIX function in the biogenesis of ARMMs, though the enrichment of multiple ESCRT proteins, coupled with the previously described recruitment identified in exosomes certainly suggests it is possible.
Additional understanding of cargo enrichment within shed MVs, has come from understanding responses to cellular stress. Hypoxia, a hallmark feature of the tumor microenvironment, leads to alterations in gene expression mediated by the hypoxia‐inducible factor family of transcription factors. 61 , 62 As described previously, hypoxia results in an increase in MV shedding, stemming from elevated expression of RAB22A. However, immunofluorescence studies demonstrate that under hypoxic conditions, RAB22A also colocalizes with enriched pools of transglutaminase‐2 (TGM2) at sites of MV budding. Together with data indicating that RAB22 knockdown only abrogates hypoxia‐induced MV shedding, these results suggest that RAB22A actively recruits protein cargo to sites of MV release. 19 Furthermore, cell stress or injury can also lead to the release of endogenous signaling molecules, including ATP. Macrophage stimulation with ATP was recently reported to result in a shift from soluble TNF release to packaging of membrane TNF within shed MVs. This shift bypasses the traditional ER and Golgi‐dependent transport pathway, and instead is dependent upon acid sphingomyelinase. 63
4.2. Nucleic acid cargo
Counted among the myriad bioactive cargos identified within shed MVs are multiple forms of nucleic acids. 1 , 64 While significant work has shed light on that cargo content, to date, we are only beginning to tease apart the trafficking mechanisms utilized to deliver nucleic acid cargo to sites of EV release. A pair of recent studies highlight the genomic (gDNA) content of shed EVs, and outline a possible trafficking mechanism enabling its release within EVs. Vagner et al. demonstrated that large EVs (L‐EVs) derived from prostate cancer cell lines or patient plasma contain large fragments (up to 2 million base pairs) of chromosomal DNA. 65 Whole genome sequencing of prostate cancer L‐EV DNA revealed that the gDNA within L‐EVs can be interrogated for the same genomic aberrations found in the shedding cell. In parallel, Reis‐Sobriero et al. showed that dysregulation of the nuclear envelope protein emerin in aggressive amoeboid tumor cells leads to nuclear shape instability. Alterations in nuclear shape were associated with disruption of the nuclear envelope; the formation of nuclear blebs and cytosolic nuclear fragments (micronuclei); and the release of large extracellular vesicles containing nuclear material. 66 While the authors demonstrate the formation of micronuclei within the cytosol, and the formation of nascent EVs containing nuclear material, precisely how the material moves from the juxtanuclear region to the cell periphery is yet to be determined.
Like DNA, extracellular vesicles, and particularly exosomes, have been reported to carry miRNA cargo. 1 While multiple trafficking pathways have been reported in exosomes, including the sequence‐specific mechanisms for targeting miRNA, 67 , 68 , 69 , 70 current understanding of miRNA loading into MVs has only recently begun to be elucidated with the publication of two manuscripts linking components of the endocytic machinery to miRNA trafficking. In the first report, Clancy and colleagues outlined a trafficking pathway linking Exportin‐5, the indispensable transport receptor for pre‐miRNA, to the ARF6‐regulated transport pathways. 71 The authors report that activation of ARF6 signals through casein kinase 2 (CK2) resulting in the phosphorylation of RanGAP1, which allows for the transfer of cargo exported from the nucleus into the ARF6‐regulated shuttling complex (consisting of ARF6‐GTP and GRP1, a member of cytohesin family of ARF GEFs) for anterograde movement (Figure 1C). Interestingly, the authors found that the exchange factor functions of GRP1 were dispensable, and its function instead was primarily as a scaffold.
