

Abstract
Various allogeneic (allo) stem cell transplantation platforms have been developed over the last 2 decades. In this review we focus on the impact of in vivo and ex vivo graft manipulation on immune reconstitution and clinical outcome. Strategies include anti-thymocyte globulin- and post-transplantation cyclophosphamide-based regimens, as well as graft engineering, such as CD34 selection and CD19/αβT cell depletion. Differences in duration of immune suppression, reconstituting immune repertoires, and associated graft-versus-leukemia effects and toxicities mediated through viral reactivations are highlighted. In addition, we discuss the impact of different reconstituting repertoires on donor lymphocyte infusions and post allo pharmacological interventions to enhance tumor control. We advocate for precisely counting all graft ingredients and therapeutic drug monitoring during conditioning in the peripheral blood, and for adjusting dosing accordingly on an individual basis. In addition, we propose novel trial designs to better assess the impact of variations in transplantation platforms in order to better learn from our diversity of “counts” and potential “adjustments.” This will, in the future, allow daily clinical practice, strategic choices, and future trial designs to be based on data guided decisions, rather than relying on dogma and habits.
Neglected basic principles of transplantation: count!
αβT cells are considered to be the major driver of the curative graft-versus-leukemia (GVL) effect, as well as graft-versus-host disease (GVHD), a life-threatening complication that limits the widespread use of allogeneic stem cell transplantations (allo-SCTs).1–3 Retrospective studies analyzing real world stem cell transplantation data and graft compositions from registries and larger centers suggest that the dose of αβT cells is not well balanced when infused into patients, with a substantial fraction of patients receiving too many αβT cells. The surplus of αβT cells per body weight seems to mainly result in increased incidences of both acute and chronic GVHD, without improving GVL effects or engraftment.4,5 Within this context, approximately 25% of all individuals in T cell repleted allo-SCT with matched unrelated donors (MUDs)4 and 50% from haploidentical donors would benefit from infusing fewer donor cells5 (Figure 1). This observation emphasizes that grafts differ substantially in immune compositions, and these variations need to be taken into consideration when treating patients. Limiting T cell numbers rarely interferes with stem cell numbers needed for a sufficient engraftment.4,5 In addition to qualitative and quantitative variations of cell types in the stem cell product, chemotherapeutic drugs used during conditioning can also impact complications and efficacy after allo-SCT. This is a consequence of the fact that concentration of a defined drug, for example, in the blood stream, cannot be precisely predicted based on body weight, body surface area, or kidney or liver function. Active drug levels in the peripheral blood interfere, however, with acute and late toxicity and immune reconstitution, drug dosage needs to be better individualized for patients.6–10 Therapeutic monitoring chemotherapeutic drugs would allow for the creation of an optimized balance between tumor reduction, space for a new hematopoietic stem cell system, inflammation, as well as immune reconstitution. A first step to overcome inter-individual variations and progress towards the generation of personalized transplantation care was the therapeutic drug monitoring of busulfan, which has been shown to reduce toxicity and has entered clinical practice in many centers across the globe.6–9 Variations in fludarabine levels have been accounted to impact T cell reconstitutions,10 and prospective studies are under way to test whether fine-tuning fludarabine levels for each patient will better synchronize immune reconstitution and improve clinical outcomes (NL6940).
Figure 1.
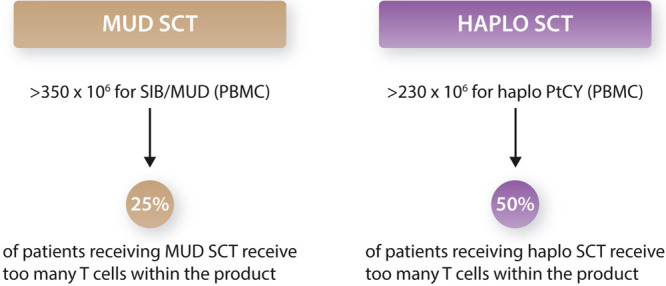
Overdosing of T cells during stem cell transplantation in T cell replete transplantations from matched unrelated and haploidentical grafts. We illustrate different T cell dosage within the context of 2 key studies.4,5 Haplo = haploidentical donor; MUD = matched unrelated donor; PBMC = peripheral mononuclear cells; PTCy = post-transplantation cyclophosphamide; SCT = stem cell transplantation; SIB = sibling.
Balancing anti-thymocyte globulin in T cell replete and deplete transplantations
Anti-thymocyte globulin (ATG) is a polyclonal antibody composite, raised by animal immunization with human T cells and as such, recognizing many different targets expressed in the hematopoietic system.11–13 Different types of ATG and different batches are currently used world-wide in sibling and in unrelated donor transplantations (eg, ATG-Thymoglobulin and ATG-Fresenius; Table 1 and14–19). The impact of clinical outcome differs across the globe. Reduced incidences of GVHD have been reported when ATG was added to conditioning regimens in Europe, which translated into an increased GVHD-relapse free survival.12 However, ATG did not show improved composite endpoints in US-based prospective clinical trials or retrospective studies.13 To understand these different clinical outcomes, it is important to acknowledge that the timing of ATG before infusion of the graft is crucial in determining the impact of ATG on the infused graft and subsequent immune reconstitution. When ATG is administered very early before transplantation (eg, from day –12) it mainly acts on host T cells and host-derived antigen presenting cells in order to facilitate engraftment and reduce GVHD by preventing cross-presentation. If ATG is administered shortly before transplantation (from day –7 or later), most ATG types will, because of their rather long half-life, affect the graft. This results in an additional in vivo T cell depletion of the infused stem cell product by circulating active ATG (Figure 2). US-based clinical trials showed no benefit of ATG on relapse and GVHD-free survival after allo-SCT,19 most likely caused by the usage of irradiation during the conditioning of the patient and ATG administration, which was placed directly after irradiation. Irradiation of patients resulted in a higher in vivo T cell depletion when compared to chemotherapy-based regimens and thus more active ATG remained in the peripheral blood when the graft was infused. Consequently, a stronger donor T cell depletion was accomplished. The net effect was that GVHD was substantially reduced in this clinical trial, but at a cost of substantially more infectious-related deaths.45 Active ATG at the moment of stem cell infusion is a highly uncontrolled mechanism where inter-individual variations in active ATG levels have been acknowledged as a challenge. Within the context of cord blood transplantations, overexposure of active ATG after allografting has been reported to negatively impact immune reconstitution and clinical outcomes.46,47 A major development to master these variations in active ATG is the forthcoming practice to determine active levels of ATG, both pre- and post-allo-SCT,48,49 as well as the effort to generate predictive models to minimize large individual variations in pharmacokinetics (PK) and pharmacodynamics (PD) of ATG.50 Within this context, inter-individual variations of active ATG have been also reported for a T cell replete reduced intensity conditioning cohort in adult patients to substantially impact clinical outcomes in terms of event-free and overall survival.51 We have proposed models which indicate that inter-individual variations in active ATG might be overcome by dosing ATG on lymphocyte counts prior to transplantation instead of on body weight, although this approach needs to be validated for other transplantation regimens and different types of ATG.52 Despite this rather limited knowledge, recommendations have been suggested by the European Society for Blood and Marrow Transplantation (EBMT) on dosing ATG based on individual lymphocyte counts in order to avoid too deep of a T cell depletion of the patient in the first months after allo-SCT.53 These insights also have a major impact on T cell deplete transplantations. For example, within the context of graft engineering, where ATG is used to allow a sufficient engraftment of T cell depleted grafts, ATG needs to be given very early before graft infusion (from day –12, thymoglobulin 1.5 mg/kg IV days –12 to –9) in order to prevent further disturbance of the precisely ex vivo designed graft composition.38 Again, the type of ATG will also impact its timing and dosing of ATG-Fresenius is different (12 mg/kg).29,39
Table 1.
