

Abstract
Bicyclo[1.1.0]butanes (BCBs) are increasingly valued as intermediates in ‘strain release’ chemistry for the synthesis of substituted four membered rings and bicyclo[1.1.1]pentanes, with applications including bioconjugation processes. Variation of the BCB bridgehead substituents can be challenging due to the inherent strain of the bicyclic scaffold, often necessitating linear syntheses of specific BCB targets. Here we report the first palladium catalyzed cross-coupling on pre-formed BCBs which enables a ‘late stage’ diversification of the bridgehead position, and the conversion of the resultant products into a range of useful small ring building blocks.
Bicyclo[1.1.0]butanes (BCBs) are valuable precursors to four-membered rings and bicyclo[1.1.1]pentanes, and useful bioconjugation agents. We describe a versatile approach to access 1,3-disubstituted BCBs, which are otherwise challenging to prepare.
Introduction
Bicyclo[1.1.0]butanes (BCBs 1, Fig. 1a) are a class of hydrocarbons consisting of two cyclopropane rings fused through a common C–C bond. The fused bicyclic structure is highly strained (ring strain energy ∼66 kcal mol−1, more than twice that of cyclopropane),1 which renders BCBs valuable intermediates in ‘strain release’ chemistry for the synthesis of four membered ring systems.2,3 In this context, BCBs that are monosubstituted at the bridgehead positions have been widely explored due to their ready accessibility4–10 and facility of ring opening reactions with nucleophiles,4,11–14 radicals15,16 and electrophiles17–19 to give cyclobutanes and cyclobutenes (2). BCBs have also recently found applications as tools for protein bioconjugation, where they exhibit high chemoselectivity for the alkylation of cysteine residues under mild conditions (e.g. BCB Ibrutinib, 3).10,20,21 A BCB-containing natural product (4) has even been isolated and synthesised.22,23
Fig. 1. (a) Reactivity/applications of BCBs. (b) Existing routes to 1,3-disubstituted BCBs. (c) This work: Bridgehead cross-coupling.

1,3-disubstituted BCBs are attractive precursors to bicyclo[1.1.1]pentanes (BCPs, 5), which are valuable motifs in drug design,24–26via a formal carbene insertion into the C1–C3 bond.26–30 However, access to 1,3-disubstituted BCBs is more challenging than their monosubstituted counterparts: the predominant routes consist of transannular 1,3-cyclization of a cyclobutyl carbanion onto a tertiary halide (6, Fig. 1b),25,28,31–33 which can suffer from competing elimination to a cyclobutene, or 3-exo-tet cyclization of a cyclopropyl carbanion onto a halomethyl substituent (7).34 Both require multistep synthetic sequences which limits facile bridgehead diversification.
We questioned whether direct introduction of a bridgehead substituent might be possible from a pre-formed mono-substituted BCB (Fig. 1c). Previous work has shown that selective deprotonation of this position is possible,12,35 and in specific tricyclic systems has been used for cross-coupling reactions,36–38 however the use of this strategy for the general synthesis of 1,3-difunctionalized BCBs is, to our knowledge, unknown. Here we report the development of a directed metalation/Negishi cross-coupling39,40 route to access these useful building blocks. The reaction proceeds under mild conditions and displays broad scope and functional group tolerance with respect to the electrophile, delivering an array of bio-relevant BCBs. These products can be readily transformed into a range of other polysubstituted small ring building blocks.
Results and discussion
BCB metalation
We first investigated deprotonation/deuterium quenching of the C3-bridgehead position of BCBs 8a–d, featuring various directing groups (Table 1). Pleasingly, sulfone 8a could be fully deuterated using PhLi or n-BuLi as base (Table 1, entries 1 and 2). While sulfoxide 8b could direct deprotonation using lithium amide bases (entries 3 and 4), lithium-sulfoxide exchange (to form 9) was found to be a competing pathway. Reaction of amide 8c proved more fruitful: complete deprotonation was achieved using PhLi or n-BuLi at −78 °C (entries 5 and 6). However, metalation of ester 8d was unsuccessful or resulted in nucleophilic addition to the carbonyl to give 10 (entries 7 and 8).