In the second study, Lee et al. uncovered a link between caveolin‐1 phosphorylation and incorporation of miRNA into MVs in response to oxidative stress (hyperoxia). 72 Oxidative stress induces the O‐GlcNAcylation of the RNA‐binding protein hnRNPA2B1 in a cav‐1 pY14‐dependent manner, which leads to a robust interaction between the two proteins, and their co‐trafficking together into MVs (Figure 1C). The O‐GlcNAcylation modification of hnRNPA2B1 resulted in alterations to associated miRNA, and increased the miRNA content in shed MVs. It is worth noting that both of these reports uncovered a miRNA trafficking pathway which was dependent on an RNA‐binding protein serving as a chaperone. This is also in line with previous reports detailing miRNA loading into exosomes. It remains to be determined whether these or other as yet unidentified pathways are disease, cell type, or even EV‐type specific. If the exosome field is any indication, it is likely that as we continue to clarify the heterogeneity of MV populations we will similarly decode distinct mechanisms of miRNA sorting into the respective vesicles. 67
5. LOOKING AHEAD
The study of shed microvesicles is an active area of research and uncovering the defining and distinct characteristics of each subpopulation of MVs is critical to advancing our understanding of the field. Unfortunately, due to the overlapping physical properties which directly influence the commonly utilized methods of MV isolation, there remain outstanding questions regarding whether and/or how the various subpopulations of vesicles pinched from the surface of cells relate to one another. While MV formation and release governs several physiological processes, it is now clear that under pathophysiological conditions such as cancer, diseased cells usurp these processes. Yet, we have minimal understanding of the intrinsic signals that ramp up MV biogenesis in tumor cells. Although there is some evidence in the literature, it is unclear if oncogenesis is always accompanied by a shift in MV subtype or cargo recruitment and the nature of the signals that may drive these changes. The activation of oncogenes or loss of tumor suppressors are key drivers of tumor initiation and are likely linked to alterations in EV profiles during disease onset and progression. In this regard, EGFRvIII, mutant K‐Ras, B‐Raf, and loss of p53 have all been coupled to altered EV biogenesis and/or composition. 10 , 73 , 74 , 75 , 76
Much of the interest in EV biology stems from their untapped clinical potential. A growing body of evidence, predominantly utilizing exosomes, highlights the potential for EV‐based liquid biopsies for the identification and longitudinal monitoring of cancer, and more recently the development of clinical grade exosomes for use as therapeutics. 12 , 77 , 78 While much of the research into liquid biopsy technology has centered around examination of protein, DNA, or RNA cargo a pair of recent publications highlight the expanding repertoire of technology which can be deployed against disease. Researchers utilized microfluidic devices to examine phenotypic changes in melanoma extracellular vesicles to monitor treatment response, or to quantify the proteolytic activity of MT1‐MMP for monitoring of in vivo tumor progression and metastasis. 79 , 80 As such, it is tempting to speculate on the future of EV clinical applications in exploiting the biochemical, physical, and functional makeup of EVs to improve diagnostics and real‐time monitoring of disease.
This article is part of the Extracellular Vesicles and Homeostasis Special Collection.
REFERENCES
- 1. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213‐228. [DOI] [PubMed] [Google Scholar]
- 2. Deatherage BL, Cookson BT. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80:1948‐1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jeppesen DK, Fenix AM, Franklin JL, et al. Reassessment of exosome composition. Cell. 2019;177(428–445):e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ratajczak MZ, Ratajczak J. Extracellular microvesicles/exosomes: discovery, disbelief, acceptance, and the future? Leukemia. 2020;34:3126‐3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Q, Zhuang X, Mu J, et al. Delivery of therapeutic agents by nanoparticles made of grapefruit‐derived lipids. Nat Commun. 