

Abstract
Photoredox catalysts are integral components of artificial photosystems, and have recently emerged as powerful tools for catalysing numerous organic reactions. However, the development of inexpensive and efficient earth-abundant photoredox catalysts remains a challenge. We here present the photochemical and photophysical properties of a Ni–Mabiq catalyst ([NiII(Mabiq)]OTf (1); Mabiq = 2-4:6-8-bis(3,3,4,4-tetramethyldihydropyrrolo)-10-15-(2,2-biquinazolino)-[15]-1,3,5,8,10,14-hexaene1,3,7,9,11,14-N6)—and of a Zn-containing analogue ([ZnII(Mabiq)OTf] (2))—using steady state and time resolved optical spectroscopy, time-dependent density functional theory (TDDFT) calculations, and reactivity studies. The Ni and Zn complexes exhibit similar absorption spectra, but markedly different photochemical properties. These differences arise because the excited states of 2 are ligand-localized, whereas metal-centered states account for the photoactivity of 1. The distinct properties of the Ni and Zn complexes are manifest in their behavior in the photo-driven aza-Henry reaction and oxidative coupling of methoxybenzylamine.
The development of earth-abundant photoredox catalysts remains a challenge. Studies of Ni- and Zn-Mabiq complexes demonstrate how the coordinating metal ion influences the photochemistry, photodynamics and reactivity of photocatalysts.
Introduction
The breadth of light-driven chemical transformations has rapidly expanded over the last decade, due to a surge of research in photoredox catalysis.1–8 Photoredox catalysts are used not only for solar fuel applications,9–13 but also as components of complex catalytic processes that couple photosensitizers with secondary organic, transition metal or Lewis acid catalysts.14–16 Consequently, contemporary developments in light mediated catalysis have enabled unique reactivities beyond what organometallic catalysts alone can achieve. Photocatalysts thus have emerged as important tools for sustainable chemistry and renewable energy needs.
An ensuing challenge within the field is to develop an array of photosensitizers for these varied catalytic processes. The conventional noble metal complexes, Ru and Ir polypyridyl complexes, remain the ‘workhorses’ of photoredox catalysis.1,17–19 However, the scarcity and cost of these elements significantly limits their applications. Alternative catalysts based on low-cost, earth-abundant elements are clearly needed.
While chemical behavior is generally similar within a given group of elements, the simple approach of replacing 2nd and 3rd row transition metal complexes with earth-abundant 1st row analogues is not straightforward. A major complicating factor is the often dramatically different photophysics of the lighter elements. A striking example is provided by the RuII polypyridyl complexes, which tend to form metal-to-ligand charge transfer (MLCT) states with lifetimes approaching microseconds.17 In contrast, ferrous analogues are notoriously short lived, with relaxation to high-spin metal-centered (MC) states often proceeding on the sub-picosecond timescale.20 Nevertheless, there have been several exciting developments with respect to earth-abundant photoredox catalysts, including first-row transition metal complexes based on Zr, and Cr through Zn.21–24 Moreover, sophisticated efforts are being made to design and tailor ligand scaffolds and impose desirable electronic structure features on the coordinated metal. Variation of the transition metal clearly can also alter the photophysical and chemical properties among complexes based on a given ligand system. However, systematic investigations with a broad range of M–ligand complexes in this context are limited.
Our own efforts in this regard center on the development of photosensitizers based on the macrocyclic biquinazoline ligand, Mabiq. We recently demonstrated that both [CuII(Mabiq)OTf] and [NiII(Mabiq)]OTf (1, Fig. 1) can be photoreduced in the presence of a sacrificial donor to the corresponding neutral [MII(Mabiq˙)]0—formally ‘MI’§—forms.25,26 Likewise, we have now verified the ability to photoreduce the Fe–, Co–, and Zn–Mabiq complexes (vide infra). Furthermore, 1 acts as an effective photoredox catalyst for the cyclization of a bromoalkyl-substituted indole.25 To our knowledge, no other ligand system can support similar photoredox behavior with such a wide range of metal ions, but the excited state mechanisms involved in these processes remain elusive.
Fig. 1. (Top) Chemical structure of 1 and 2. (Bottom) Molecular structure of the newly synthesized 2 (50% ellipsoids; solvent molecules and hydrogen atoms omitted for clarity).

In this work, we have focused our attention on the detailed photochemical and photophysical studies of the Ni complex 1, a confirmed photocatalyst. We aimed to obtain a deeper understanding of the root of its photoactivity by probing the excited state structure and dynamics of this compound, using spectroscopic and computational methods. As a benchmark, we compare the results with the zinc–Mabiq analogue ([ZnII(Mabiq)OTf], 2, Fig. 1). In contrast to the Ni compound, the fully occupied d-orbitals of the redox-inert Zn ion are not expected to contribute significantly to the spectroscopic and redox properties of the complex. Thus, a comparative study of these complexes is expected to yield valuable information about how the overall properties are altered by varied metal–ligand interactions. We present results for both complexes using a combination of steady state and transient optical spectroscopy, time-dependent density functional theory (TDDFT) calculations, and reactivity studies. In addition, we examined the comparative activities of the two compounds in select photocatalytic reactions. The comparative properties and reactivity of 1 and 2—alongside select supporting studies of the Fe– and Co–Mabiq complexes—provide insight into the unique characteristics and potential applications of the metal–Mabiq (M–Mabiq) system.
Results and discussion
Compound 2 was generated according to an established procedure for MII–Mabiq synthesis,27 and its composition was verified by mass spectrometry, 1H NMR spectroscopy and CHN analysis (see ESI and Fig. S1† for details).
In the solid state, complex 2 adopts a square pyramidal geometry (Fig. 1), with the triflate ligand in the apical position (Zn–O = 2.0661(15) Å). The ligand is significantly less buckled than the uncomplexed HMabiq,28 as also observed in the structure of 1.25 The 19F NMR spectra (Fig. S2 and S3†) of 2 show a single resonance in both DCM-d2 (−79 ppm) and MeCN-d3 (−78 ppm), similar to the spectrum of 1 (−79 ppm, MeCN-d3; Fig. S4†). Thus, the triflate molecule of 2 may dissociate in solution. The 1H NMR of the complex in MeCN-d3 with added Et3N (500 equiv., Fig. S6†) shows two species, which indicates that the amine also may coordinate at high concentrations.
Optical properties: steady-state spectra
The absorption spectra of 1 and 2 (Fig. 2, top) are overall similar in structure. Both feature a moderately intense progression of bands in the blue-green spectral region (λmax = 489 nm/2.53 eV and 457 nm/2.71 eV, respectively), followed by more intense transitions in the near-UV. These characteristic absorption features also dominate the visible spectra of CoIII– and CuII–Mabiq complexes,26,29 and are recognizable in the spectra of other Mn–Mabiq compounds.27 The similarity among a fairly large range of coordination compounds suggests that these features predominantly involve ligand-localized (singlet) states. In support of this, we note that for both 1 and 2 (Fig. S8 and S9†) the absorption spectra are essentially solvent-insensitive, implying that at least the initial excitation has negligible charge-transfer (CT) character. Despite the similarities among the absorption spectra, the photo-chemical and -physical behavior of the complexes differ markedly and are strongly dependent on the metal center, as illustrated by our studies with 1 and 2 (vide infra).
Fig. 2. Top: Absorption spectra (1 : 1, THF : MeCN) of 1 (red) and 2 (blue). The emission spectrum of 2 (λmax = 505 nm) is shown by the dotted trace. Bottom: Absorption spectra of the photoreduced forms of 1 (1red; red) and of 2 (2red; blue); 1 : 1, THF : MeCN (see Fig. S13 and S16† for photoreduction data).