Directed metalation studies on BCBs 8a–da.
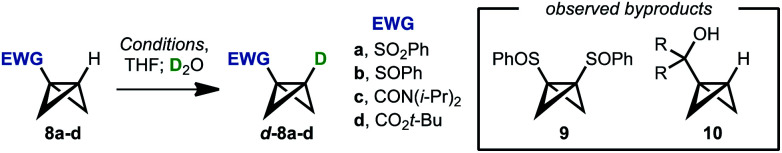
| ||||
|---|---|---|---|---|
| Entry | BCB | Conditions | Equiv. base | d-8 (conversion, %)b |
| 1 | 8a | PhLi, −78 °C | 1.1 | 100 |
| 2 | 8a | n-BuLi, −78 °C | 1.1 | 100 |
| 3 | 8b | LiTMP, 0 °C or −78 °C | 3.0 | 65–72c |
| 4 | 8b | LDA, −78 °C | 3.0 | 75c |
| 5 | 8c | PhLi, −78 °C | 1.1 | 100 |
| 6 | 8c | n-BuLi, −78 °C | 1.1 | 100 |
| 7 | 8d | Mg(TMP)2, LiCl, 0 °C | 1.1 | n.r.d |
| 8 | 8d | n-BuLi, −78 °C | 1.0 | 50e |
Reactions conducted on 0.1 mmol scale, quenched with D2O at the indicated temperature.
Conversion based on ratio of d-8 : 8 as determined by 1H NMR spectroscopic analysis of the crude reaction mixture.
Conversion as judged by integration of C3 in 8bvs. C2/C4 integration of 8b, d-8b and 9 in the 1H NMR spectrum of the crude reaction mixture; analysis complicated by diastereomers of 9.
n.r. = no reaction.
1 : 1 mixture of 8d and 10. See the ESI for full details of optimization.
BCB cross-coupling
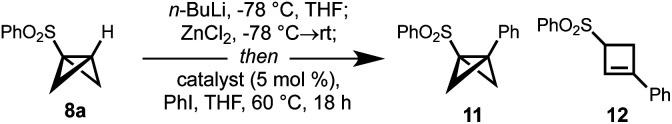
With sulfone and amide directing groups successfully identified, cross-coupling of the resultant carbanions was addressed. The use of palladium catalysis for the synthesis of functionalized BCBs could potentially be problematic due to competing rearrangements in which Pd(ii) species promote electrophilic cleavage of the C1–C3 bond to give ring-opened diene products,41–43 a process that is of value in the synthesis of functionalized cyclobutanes.17 We anticipated that the pi-acidity of palladium complexes could be lowered by the use of electron-rich ligands, enabling the selective cross-coupling of metalated BCBs while avoiding rearrangement. We also conjectured that rapid transmetalation might further limit the potential of Pd(ii) intermediates to effect ring-opening processes, and thus targeted the use of a BCB-zinc species, formed by treatment of lithiated 8a (generated using n-BuLi) with zinc chloride. The organozinc reagent, which could be isolated and is stable at room temperature for several hours,44 was then heated to 60 °C with Pd(dba)2 (5 mol%), trifurylphosphine (tfp, 10 mol%) and iodobenzene in THF. Pleasingly, the desired 1,3-disubstituted BCB 11 was obtained in excellent yield (Table 2, entry 1, 72%), with no isomerization to the cyclobutene 12 observed in the 1H NMR spectrum of the crude reaction mixture. However, substantial isomerization to 12 (>50%, <1 h) occurred on standing in deuterated chloroform which had not been pre-treated with potassium carbonate, indicating that acid-promoted isomerization of 11 is facile, in particular when a benzylic cation is generated. The use of Pd(PPh3)4, or the less electron-rich SPhos ligand proved less effective unless employed at higher catalyst loading (entry 3), while Pd(ii) catalysts indeed resulted in low conversion and/or competing isomerization of 11 to 12 (entries 5 and 6). Variation of solvent and temperature (entries 8–11) showed optimal conversion was achieved in THF at 40 °C. The use of PhLi in the metalation/coupling sequence further improved the yield (98%, entry 12), and was taken forward as the optimized conditions due to its less hazardous and more functional group tolerant nature.