Different Types of Transplantation Platforms, Clinical Outcomes, Viral, Infections and Immune Repertoires.
| Study | Patients | Donor | Intervention | Numbers (n) | Acute GVHD | Chronic GVHD | EFS/OS | CRFS | Relapse | NRM | CMV | EBV | BK | Adeno |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATG | ||||||||||||||
| Chang et at14 | Adult hematological malignancies | MRD; PBSC/BM | ATG-T 1.5 mg/kg on day –3 to –1 | 263 | II–IV: 13.7% | 2 y 27.9% (ext 8.5%) | 3 y OS 69% | 3 y 38.7% | 3 y 20.8% | 3 y 9.9% | Day 100: 22.7% | Day 180: 7.8% | NA | NA |
| III–IV: 8.4% | LFS 65.7% | |||||||||||||
| Walker et al15 | Adult hematological malignancies | MUD/MMUD; PBSC/BM | ATG-T 5 mg/kg on day –2, and 2 mg/kg on days –1 and +1 | 101 | NA | 2 y 26.3% | 2 y OS 70.6% | 1 y 57.6% | 2 y 16.7% | 2 y 21.2% | NA | 20% DNAemia requiring therapy | NA | NA |
| Socié et al16 | Adult hematological malignancies | MUD; BM/PBSC | ATG-F 20 mg/kg day –3 to –1 | 103 | NA | 3 y 12.2% | 3 y OS 55% | NA | 3 y 33% | 3 y 19% | NA | NA | NA | NA |
| Baron et al17 | Adult AML | MRD/MUD; PBSC | ATG-F 30 mg/kg or ATG-T 5 mg/kg; days not known | 569 ATG-fresenius; 249 ATG-thymoglobulin | II–IV: 18%–24% | 2 y 30% | 2 y | 2 y 50%–52% | 2 y 24%–28% | 2 y | NA | NA | NA | NA |
| III–IV: 6%–7%; | OS 67%–68% | 11.5%–16% | ||||||||||||
| LFS 60%–61% | ||||||||||||||
| Finke et al18 | Adult hematological malignancies | MRD/MUD; PBSC/BM | ATG-F 20 mg/kg on day –3 to –1 | 103 | I–IV: 56.3% | 2 y 30.8% (ext 12.2%) | 2 y | NA | 2 y 28.9% | 2 y 19.6% | 53.8% DNAemia; 5.7% CMV disease | 5% PTLD | NA | NA |
| II–IV: 33% | OS 59.2% | |||||||||||||
| III–IV: 11.7% | EFS 51.6% | |||||||||||||
| Soiffer et al19 | Adult AML, MDS, ALL | MUD; PBSC/BM | ATG-F 20 mg/kg –3 to –1 | 126 | II–IV: 23% | 2 y 16% | 2 y | 2 y 38% | 2 y 32% | 2 y 21% | 62% (R+) DNAemia | 1.6% PTLD | NA | NA |
| III–IV: 4.3% | Moderate-severe 12% | OS 59% | ||||||||||||
| PFS 47% | ||||||||||||||
| Alemtuzumab | ||||||||||||||
| Green et al20 | Adult hematological malignancies | Matched/mismatched; PBSC/BM | Alemtuzumab dose 50–100 m | 313 | II–IV: 32%–40% | 2 y 32%–41% | NA | NA | 2 y 24%–36% | 2 y 16%–24% | >80% (R+) DNAemia | NA | NA | NA |
| III–IV: 2%–15% | ||||||||||||||
| van Besien et al21 | Adult AML/MDS | MRD/MUD/MMUD PBSC/BM | Alemtuzumab | 95 | II–IV: 23.3% | 2 y 16% | 2 y | NA | 1 y 23.7% | 1 y 24.6% | NA | NA | NA | NA |
| OS 40.5% | ||||||||||||||
| III–IV: 8.6% | PFS 33% | |||||||||||||
| Carpenter et al22 | Adult hematological malignancies | MRD/MMRD/MUD/MMUD; PBSC/BM | Alemtuzumab (75 in vivo, 36 ex vivo) | 111 | NA | NA | NA | NA | NA | NA | NA | 2 y | NA | NA |
| 40.3% DNAemia; 1% PTLD | ||||||||||||||
| PTCy | ||||||||||||||
| Kanakry et al23 | Adult leukemia/MDS | MRD/MUD; BM | PTCy | 209 | II–IV: 45% | 3 y 13% | 3 y OS 58% | 3 y 39% | 3 y 36% | 3 y 17% | NA | NA | NA | NA |
| III–IV: 11% | EFS 46% | |||||||||||||
| Cieri et al24 | Adult high-risk hematological malignancy | Haplo; PBSC | PTCy | 40 | II–IV: 15% | 1 y 20%; severe 5.1% | 1 y OS 56% | NA | 1 y 35% | 1 y 17% | 63% DNAemia | 15% DNAemia (66% of these pts treated). No PTLD | 18% | NA |
| III–IV: 7.5% | EFS 48% | 17% CMV disease | ||||||||||||
| Berger et al25 | Pediatric; high-risk hematological malignancy | Haplo; BM/PBSC | PTCy | 33 | II–IV: 22% | 1 y 4% | 1 y OS 72% | NA | 1 y 24% | 1 y 9% | 36% DNAemia | 3% DNAemia | 17% | 3% DNAemia; not symptomatic |
| III–IV: 3% | EFS 61% | No CMV disease | No PTLD | |||||||||||
| Devillier et al26 | AML/high-risk MDS | Haplo; BM/PBSC | PTCy | 60 | II–IV: 18% | 2 y 14%; severe 4% | 1 y | 1 y 37% | 1 y 34% | 1 y 27% | NA | NA | NA | NA |
| OS 50% | ||||||||||||||
| III–IV: 2% | EFS 39% | |||||||||||||
| Mehta et al27 | Adult hematological malignancies | MMUD; BM/PBSC | PTCy | 41 | II–IV: 37% | 2 y 30% | 2 y OS 52% | NA | 2 y 20% | 2 y 35% | NA | NA | NA | NA |
| III–IV: 17% | EFS 42% | |||||||||||||
| Mielcarek et al28 | Adult hematological malignancies | MRD/MUD/MMUD; PBSC | PTCy | 43 | II–IV: 77% | 1 y 16% requiring IS; ext 30% | 2 y OS 70% | 2 y 50% | 2 y 17% | 2 y 14% | NA | NA | NA | NA |
| III–IV: 0% | 2 y EFS 69% | |||||||||||||
| PTCy vs ATG | ||||||||||||||
| Battipaglia et al29 | Adult AML | MMUD; BM/PBSC | PTCy vs ATG-F/ATG-T | 93 PTCY, 179 ATG | PTCY | PTCY | PTCY | PTCY: | PTCY: | PTCY: | NA | NA | NA | NA |
| II–IV: 30% | Any: 39% | 2 y OS: 56% | 2 y 37% | 2 y 29% | 2 y 16% | |||||||||
| III–IV: 9% | Ext: 17% | 2 y EFS: 55% | ATG: | ATG: | ATG: | |||||||||
| ATG: | ATG | ATG | 2 y 28% | 2 y 37% | 2 y 29% | |||||||||
| II–IV: 32% | Any: 36% | 2 y OS: 38% | ||||||||||||
| III–IV: 19% | Ext: 20% | 2 y EFS: 34% | ||||||||||||
| Battipaglia et al30 | Adult AML | MRD; BM/PBSC | PTCy vs ATG-F (26%)/ATG-T (74%) | 197 PTCY, 1913 ATG | PTCY | PTCY | PTCY | PTCY: | PTCY: | PTCY: | NA | NA | NA | NA |
| II–IV: 19% | Any: 37% | 2 y OS: 64% | 2 y 44% | 2 y 36% | 2 y 8% | |||||||||
| III–IV: 6% | Ext: 16% | 2 y PFS: 55% | ATG: | ATG: | ATG: | |||||||||
| ATG: | ATG | ATG | 2 y 49% | 2 y 32% | 2 y 10% | |||||||||
| II–IV: 17% | Any: 30% | 2 y OS: 65% | ||||||||||||
| III–IV: 6% | Ext: 12% | 2 y PFS: 58% | ||||||||||||
| Brissot et al31 | Adult AML | MUD; BM/PBSC | PTCy vs ATG-T 5 mg/kg | 174 PTCY, 1452 ATG | PTCY | PTCY | PTCY | PTCY: | PTCY: | PTCY: | NA | NA | NA | NA |
| II–IV: 28.8% | Any: 31.4% | 2 y OS: 62.7% | 2 y 41.6% | 2 y 25.2% | 2 y 15.2% | |||||||||