Appreciable luminescence is only observed for 2, which has an emission quantum yield approaching 0.7 in DCM. The Stokes shift is modest and the emission spectrum (Fig. 2, top, dotted line) exhibits mirror-image symmetry with the absorption. The emission wavelength is close in energy to the very weak emission observed for the [CuII(Mabiq)OTf] (513 nm),26 as well as of the uncoordinated Mabiq ligand (501 nm; Fig. S7b†), as a further indication of the ligand-centered character of the transitions. In stark contrast to this dye-like optical behaviour, the Ni-containing 1—as well as the other open d-shell Mn–Mabiq compounds that we have examined (vide infra)—is effectively non-emissive (quantum yield < 10−4). This might be expected, as the open-shell configuration of 1 allows for relaxation pathways involving optically dark MC and MLCT/LMCT states.
O2 sensitization
Structurally related Zn pyrrin- and pyrrole-based complexes act as singlet oxygen photosensitizers, which has prompted interest in such compounds for photodynamic therapy.30–32 To probe the potential formation of triplet states in our system, we examined singlet oxygen generation by 1 and 2, using dihydroxynapthalene (DHN) as a 1O2 scavenger (Fig. S28 and S30†). Based on the absorption of the oxidized product, juglone, a reaction yield of 10% was determined using 2 as the photosensitizer. In contrast to 2, 1 does not act as a 1O2 photosensitizer: no changes were observed in the absorption spectrum upon irradiation of an aerated 1/DHN solution (Fig. S30†). This could suggest that intersystem crossing (ISC) is not the primary luminescence quenching route for 1. However, other factors should be taken into consideration: the triplet excited state may be too short-lived to efficiently participate in diffusion-controlled reactions with O2; the 3NiII* energy may be insufficient for the sensitization; or triplet–triplet annihilation may be sterically hindered. Our work (vide infra), as well as recent work on other NiII photocatalysts,33 implies that this lack of sensitization is predominantly due to the low energy of the 3NiII* state.
Photoreduction
Compound 2 can be photoreduced in the presence of Et3N as a sacrificial donor, in accord with the photochemical behaviour of 1 and the Cu–Mabiq complexes (Scheme 1).25,26 Upon irradiation of a solution of 2 (λ = 455 nm), a series of new bands appears in the red spectral region, coincident with a decrease in the higher energy absorption bands of [Zn(Mabiq)OTf] (Fig. 2, S15 and S16†). The product spectrum is identical to that of the chemically generated [ZnII(Mabiq˙)] (2red),27 confirming that 2 undergoes one-electron reduction during the photochemical reaction. The photoreduction to 2red is both fast, reaching completion within seconds (at [2] = 77 μM, Pref = 248 mW, [Et3N] = 38.5 mM, MeCN : THF = 1 : 1; Fig. S15†), and efficient (quantum yield of 0.79 as determined at Pref = 1.4 mW, [Et3N] = 345 μM; Fig. S16 and S17†). The yield is three orders of magnitude larger than the photoreduction yield of 1 (see ref. 25 and Fig. S13 and S14;†Φ = 2.9 × 10−4 at [1] = 67 μM, Pref = 245 mW, [Et3N] = 33.5 mM). However, the photogenerated 2red decays shortly after its formation, as seen by the disappearance of its 550–800 nm features, and the concomitant emergence of new bands below 500 nm. These latter bands also subsequently decrease in intensity. We note that the ability to chemically synthesize and isolate [ZnII(Mabiq˙)]27 indicates that the reduced form of the Zn complex is not inherently unstable, but that the decomposition is light-driven under the photoreduction conditions. The photo-decomposition of 2red is unique in the Mn–Mabiq series, as the corresponding Ni and Cu complexes are stable indefinitely under analogous conditions.25,26 Similarly, the photoreduction of the two open-shell FeII– and CoII–Mabiq complexes, [FeII(Mabiq)(MeCN)2]PF6 and [CoII(Mabiq)(THF)]PF6, also leads to photostable reduced products (Fig. S18 and S21†) with quantum yields comparable to that of 1 (Fe: 1.8 × 10−4; Co: 3.6 × 10−4). The ability of Et3N to coordinate to 2, as evidenced by 1H NMR, may contribute to the rapid photoreduction of this particular complex in the series, as well as the subsequent instability of 2red.
Scheme 1. The series of M–Mabiq complexes (M = Fe, Co, Ni, Cu, Zn) can be photoreduced upon irradiation at λ = 455 nm, in the presence of a sacrificial donor.

In order to assess potential specific interactions between the reducing agent and the M–Mabiq compounds, we additionally examined the photoreduction of 1 by two other sacrificial donors: dimethylaniline (DMA) and N-(2,4,6-trimesitylbenzyl)dicyclohexylamine (N(CH2Mes)Cy2). Structurally, the donors differ significantly from Et3N. The nitrogen atom of N(CH2Mes)Cy2 is hindered by the bulky cyclohexyl and mesityl groups, which might (partially) inhibit collisional quenching and ground-state pre-association. The aniline moiety of DMA also is a poor ligand relative to triethylamine. However, these molecules have comparable oxidation potentials to Et3N (Eox (vs. SCE, MeCN): DMA = ca. 0.7 V;34 N(CH2Mes)Cy2 = 0.78 V (ref. 25)). Therefore, the amines should readily reduce NiII* if photoreduction occurs in an outer-sphere, diffusion-controlled manner.
The NMR spectrum of the Ni complex was unaltered upon addition of >100 equiv. (>0.5 M) of any of the three reductants to solutions of 1 (see ESI and Fig. S5a and b†). Thus, as expected for the square planar d8 complex we find no evidence of ground-state coordination or non-covalent interactions of the reductants to 1. Nevertheless, we observe substantial differences in the photoreduction efficiency. The photoreduction of 1 by N(CH2Mes)Cy2 proceeds with a yield that is nearly identical to that using triethylamine (Φ = 2.1 × 10−4 at [1] = 67 μM, Pref = 245 mW, [(N(CH2Mes)Cy2)] = 33.5 mM; Fig. S23†). However, only low yields of 1red were obtained using DMA, even with 10-fold higher concentrations of the reductant (Φ = 5 × 10−5 at [1] = 51 μM, Pref = 245 mW, [DMA] = 255 mM; Fig. S24†). The oxidized forms of all three donor molecules examined here should have limited chemical stability; a factor which is critical to efficient photoreduction.34 Nonetheless, factors beyond the redox potential clearly have an impact on the photoreduction.
Excited-state relaxation dynamics
The steady state absorption/emission spectroscopy, oxygen sensitization, and photoreduction experiments described above imply that, although the initial excitation of 1 and 2 is to states of similar character, the subsequent relaxation dynamics are substantially different. To elucidate these relaxation processes, time-resolved optical spectroscopies are the method of choice.
