

Multiple myeloma (MM) is the second most common hematologic malignancy, characterized by clonal expansion of malignant plasma cells in the bone marrow (BM). Together with genetic changes promoting cell survival, MM is associated with progressive immune dysregulation that results in a tumor microenvironment promoting disease progression and loss of immune surveillance.1 Although the advent of novel therapies has improved the outcomes of MM patients, the disease relapses in most cases and new treatment options are urgently needed. In this context, the development of cancer immunotherapeutic strategies has allowed immunotherapy to become a promising new treatment option for MM patients.2 These innovative immune-based therapies have shown encouraging results, but at present, they are limited by the ability to generate durable anti-MM responses. This article briefly reviews the status of T cell-based immunotherapies in MM and analyzes the potential mechanisms of resistance to these therapies to provide a perspective on the strategies used to generate more favorable therapies in MM.
Adoptive cellular therapy using chimeric antigen receptor-transduced T cells (CAR-T) has emerged as a remarkable treatment strategy against B-cell neoplasms. The antigen-binding counterpart of the CAR is an antibody fragment, single chain variable fragment (scFv), which recognizes a membrane protein associated to a cancer cell.3 Promising results have been described for B-cell maturation antigen (BCMA)-targeted therapies in relapsed refractory MM (RRMM).4 The restricted expression of BCMA and its role in MM pathogenesis have made BCMA an attractive target for CAR-T therapies. However, to date, the different BCMA-CAR-T cell therapies have limited efficacy due to the lack of T cell persistence and durability.
To facilitate an immune synapse formation between immune effectors and malignant cells, bispecific antibodies (BsAbs) have been designed as recombinant bispecific proteins with two linked scFvs from two different antibodies, one targeting a cell-surface molecule on T cells (CD3) and the other targeting antigens present on cancer cells.5 Since BsAbs directly stimulate CD3 they activate T cells independently from antigen presentation on major histocompatibility complex (MHC) class I and they have the ability to activate T cells in the absence of co-stimulation reducing the risk of anergy.5 Early clinical data show the promising efficacy of anti-BCMA BsAbs in RRMM.2 However, longer follow-up is required to evaluate the durability of responses and safety of these approaches.
Responses to cancer immunotherapies largely depend on the status of the immune system and its ability of inducing durable antitumor responses. Available data from clinical studies indicate that resistance to T cell-based therapies can stem from T cell intrinsic factors, immunosuppressive BM tumor microenvironment (TME), and loss of target antigen due to tumor cell mutations. It has been demonstrated that a peak expansion of BCMA-directed CAR-T cells and a preserved CD4:CD8 ratio with a higher number of naïve/stem cell memory T cells generally correlates with clinical responses.6 In contrast, T cell exhaustion characterized by increased expression of inhibitory receptors, reduced proliferation, and distinct transcriptional signature has been associated with insufficient and short anti-tumor activity of cancer immunotherapies.7 Our group has recently shown that enrichment of exhausted T cells correlates with lack of response to BCMA-targeted therapies and is associated with disease progression.8
The immunosuppressive BM TME is also an important factor impairing the efficacy of immunotherapy in MM. The interactions between MM and stromal cells along with associated soluble factors (IL-6, IL-10, and TGF-β) drive a tolerogenic and suppressive environment.1 Inhibitor myeloid cells such the myeloid-derived suppressor cells (MDSC), plasmacytoid dendritic cells (pDCs), and regulatory T cells (Treg) have been reported in MM patients. They play a role in disease progression and inhibit T cell proliferation and activation, leading to the failure of the anti-MM immune response.1
Antigen escape due to tumor heterogeneity and selection pressure, especially under highly effective immunotherapies, is also an important point that needs to be considered in MM patients receiving targeted-immunotherapies. Studies have shown that MM is characterized by interpatient and intratumor heterogeneity closely related to progression and resistance to therapy. Recent studies have reported resistance to anti-BCMA-targeted therapies due to the rare existence of chromosome 16 aberrations (focal biallelic loss of BCMA) as a result of tumor evolution and selection under the pressure of therapy.8,9 Relapses caused by low BCMA expressing cells due to its cleavage from tumor cells by γ-secretase (GS) have also been described. Therefore, strategies to increase the amount of BCMA on tumor cells such as the use of GS inhibitors have been explored to increase the susceptibility of BCMA-low tumor cells to CAR-T cells.10 These approaches, together with a new interest in multi-antigen targeting, could prevent relapse due to BCMA loss or downregulation.