| III–IV: 8.8%; | Ext: 18.5% | 2 y PFS: 59.7% | ATG: | ATG: | ATG: | |||||||||
| ATG: | ATG | ATG | 2 y 49.3% | 2 y 23.7% | 2 y 16.7% | |||||||||
| II–IV: 29.2% | Any: 33.6% | 2 y OS: 64.8% | ||||||||||||
| III–IV: 9% | Ext: 13.1% | 2 y PFS: 59.6% | ||||||||||||
| Ruggeri et al32 | Adult AML | MUD; BM/PBSC | PTCy vs ATG-F/ATG-T | 193 PTCY, 115 ATG | PTCY | PTCY | PTCY | PTCY: | PTCY: | PTCY: | NA | NA | NA | NA |
| III–IV: 4.7% | Any: 33.7% | 2 y OS: 58% | 2 y 50.9% | 2 y 21.6% | 2 y 22.4% | |||||||||
| ATG: | Ext: 8.6% | 2 y PFS: 56% | ATG: | ATG: | ATG: | |||||||||
| III–IV: 12.5% | ATG | ATG | 2 y 38.9% | 2 y 22.3% | 2 y 30.5% | |||||||||
| Any: 28.3% | 2 y OS: 54.2% | |||||||||||||
| Ext: 12.6% | 2 y PFS: 47.2% | |||||||||||||
| Retière et al33 | Adult hematological malignancies | MRD/MUD/MMUD/haplo; BM/PBSC | PTCy vs ATG-T 2.5 mg/kg on day –1 or days –2 and –1 | 30 PTCY, 15 ATG | PTCY | NA | PTCY | NA | PTCY: | NA | DNAemia | DNAemia requiring treatment | PTCY 3% | PTCY 15% |
| II–IV: 47% | 2 y OS: 79% | 2 y 17% | PTCY 27% | PTCY 0% | ATG 0% | ATG 20% | ||||||||
| III–IV: 10% | 2 y PFS: 63% | ATG: | ATG 40% | ATG 33% | ||||||||||
| ATG: | ATG | 2 y 33% | ||||||||||||
| II–IV: 47% | 2 y OS: 73% | |||||||||||||
| III–IV: 20% | 2 y PFS: 60% | |||||||||||||
| Ex vivo graft engineering | ||||||||||||||
| Pasquini et al34 | Adult AML | MRD; PBSC | CD34+ T cell depletion | 44 | II–IV: 23% | 2 y 19% | 2 y OS 59% | 2 y 42% | 2 y 24% | 2 y 21% | NA | NA | NA | NA |
| III–IV: 4.5% | EFS 55% | |||||||||||||
| Barba et al35 | Adult leukemia | MRD/MUD/MMUD; PBSC | CD34+ T cell depletion | 241 | II–IV: 16% | 3 y 5% | 3 y OS 57% | 1 y 65% | 3 y 22% | 3 y 24% | NA | NA | NA | NA |
| III–IV: 5% | EFS 54% | 3 y 52% | ||||||||||||
| Bayraktar et al36 | Adult AML | MRD/MUD/MMUD; PBSC/BM | CD34+ T cell depletion | 115 | II–IV: 5% | 3 y 13% | 1 y OS 68% | NA | 1 y 17% | 1 y 18% | NA | NA | NA | NA |
| III–IV: 1% | EFS 62% | 3 y 18% | 3 y 24% | |||||||||||
| 3 y OS 57% | ||||||||||||||
| EFS 58% | ||||||||||||||
| van Esser et al37 | Adult hematological malignancies | SIB/MUD; BM/PBSC | CD34+ T cell depletion or sheep erythrocyte rosetting | 85 | II–IV: 57% | 1 y 38% | NA | NA | NA | 1 y 29% | NA | 54% (R+) DNAemia | NA | NA |
| 12% PTLD | ||||||||||||||
| de Witte et al38 | Adult hematological malignancies | MRD/MUD/MMUD; PBSC | αβT cell depletion | 35 | II–IV: 37% | 2 y | 2 y | NA | 2 y 29% | 2 y 32% | 64% (R+) DNAemia, CMV disease 6% | 44% | NA | NA |
| III–IV: 17% | 23% | OS 52% | ||||||||||||
| Moderate: 17% | EFS 40% | |||||||||||||
| Locatelli et al39 | Pediatric AML/ALL | Haplo; PBSC | αβT cell/CD19 depletion | 80 | I–II skin only: 30% | 0% | 5 y: | NA | 5 y 24% | 5 y 5% | NA | NA | NA | NA |
| >II or visceral: 0% | OS 72% | |||||||||||||
| LFS 71% | ||||||||||||||
| Laberko et al40 | pediatric malignant (114) + nonmalignant (68) | MUD/haplo; PBSC | αβT cell/CD19 depletion | 182 | Malignant: II–IV: 40% | NA | 2 y OS 68% | NA | 2 y 37% | 2 y 13% | 51% | 33% | NA | NA |
| Nonmalignant II–IV: 27% | 2 y malignant 58% | |||||||||||||
| 2 y nonmalignant 78% | ||||||||||||||
| Lang et al41 | Pediatric AML/MDS/nonmalignant | Haplo; PBSC | αβT cell/CD19 depletion | 41 | II: 10% | 1.6 y average FU: 18% limited; 9% ext | 1.6 y average FU: OS 52%; EFS NA | NA | 1.6 y average FU: 42% | 1.6 y average FU: 7% | NA | NA | NA | NA |
| III–IV: 15% | ||||||||||||||
| Maschan et al42 | Pediatric high-risk AML | MUD/MMUD/haplo; PBSC | αβT cell/CD19 depletion | 33 | II: 23% | 2 y 30% | 2 y | NA | 2 y 31% | 2 y 10% | 52% DNAemia; 6% CMV disease | 50% DNAemia; 6% rituximab | NA | NA |
| III: 16% | OS 67% | |||||||||||||
| IV: 0% | EFS 60% | |||||||||||||
| Bertaina et al43 | Pediatric nonmalignant | Haplo; PBSC | αβT cell/CD19 depletion | 23 | I–II: 13.1% | 0% | 2 y | NA | NA | 9.3% | 38% DNAemia CMV/adeno | 50% DNAemia; 6% rituximab | NA | 38% DNAemia CMV/adeno |
| III–IV: 0% | OS 91% | |||||||||||||
| EFS 74% | ||||||||||||||
| Balashov et al44 | Pediatric nonmalignant | MUD/MMRD/haplo; PBSC | αβT cell/CD19 depletion | 37 | II: 21.5% | Any 3% | 2 y | NA | NA | NA | NA | NA | NA | NA |
| IV: 2.8% | Ext 3% | OS 96.7% | ||||||||||||
| EFS 67.7% | ||||||||||||||
Depicted is a selection of studies.
Adeno = adenovirus; ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; ATG = anti-thymocyte globulin; ATG-F = anti-thymocyte globulin-fresenius; ATG-T = anti-thymocyte globulin-thymoglobulin; BK = BK virus; BM = bone marrow; CMV = cytomegalovirus; CRFS = cGVHD-free relapse-free survival; cGVHD = chronic graft vs host disease; EBV = Epstein-Barr virus; EFS = event-free survival; ext = extensive; FU = follow up; GVHD = graft vs host disease; haplo = haploidentical donor; IS = immune suppression; LFS = leukemia-free survival; MDS = myelodysplastic syndrome; MMRD = mismatched related donor; MMUD = mismatched unrelated donor; MRD = matched related donor; MUD = matched unrelated donor; NA = not available; NRM = nonrelapse mortality; OS = overall survival; PBSC = peripheral blood stem cell; PFS = progression-free survival; PTCY = post-transplantation cyclophosphamide; PTLD = posttransplant lymphoproliferative disease; pts = patients; R+ = cytomegalovirus positive recipient; SIB = sibling; y = year.
Figure 2.
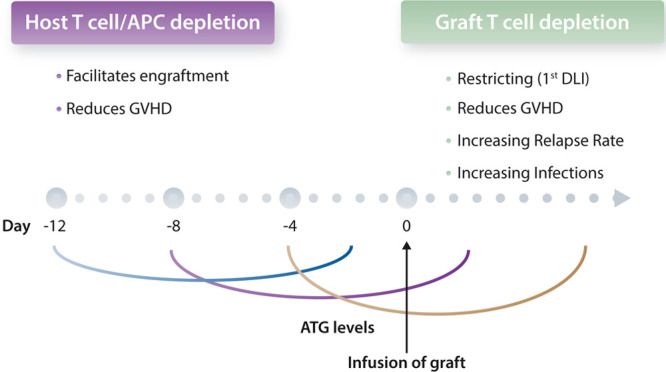
The dual sword of ATG, it is all about timing. APC = antigen presenting cell; ATG = anti-thymocyte globulin; DLI = donor lymphocyte infusion; GVHD = graft vs host disease.