Using time-resolved fluorescence spectroscopy, only the excited state dynamics of 2 can be addressed conveniently (Fig. S27†). As 1 is essentially non-emissive, its relaxation dynamics must be followed by ultrafast transient absorption (TA) spectroscopy.35,36 To investigate both 1 and 2 on common footing, and to monitor dynamics beyond the relaxation of the Mabiq-localized excited singlet state, we recorded their fs/ps TA spectra in the 1 : 1 MeCN : THF solvent system relevant to the photocatalytic experiments (Fig. 3; noteworthy delay times and spectral positions are indicated by the horizontal and vertical lines in (a) and (d)). Spectra for both complexes during the subsequent ns/μs time range are shown in Fig. S35 and S38.† All decay constants determined from a comprehensive analysis, along with our assignments, are summarized in Table 1. While the statistical errors (see Table S11†) in the multi-exponential fit model are small, multi-component models are delicate. We therefore supply this uncertainty estimate with an estimate of the upper bound of possible parameter ranges from the peak-widths of a maximum entropy analysis (vide infra). This analysis yields ranges of approximately 50% of the mean value. Notably, even using this highly conservative upper bound the time constants remain well separated and we can safely consider the reported time-constants as meaningful.
Fig. 3. (a) and (d) Transient absorption data of 1 and 2 in degassed 1 : 1 MeCN : THF after excitation at 460 nm (70 fs pulse) and 493 nm (49 fs pulse), respectively. Red-hued colors denote increased absorption due to, e.g., excited absorption or product absorption, blue-hued colors denote decreased absorption due to, e.g., ground state bleach or stimulated emission. (b) and (e) Kinetic traces extracted along the areas marked by the vertical lines. (c) and (f) Transient spectra extracted at selected probe-delays according to the horizontal lines. Note that in (c), the ground state bleach was subtracted for clarity (see text for details), while the cuts in (f) were unaltered. The ESA features marked by the vertical dashed lines in (c) are discussed in detail in the text.

Summary of the dynamics observed for 1 and 2 in the transient spectroscopya.
| Assignment | |
|---|---|
| 1 (Ni II ) | |
| 0.37 ps | Solvation dynamics |
| 1.1 ps | 1NiII* → MC/CT |
| 2.5 ps | MC/CT → MC |
| 14 ps | Spectral narrowing, e.g., vibrational relaxation |
| 81 ps | Combined MC → GS and MC → MC/CT |
| 772 ps | MC/CT → ground state (GS) |
| 2 (Zn II ) | |
| 2 ps | Solvation dynamics |
| 62 ps | Geometric relaxation |
| 1.1 nsb | SE loss/1ZnII* → 3ZnII |
| 580 ns | Relaxation within 3ZnII manifold |
| 1 μs | Decay of ligand-centered 3ZnII state |
| 5 μs | Ligand dissociation (tentative) |
| 140 μs | Ligand re-association (tentative) |
Fit statistics, selected traces and residuals can be found in the ESI, Fig. S66, S67 and Table S11.
Time constant determined from fluorescence decay. The same constant as determined from the fs–ps TA measurements is 770 ps with a large error, since this is close to the full scanning range. For the ns–μs experiment a value of 1.5 ns is obtained, shorter than the 2.7 ns excitation pulse.
A number of significant differences in the spectral evolution of the two complexes are readily apparent—in particular, the dynamics of 1 are faster and more complicated on the fs/ps time scale. We outline these differences in detail in the following sections.
Time-evolution of transient spectra
The time-resolved fluorescence of 2 shows that the initially populated state—a ligand-centered singlet—decays mono-exponentially with a 1.1 ns lifetime (Fig. S27†). While fluorescence provides only limited information about the excited-state behaviour, it suggests that at least the initial photophysics is relatively simple.
The fluorescence properties can be compared with observations from TA. Immediately on excitation, both 1 and 2 feature stimulated emission (SE) to the red of the excitation wavelength—i.e., the longest wavelength absorption band—and a broad featureless excited-state absorption (ESA) band extending into the near-IR. These early time spectra can be assigned to ligand-localized singlet states, referred to as 1NiII* and 1ZnII* respectively from here on (singlet and triplet notations refer to the overall spin state of the complex).
We can follow the dynamics of these initial singlet excited states by the transient kinetics around the excitation wavelength and in the long-wavelength region (magenta and brown vertical cuts in Fig. 3a and d, respectively).
The initial TA kinetics of 2 semi-quantitatively matches the time constant of the fluorescence decay, as the SE decays with a time-constant close to the 1 ns length of the optical delay stage. Beyond this loss of stimulated emission, only subtle changes occur in the spectra during this time period, as illustrated by both vertical (Fig. 3e) and horizontal cuts (Fig. 3f) through the TA data. These changes can be attributed to solvation dynamics and small-scale geometric relaxation.
The corresponding kinetic traces of 1 (Fig. 3b), on the other hand, exhibit much faster and more complicated behaviour, with a concomitant decay of the long-wavelength ESA and SE on a timescale of only ∼1 ps. This rapid decay of the SE signal of 1 explains the negligible luminescence: the population transfers out of the initial, optically bright, 1NiII* state with a rate constant far larger than the radiative relaxation rate.
After the decay of the initial 1ZnII* excited state, the relaxation of 2 continues at longer timescales (see Fig. S38 and S39†). We assign these slower dynamics, as described in Table 1, to processes within a ligand-centered 3ZnII state. This triplet state decays on timescales of 580 ns and 1 μs, followed by formation of lower-amplitude signals that decay on timescales up to 140 μs. A possible explanation of the latter processes is ligand dissociation and finally non-geminate re-association; unlike 1, the Zn complex can coordinate a fifth ligand in the axial position.
In contrast to 2, 1 displays substantial spectral dynamics over much of the sub-ns range—also after the initial ∼1 ps decay phase. The complexity of the spectral time-evolution clearly demonstrates that the 1NiII* state relaxes via multiple distinct intermediates before the ground state is recovered. This complexity contrasts with the essentially two-state relaxation dynamics observed for bipyridyl (bpy) based NiII photocatalysts.33
To shed light on these dynamics, we show the transient spectra of 1 at selected delay times in Fig. 3c. As the spectra demonstrate significant time-dependent changes in the excited states, we have subtracted the ground state bleach contribution using a published weighting procedure37,38 to clearly illustrate these changes. As such, these spectra directly show the time-evolution of the excited-state absorption spectra, distorted only at early times by the stimulated emission contribution.
The excited-state spectrum we observe at the earliest times (Fig. 3c, red trace, 0.2 ps) is, as noted above, typical for locally excited organic molecules—with a largely featureless photo-induced absorption extending into the near-infrared (NIR). Over the next ten ps, well-defined spectral structures emerge. Two features of particular interest are highlighted with vertical dashed lines in Fig. 3c: First, a relatively narrow ESA feature appears at approximately 465 nm concomitant with the decay of the ligand-localized 1NiII* state. This is followed by the emergence of a feature at 490 nm. Accordingly, kinetic traces from the main ESA bands at 487 and 364 nm, which capture both of these contributions (Fig. 3b, violet and green traces), are relatively complex; an indication of the formation and decay of several distinct spectral species. Such dynamics—fast decay of SE and broad ESA features, followed by well-defined ESA bands slightly red-shifted relative to the absorption spectrum—are often seen in association with energy-transfer from optically bright ligand-localized states to dark metal-centered states in open d-shell transition metal complexes (exemplified, e.g., by the photoinduced dynamics of nickel porphyrin).39,40 The time-evolution of the spectra here shows, however, that this transition from ligand-localized to metal-centered states involves at least one short-lived intermediate state.
Global kinetic analysis
A more complete picture of the spectral evolution of 1, and thus the overall relaxation dynamics, requires global analysis of the whole dataset. In order to ensure maximal reliability of the extracted model, we performed two independent analyses: a model independent maximum entropy analysis (Fig. S34†),41,42 as well as singular-value decomposition followed by a global fit to a sum-of-exponentials model.43 We find good agreement of timescales from both approaches, implying good consistency of the extracted physical model. In agreement with the complex dynamics suggested by the single-wavelength kinetic traces, we find that six components are necessary to describe the observed spectra within an exponential decay model.