BCB cross-coupling optimizationa.
| ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | 11 b (%) |
| 1 | Pd(dba)2/2 tfp | THF | 60 | 72 (75) |
| 2 | Pd(PPh3)4 | THF | 60 | 47 (50) |
| 3c | Pd(PPh3)4 | THF | 60 | 81 (83) |
| 4 | Pd(dba)2/2 SPhos | THF | 60 | n.d.d (13) |
| 5 | Pd(dppf)Cl2 | THF | 60 | n.d. (5) |
| 6 | Pd(PhCN)2Cl2 | THF | 60 | n.d. (6)e |
| 7 | Pd(t-Bu3P)2 | THF | 60 | n.d. (18)f |
| 8 | Pd(dba)2/2 tfp | 1,4-Dioxane | 60 | n.d. (23) |
| 9 | Pd(dba)2/2 tfp | DMF | 60 | n.d. (47) |
| 10 | Pd(dba)2/2 tfp | THF | 40 | 89 (n.d.) |
| 11 | Pd(dba)2/2 tfp | THF | rt | n.d. (67) |
| 12 g | Pd(dba) 2 /2 tfp | THF | 40 | 98 (100) |
Cross-couplings run on 0.1 mmol scale with 1.0 equiv. of PhI.
Isolated yields. Values in parentheses indicate conversion based on the ratio of 11 to 8a and 12 as determined by 1H NMR spectroscopic analysis of the crude reaction mixture.
10 mol% catalyst.
n.d. = not determined.
29% conversion to 12.
9% conversion to 12.
0.25 mmol scale using 8a (1.2 equiv.), PhLi (1.2 equiv.) and PhI (1.0 equiv.). tfp = 2-trifurylphosphine. rt = room temperature.
Having identified optimal conditions, we next assessed the scope of the reaction (Scheme 1). A series of aryl iodides bearing electron withdrawing and donating groups were first investigated; to our delight, these couplings proceeded in good to excellent yields (70–96%, 13–25), irrespective of the nature of the arene substituent or its position (para, meta, ortho), with the exception of o-CF3 product 23 (51%).45 Many of these aryl-BCBs are hard to access via traditional routes, in particular for ortho-substituted aryl BCB products.26,29 The scalability of the chemistry was demonstrated through the successful synthesis of BCB 11 on >gram scale, with no decrease in yield (1.89 g, 96%). These disubstituted BCB products were found to be relatively stable, with resistance to rearrangement increasing with electron withdrawing character of the coupled aryl group; as noted above, little to no isomerization took place under the reaction conditions.
Scheme 1. Synthesis of 1,3-disubstituted BCBs: reaction scope. Reaction conditions unless stated otherwise: 8a or 8c (0.30 mmol, 1.2 equiv.), PhLi (0.30 mmol, 1.2 equiv. 1.6–1.8 M in Bu2O), THF, −78 °C, 15 min; ZnCl2 (0.30 mmol, 1.2 equiv.), THF, −78 °C to rt; Pd(dba)2 (5 mol%), tfp (10 mol%), R–I (0.25 mmol, 1.0 equiv.) THF, 40 °C, 16 h. aReaction conducted with 8.76 mmol (1.70 g) of 8a. b0.48 equiv. of heteroaryl iodide. cReaction run using the aryl triflate/10 mol% Pd(PPh3)4 as catalyst. 11, 14, 16 and 18 were determined by single crystal X-ray diffraction studies.45 See the ESI† for details of unsuccessful substrates.