Alternative ex vivo and in vivo T cell depletion platforms: campath and post-transplantation cyclophosphamide
To overcome the high variety in ATG products in terms of specificity and numbers used for in vivo T cell depletion, many centers have explored and still use campath. Campath is an anti-CD52 antibody which associates in clinical outcomes with low incidences of GVHD and can be given ex vivo “in the bag” or in vivo (Table 1 and20–22). A similar impact on interindividual variations in lymphocyte counts on active campath in the peripheral blood was reported.54 However, to the best of our knowledge, there have been no published transplantation studies based on this information. Disease-specific properties also need to be taken into consideration. For example, patients suffering from myelofibrosis have been reported to suffer from higher incidences of GVHD, and adding ruxolitinib before and shortly after transplantation has been reported to dampen GVHD, most likely by reducing cytokine storms.55,56 Another interesting transplantation platform is in vivo T cell depletion by administration of high-dose post-transplantation cyclophosphamide (PTCy) shortly after transplantation to eradicate allo-reactive αβT cells, which was first reported in transplantations using haploidentical donors. PTCy was also used more recently in HLA-matched donors (Table 1).23–28 Although PTCy allowed for a rapid increase in the use of haploidentical donors,57 most recent EBMT registry studies suggest that there is no substantial difference in composite endpoints when comparing ATG to PTCy in MUD donors (Table 1).29–33 Given that PTCy will be difficult to individualize in terms of PK and PD because of its complex chemistry,58 based on the retrospective analysis of registry data, one could even argue that dose adjusted ATG45,51 might be the preferred choice to date in fully matched MUD donors, while PTCy appears to be superior when 9 out of 10 MUD donors29 or haploidentical donors32 are used.
Ex vivo T cell engineering strategies focusing on defined subsets
Ex vivo T cell engineering by means of CD34 selection (Table 1) 34–37 or αβ T cell depletion (Table 1) 38–44 is the most controlled way to define graft composition to date, and has been shown to be at least as successful as other optimized platforms in reducing the incidence of GVHD while maintaining graft-versus-leukemia effects (Table 1). Pasquini et al34 reported that CD34 selection of peripheral derived blood stem cells of HLA-matched sibling donors results in a well-defined allograft, with 0.01–1 × 105 αβ T cells/kg, and is associated with a low incidence of chronic GVHD. Subsequently, αβ T cell/CD19 depletion entered clinical practice in haploidentical-SCT,59 matched related donors, and MUD.42 These depletion techniques restrict αβ T cells to around 0.1–1 × 105 αβ T cells/kg59 (comparable with CD34 selection) while preserving natural killer (NK) cells (CD3 depletion) or NK and γδ T cells (αβ T cell depletion). αβ T cell depletion associated with a low incidence of acute GVHD in a cohort of pediatric patients with both malignant and nonmalignant diseases43,60 was reported also for PTCy (Table 1). In a very recent study of adult patients with malignant disease, with a longer follow-up, chronic GVHD rate was also surprisingly low.38 Because graft engineering is more expensive and cumbersome compared to the application of ATG or PTCy, at this stage, most centers use ATG- or PTCy-based regimens. However, as engineering chimeric antigen receptor T cells increases in availability and prevalence, the complexity and costs of graft engineering will become negligible for centers when assessing its potential strategic advantages.3
The use of more complex engineering techniques will largely depend on reimbursement strategies. Beginning in 2020 in the Netherlands, reimbursement for graft engineering should facilitate its implementation to a broader patient population. In addition, as randomized studies comparing different transplantation platforms strategies will either not be performed at a larger scale, or are quickly outdated given the rapid pace of developments, creation of a new network of studies, like those designed for coronavirus disease (COVID)-related research, seems to be timely.61 The EBMT registry covering transplantations and cellular immune therapies like CAR T cells will be well-suited to serve as a potential backbone for these studies, but would need to be extended.62,63 These studies should also include inquiries into the socio-economic impacts and quality of life for patients in order to cover all aspects of earlier financial investments and future societal gains.62
T cell depletion and viral reactivations
The 2 clinical measurements of a well-balanced T cell reconstitution are the incidence of GVHD and viral reactivations. While data on incidence of GVHD are usually decently reported in registries and clinical trials, data on different viral reactivations or infections after allo-SCT in the different platforms are scarce (Table 1). Lack of reporting does not necessarily indicate the absence of the event. For example, initial reports on PTCy did not report on BK virus (BK)-reactivation, while later studies indicate that BK-reactivations are a substantial clinical problem in up to one fifth of all patients.24,25 Also, reporting on cytomegalovirus (CMV) reactivations is rather heterogenous. While incidences of infections are related to the overall cohort (frequently around 30%–40%) most of the time, incidences can be underestimated, as reactivation occurs mainly in CMV-positive recipients. Reporting incidence in relation to recipient positivity would be more appropriate38 and allow for a better comparison of incidences between different trials and retrospective cohorts. The difference between preemptive CMV treatment and actual CMV disease is important information for assessing the strength of an immune system and more important than diving into different techniques and thresholds of CMV reporting, which are rather heterogenous and do not inform us about immune competence.64 Incidences on Epstein-Barr virus (EBV) infections and reactivations are also not indicated in most reports (Table 1). Initial studies in 2001 showed an incidence of EBV reactivation of around 30% in T cell replete transplantation platforms, while after CD34 graft engineering with different techniques, 65% of EBV reactivations were reported.37 EBV reactivations were, at the time of the initial reports, a substantial clinical problem, as anti-CD20 antibodies were only approved in 1998 by the European Medicines Agency. In 2020, the use of ATG or haploidentical donors were still identified as risk factors for EBV reactivations.65 Also in the absence of CD19 depletion within the context of an αβT cell depletion, incidences of EBV reactivation have been reported to be around 40%,38 frequencies which are in line with what was reported after campath conditioning.22 However, as these reports are from patients treated after anti-CD20 antibodies were approved, no substantial increases in posttransplant lymphoproliferative disorders are observed in either platform to date. Thus, reactivation of a virus does not necessarily affect clinical outcomes, as evidenced by the history of EBV reactivation over the last 2 decades. Adding CD19 depletion during graft engineering to the αβT cell depletion platform, however, nearly abolished EBV reactivations in an interim analysis of an ongoing clinical trial (NL6940, de Witte and Kuball, unpublished observation, 2021). Thus, filling the knowledge gap on viral reactivations in relation to the transplantation regimen in a registry would allow for better assessment of immune competence across different platforms. Viral reactivations do not always need to be harmful, and some viral reactivations might be even beneficial. For example, it has been suggested that CMV reactivations could assist in certain, but not all, transplantation platforms to improve leukemia control.64,66–71 Novel drugs that prevent CMV reactivation and have been introduced recently to the market72 could, within this context, be counterproductive for certain transplantation platforms.
Immune reconstitution impacts toxicity and efficacy of post-transplantation viral reactivations and maintenance therapies
Acknowledging that different transplantation platforms associate with different immune repertoires early after transplantation1 increases our understanding of the different biological impacts of CMV reactivation on toxicity and leukemia control.68 These different immune repertoires can also provide a strategic reason to choose 1 transplantation platform over the other, as different options arise for maintenance therapies after transplantation. αβ T cell depletion is, in contrast to all other platforms, dominated by an early NK cell and γδT cell recovery, most likely derived from the infused product (Figure 3),38,39,43,60 which usually does not require immune suppression beyond 1 month. ATG-based platforms favor a strong recovery of αβT cells, which associate with the need of additional immune suppression for at least 4 months.51 PTCy has been suggested to selectively deplete rapidly proliferating allo-reactive αβT cells. Rapidly reconstituting NK cells and γδT cells are most likely also diminished as collateral damage of PTCy. However most recent data suggest that depletion of allo-reactive αβT cells is less important than creating a αβT cell dysfunction and suppression.73,74 This would explain why a prolonged immune suppression is needed after PTCy. The increased immune suppressive environment after PTCy would also better explain the conflicting data reported on numbers between different immune reconstitution patterns observed after ATG and PTCy-based transplantation regimens.33,75 The described substantial differences in immune reconstitution patterns depending on transplantation regimens most likely explain the aforementioned conflicting data on the impact of CMV reactivation on leukemia control. NK and γδT cells, particularly after αβT cell depletion, are dominant subsets that will be protective as they both expand upon CMV reactivation and also cross-react with leukemic cells.67,68,76–82 We, and others, have shown that ex vivo T cell depletion strategy regimens favor memory cells like NK cells and γδT cells.38,39,60 However, a major challenge remains sporadic reporting and lack of harmonization in immune monitoring (eg, Table 2 and, 25,38,39,41,42,83 for review53).