The wavelength-dependence of this multi-component fit is best represented by the spectra associated with the decay components. The two most common representations of these “global” spectra are the Decay Associated Spectra (DAS) and the Evolutionary Associated difference Spectra (EAS), where the former is simply a plot of the amplitude of each decay component as a function of wavelength, and the model can be interpreted as representing the parallel decay of six independent species. In the EAS model, we rather assume a strictly sequential organization of compartments—or “states” (not necessarily electronic eigenstates of the system)—and the time-constants refer to the (unidirectional) population transfer between these. Hence, the EAS can be considered the spectra of these individual “states”. In Fig. 4 we plot the DAS and EAS corresponding to the decay-component of 1. The corresponding global component spectra for 2 can be found in Fig. S41.† The effective picture emerging from the analysis of these global spectra is summarized in Table 1 and depicted in Fig. 6.
Fig. 4. Decay associated- (DAS, left column) and evolutionary associated- (EAS, right column) decay spectra extracted from a global fit of the relaxation dynamics of 1. DAS amplitudes are shown on absolute scale, while the EAS are normalized to their maximum for easier comparison of spectral shapes. The component spectra are organized to highlight similarities in EAS component spectra. The time-constants are component lifetimes extracted from the relaxation dynamics of 1. SE contributions to the spectra are found only around the 460 nm excitation wavelength at the earliest two times.

Fig. 6. Effective relaxation model of the compounds 1 (a) and 2 (b) as extracted from the time-resolved absorption data. Components assigned to solvation and vibrational cooling omitted for clarity. Metal-centered states are depicted in red; ligand-centered states are indicated in black.

At early times, the dynamics are dominated by components with characteristic times of 370 fs and 1.1 ps (Fig. 4, top). The faster component can be assigned to solvation, as it occurs with the average solvation times of the two pure solvents.44 The 1.1 ps spectra on the other hand prominently contain the decay of the SE contribution as well as the appearance of a characteristic ESA feature close to the excitation wavelength. Thus, these spectral dynamics reveal the transfer of the initially excited ligand-localized 1NiII* state to states that are optically dark from the ground state.
This energy-transfer step is accompanied by the (quasi-) ground state recovery of the Mabiq ligand: i.e., after this initial relaxation phase, the excited population resides almost completely on the metal center, while the ligand is in a ground state distorted by the excited metal center. Consequently, we can assign the spectra of the intermediate time components (14 and 81 ps lifetimes. Fig. 4, central panels) to transitions of ligand-localized π–π* character. The 14 ps component is primarily associated with spectral narrowing—as demonstrated by the second-derivative profile of the DAS and slightly broadened EAS spectral profile—which suggests vibrational cooling45 or heat dissipation.46
Interestingly, the fast appearance and subsequent decay of a 465 nm feature highlighted with a vertical dashed line in Fig. 3c is clearly mirrored in the global analysis: immediately after the 1NiII* decay, the transient spectrum in Fig. 3 takes on a shape similar to—although strongly distorted—the 81 ps EAS. The global analysis captures this behaviour as a corresponding EAS with 1.1 ps growth and 2.5 ps decay (Fig. 4, bottom panel). The presence of a short-lived intermediate in the ligand → MC transfer demonstrated by this component is by itself interesting; in addition, we find that a state with an essentially identical spectrum re-emerges at long times as the MC states decay. This state subsequently decays on a much longer timescale, with a time constant of approximately 770 ps.
While short-lived intermediates as observed here can be assigned to, e.g., vibrational cooling, we stress that the re-emergence of long-lived states with essentially identical spectral characteristics—as shown by the strong resemblance between the 2.5 ps and 770 ps EAS—makes such an assignment here highly implausible.
Earlier studies on macrocyclic nickel systems instead point to contributions from charge-transfer (CT) states in the electronic structure—both indirectly through the relaxation rate dependency on solvent polarity,39 and through spectroscopic signatures in ultrafast XANES experiments.47 While a complete one-electron ligand-to-metal charge transfer in 1 is unlikely, as one would then expect to observe the Mabiq cation spectrum, we find admixture of states with CT character to the MC states to be plausible in the relatively polar solvent environment in these experiments. This contrasts with photocatalytically active NiII–bpy complexes, where the long-lived lowest energy states appear to be of purely d–d character.33,48
Comparing the profiles of the DAS spectra, we find that both the 80 ps component assigned to population of MC states and the 772 ps component clearly contain the GSB signature in the 400–460 nm range. In other words, the ground state of the complex recovers bi-exponentially, implying a branching in the relaxation pathway: only a fraction of the initial excited state population reaches the long-lived states with admixture of CT states, while the remainder presumably relaxes directly from the fully metal-centered states.
We can compare the observed excited-state dynamics with those inferred from the photoreduction of 1. The dependence of the reaction quantum yield on TEA concentration was determined in our previous study.25 Based on analysis of the results using a standard diffusion model49 and a diffusion-limited quenching rate typical of molecules comparable to our system (∼1010 M−1 s−1),50 the lifetime of the reactive excited state is predicted to be between 580 and 900 ps. This is in excellent agreement with the measured lifetime of 770 ps, which indicates that the outer-sphere electron transfer (ET) process with TEA can occur from this mixed MC/CT state.
Electronic structure
To correlate the spectroscopic signals with the electronic states involved in the optical response of the complexes, we employed electronic structure calculations at the linear response time-dependent density functional theory (DFT) level. The TDDFT-derived energy level diagrams for 1 and 2 are shown in Fig. 5a (see also Fig. S42 and S43†). To highlight the electronic structure of the states—i.e., the parts of the complexes involved in the photo-excitation and relaxation—we computed the electron density differences between the ground and excited states (the density differences; Fig. 5b, S44–S47†), the spin density distributions of the lowest energy triplet states (Fig. 5c), and performed a Mulliken population analysis (Table S5†).
Fig. 5. (a) Vertical excitation energies (VEEs) derived at the TDDFT/B3LYP level for 1 and 2 at the optimized singlet (S0) and lowest energy triplet (T1) geometries. Optically allowed states are shown with solid lines, optically dark states are denoted by dashed lines; MC states are shown in red, and ligand-centered states in black. Note that the lowest energy optically allowed state in 1 is the ligand-centered S4 state, whereas in 2 the lowest optically active singlet is the ligand-centered S1 state. (b) Total electron density difference upon photo-excitation into the lowest energy optically bright state for 1 (S4, top) and 2 (S1, bottom), with positive and negative differences between the excited and ground states marked in blue and red, respectively. The charge migration takes place almost exclusively on the ligand for both species. (c) Spin density (α–β) for the lowest energy triplet state of 1 (top) and 2 (bottom), with positive and negative differences between alpha and beta spin densities marked in red and blue, respectively. Note that the excess spin is almost entirely metal localized for 1, while it is distributed on the ligand for 2.

Our interpretation that the absorption consists of π–π* singlet transitions between Mabiq localized states is supported by the TDDFT calculations. This is also consistent with the observation that the optically bright transitions for 1 and 2 are of similar character in both complexes, as well as in other transition metal–Mabiq complexes.27–29 The calculated excitation energies of 1 and 2 are of similar magnitude (Fig. 5a; 1: 2.85 eV S0 → S4; 2: 2.77 eV S0 → S1), and the density differences associated with these transitions (Fig. 5b; corresponding natural transition orbitals (NTO) in Fig. S48†) demonstrate ligand-localized motion of charge. Similar vertical excitation energies are also obtained at the second-order correlated (ADC(2)/TZVP) level (Table S4†).