Trisubstituted aryl (26) and naphthyl (27) iodides also served as effective coupling partners, the former of which demonstrated useful selectivity for coupling of the aryl iodide (both 82% yield). Alkenyl iodides were similarly competent substrates, using either electron-poor (28, 54%) or electron-rich alkenes (E/Z-29, 61–74%). Interestingly, the product of coupling 5-iodo-1,3-dimethyluracil underwent rearrangement to cyclobutene 30 (53%). sp3–sp coupling using a 1-iodoalkyne proved more challenging, proceeding in low yield (31, 17%).
Heterocycle-substituted BCBs are highly appealing from a medicinal chemistry perspective. We were delighted to find that a range of azacycles could be successfully installed at the BCB bridgehead, including 2-, 3-, and 4-substituted pyridines (32–34), quinoline (35), isoquinoline (36), and indole (37) motifs. The yields for these couplings were uniformly high (83–93%) with the exception of isoquinoline 36 (38%). A double cross-coupling could even be achieved in relatively high yield using 2,6-diiodopyridine (38, 59%).
The use of other directing groups was briefly tested. The metalation/coupling chemistry was successfully applied to N,N-diisopropylamide BCB 8c, which gave products 39 and 40 in good yields (73% and 93% respectively). Coupling of commercially available 4-trifluoromethylphenyl BCB sulfone (41, 76%) further underlined the potential for variation at this position. Finally, the use of more complex, bio-relevant coupling partners was explored: this includes the coupling of phenylalanine and serine derivatives (42 and 43 respectively), and a galactose aryl ester (44). The aryl triflate of estrone also underwent coupling using Pd(PPh3)4 as the catalyst, albeit in low yield.
BCB diversification
1,3-disubstituted BCBs should offer useful opportunities in synthesis due to the potential for strain release. For example, the rapid BCB-to-cyclobutene rearrangement noted above for compound 30 could be effected in quantitative yield on subjection of chloroform solutions of BCBs to a catalytic amount of 1 M HCl (6, 46, 47, Scheme 2). Cyclobutene 12 could be further converted to [2.1.0]housane 48via Rh-catalyzed cyclopropanation. The central C1–C3 bond could also be reduced using LiAlH4 (49), or oxidised (NCS, MeOH, 50) to cyclobutane derivatives. While the former transformation afforded a 1 : 1 mixture of diastereomers of 49, presumably due to the formation of a configurationally labile α-sulfone carbanion intermediate, the latter gave exclusively syn addition of the two heteroatoms. The reasons for this are unclear, but may reflect a concerted addition process or single electron transfer mechanism.46,47
Scheme 2. (a) Further transformations of 1,3-disubstituted BCBs. (b) Application to synthesis of the BCP-F2-darapladib sidechain.

Having enabled the initial bridgehead functionalization, the sulfone functionality was found to be capable of directing further lithiation chemistry: treatment with n-BuLi led to metalation of the sulfone phenyl substituent (51), which could be trapped on reaction with electrophiles (NFSI, 52) or subjected to transmetalation/Negishi cross-coupling (53). These transformations enable fine-tuning of the electronic properties of the sulfone substituent, which have been demonstrated to be important in strain release reactivity.14 We were also able to access the difluorobicyclo[1.1.1]pentane (BCP-F2) 54 on treatment with difluorocarbene precursor 55, developed in our group. Collectively, these reactions show how BCBs represent a source of numerous small ring scaffolds.