Figure 3.
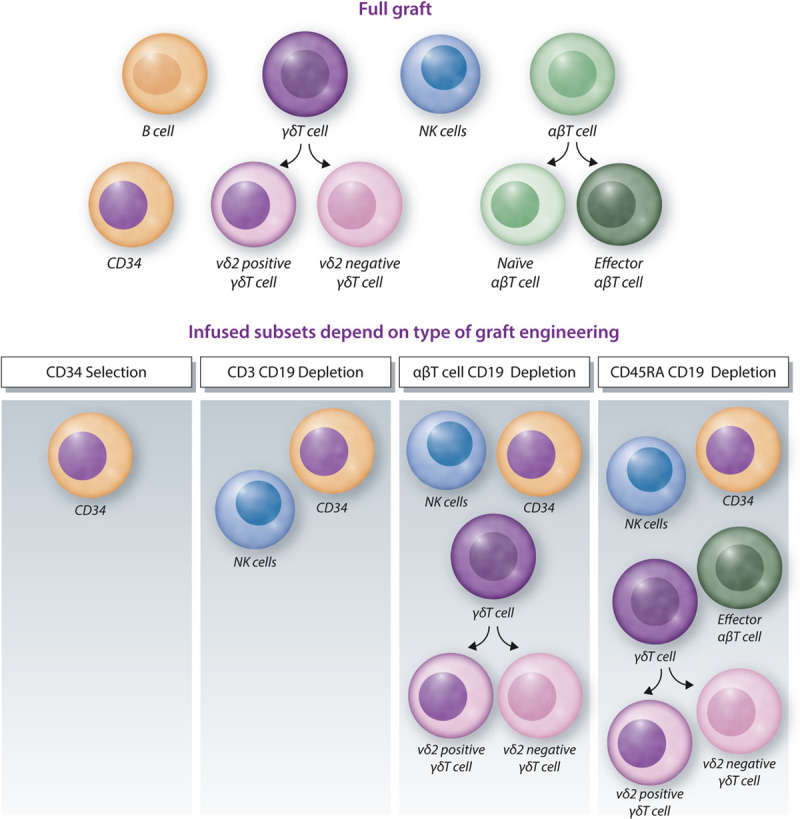
Graft engineering. Different graft compositions are depicted from different graft sources after different types of graft engineering. NK = natural killer.
Table 2.
Immunological Recovery After Allo-SCT With T Cell Depletion.
| Study | Intervention | T Cell IR | γδT Cell IR | αβT Cell IR | B Cell IR | NK Cell IR |
|---|---|---|---|---|---|---|
| Berger et al25 | PTCy | Day 60 | NA | NA | Day 60 | Day 60 |
| CD4 132/μL | 46/μL | 154/μL | ||||
| CD8 491/μL | ||||||
| de Witte et al38 | αβT cell depletion | Day 100 | Day 100 | Day 100 | Day 100 | |
| 43/μL | CD8 148/μL; CD4 60/μL | 130/μL | 182/μL | |||
| vd2+ 21/μL | ||||||
| vd2- 22/μL | ||||||
| Locatelli et al39 | αβT cell/CD19 depletion | Day 100 | Day 100 | Day 100 | Day 100 | Day 100 |
| CD3 254/μL | 46/μL | 186/μL | 2/μL | 196/μL | ||
| CD4 89/μL | ||||||
| CD8 98/μL | ||||||
| Lang et at41 | αβT cell/CD19 depletion | Day 90 | NA | NA | Day 90 | Day 30 |
| CD3 374–479/μL | 24–83/μL | 352–430/μL | ||||
| CD4 120–159/μL | ||||||
| Maschan et al42 | αβT cell/CD19 depletion | Day 30 | NA | NA | NA | Day 30 |
| CD3 245/μL | 385/μL | |||||
| CD4 40/μL | ||||||
| CD8 135/μL | ||||||
| Laberko et al83 | αβT cell/CD19 depletion | Day 120 | Day 120 | Day 120 | Day 120 | NA |
| CD3 220–384/μL | 38/μL | 262/μL | 18–44/μL |
Examples representing diversity in analyses.
allo-SCT = allogeneic stem cell transplantation; IR = immune reconstitution; NA = not available; NK = natural killer; PTCy = post-transplantation cyclophosphamide; μL = microliter.
Not only infections, but drugs can also act differently, depending on the current immune environment (Figure 4). After T cell replete transplantation, lenalidomide has detrimental effects by inducing lethal GVHD if given shortly after transplantation.84 In contrast, tyrosine kinase inhibitors, currently used in clinical trials as maintenance therapies for leukemia control, most likely do not substantially depend on the transplantation platform in terms of toxicity. However, part of their activity was suggested to be mediated via interleukin-15 on CD8 positive αβT cells,85 and a strong immune suppression might therefore impair efficacy. However, more data are needed to thoroughly investigate the impact of post-transplantation pharmacological interventions on clinical outcomes. Checkpoint inhibitors are another example of a drug where the currently available reports suggest that they are most likely harmful if administered too early after transplantation in T cell replete platforms. Toxicity in T cell deplete platforms might be less due to the reduced size of the αβT cell repertoire (Figure 4).2,86 Also the choice of bispecific molecules could be driven by the transplantation platform. Anti-CD3 engagers could bare the risk of inducing GVHD if used too early in T cell replete platforms, and trispecific killer engager molecules, which mainly act in NK cells, could be the preferred choice for transplantation platforms favoring a strong NK cell reconstitution.87
Figure 4.
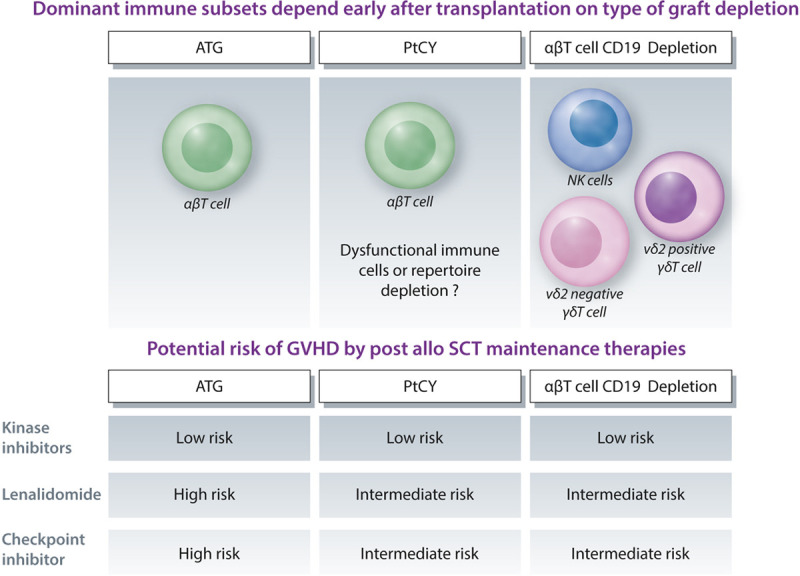
Transplantation platforms, associated dominant immune repertoires early after transplantation, and potential risk of GVHD induced by maintenance therapy. ATG = anti-thymocyte globulin; GVHD = graft vs host disease; NK = natural killer; PTCy = post-transplantation cyclophosphamide; SCT = stem cell transplantation.
Donor lymphocyte infusions and engineered immune effector cells within the context of different transplantation platforms
Donor lymphocyte infusion (DLI) are considered as the cornerstone of allo-SCT, either used as prophylactic intervention (thus in patients with a priori high risk of relapse) preemptively (thus triggered by minimal residual disease or a reduced donor chimerism), or as therapy once relapse occurs.1,2 In particular, the preemptive and prophylactic usage of DLIs depends on the transplantation platform in terms of timing and dosing (Figure 5) (modified and updated from Yun and Waller88). After αβT cell depletion, we have been able to administer a DLI to the majority of patients 3 months after allo-SCT, while preemptive or prophylactic DLI after other types of transplantations are usually administered later, and for T cell replete transplantations are only an option for a minority of patients (for review see1). T cell replete platforms including ATG and PTCy require per default ongoing immune suppression for many months to keep co-infused graft T cells under control. In the case of GVHD, immune suppression needs to be prolonged. However, the drawback of acute GVHD is extensive chronic GVHD in a majority of patients, at least when treated with ATG, and will severely impact quality of life and overall survival. An additional important consideration is the upcoming combination therapy of allo-SCT beyond DLI, but with genetically modified T, NK, γδT cells.3,77,89–93 These strategies favor platforms that do not depend on prolonged immune suppressions.