2013;4:1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang H, Freitas D, Kim HS, et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field‐flow fractionation. Nat Cell Biol. 2018;20:332‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Q, Yu J, Kadungure T, Beyene J, Zhang H, Lu Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat Commun. 2018;9:960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q. Formation and release of arrestin domain‐containing protein 1‐mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci U S A. 2012;109:4146‐4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Vizio D, Kim J, Hager MH, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601‐5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kerr J, Wyllie A, Currie A. Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br J Cancer. 1972;26:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367.640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McAndrews KM, Kalluri R. Mechanisms associated with biogenesis of exosomes in cancer. Mol Cancer. 2019;18:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tricarico C, Clancy J, D'Souza‐Schorey C. Biology and biogenesis of shed microvesicles. Small GTPases. 2017;8:220‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blanc L, Vidal M. New insights into the function of Rab GTPases in the context of exosomal secretion. Small GTPases. 2018;9:95‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schorey JS, Cheng Y, Singh PP, Smith VL. Exosomes and other extracellular vesicles in host‐pathogen interactions. EMBO Rep. 2015;16:24‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hager MH, Morley S, Bielenberg DR, et al. DIAPH3 governs the cellular transition to the amoeboid tumour phenotype. EMBO Mol Med. 2012;4:743‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li R, Peng C, Zhang X, Wu Y, Pan S, Xiao Y. Roles of Arf6 in cancer cell invasion, metastasis and proliferation. Life Sci. 2017;182:80‐84. [DOI] [PubMed] [Google Scholar]
- 19. Wang T, Gilkes DM, Takano N, et al. Hypoxia‐inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci U S A. 2014;111:E3234‐3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clancy JW, Sedgwick A, Rosse C, et al. Regulated delivery of molecular cargo to invasive tumour‐derived microvesicles. Nat Commun. 2015;6:6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deregibus MC, Cantaluppi V, Calogero R, et al. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007;110:2440‐2448. [DOI] [PubMed] [Google Scholar]
- 22. Ginestra A, La Placa MD, Saladino F, Cassarà D, Nagase H, Vittorelli ML. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res. 1998;18:3433‐3437. [PubMed] [Google Scholar]
- 23. Lima LG, Oliveira AS, Campos LC, et al. Malignant transformation in melanocytes is associated with increased production of procoagulant microvesicles. Thromb Haemost. 2011;106:712‐723. [DOI] [PubMed] [Google Scholar]
- 24. Al‐Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619‐624. [DOI] [PubMed] [Google Scholar]
- 25. Kreger BT, Dougherty AL, Greene KS, Cerione RA, Antonyak MA. Microvesicle cargo and function changes upon induction of cellular transformation. J Biol Chem. 2016;291:19774‐19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muralidharan‐Chari V, Clancy J, Plou C, et al. ARF6‐regulated shedding of tumor cell‐derived plasma membrane microvesicles. Curr Biol. 2009;19:1875‐1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bi J, Ichu TA, Zanca C, et al. Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab. 2019;30(525–538):e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue‐factor‐bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604‐1611. [DOI] [PubMed] [Google Scholar]
- 29. Lima LG, Chammas R, Monteiro RQ, Moreira MEC, Barcinski MA. Tumor‐derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine‐dependent manner. Cancer Lett. 2009;283:168‐175. [DOI] [PubMed] [Google Scholar]
- 30. Bennett R Jr, Jarvela T, Engelhardt P, et al. Mucin MUC1 is seen in cell surface protrusions together with ezrin in immunoelectron tomography and is concentrated at tips of filopodial protrusions in MCF‐7 breast carcinoma cells. J Histochem Cytochem. 2001;49:67‐77. [DOI] [PubMed] [Google Scholar]
- 31. Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol. 2008;70:431‐457. [DOI] [PubMed] [Google Scholar]
- 32. Shurer CR, Kuo JC, Roberts LM, et al. Physical principles of membrane shape regulation by the glycocalyx. Cell. 2019;177(1757–1770):e1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266‐276. [DOI] [PubMed] [Google Scholar]