As a final demonstration of potential utility in pharmaceutical research, we applied this chemistry to the synthesis of the BCP-F2 analogue of the atherosclerosis candidate darapladib, a compound with an existing BCP analogue.25 Gram scale lithiation of 8c/cross-coupling delivered BCP 40 in 90% yield (1.35 g), treatment of which with difluorocarbene source 55 afforded the desired BCP-F256 in 33% yield (60% brsm) along with 40% of the undesired rearrangement product 57. Notably, other difluorocarbene sources26,29 failed to deliver 56. The N,N-diisopropylamide group in 56 could be transformed to the corresponding BCP-F2 acid 58via a two step reduction/oxidation sequence;48 the BCP analogue of this compound has been converted to BCP-darapladib in previous work.25
Conclusions
In summary, we have developed a general cross-coupling strategy to access 1,3-disubstituted bicyclo[1.1.0]butanes featuring a wide variety of substituents at the 3-position. This approach minimizes the degree of BCB isomerization typically seen with transition metals through use of an electron-rich Pd(0) catalyst, and generally proceeds in high yields. The methodology enables the introduction of arenes and heteroarenes, and can be extended to scaffolds of importance in biology and medicine. In addition to the diversity of coupling partners, the functional group tolerance is notable: nitro, ester, ketone, halide, triflate, silyl, nitrile, ether, carbamate and acetal groups are all successfully accommodated, which augurs well for applications in medicinal chemistry. Further diversifications of the BCB products into other small ring systems, and synthesis of the BCP-F2 analogue of a darapladib fragment, highlight the utility of these ‘strain release’ precursors in synthesis.
Author contributions
R. E. M. and E. A. A. conceived the work. R. E. M., M. M. H., J. N. and R. C. carried out the experimental work. K. E. C. acquired and solved the X-ray crystal structure. E. A. A. directed the work. All authors co-wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
R. E. M. thanks the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for a studentship (EP/L015838/1) generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Jannsen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. J. N. thanks the Marie Skłodowska-Curie actions for an Individual Fellowship (GA No 786683). E. A. A. thanks the EPSRC for support (EP/S013172/1). The authors thank Felix Urbitsch for helpful discussions on difluorocarbene sources.
Electronic supplementary information (ESI) available: Experimental procedures and data for novel compounds, copies of 1H and 13C NMR spectra, cif file for single crystal X-ray spectrum of compounds 11, 14, 16 and 18. CCDC 2074459–2074463. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01836a
Notes and references
- Khoury P. R. Goddard J. D. Tam W. Tetrahedron. 2004;60:8103–8112. [Google Scholar]
- Turkowska J. Durka J. Gryko D. Chem. Commun. 2020;56:5718–5734. doi: 10.1039/d0cc01771j. [DOI] [PubMed] [Google Scholar]
- Pramanik M. M. D. Qian H. Xiao W.-J. Chen J.-R. Org. Chem. Front. 2020;7:2531–2537. [Google Scholar]
- Panish R. Chintala S. R. Boruta D. T. Fang Y. Taylor M. T. Fox J. M. J. Am. Chem. Soc. 2013;135:9283–9286. doi: 10.1021/ja403811t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C. Davies H. M. L. Org. Lett. 2013;15:310–313. doi: 10.1021/ol303217s. [DOI] [PubMed] [Google Scholar]