A summary of the factors mentioned above is presented in Figure 1.
Figure 1.
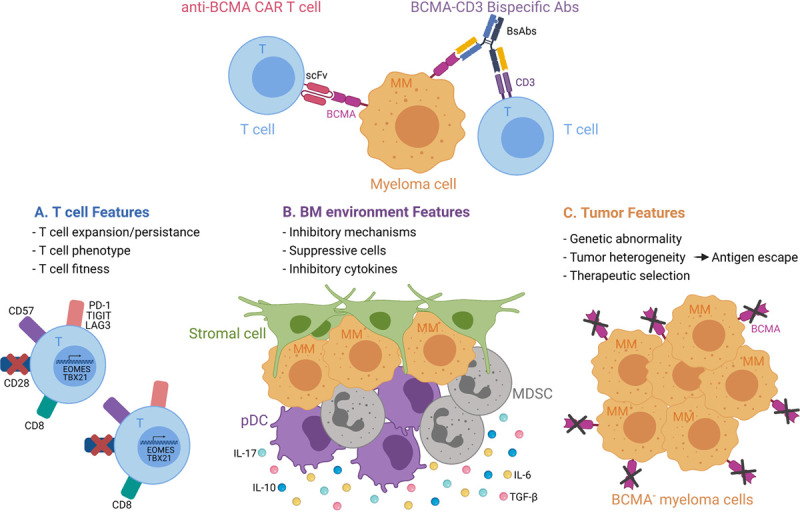
Summary of factors involved in mechanisms of resistance. The resistance can stem from T cell, BM, and/or tumor features. BCMA = B-cell maturation antigen; BM = bone marrow; CAR-T = chimeric antigen receptor-transduced T cells; DC = dendritic cell; MDSC = myeloid-derived suppressor cell. The figure has been created using bioRender.
Strategies to generate more favorable T cell-based therapies
The clinical success of the T cell-based therapies depends upon the overall fitness of T cells. The starting quality and expansion of T cells dictate CAR-T cell persistence and cytotoxicity, and correlate with durable responses.6 Therefore, several strategies have been explored to improve the efficacy and durability of these cells. These consist of newer transduction systems including non-viral methods such as the piggyBac transposon system and alternatives to scFvs and co-stimulatory domains.2 In addition, the use of products such as the phosphoinositide 3 kinase (PI3K) inhibitor bb007 and genetic modifications of TET2 are being evaluated to generate T cell products with increased memory-like phenotype.11 Further promising approaches include the use of cytokines (IL-7 and IL-15) in the culture media during CAR-T manufacturing to enrich for memory-like T cells, and the development of engineered CAR-T cells able to secrete pro-inflammatory cytokines (IL-12) to improve their efficacy.12
Another mechanism of resistance to T cell-based therapies is due to a dysfunctional/exhausted state of T cells caused by either intrinsic T cell defects or tolerogenic BM microenvironment. It is characterized by sustained expression of inhibitory receptors and a distinct transcriptional state that can be partially reversed through disruption of inhibitory receptors.7 However, immune checkpoint inhibition does not prevent the suppressive imprinting of exhaustion.13 Therefore, given the complex interactions existing between epigenetic modifying proteins and lineage-specific transcription factors regulating T cell differentiation and function, a better knowledge of these regulators is necessary to explore its therapeutic possibilities.
Targeting the BM TME is also an appealing strategy to prevent immune escape and limit T cell exhaustion; several strategies are currently under investigation. These include the use of agents able to deactivate MDSCs (COX2 and ARG1 inhibitors), differentiate MDSCs into mature cells (ATRA or Vitamins A or D3), and prevent MDSC development (tyrosine kinases and STAT3 inhibitors).11 Dendritic cells (DCs) can also influence responsiveness to immunotherapies, and several strategies are currently being pursued to reverse DC-mediated tolerance. These include the use of agents promoting DCs activation/mobilization (GM-CSF and FLT3L), DCs immunogenic functions (TLR7/TLR8 agonists), and preventing immune-suppressive DCs functions (IDO and STAT3 inhibitors).7 Combining T cell-based therapies with other immunotherapies (immune checkpoint inhibitors and immunomodulatory drugs) and small molecules able to modulate the BM microenvironment is also currently under investigation. These combinations could effectively target both T cell exhaustion and BM-driven immunosuppression, and restore T cell immunity against MM leading to better outcomes.