Figure 5.
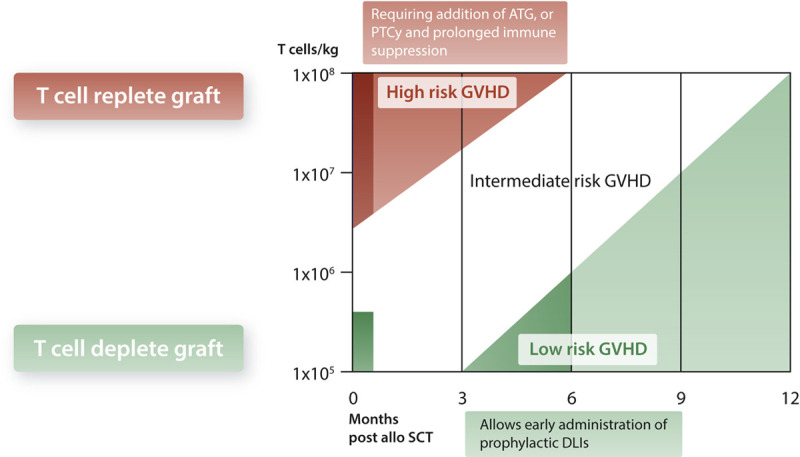
Dosing and timing of DLI. Risk of GVHD is plotted against T cell numbers received either during graft infusion, or later as donor lymphocyte infusion (modified and updated from Yun and Waller88). ATG = anti-thymocyte globulin; DLI = donor lymphocyte infusion; GVHD = graft vs host disease; PTCy = post-transplantation cyclophosphamide; SCT = stem cell transplantation.
Conclusions: adjust!
Substantial variations in terms of immune reconstitution are observed within and between different allo-SCT platforms. However, it is striking that the vast majority of allo-SCT studies only report clinical outcomes as parameters of success, and that information on the actual “drug delivered”—the infused donor cells and their capacity to execute a GVL reaction—is often lacking. Better capturing, understanding, and mastering a well-balanced immune recovery for all platforms will be key for increasing the overall success of transplantation, as well as for novel posttransplantation interventions. This knowledge will allow for better assessment of the habits and strategic choices of different centers, can guide daily practice, and create a basis for novel trial designs. The benchmarking initiative started by EBMT can assist in increasing data completeness and quality63 to develop COVID-inspired trial designs such as “a randomized embedded multifactorial adaptive platform”61 for the entire community to create more data evaluating “counts” and “adjustments.”
Practical advice of count and adjust
One might argue that our proposed strategy of individualized allo-transplantation platforms and counting of all graft ingredients is either not applicable in daily routine due to its complexity, or should be first further evaluated in clinical studies. While clinical studies are, of course, of great importance for future insights into individualized transplantation platforms, we provided vast evidence that many steps should be introduced today into clinical routines. We advocate for 4 different pillars, with different levels of readiness. The first pillar, which is ready for all centers today, is overcoming the vast overshoot of T cells in grafts for all T cell replete transplantations. This would only require the addition of CD3 to CD34 counts as a quality control before infusion at a center, as not all donor centers provide grafts with T cell counts (Figure 1). Donor centers are at the same time encouraged to add CD3 counts to CD34 counts during their daily routine. The second pillar is adjusting the dosing of ATG based on lymphocyte counts when using ATG-Thymoglobulin, as advised in the recent EBMT handbook.53 In contrast, more data will be needed for lymphocyte count-adjusted ATG-Fresenius before providing specific advice. The third pillar is graft engineering as an additional intervention that can be introduced into daily clinical practice if strategically interesting, although associated with expertise of the stem cell laboratory and also with additional costs during the beginning of the transplantation. Whether graft engineering is superior as compared to T cell replete transplantation strategies when carefully restricting dosing of T cells and ATG remains to be clinically studied. The fourth pillar is therapeutic drug monitoring during conditioning, which is well-established for busulfan. Dose adjustments of additional chemotherapy agents used during conditioning have been studied less extensively and need further evaluation before being implemented into daily clinical routine. Thus, we conclude that the 2 main points of advice, either infusing less graft cells and when administering ATG-Fresenius adjusting timing, or dosing for patients with low lymphocyte counts, can be implemented at all centers today without any additional costs.
Disclosures
JK reports grants from Gadeta, Novartis, and Miltenyi Biotech and is the inventor on patents dealing with γδT cell-related aspects, as well as the co-founder and shareholder of Gadeta. All the other authors have no conflicts of interest to disclose.
Sources of funding
This study was provided by Stichting Koningin Wilhelmina Fonds (KWF) UU 2015-7553 to MdW; Nederlandse organisatie voor gezondheidsonderzoek en zorginnovatie (ZonMW) 43400003 and VIDI-ZonMW 917.11.337, KWF UU 2010-4669, UU 2013-6426, UU 2014-6790, and UU 2015-7601, UU 2018-11393, UU 2018-11979, UU 2020-12586, and UU 2021-13043 to JK.
References
- 1.Schmid C, Kuball J, Bug G. Defining the role of donor lymphocyte infusion in high-risk hematologic malignancies. J Clin Oncol. 2021; 39:397–418. [DOI] [PubMed] [Google Scholar]
- 2.Frederik Falkenburg JH, Schmid C, Kolb HJ, Locatelli F, Kuball J. Delayed transfer of immune cells or the art of donor lymphocyte infusion. In: Carreras E, Dufour C, Mohty M, Kroger N, eds. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. 7th ed. Cham, Switzerland: Springer; 2019:443–448. [PubMed] [Google Scholar]
- 3.Chabannon C, Kuball J, Bondanza A, et al. Hematopoietic stem cell transplantation in its 60s: a platform for cellular therapies. Sci Transl Med. 2018; 10:eaap9630. [DOI] [PubMed] [Google Scholar]
- 4.Czerw T, Labopin M, Schmid C, et al. High CD3+ and CD34+ peripheral blood stem cell grafts content is associated with increased risk of graft-versus-host disease without beneficial effect on disease control after reduced-intensity conditioning allogeneic transplantation from matched unrelated donors for acute myeloid leukemia - an analysis from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Oncotarget. 2016; 7:27255–27266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mussetti A, De Philippis C, Carniti C, et al. CD3+ graft cell count influence on chronic GVHD in haploidentical allogeneic transplantation using post-transplant cyclophosphamide. Bone Marrow Transplant. 2018; 53:1522–1531. [DOI] [PubMed] [Google Scholar]
- 6.Langenhorst JB, Boss J, van Kesteren C, et al. A semi-mechanistic model based on glutathione depletion to describe intra-individual reduction in busulfan clearance. Br J Clin Pharmacol. 2020; 86:1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartelink IH, Lalmohamed A, van Reij EM, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. 2016; 3:e526–e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartelink IH, van Reij EM, Gerhardt CE, et al. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol Blood Marrow Transplant. 2014; 20:345–353. [DOI] [PubMed] [Google Scholar]
- 9.Bartelink IH, Boelens JJ, Bredius RG, et al. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. 2012; 51:331–345. [DOI] [PubMed] [Google Scholar]
- 10.Langenhorst JB, Dorlo TPC, van Maarseveen EM, et al. Population pharmacokinetics of fludarabine in children and adults during conditioning prior to allogeneic hematopoietic cell transplantation. Clin Pharmacokinet. 2019; 58:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohty M. Mechanisms of action of antithymocyte globulin: T-cell depletion and beyond. Leukemia. 2007; 21:1387–1394. [DOI] [PubMed] [Google Scholar]
- 12.Bacigalupo A. ATG in allogeneic stem cell transplantation: standard of care in 2017? Point. Blood Adv. 2017; 1:569–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baron F, Mohty M, Blaise D, et al. Anti-thymocyte globulin as graft-versus-host disease prevention in the setting of allogeneic peripheral blood stem cell transplantation: a review from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. 2017; 102:224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang YJ, Wu DP, Lai YR, et al. Antithymocyte globulin for matched sibling donor transplantation in patients with hematologic malignancies: a multicenter, open-label, randomized controlled study. J Clin Oncol. 2020; 38:3367–3376. [DOI] [PubMed] [Google Scholar]
- 15.Walker I, Panzarella T, Couban S, et al. Addition of anti-thymocyte globulin to standard graft-versus-host disease prophylaxis versus standard treatment alone in patients with haematological malignancies undergoing transplantation from unrelated donors: final analysis of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2020; 7:e100–e111. [DOI] [PubMed] [Google Scholar]
- 16.Socié G, Schmoor C, Bethge WA, et al. Chronic graft-versus-host disease: long-term results from a randomized trial on graft-versus-host disease prophylaxis with or without anti-T-cell globulin ATG-Fresenius. Blood. 2011; 117:6375–6382. [DOI] [PubMed] [Google Scholar]