- 34. Kang F, Zhu J, Wu J, et al. O(2)‐3‐Aminopropyl diazeniumdiolates suppress the progression of highly metastatic triple‐negative breast cancer by inhibition of microvesicle formation via nitric oxide‐based epigenetic regulation. Chem Sci. 2018;9:6893‐6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol. 2016;29:1‐6. [DOI] [PubMed] [Google Scholar]
- 36. Pasquet JM, Dachary‐Prigent J, Nurden AT. Microvesicle release is associated with extensive protein tyrosine dephosphorylation in platelets stimulated by A23187 or a mixture of thrombin and collagen. Biochem J. 1998;333(Pt 3):591‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pasquet JM, Dachary‐Prigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem. 1996;239:647‐654. [DOI] [PubMed] [Google Scholar]
- 38. Taylor J, Azimi I, Monteith G, Bebawy M. Ca(2+) mediates extracellular vesicle biogenesis through alternate pathways in malignancy. J Extracell Vesicles. 2020;9:1734326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taylor J, Jaiswal R, Bebawy M. Calcium‐calpain dependent pathways regulate vesiculation in malignant breast cells. Curr Cancer Drug Targets. 2017;17:486‐494. [DOI] [PubMed] [Google Scholar]
- 40. Monteith GR, Prevarskaya N, Roberts‐Thomson SJ. The calcium‐cancer signalling nexus. Nat Rev Cancer. 2017;17:367‐380. [DOI] [PubMed] [Google Scholar]
- 41. Crawford S, Diamond D, Brustolon L, Penarreta R. Effect of increased extracellular ca on microvesicle production and tumor spheroid formation. Cancer Microenviron. 2010;4:93‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sedgwick AE, Clancy JW, Olivia Balmert M, D'Souza‐Schorey C. Extracellular microvesicles and invadopodia mediate non‐overlapping modes of tumor cell invasion. Sci Rep. 2015;5:14748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muralidharan‐Chari V, Hoover H, Clancy J, et al. ADP‐ribosylation factor 6 regulates tumorigenic and invasive properties in vivo. Can Res. 2009;69:2201‐2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. D'Souza‐Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347‐358. [DOI] [PubMed] [Google Scholar]
- 45. Li B, Antonyak MA, Zhang J, Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012;31:4740‐4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clancy JW, Tricarico CJ, Marous DR, D'Souza‐Schorey C. Coordinated regulation of intracellular fascin distribution governs tumor microvesicle release and invasive cell capacity. Mol Cell Biol. 2019;39:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Di Vizio D, Morello M, Dudley AC, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;181:1573‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oppel F, Muller N, Schackert G, et al. SOX2‐RNAi attenuates S‐phase entry and induces RhoA‐dependent switch to protease‐independent amoeboid migration in human glioma cells. Mol Cancer. 2011;10(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Q, Lu Q. Plasma membrane‐derived extracellular microvesicles mediate non‐canonical intercellular NOTCH signaling. Nat Commun. 2017;8:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao F, Sun L, Yang N, et al. Increased release of microvesicles containing mitochondria is associated with the myeloid differentiation of AML‐M5 leukaemia cells. Exp Cell Res. 2020;395:112213. [DOI] [PubMed] [Google Scholar]
- 51. Malkin EZ, Bratman SV. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020;11:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. D'Souza‐Schorey C, Clancy JW. Tumor‐derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012;26:1287‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol. 2018;19:679‐696. [DOI] [PubMed] [Google Scholar]
- 54. Johnstone RM, Bianchini A, Teng K. Reticulocyte maturation and exosome release: transferrin receptor containing exosomes shows multiple plasma membrane functions. Blood. 1989;74:1844‐1851. [PubMed] [Google Scholar]
- 55. Vidal M, Mangeat P, Hoekstra D. Aggregation reroutes molecules from a recycling to a vesicle‐mediated secretion pathway during reticulocyte maturation. J Cell Sci. 1997;110(Pt 16):1867‐1877. [DOI] [PubMed] [Google Scholar]
- 56. Baietti MF, Zhang Z, Mortier E, et al. Syndecan‐syntenin‐ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14:677‐685. [DOI] [PubMed] [Google Scholar]