To prevent tumor escape due to antigen loss, targeting new antigens either alone or in combination with BCMA is also being investigated. The G protein-coupled receptor, class C group 5 D (GPRC5D) and Fc receptor-homolog 5 (FcRH5) are highly expressed on MM cells and have been recently tested with promising results as new targets. Dual CAR-T cell targeting strategies (BCMA with CD19, GPRC5D, and SLAMF7) have also been investigated and are currently in early clinical trials to demonstrate their activity.14 Lastly, the use of pharmacological agents able to increase target antigen density on MM cells (GS inhibitors) for BCMA has also been explored to reduce the risk of relapse.10
Additional challenges that need to be considered in this context are related to the timing of these strategies in the disease course. While most of the studies were performed in the relapsed setting, recent studies reported that triple-refractory patients have a distinct immune microenvironment characterized by reduction of CD4 T cells, lower levels of naive T cell, and decreased numbers of central memory T cells when compared to newly diagnosed (NDMM) patients.15 Taken together, these data suggest that earlier use of these therapies in the disease course may be more appropriate. Two multicenter clinical trials, KarMMa-4 and CARTITUDE-4, are now exploring the use of CAR-T cell therapies in combination with standard MM regimens in earlier lines of treatment.2
In conclusion, the development of T cell-based therapies has provided new treatment options for MM patients, but at present, their use is limited by the ability to generate durable anti-MM T cell responses. Immune dysregulation caused by intrinsic T cell defects or tolerogenic BM microenvironment and/or antigen escape due to antigen loss have been associated with limited activity and development of relapse. Further studies will help determine whether these factors remain important with other multiantigen–targeting strategies, and when these approaches are used earlier in the disease course. An increased understanding of the complex interactions between the malignant clone and immune cells is also critical to optimize these therapies and achieve more durable remissions. As such, a comprehensive genomic and transcriptomic analysis of the tumor together with the immune cell repertoire analysis can help to identify patients with the highest likelihood of relapse and lead to the development of prevention strategies. Such approaches can guide the development of dynamic biomarkers of response to T cell-based therapies that capture tumor and immune response evolution and predict clinical outcomes.
Disclosures
The authors have no conflicts of interests to disclose.
Acknowledgments
As a result of the space constraints, we apologize that we were unable to cite all our colleagues that have made an impact in defining our current knowledge on this topic.
References
- 1.Leblay N, Maity R, Hasan F, et al. Deregulation of adaptive T cell immunity in multiple myeloma: insights into mechanisms and therapeutic opportunities. Front Oncol. 2020; 10:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah UA, Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ. 2020; 370:m3176. [DOI] [PubMed] [Google Scholar]
- 3.Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. 2020; 38:473–488. [DOI] [PubMed] [Google Scholar]
- 4.Rodríguez-Otero P, Prósper F, Alfonso A, Paiva B, Miguel JFS. CAR T-cells in multiple myeloma are ready for prime time. J Clin Med. 2020; 9:3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahlén E, Veitonmäki N, Norlén P. Bispecific antibodies in cancer immunotherapy. Ther Adv Vaccines Immunother. 2018; 6:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen AD, Garfall AL, Stadtmauer EA, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019; 129:2210–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z, Liu S, Zhang B, et al. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol. 2020; 8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leblay N, Maity R, Barakat E, et al. Cite-seq profiling of T cells in multiple myeloma patients undergoing BCMA targeting CAR-T or bites immunotherapy. Blood. 2020; 136(suppl 1):11–12.32276273 [Google Scholar]
- 9.Da Vià MC, Dietrich O, Truger M, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021; 27:616–619. [DOI] [PubMed] [Google Scholar]
- 10.Pont MJ, Hill T, Cole GO, et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019; 134:1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alsina M, Shah N, Raje NS, et al. Updated results from the phase I CRB-402 study of anti-bcma CAR-T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma: correlation of expansion and duration of response with T cell phenotypes. Blood. 2020; 136(suppl 1):25–26. [Google Scholar]
- 12.Yeku OO, Brentjens RJ. Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem Soc Trans. 2016; 44:412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol. 2018; 18:340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang L, Zhang J, Li M, et al. Characterization of novel dual tandem CD19/BCMA chimeric antigen receptor T cells to potentially treat multiple myeloma. Biomark Res. 2020; 8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visram A, Dasari S, Anderson E, et al. Relapsed multiple myeloma demonstrates distinct patterns of immune microenvironment and malignant cell-mediated immunosuppression. Blood Cancer J. 2021; 11:45. [DOI] [PMC free article] [PubMed] [Google Scholar]