The DFT results also suggest an explanation for the experimentally observed differences in the relaxation dynamics of the two complexes. The spin densities of the 3ZnII triplet excited states (Fig. 5c) show that the excitation remains on the ligand, regardless of spin state, upon excited-state relaxation of 2. The minimal contribution of MC states to the electronic structure and dynamics again suggests that 2 can be considered as an organic chromophore (i.e., Mabiq) confined to a limited conformational range by the coordinating Zn2+ ion. The ∼1 ns intersystem crossing to the Mabiq triplet state is reasonable for an organic chromophore with an associated metal.
The structure and relaxation effects of 1 contrast with this simple behaviour. The 1NiII*–3NiII* energy gap is substantially larger than the corresponding singlet-triplet energy difference of 2. While the electronic structure of the 1ZnII* and 3ZnII states are both predominantly ligand-localized, the corresponding 1NiII* and 3NiII states are clearly different (Fig. 5b and c), with a 3NiII state that is essentially metal-centered. Thus, the computational data suggest that intersystem crossing in 1 involves energy transfer from the Mabiq ligand to the metal center, in agreement with our experimental observations. A large number of additional states are located between the ground and the optically bright 1NiII* state, many of which are also metal-centered, of both singlet and triplet character. The large number of states facilitates the observed rapid ligand-to-metal energy transfer. A similar electronic picture is also obtained at the TDDFT/CAM-B3LYP level (Fig. S43†), suggesting that the results are robust and do not arise from, e.g., DFT charge transfer problems. The calculations also suggest that spin–orbit coupling (SOC) effects are small for the Zn–Mabiq complex, but could contribute into transition between singlet and triplet states for Ni–Mabiq (Fig. S49†).
The implicit assumption that intersystem crossing is an efficient quenching channel for the 1NiII* state is well-founded, given the typical behaviour of open-shell transition metals.21 The optical spectra themselves only reveal that the final state in the relaxation cascade is (i) optically dark from the ground state (at least in the UV/Vis spectral region), and (ii) has a prominent ESA band slightly redshifted from, but close in energy to, the (1NiII → 1NiII*) ground state absorption band. In this regard, the calculations support our expectation of relaxation via metal centered states. All MC transitions with energies in the range of the TA measurements, either from the singlet ground state or from the triplet state, are dark; any optically active transitions primarily involve the Mabiq ligand.
With the assignment of triplet character to the low-lying energy levels of 1 and 2, we can reconcile and at least semi-quantitatively explain the photoreduction quantum yields and the time constants from the TA measurements. The quantum yield, QY, of the photoinduced reduction of the M–Mabiq catalysts by Et3N is given by: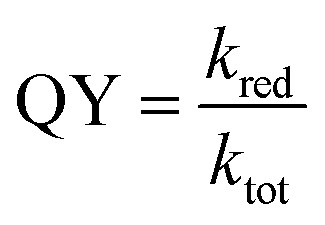 with the effective quenching rate, kred, and the total relaxation rate, ktot, as determined from the TA measurements. With a QY of 2.9 × 10−4 for Ni–Mabiq and ktot = 1/772 ps, we obtain a surprisingly long quenching time of t = 1/kred = 2.7 μs. The low QY is therefore the consequence of a low electron transfer rate and the rapid decay of the excited 1. Using the same value of kred as a first guess for 2, we predict a total decay time of 10 μs, which is well within the range of the slow time constants obtained from the TA data for the Zn complex. In summary, the reduction is rather slow, and the QY is limited by the lifetime of the triplet states.
with the effective quenching rate, kred, and the total relaxation rate, ktot, as determined from the TA measurements. With a QY of 2.9 × 10−4 for Ni–Mabiq and ktot = 1/772 ps, we obtain a surprisingly long quenching time of t = 1/kred = 2.7 μs. The low QY is therefore the consequence of a low electron transfer rate and the rapid decay of the excited 1. Using the same value of kred as a first guess for 2, we predict a total decay time of 10 μs, which is well within the range of the slow time constants obtained from the TA data for the Zn complex. In summary, the reduction is rather slow, and the QY is limited by the lifetime of the triplet states.
Catalysis
Given the distinctive photophysics, differing behaviour with respect to photoreduction, and activity toward O2, we examined the comparative ability of 1 and 2 to act as a photoredox catalyst in a series of photo-driven reactions. The first of these is the previously reported cyclization of an N-(ω-bromoalkyl)-substituted indole (dimethyl 2-(3-(1H-indol-1-yl)propyl)-2-bromomalonate) by 1 (Table 2, top scheme; mechanism in Scheme S1†), which gave the product in high yield.25 In this case, irradiation of the reaction mixture facilitates reduction of 1 by Et3N, and the resultant 1red triggers the ring closing in a diffusive manner. A catalytic cycle for the formation of the cyclized indole should also be feasible using 2, based on its redox properties alone. However, low product yields were obtained and degradation of the metal complex occurred, as evidenced by bleaching of the solution throughout the reaction.51 Conversely, while the photoreduction quantum yield is relatively low, 1 is an effective photoredox catalyst, as it appears resistant to degradation and remains active throughout the extended reaction period.25 In related preliminary work, we observed that the FeII–, CoII– and CuII–Mabiq complexes all exhibit varying reaction yields and selectivities,51 further demonstrating that the choice of metal center clearly influences the activity.
Photoredox-catalysed cyclization of an N-(ω-bromoalkyl)-substituted indole (top), aza-Henry reaction (middle) and oxidative coupling of 4-methoxybenzylamine (bottom).
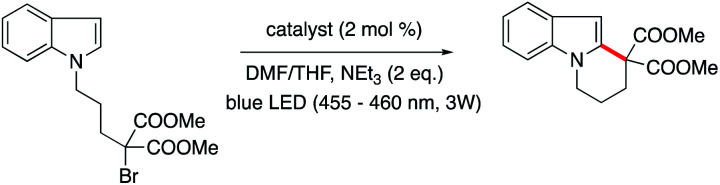
| ||
|---|---|---|
| Entry | Catalyst | % yielda |
| 1 | 1 | 86 ± 5 (13 h) |
| 2 | 2 | 35 (13 h) |
Yields are mean ± standard deviation of measurements performed at least in duplicate. Reactions were run on a 0.05 mmol scale. Product yields were determined by 1H NMR using mesitylene as an internal standard.
For the oxidative coupling of benzylamine, ESI-MS was further used to confirm the presence of the desired product ([M + H]+m/z = 256.09).
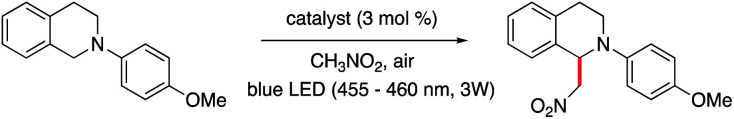
| ||
|---|---|---|
| Entry | Catalyst | % yieldb (time) |
| 1 | 1 | 82 ± 7 (5 h) |
| 2 | 1/without irradiation | — |
| 3 | None | 14 ± 6 (5 h) |
| 4 | 2 | 36 ± 13 (30 min) |
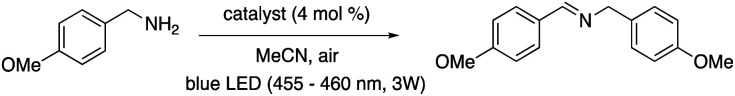
| ||
|---|---|---|
| Entry | Catalyst | % yieldc (time) |
| 1 | 1 | 27 ± 0 (17 h) |
| 2 | 1/without irradiation | — (12 h) |
| 3 | None | — (12 h) |
| 4 | 2 | 73 ± 12 (30 min) |
We next examined the photocatalytic ability of 1 and 2 in two reactions that do not rely on initial reduction of the catalyst by a sacrificial donor: the oxidative C–H functionalization of N-phenyl-tetrahydroisoquinoline (THIQ), and the oxidative coupling of 4-methoxybenzylamine (Table 2; see ESI† for details).