- 57. Hikita T, Kuwahara A, Watanabe R, Miyata M, Oneyama C. Src in endosomal membranes promotes exosome secretion and tumor progression. Sci Rep. 2019;9:3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Larios J, Mercier V, Roux A, Gruenberg J. ALIX‐ and ESCRT‐III‐dependent sorting of tetraspanins to exosomes. J Cell Biol. 2020;219:e201904113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Indira Chandran V, Welinder C, Mansson AS, et al. Ultrasensitive immunoprofiling of plasma extracellular vesicles identifies syndecan‐1 as a potential tool for minimally invasive diagnosis of glioma. Clin Cancer Res. 2019;25:3115‐3127. [DOI] [PubMed] [Google Scholar]
- 60. Zimmermann P, Zhang Z, Degeest G, et al. Syndecan recycling [corrected] is controlled by syntenin‐PIP2 interaction and Arf6. Dev Cell. 2005;9:377‐388. [DOI] [PubMed] [Google Scholar]
- 61. Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies Hypoxia Inducible Factor‐1 (HIF‐1)‐target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37:4587‐4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bhandari V, Hoey C, Liu LY, et al. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet. 2019;51:308‐318. [DOI] [PubMed] [Google Scholar]
- 63. Soni S, O'Dea KP, Tan YY, et al. ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J. 2019;33:6442‐6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sedgwick AE, D'Souza‐Schorey C. The biology of extracellular microvesicles. Traffic. 2018;19:319‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vagner T, Spinelli C, Minciacchi VR, et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J Extracell Vesicles. 2018;7:1505403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Reis‐Sobreiro M, Chen JF, Novitskaya T, et al. Emerin deregulation links nuclear shape instability to metastatic potential. Cancer Res. 2018;78:6086‐6097. [DOI] [PubMed] [Google Scholar]
- 67. Temoche‐Diaz MM, Shurtleff MJ, Nottingham RM, et al. Distinct mechanisms of microRNA sorting into cancer cell‐derived extracellular vesicle subtypes. Elife. 2019;8. 10.7554/eLife.47544.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shurtleff MJ, Temoche‐Diaz MM, Karfilis KV, Ri S, Schekman R. Y‐box protein 1 is required to sort microRNAs into exosomes in cells and in a cell‐free reaction. Elife. 2016;5:e19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McKenzie AJ, Hoshino D, Hong NH, et al. KRAS‐MEK signaling controls Ago2 sorting into exosomes. Cell Rep. 2016;15:978‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Melo SA, Sugimoto H, O'Connell JT, et al. Cancer exosomes perform cell‐independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Clancy JW, Zhang Y, Sheehan C, D'Souza‐Schorey C. An ARF6‐Exportin‐5 axis delivers pre‐miRNA cargo to tumour microvesicles. Nat Cell Biol. 2019;21:856‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee H, Li C, Zhang Y, Zhang D, Otterbein LE, Jin Y. Caveolin‐1 selectively regulates microRNA sorting into microvesicles after noxious stimuli. J Exp Med. 2019;216:2202‐2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Choi D, Montermini L, Kim DK, Meehan B, Roth FP, Rak J. The impact of oncogenic EGFRvIII on the proteome of extracellular vesicles released from glioblastoma cells. Mol Cell Proteomics. 2018;17:1948‐1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lunavat TR, Cheng L, Einarsdottir BO, et al. BRAF(V600) inhibition alters the microRNA cargo in the vesicular secretome of malignant melanoma cells. Proc Natl Acad Sci U S A. 2017;114:E5930‐E5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee TH, Chennakrishnaiah S, Audemard E, Montermini L, Meehan B, Rak J. Oncogenic ras‐driven cancer cell vesiculation leads to emission of double‐stranded DNA capable of interacting with target cells. Biochem Biophys Res Commun. 2014;451:295‐301. [DOI] [PubMed] [Google Scholar]
- 76. Zhang S, Wang C, Ma B, et al. Mutant p53 drives cancer metastasis via RCP‐mediated Hsp90alpha secretion. Cell Rep. 2020;32:107879. [DOI] [PubMed] [Google Scholar]
- 77. Melo SA, Luecke LB, Kahlert C, et al. Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mendt M, Kamerkar S, Sugimoto H, et al. Generation and testing of clinical‐grade exosomes for pancreatic cancer. JCI Insight. 2018;3:e99263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang P, Wu X, Gardashova G, et al. Molecular and functional extracellular vesicle analysis using nanopatterned microchips monitors tumor progression and metastasis. Sci Transl Med. 2020;12:eaaz2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang J, Wuethrich A, Sina AA, et al. Tracking extracellular vesicle phenotypic changes enables treatment monitoring in melanoma. Sci Adv. 2020;6:eaax3223. [DOI] [PMC free article] [PubMed] [Google Scholar]