- Chen K. Huang X. Kan S. B. J. Zhang R. K. Arnold F. H. Science. 2018;360:71. doi: 10.1126/science.aar4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramazanov I. R. Yaroslavova A. V. Dzhemilev U. M. Russ. Chem. Rev. 2012;81:700–728. [Google Scholar]
- Weber J. Haslinger U. Brinker U. H. J. Org. Chem. 1999;64:6085–6086. [Google Scholar]
- Nilsen N. O. Skattebøl L. Baird M. S. Buxton S. R. Slowey P. D. Tetrahedron Lett. 1984;25:2887–2890. [Google Scholar]
- Schwartz B. D. Zhang M. Y. Attard R. H. Gardiner M. G. Malins L. R. Chem.–Eur. J. 2020;26:2808–2812. doi: 10.1002/chem.201905539. [DOI] [PubMed] [Google Scholar]
- Guo L. Noble A. Aggarwal V. K. Angew. Chem., Int. Ed. 2021;60:212–216. doi: 10.1002/anie.202011739. [DOI] [PubMed] [Google Scholar]
- Gaoni Y. Tomazic A. J. Org. Chem. 1985;50:2948–2957. [Google Scholar]
- Gianatassio R. Lopchuk J. M. Wang J. Pan C.-M. Malins L. R. Prieto L. Brandt T. A. Collins M. R. Gallego G. M. Sach N. W. Spangler J. E. Zhu H. Zhu J. Baran P. S. Science. 2016;351:241–246. doi: 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopchuk J. M. Fjelbye K. Kawamata Y. Malins L. R. Pan C.-M. Gianatassio R. Wang J. Prieto L. Bradow J. Brandt T. A. Collins M. R. Elleraas J. Ewanicki J. Farrell W. Fadeyi O. O. Gallego G. M. Mousseau J. J. Oliver R. Sach N. W. Smith J. K. Spangler J. E. Zhu H. Zhu J. Baran P. S. J. Am. Chem. Soc. 2017;139:3209–3226. doi: 10.1021/jacs.6b13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvi M. Aggarwal V. K. J. Am. Chem. Soc. 2019;141:9511–9515. doi: 10.1021/jacs.9b03653. [DOI] [PubMed] [Google Scholar]
- Ociepa M. Wierzba A. J. Turkowska J. Gryko D. J. Am. Chem. Soc. 2020;142:5355–5361. doi: 10.1021/jacs.0c00245. [DOI] [PubMed] [Google Scholar]
- Fawcett A. Biberger T. Aggarwal V. K. Nat. Chem. 2019;11:117–122. doi: 10.1038/s41557-018-0181-x. [DOI] [PubMed] [Google Scholar]
- Bennett S. H. Fawcett A. Denton E. H. Biberger T. Fasano V. Winter N. Aggarwal V. K. J. Am. Chem. Soc. 2020;142:16766–16775. doi: 10.1021/jacs.0c07357. [DOI] [PubMed] [Google Scholar]
- Walczak M. A. A. Krainz T. Wipf P. Acc. Chem. Res. 2015;48:1149–1158. doi: 10.1021/ar500437h. [DOI] [PubMed] [Google Scholar]
- Tokunaga K. Sato M. Kuwata K. Miura C. Fuchida H. Matsunaga N. Koyanagi S. Ohdo S. Shindo N. Ojida A. J. Am. Chem. Soc. 2020;142:18522–18531. doi: 10.1021/jacs.0c07490. [DOI] [PubMed] [Google Scholar]
- Sletten E. and Pengshung M., Disulfide Bioconjugation, US pat., 2020181192 (A1), 2020
- DeGuire S. M. Ma S. Sulikowski G. A. Angew. Chem., Int. Ed. 2011;50:9940–9942. doi: 10.1002/anie.201104366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. Niisuke K. Boeglin W. E. Voehler M. Stec D. F. Porter N. A. Brash A. R. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18941–18945. doi: 10.1073/pnas.0707148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepan A. F. Subramanyam C. Efremov I. V. Dutra J. K. O'Sullivan T. J. DiRico K. J. McDonald W. S. Won A. Dorff P. H. Nolan C. E. Becker S. L. Pustilnik L. R. Riddell D. R. Kauffman G. W. Kormos B. L. Zhang L. Lu Y. Capetta S. H. Green M. E. Karki K. Sibley E. Atchison K. P. Hallgren A. J. Oborski C. E. Robshaw A. E. Sneed B. O'Donnell C. J. J. Med. Chem. 2012;55:3414–3424. doi: 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]