The THIQ functionalization represents a photoredox catalysed aza-Henry reaction (Table 2, middle scheme; mechanism in Scheme S2†).52 In the initial step, reductive quenching of the excited state of the catalyst by the isoquinoline leads to formation of a cationic amine radical. As discussed above, this step is expected to proceed on the low μs timescale. The solvent nitromethane (CH3NO2) can serve to regenerate the catalyst, and facilitates iminium ion formation. The resultant reduced oxidant and amine cation radical will likely remain in close proximity due to the lack of time for diffusion; alternatively the reduced oxidant readily combines with CH3NO2via a hopping process. Either way, catalyst reoxidation and iminium ion formation via a quasi-geminate step should be fast and efficient.
The final product of the aza-Henry reaction is generated upon addition of the nitromethane-derived CH2NO2 to the iminium. Thus, nitromethane also acts as the co-substrate. The aza-Henry reaction exploits the direct reaction of MII* with the substrate, unlike the previously examined indole cyclization, which reflected the interaction of a substrate with the reduced ‘MI’–Mabiq forms. The reaction also allowed us to probe the ability to regenerate the reduced catalyst forms in the presence of an oxidant, which would allow application of the Mabiq complexes in a broader range of chemistry.
The product yields obtained in the photoredox catalysed aza-Henry reaction by 1 and 2, using nitromethane as the solvent, were 82% (1, after 5 h) and 36% (2, after 30 min). With the illumination power and total time, we can estimate a lower limit of the overall reaction quantum yield of 1.5 × 10−3. The yields obtained for the Ni-complex are comparable to those reported using [Ru(bpy)2]2+ and [Ir(ppy)2(dtbbpy)]+.52 Thus, 1 shows promise as an effective catalyst for such C–H functionalization reactions. The lower yields of 2 in the reaction are again due to degradation of the Zn-complex. A significant decrease in the absorption bands of 2 was already observed within the first 30 minutes reaction time (Fig. S55†). This contrasts with the behaviour of 1, which remained intact throughout the reaction, as evidenced by both absorption and NMR spectroscopies (Fig. S56 and S57†). We note that irradiation of a solution of 1 and the isoquinoline in the absence of a suitable oxidant also leads to rapid decomposition of the Ni-complex (Fig. S58 and S59†), which we speculate is due to attack of 1red by the isoquinoline radical.
The final reaction that was examined, the oxidative coupling of 4-methoxybenzylamine (Table 2, bottom scheme), depends on the participation of O2 in the catalytic cycle (Scheme S3†). Both energy transfer and photoredox mechanisms have been proposed for this reaction—in the latter case, O2 would serve as an oxidant and to generate an imine intermediate.53 The Ni–Mabiq complex was able to catalyse the coupling reaction, albeit with only 27% product yield even after 17 h. No significant amount of side product was detected. Nevertheless, 1 remained stable under these conditions (Fig. S60†). Using 2 as the photocatalyst, a larger amount of N-(4-methoxybenzyl)-1-(4-methoxyphenyl)methanimine was obtained (73% yield, 30 min). However, complete degradation of the Zn compound was again observed after 30 min (Fig. S61†).
To supplement the basic reactivity studies, we examined the ability of 4-methoxybenzylamine to photoreduce the Mabiq compounds—the first step in the photoredox pathway. Both 1 and 2 can be photoreduced using 4-methoxybenzylamine under inert atmosphere, indicating that an electron transfer pathway is feasible (Fig. S62 and S63†). However, after the initial formation of some 2red by the amine, the spectrum of the Zn-complex is further altered. Evaluation of the photoreduction data shows that the reduction of NiII* by methoxybenzylamine is photo-reversible, with comparable quantum yield values for the forward and back electron transfer processes (ca. 1.7 × 10−5; Fig. S62†). The low yield is consistent with the low coupling product yields obtained with 1, even after 17 h. From the illumination conditions of 4-methoxybenzylamine with 1 we can estimate a lower limit of the overall reaction quantum yield of 4.3 × 10−4. In comparison, for the quenching QY—taking into account the concentration of methoxybenzylamine under the reaction conditions (25 mM), and the linear dependence of the process on the quencher concentration25—we obtain a value of 8.5 × 10−4. Within the uncertainty of this estimate the two yields are equal, showing that indeed all steps after the first ET occur with near unity.
The quenching of the photo-excited 2 is associated with QYs of 0.12 to the reduced form, and 0.20 to an unidentified side product, both of which rapidly convert further to some final species with an optical spectrum that is distinct from that of 2 or 2red (Fig. S63†). Clearly any accumulation of 2red also leads to degradation of the original form of the Zn catalyst. A slight shift in both the 1H NMR resonances and absorption spectrum of 2 in the presence of the substrate is observed (Fig. S64 and S65†), which suggests that the ability of the amine to coordinate to the Zn may contribute to the activity and degradation of 2. However, overall, the high reduction yields together with the ability of O2 to reoxidize any accumulated 2red enables significant product formation before the catalyst is fully consumed.
While the above reactions are quite distinct, the photoredox catalytic activity of the M–Mabiq complexes in all of the reactions hinges on the ability of the excited states to engage in single electron transfer with a sacrificial donor or the substrate. Our model for the excited state relaxation pathways for 1 and 2, based on spectroscopic characteristics and the calculated electronic structure, is summarized in Fig. 6. ET involving the singlet states is precluded by the short lifetimes—the close vicinity of the resulting ion pair also would likely lead to a rapid back-ET and therefore be unproductive. Single electron transfer between the amine substrates and M–Mabiq will occur via the triplet states, where back ET is consequently blocked by the Pauli principle.54 In the Zn-containing 2, all relevant excited states, including the triplet states, are ligand-localized. As a result, photoreduction results in formation of what is essentially a Mabiq radical anion coordinated by a ZnII; decomposition proceeds as is typical for organic radical ions. The longer excited state lifetime of 2 should favor catalytic activity, but the instability of 2 hinders its use as a photoredox catalyst. Oxidants, as used in the light-driven aza-Henry and oxidative coupling reactions, regenerate 2 to some degree. However, even in the presence of excess oxidant the degradation of 2red appears to outcompete its reoxidation. In contrast, the initial ligand-centered excited state of 1 relaxes rapidly towards states of metal-centered and partial CT character. The involvement of metal-centered states in 1red appears to make this complex more robust, further highlighting the synergy between the metal and redox-active ligand.
Our preliminary studies with alternate electron donors and select substrates demonstrate, however, that the nature of the donor is crucial. Electron transfer between 1 and either DMA or 4-methoxybenzylamine was sluggish; in the latter case this significantly limited the catalysis. Moreover, the isoquinoline radical can destroy the complex if subsequent transformations of the intermediate are not fast enough (Fig. S58 and S59†). More detailed studies will be required to determine the specific donor/M–Mabiq interactions and their importance in the mechanism of excited state electron transfer. Finally, the generally greater stability of 1red allows this species to be more effectively regenerated by oxidants, allowing 1 to engage in various stages of a photoredox cycle.