- 17.Baron F, Galimard JE, Labopin M, et al. Allogeneic peripheral blood stem cell transplantation with anti-thymocyte globulin versus allogeneic bone marrow transplantation without anti-thymocyte globulin. Haematologica. 2020; 105:1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finke J, Bethge WA, Schmoor C, et al. Standard graft-versus-host disease prophylaxis with or without anti-T-cell globulin in haematopoietic cell transplantation from matched unrelated donors: a randomised, open-label, multicentre phase 3 trial. Lancet Oncol. 2009; 10:855–864. [DOI] [PubMed] [Google Scholar]
- 19.Soiffer RJ, Kim HT, McGuirk J, et al. Prospective, randomized, double-blind, phase III clinical trial of anti-T-lymphocyte globulin to assess impact on chronic graft-versus-host disease-free survival in patients undergoing HLA-matched unrelated myeloablative hematopoietic cell transplantation. J Clin Oncol. 2017; 35:4003–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green K, Pearce K, Sellar RS, et al. Impact of alemtuzumab scheduling on graft-versus-host disease after unrelated donor fludarabine and melphalan allografts. Biol Blood Marrow Transplant. 2017; 23:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Besien K, Kunavakkam R, Rondon G, et al. Fludarabine-melphalan conditioning for AML and MDS: alemtuzumab reduces acute and chronic GVHD without affecting long-term outcomes. Biol Blood Marrow Transplant. 2009; 15:610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpenter B, Haque T, Dimopoulou M, et al. Incidence and dynamics of Epstein-Barr virus reactivation after alemtuzumab-based conditioning for allogeneic hematopoietic stem-cell transplantation. Transplantation. 2010; 90:564–570. [DOI] [PubMed] [Google Scholar]
- 23.Kanakry CG, Tsai HL, Bolaños-Meade J, et al. Single-agent GVHD prophylaxis with posttransplantation cyclophosphamide after myeloablative, HLA-matched BMT for AML, ALL, and MDS. Blood. 2014; 124:3817–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cieri N, Greco R, Crucitti L, et al. Post-transplantation cyclophosphamide and sirolimus after haploidentical hematopoietic stem cell transplantation using a treosulfan-based myeloablative conditioning and peripheral blood stem cells. Biol Blood Marrow Transplant. 2015; 21:1506–1514. [DOI] [PubMed] [Google Scholar]
- 25.Berger M, Lanino E, Cesaro S, et al. Feasibility and outcome of haploidentical hematopoietic stem cell transplantation with post-transplant high-dose cyclophosphamide for children and adolescents with hematologic malignancies: an AIEOP-GITMO retrospective multicenter study. Biol Blood Marrow Transplant. 2016; 22:902–909. [DOI] [PubMed] [Google Scholar]
- 26.Devillier R, Bramanti S, Fürst S, et al. T-replete haploidentical allogeneic transplantation using post-transplantation cyclophosphamide in advanced AML and myelodysplastic syndromes. Bone Marrow Transplant. 2016; 51:194–198. [DOI] [PubMed] [Google Scholar]
- 27.Mehta RS, Saliba RM, Chen J, et al. Post-transplantation cyclophosphamide versus conventional graft-versus-host disease prophylaxis in mismatched unrelated donor haematopoietic cell transplantation. Br J Haematol. 2016; 173:444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mielcarek M, Furlong T, O’Donnell PV, et al. Posttransplantation cyclophosphamide for prevention of graft-versus-host disease after HLA-matched mobilized blood cell transplantation. Blood. 2016; 127:1502–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Battipaglia G, Labopin M, Kröger N, et al. Posttransplant cyclophosphamide vs antithymocyte globulin in HLA-mismatched unrelated donor transplantation. Blood. 2019; 134:892–899. [DOI] [PubMed] [Google Scholar]
- 30.Battipaglia G, Labopin M, Hamladji RM, et al. Post-transplantation cyclophosphamide versus antithymocyte globulin in patients with acute myeloid leukemia undergoing allogeneic stem cell transplantation from HLA-identical sibling donors: a retrospective analysis from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Cancer. 2021; 127:209–218. [DOI] [PubMed] [Google Scholar]
- 31.Brissot E, Labopin M, Moiseev I, et al. Post-transplant cyclophosphamide versus antithymocyte globulin in patients with acute myeloid leukemia in first complete remission undergoing allogeneic stem cell transplantation from 10/10 HLA-matched unrelated donors. J Hematol Oncol. 2020; 13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruggeri A, Sun Y, Labopin M, et al. Post-transplant cyclophosphamide versus anti-thymocyte globulin as graft- versus-host disease prophylaxis in haploidentical transplant. Haematologica. 2017; 102:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Retière C, Willem C, Guillaume T, et al. Impact on early outcomes and immune reconstitution of high-dose post-transplant cyclophosphamide vs anti-thymocyte globulin after reduced intensity conditioning peripheral blood stem cell allogeneic transplantation. Oncotarget. 2018; 9:11451–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasquini MC, Devine S, Mendizabal A, et al. Comparative outcomes of donor graft CD34+ selection and immune suppressive therapy as graft-versus-host disease prophylaxis for patients with acute myeloid leukemia in complete remission undergoing HLA-matched sibling allogeneic hematopoietic cell transplantation. J Clin Oncol. 2012; 30:3194–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barba P, Hilden P, Devlin SM, et al. Ex vivo CD34+-selected T cell-depleted peripheral blood stem cell grafts for allogeneic hematopoietic stem cell transplantation in acute leukemia and myelodysplastic syndrome is associated with low incidence of acute and chronic graft-versus-host disease and high treatment response. Biol Blood Marrow Transplant. 2017; 23:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayraktar UD, de Lima M, Saliba RM, et al. Ex vivo T cell-depleted versus unmodified allografts in patients with acute myeloid leukemia in first complete remission. Biol Blood Marrow Transplant. 2013; 19:898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Esser JW, van der Holt B, Meijer E, et al. Epstein-Barr virus (EBV) reactivation is a frequent event after allogeneic stem cell transplantation (SCT) and quantitatively predicts EBV-lymphoproliferative disease following T-cell–depleted SCT. Blood. 2001; 98:972–978. [DOI] [PubMed] [Google Scholar]
- 38.de Witte MA, Janssen A, Nijssen K, et al. αβ T-cell graft depletion for allogeneic HSCT in adults with hematological malignancies. Blood Adv. 2021; 5:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Locatelli F, Merli P, Pagliara D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after αβ T-cell and B-cell depletion. Blood. 2017; 130:677–685. [DOI] [PubMed] [Google Scholar]
- 40.Laberko A, Sultanova E, Gutovskaya E, et al. Mismatched related vs matched unrelated donors in TCRαβ/CD19-depleted HSCT for primary immunodeficiencies. Blood. 2019; 134:1755–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lang P, Feuchtinger T, Teltschik HM, et al. Improved immune recovery after transplantation of TCRαβ/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transplant. 2015; 50(suppl 2):S6–S10. [DOI] [PubMed] [Google Scholar]
- 42.Maschan M, Shelikhova L, Ilushina M, et al. TCR-alpha/beta and CD19 depletion and treosulfan-based conditioning regimen in unrelated and haploidentical transplantation in children with acute myeloid leukemia. Bone Marrow Transplant. 2016; 51:668–674. [DOI] [PubMed] [Google Scholar]
- 43.Bertaina A, Merli P, Rutella S, et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. 2014; 124:822–826. [DOI] [PubMed] [Google Scholar]
- 44.Balashov D, Shcherbina A, Maschan M, et al. Single-center experience of unrelated and haploidentical stem cell transplantation with TCRαβ and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. 2015; 21:1955–1962. [DOI] [PubMed] [Google Scholar]
- 45.Boelens JJ, Admiraal R, Kuball J, et al. Fine-tuning antithymocyte globulin dosing and harmonizing clinical trial design. J Clin Oncol. 2018; 36:1175–1176. [DOI] [PubMed] [Google Scholar]
- 46.Admiraal R, Lindemans CA, van Kesteren C, et al. Excellent T-cell reconstitution and survival depend on low ATG exposure after pediatric cord blood transplantation. Blood. 2016; 128:2734–2741. [DOI] [PubMed] [Google Scholar]