- Measom N. D. Down K. D. Hirst D. J. Jamieson C. Manas E. S. Patel V. K. Somers D. O. ACS Med. Chem. Lett. 2017;8:43–48. doi: 10.1021/acsmedchemlett.6b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bychek R. M. Hutskalova V. Bas Y. P. Zaporozhets O. A. Zozulya S. Levterov V. V. Mykhailiuk P. K. J. Org. Chem. 2019;84:15106–15117. doi: 10.1021/acs.joc.9b01947. [DOI] [PubMed] [Google Scholar]
- Applequist D. E. Wheeler J. W. Tetrahedron Lett. 1977;18:3411–3412. [Google Scholar]
- Applequist D. E. Renken T. L. Wheeler J. W. J. Org. Chem. 1982;47:4985–4995. [Google Scholar]
- Ma X. Sloman D. L. Han Y. Bennett D. J. Org. Lett. 2019;21:7199–7203. doi: 10.1021/acs.orglett.9b02026. [DOI] [PubMed] [Google Scholar]
- Ma X. Pinto W. Pham L. N. Sloman D. L. Han Y. Eur. J. Org. Chem. 2020:4581–4605. [Google Scholar]
- Hall H. K. Smith C. D. Blanchard E. P. Cherkofsky S. C. Sieja J. B. J. Am. Chem. Soc. 1971;93:121–130. [Google Scholar]
- Hoz S. Livneh M. J. Am. Chem. Soc. 1987;109:7483–7488. [Google Scholar]
- Wiberg K. B. Ciula R. P. J. Am. Chem. Soc. 1959;81:5261–5262. [Google Scholar]
- Gaoni Y. J. Org. Chem. 1982;47:2564–2571. [Google Scholar]
- Gaoni Y. Tetrahedron. 1989;45:2819–2840. [Google Scholar]
- Kottirsch G. Szeimies G. Chem. Ber. 1990;123:1495–1506. [Google Scholar]
- Stephen A. Hashmi K. Vollmer A. Szeimies G. Liebigs Ann. 1995;1995:471–475. [Google Scholar]
- Kobayashi Y. Nakamoto M. Inagaki Y. Sekiguchi A. Angew. Chem., Int. Ed. 2013;52:10740–10744. doi: 10.1002/anie.201304770. [DOI] [PubMed] [Google Scholar]
- For related cross-coupling of small-ring bridgehead organozincs, see: ; Makarov I. S. Brocklehurst C. E. Karaghiosoff K. Koch G. Knochel P. Angew. Chem., Int. Ed. 2017;56:12774–12777. doi: 10.1002/anie.201706799. [DOI] [PubMed] [Google Scholar]
- Schwärzer K. Zipse H. Karaghiosoff K. Knochel P. Angew. Chem., Int. Ed. 2020;59:20235–20241. doi: 10.1002/anie.202009340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauben W. G. Kielbania A. J. J. Am. Chem. Soc. 1972;94:3669–3671. [Google Scholar]
- Gassman P. Atkins T. J. Am. Chem. Soc. 1972;94:7748–7756. [Google Scholar]
- Bishop K. C. Chem. Rev. 1976;76:461–486. [Google Scholar]
- See the ESI† for details
- Low temperature single crystal X-ray diffraction data for 11, 14, 16 and 18 were collected using a Rigaku Oxford SuperNova diffractometer. Raw frame data were reduced using CrysAlisPro and the structures were solved using ‘Superflip [L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790.] before refinement with CRYSTALS [(a) P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30. (b) R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr. 2010, 43, 1100–1107.] as per the ESI† (CIF). Full refinement details are given in the ESI† (CIF); Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2074459–63).†
- Hoz S. Livneh M. Cohen D. J. Am. Chem. Soc. 1987;109:5149–5156. [Google Scholar]
- Saettel N. J. Wiest O. J. Org. Chem. 2003;68:4549–4552. doi: 10.1021/jo0267549. [DOI] [PubMed] [Google Scholar]
- Houston S. D. Fahrenhorst-Jones T. Xing H. Chalmers B. A. Sykes M. L. Stok J. E. Farfan Soto C. Burns J. M. Bernhardt P. V. De Voss J. J. Boyle G. M. Smith M. T. Tsanaktsidis J. Savage G. P. Avery V. M. Williams C. M. Org. Biomol. Chem. 2019;17:6790–6798. doi: 10.1039/c9ob01238a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.