The 3ZnII* state of the Zn complex may be more suited for select energy transfer reactions, as for singlet O2 sensitization. With respect to singlet-oxygen sensitization, the (1NiII–3NiII) energy gap is much smaller than the (3O2–1O2) energy gap, and consequentially the process is energetically disfavored. Thus, 1 may be less effective for energy transfer processes.
Overview of photodynamics
It is worth comparing the photophysics of our Mabiq complexes to that of M-bpy based compounds. The bpy ligand is essential to the functionality of [Ru(bpy)3]2+, whose underlying photophysical properties have been studied in great depth.17 Excitation of [Ru(bpy)3]2+ in the visible region leads to population of long-lived 3MLCT excited states, which subsequently engage in single electron transfer. The sub-ps excited state lifetimes of the 3MLCT states of [Fe(bpy)3]2+ on the other hand can be attributed to relaxation to MC states, which in contrast to those of the Ru complex lie below the MLCT states.55 This altered ordering reflects the general problem of first row transition metal complexes, where low lying d–d states lead to short excited state lifetimes and limit the photoredox capacity of the compounds. A notable exception is the behavior of the other class of NiII photoredox catalysts (tBubpy)NiII(Ar)(X); X = halide or OR−, also based on the bpy ligand.33,48,56 The low-lying 3d–d states are not only long lived, but allow this complex to undergo photoinduced Ni–aryl bond homolysis.
The Mabiq ligand likewise contains a bipyridine-like moiety—as well as a second redox-active functionality in the diketiminate. The presence of multiple redox-active units in the macrocyclic M–Mabiq systems results in photophysics that are more complex than the bipyridyl systems, with ligand-centered, CT and MC character all involved (Fig. 6). The initially accessed ligand-centered π–π* state of 2 can also engage in single electron transfer, whereas the CT and MC states become more relevant for 1. Our findings suggest that these latter states lie closer in energy than what is observed in bpy-based compounds. Interactions between CT, MC, and π–π* states account for the complex photodynamics of 1, and further underscores the synergy between the redox-active ligand and the metal centers. The nature of the bound metal center may alter the relative energies of these CT and MC states. For the Fe, Co and Cu complexes, which possess additional axial ligands, the d–d states might also enable M–L bond homolysis. Overall, the unique features of the Mabiq ligand allow for rich and varied photochemistry.
Conclusions
The nature of the coordinated metal ion clearly has a significant influence on the photophysical properties and reactivity of the Mabiq complexes, as exemplified by the Ni and Zn complexes 1 and 2. Our findings unravel contributions of the various ligand-centered, charge transfer and MC states, and provide initial insight into how the interactions between the metal and redox-active ligands influence activity and stability of photoredox catalysts. With the exception of the Zn complex, the chemical stability of the M–Mabiq series is a notable feature of this class of photocatalysts. Light-driven access to the reduced, stable forms of MII–Mabiq complexes containing Fe → Cu may also permit more diverse organic transformations.
In our system, the Mabiq system scaffold provides a highly unique degree of freedom in a second metal binding site at the complex periphery. Incorporation of a second metal ion may enable alterations to the electronic structure of the core metal complex, while circumventing the need to modify the ligand backbone. As such, the second coordination site may have an influence on stability and electron transfer efficiencies. The photochemical properties of a series of bimetallic Mabiq compounds can be systematically studied. The second coordination site also opens up entirely new possibilities for bifunctional photoredox catalysis. Overall, we believe the properties of the Mabiq system will allow these complexes to become valuable additions to the growing repertoire of earth-abundant photoredox catalysts.
Author contributions
RL carried out synthesis and characterization of compounds, and performed photoreduction experiments. RL & HD performed data analysis for photoreduction experiments. OZE performed photocatalytic addition- and coupling-reactions. RL, HD, and MB performed the time-resolved optical experiments. SLM and VRIK designed and performed the DFT calculations. RL, SLM, HD and OZE made comparable and substantial contributions to experiments and analysis. AJR carried out additional DFT calculations. CJ was responsible for X-ray crystallography. ER, ET, JH, and CRH planned, designed, and analyzed the experiments. CRH and ET conceived the project and wrote the paper with contributions from all authors.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful to Prof. Jonathan Finley and Lukas Hanschke for the use of instrumentation, and assistance with sample measurements for fluorescence lifetime data. RL thanks the TUM International Graduate School of Science and Engineering (IGSSE) for financial support. We are grateful for financial support from the DFG (Deutsche Forschungsgemeinschaft, German Research Foundation) through the excellence cluster e-conversion, under Germany's Excellence Strategy – EXC 2089/1 – 390776260, as well as funding from DFG grant 426785626.
Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic and TDDFT data, crystallographic tables and CIF file. CCDC 1949717. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06096h
Footnotes
In our previous studies, we have shown that all of the previously isolated, formal MI–Mabiq complexes are best described by [MII(Mabiq˙)], containing the reduced Mabiq radical, rather than a monovalent metal center.
Notes and references
- Prier C. K. Rankic D. A. MacMillan D. W. C. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. H. Twilton J. MacMillan D. W. C. J. Org. Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D. M. Yoon T. P. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo L. Pagire S. K. Reiser O. König B. Angew. Chem., Int. Ed. 2018;57:10034–10072. doi: 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Stephenson C. R. J., MacMillan D. W. C. and Yoon T. P., Visible light photocatalysis in organic chemistry, Wiley-VCH Verlag GmbH & Co., Weinheim, Germany, 2018 [Google Scholar]
- Narayanam J. M. R. Stephenson C. R. J. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- Xi Y. Yi H. Lei A. Org. Biomol. Chem. 2013;11:2387–2403. doi: 10.1039/c3ob40137e. [DOI] [PubMed] [Google Scholar]
- Chemical Photocatalysis, ed. B. König, De Gruyter, Berlin, 2013 [Google Scholar]
- Hammarstrom L. Acc. Chem. Res. 2015;48:840–850. doi: 10.1021/ar500386x. [DOI] [PubMed] [Google Scholar]
- Ashford D. L. Gish M. K. Vannucci A. K. Brennaman M. K. Templeton J. L. Papanikolas J. M. Meyer T. J. Chem. Rev. 2015;115:13006–13049. doi: 10.1021/acs.chemrev.5b00229. [DOI] [PubMed] [Google Scholar]
- Berardi S. Drouet S. Francas L. Gimbert-Surinach C. Guttentag M. Richmond C. Stoll T. Llobet A. Chem. Soc. Rev. 2014;43:7501–7519. doi: 10.1039/c3cs60405e. [DOI] [PubMed] [Google Scholar]
- Archer S. Weinstein J. A. Coord. Chem. Rev. 2012;256:2530–2561. [Google Scholar]
- Brennaman M. K. Dillon R. J. Alibabaei L. Gish M. K. Dares C. J. Ashford D. L. House R. L. Meyer G. J. Papanikolas J. M. Meyer T. J. J. Am. Chem. Soc. 2016;138:13085–13102. doi: 10.1021/jacs.6b06466. [DOI] [PubMed] [Google Scholar]
- Terrett J. A. Cuthbertson J. D. Shurtleff V. W. MacMillan D. W. C. Nature. 2015;524:330–334. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J. Le C. Zhang P. Shaw M. H. Evans R. W. MacMillan D. W. C. Nat. Rev. Chem. 2017;1:0052. [Google Scholar]
- Skubi K. L. Blum T. R. Yoon T. P. Chem. Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Rotondo D. M. McCusker J. K. Chem. Soc. Rev. 2016;45:5803–5820. doi: 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]
- Tucker J. W. Stephenson C. R. J. J. Org. Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- Koike T. Akita M. Inorg. Chem. Front. 2014;1:562–576. [Google Scholar]
- Carey M. C. Adelman S. L. McCusker J. K. Chem. Sci. 2019;10:134–144. doi: 10.1039/c8sc04025g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen C. B. Wenger O. S. Chem.–Eur. J. 2018;24:2039–2058. doi: 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- Wenger O. S. J. Am. Chem. Soc. 2018;140:13522–13533. doi: 10.1021/jacs.8b08822. [DOI] [PubMed] [Google Scholar]
- Hossain A. Bhattacharyya A. Reiser O. Science. 2019;364:eaav9713. doi: 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]
- Traub L. Reiser O. Phys. Sci. Rev. 2019;4:20170172. [Google Scholar]
- Grübel M. Bosque I. Altmann P. J. Bach T. Hess C. R. Chem. Sci. 2018;9:3313–3317. doi: 10.1039/c7sc05320g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark H. S. Altmann P. J. Sproules S. Hess C. R. Inorg. Chem. 2018;57:6401–6409. doi: 10.1021/acs.inorgchem.8b00471. [DOI] [PubMed] [Google Scholar]
- Banerjee P. Company A. Weyhermuller T. Bill E. Hess C. R. Inorg. Chem. 2009;48:2944–2955. doi: 10.1021/ic8020172. [DOI] [PubMed] [Google Scholar]
- Müller E. Bernardinelli G. von Zelewsky A. Inorg. Chem. 1988;27:4645–4651. [Google Scholar]
- Puttock E. V. Banerjee P. Kaspar M. Drennen L. Yufit D. S. Bill E. Sproules S. Hess C. R. Inorg. Chem. 2015;54:5864–5873. doi: 10.1021/acs.inorgchem.5b00636. [DOI] [PubMed] [Google Scholar]
- Moreira L. M. Santos F. V. d. Lyon J. P. Maftoum-Costa M. Pacheco-Soares C. Silva N. S. d. Aust. J. Chem. 2008;61:741–754. [Google Scholar]
- Linden G. Zhang L. Pieck F. Linne U. Kosenkov D. Tonner R. Vazquez O. Angew. Chem., Int. Ed. 2019;58:12868–12873. doi: 10.1002/anie.201907093. [DOI] [PubMed] [Google Scholar]
- Antoni P. M. Naik A. Albert I. Rubbiani R. Gupta S. Ruiz-Sanchez P. Munikorn P. Mateos J. M. Luginbuehl V. Thamyongkit P. Ziegler U. Gasser G. Jeschke G. Spingler B. Chem.–Eur. J. 2015;21:1179–1183. doi: 10.1002/chem.201405470. [DOI] [PubMed] [Google Scholar]
- Ting S. I. Garakyaraghi S. Taliaferro C. M. Shields B. J. Scholes G. D. Castellano F. N. Doyle A. G. J. Am. Chem. Soc. 2020;142:5800–5810. doi: 10.1021/jacs.0c00781. [DOI] [PubMed] [Google Scholar]
- Saeedi S. White T. A. ACS Appl. Energy Mater. 2020;3:56–65. [Google Scholar]
- Megerle U. Pugliesi I. Schriever C. Sailer C. F. Riedle E. Appl. Phys. B. 2009;96:215–231. [Google Scholar]
- Riedle E. Bradler M. Wenninger M. Sailer C. F. Pugliesi I. Faraday Discuss. 2013;163:139–158. doi: 10.1039/c3fd00010a. [DOI] [PubMed] [Google Scholar]
- Wilcken R. Schildhauer M. Rott F. Huber L. A. Guentner M. Thumser S. Hoffmann K. Oesterling S. de Vivie-Riedle R. Riedle E. Dube H. J. Am. Chem. Soc. 2018;140:5311–5318. doi: 10.1021/jacs.8b02349. [DOI] [PubMed] [Google Scholar]
- Kovalenko S. A. Schanz R. Hennig H. Ernsting N. P. J. Chem. Phys. 2001;115:3256–3273. [Google Scholar]
- Zamyatin A. V. Soldatova A. V. Rodgers M. A. J. Inorg. Chim. Acta. 2007;360:857–868. [Google Scholar]
- Retsek J. L. Drain C. M. Kirmaier C. Nurco D. J. Medforth C. J. Smith K. M. Sazanovich I. V. Chirvony V. S. Fajer J. Holten D. J. Am. Chem. Soc. 2003;125:9787–9800. doi: 10.1021/ja020611m. [DOI] [PubMed] [Google Scholar]
- Kutta R.-J. Langenbacher T. Kensy U. Dick B. Appl. Phys. B. 2013;111:203–216. [Google Scholar]
- Kölle P. Pugliesi I. Langhals H. Wilcken R. Esterbauer A. J. de Vivie-Riedle R. Riedle E. Phys. Chem. Chem. Phys. 2015;17:25061–25072. doi: 10.1039/c5cp02981c. [DOI] [PubMed] [Google Scholar]
- van Stokkum I. H. M. Larsen D. S. van Grondelle R. Biochim. Biophys. Acta, Bioenerg. 2004;1657:82–104. doi: 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Horng M. L. Gardecki J. A. Papazyan A. Maroncelli M. J. Phys. Chem. 1995;99:17311–17337. [Google Scholar]
- Balevicius Jr V. Wei T. Di Tommaso D. Abramavicius D. Hauer J. Polivka T. Duffy C. D. P. Chem. Sci. 2019;10:4792–4804. doi: 10.1039/c9sc00410f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani Y. Uesugi Y. Kitagawa T. J. Chem. Phys. 1999;111:8950–8962. [Google Scholar]
- Shelby M. L. Lestrange P. J. Jackson N. E. Haldrup K. Mara M. W. Stickrath A. B. Zhu D. Lemke H. T. Chollet M. Hoffman B. M. Li X. Chen L. X. J. Am. Chem. Soc. 2016;138:8752–8764. doi: 10.1021/jacs.6b02176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welin E. R. Le C. Arias-Rotondo D. M. McCusker J. K. MacMillan D. W. C. Science. 2017;355:380–385. doi: 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y. Nishimura Y. Arai T. Sakuragi H. Tokumaru K. Matsunaga D. J. Photochem. Photobiol., A. 1995;89:37–44. [Google Scholar]
- Montalti M., Credi A., Prodi L. and Gandolfi M. T., Handbook of Photochemistry, Taylor & Francis Group, LLC, Boca Raton, Florida USA, 3rd edn, 2006 [Google Scholar]
- Grübel, PhD, Chemistry M., PhD dissertation, Technical University Munich, 2019 10.14459/2020md1515118 [DOI] [Google Scholar]
- Condie A. G. González-Gómez J. C. Stephenson C. R. J. Am. Chem. Soc. 2010;132:1464–1465. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]
- Johnson J. A. Luo J. Zhang X. Chen Y.-S. Morton M. D. Echevería E. Torres F. E. Zhang J. ACS Catal. 2015;5:5283–5291. [Google Scholar]
- Megerle U. Wenninger M. Kutta R.-J. Lechner R. König B. Dick B. Riedle E. Phys. Chem. Chem. Phys. 2011;13:8869–8880. doi: 10.1039/c1cp20190e. [DOI] [PubMed] [Google Scholar]
- McCusker J. K. Science. 2019;363:484–488. doi: 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- Shields B. J. Kudisch B. Scholes G. D. Doyle A. G. J. Am. Chem. Soc. 2018;140:3035–3039. doi: 10.1021/jacs.7b13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.