- 47.de Koning C, Nierkens S, Boelens JJ. Strategies before, during, and after hematopoietic cell transplantation to improve T-cell immune reconstitution. Blood. 2016; 128:2607–2615. [DOI] [PubMed] [Google Scholar]
- 48.Admiraal R, van Kesteren C, Jol-van der Zijde CM, et al. Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haemopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol. 2015; 2:e194–e203. [DOI] [PubMed] [Google Scholar]
- 49.Lindemans CA, Te Boome LC, Admiraal R, et al. Sufficient immunosuppression with thymoglobulin is essential for a successful haplo-myeloid bridge in haploidentical-cord blood transplantation. Biol Blood Marrow Transplant. 2015; 21:1839–1845. [DOI] [PubMed] [Google Scholar]
- 50.Storek J, Mohty M, Boelens JJ. Rabbit anti-T cell globulin in allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015; 21:959–970. [DOI] [PubMed] [Google Scholar]
- 51.Admiraal R, Nierkens S, de Witte MA, et al. Association between anti-thymocyte globulin exposure and survival outcomes in adult unrelated haemopoietic cell transplantation: a multicentre, retrospective, pharmacodynamic cohort analysis. Lancet Haematol. 2017; 4:e183–e191. [DOI] [PubMed] [Google Scholar]
- 52.Storek J. Anti-thymocyte globulin dosing-per kg or per lymphocyte? Lancet Haematol. 2017; 4:e154–e155. [DOI] [PubMed] [Google Scholar]
- 53.Kuball J, Boelens JJ. Clinical and biological concepts for mastering immune reconstitution after HSCT: toward practical guidelines and greater harmonization. In: Carreras E, Dufour C, Mohty M, Kroger N, eds. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. 7th ed. Cham, Switzerland: Springer; 2019:69–74. [PubMed] [Google Scholar]
- 54.Admiraal R, Jol-van der Zijde CM, Furtado Silva JM, et al. Population pharmacokinetics of alemtuzumab (campath) in pediatric hematopoietic cell transplantation: towards individualized dosing to improve outcome. Clin Pharmacokinet. 2019; 58:1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kröger N, Shahnaz Syed Abd Kadir S, Zabelina T, et al. Peritransplantation ruxolitinib prevents acute graft-versus-host disease in patients with myelofibrosis undergoing allogenic stem cell transplantation. Biol Blood Marrow Transplant. 2018; 24:2152–2156. [DOI] [PubMed] [Google Scholar]
- 56.Shahnaz Syed Abd Kadir S, Christopeit M, Wulf G, et al. Impact of ruxolitinib pretreatment on outcomes after allogeneic stem cell transplantation in patients with myelofibrosis. Eur J Haematol. 2018; 101:305–317. [DOI] [PubMed] [Google Scholar]
- 57.Passweg JR, Baldomero H, Bader P, et al. Use of haploidentical stem cell transplantation continues to increase: the 2015 European Society for Blood and Marrow Transplant activity survey report. Bone Marrow Transplant. 2017; 52:811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Jonge ME, Huitema AD, Rodenhuis S, et al. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet. 2005; 44:1135–1164. [DOI] [PubMed] [Google Scholar]
- 59.Handgretinger R. Negative depletion of CD3(+) and TcRαβ(+) T cells. Curr Opin Hematol. 2012; 19:434–439. [DOI] [PubMed] [Google Scholar]
- 60.Airoldi I, Bertaina A, Prigione I, et al. γδ T-cell reconstitution after HLA-haploidentical hematopoietic transplantation depleted of TCR-αβ+/CD19+ lymphocytes. Blood. 2015; 125:2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angus DC, Berry S, Lewis RJ, et al. The REMAP-CAP (randomized embedded multifactorial adaptive platform for community-acquired pneumonia) study. Rationale and design. Ann Am Thorac Soc. 2020; 17:879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGrath E, Chabannon C, Terwel S, et al. Opportunities and challenges associated with the evaluation of chimeric antigen receptor T cells in real-life. Curr Opin Oncol. 2020; 32:427–433. [DOI] [PubMed] [Google Scholar]
- 63.Snowden JA, Saccardi R, Orchard K, et al. Benchmarking of survival outcomes following haematopoietic stem cell transplantation: a review of existing processes and the introduction of an international system from the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Bone Marrow Transplant. 2020; 55:681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verduyn Lunel FM, Raymakers R, van Dijk A, et al. Cytomegalovirus status and the outcome of T cell-replete reduced-intensity allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2016; 22:1883–1887. [DOI] [PubMed] [Google Scholar]
- 65.Ru Y, Zhang X, Song T, et al. Epstein-Barr virus reactivation after allogeneic hematopoietic stem cell transplantation: multifactorial impact on transplant outcomes. Bone Marrow Transplant. 2020; 55:1754–1762. [DOI] [PubMed] [Google Scholar]
- 66.Elmaagacli AH, Steckel NK, Koldehoff M, et al. Early human cytomegalovirus replication after transplantation is associated with a decreased relapse risk: evidence for a putative virus-versus-leukemia effect in acute myeloid leukemia patients. Blood. 2011; 118:1402–1412. [DOI] [PubMed] [Google Scholar]
- 67.Cichocki F, Cooley S, Davis Z, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia. 2016; 30:456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Litjens NHR, van der Wagen L, Kuball J, et al. Potential beneficial effects of cytomegalovirus infection after transplantation. Front Immunol. 2018; 9:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cichocki F, Taras E, Chiuppesi F, et al. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight. 2019; 4:125553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaito S, Nakajima Y, Hara K, et al. Heterogeneous impact of cytomegalovirus reactivation on nonrelapse mortality in hematopoietic stem cell transplantation. Blood Adv. 2020; 4:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Green ML, Leisenring W, Xie H, et al. Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre-emptive therapy: a retrospective cohort study. Lancet Haematol. 2016; 3:e119–e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marty FM, Ljungman P, Chemaly RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017; 377:2433–2444. [DOI] [PubMed] [Google Scholar]
- 73.Wachsmuth LP, Patterson MT, Eckhaus MA, et al. Post-transplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J Clin Invest. 2019; 129:2357–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rambaldi B, Kim HT, Reynolds C, et al. Impaired T- and NK-cell reconstitution after haploidentical HCT with posttransplant cyclophosphamide. Blood Adv. 2021; 5:352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dekker L, de Koning C, Lindemans C, et al. Reconstitution of T cell subsets following allogeneic hematopoietic cell transplantation. Cancers (Basel). 2020; 12:E1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scheper W, van Dorp S, Kersting S, et al. γδT cells elicited by CMV reactivation after allo-SCT cross-recognize CMV and leukemia. Leukemia. 2013; 27:1328–1338. [DOI] [PubMed] [Google Scholar]
- 77.Sebestyen Z, Prinz I, Déchanet-Merville J, et al. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat Rev Drug Discov. 2020; 19:169–184. [DOI] [PubMed] [Google Scholar]
- 78.de Witte MA, Kuball J, Miller JS. NK cells and γδT cells for relapse protection after allogeneic hematopoietic cell transplantation (HCT). Curr Stem Cell Rep. 2017; 3:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johanna I, Straetemans T, Heijhuurs S, et al. Evaluating in vivo efficacy - toxicity profile of TEG001 in humanized mice xenografts against primary human AML disease and healthy hematopoietic cells. J Immunother Cancer. 2019; 7:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sebestyen Z, Scheper W, Vyborova A, et al. RhoB mediates phosphoantigen recognition by Vγ9Vδ2 T cell receptor. Cell Rep. 2016; 15:1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scheper W, Gründer C, Straetemans T, et al. Hunting for clinical translation with innate-like immune cells and their receptors. Leukemia. 2014; 28:1181–1190. [DOI] [PubMed] [Google Scholar]
- 82.Scheper W, Sebestyen Z, Kuball J. Cancer immunotherapy using γδT cells: dealing with diversity. Front Immunol. 2014; 5:601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laberko A, Bogoyavlenskaya A, Shelikhova L, et al. Risk factors for and the clinical impact of cytomegalovirus and Epstein-Barr virus infections in pediatric recipients of TCR-α/β- and CD19-DEPLETED GRAFTS. Biol Blood Marrow Transplant. 2017; 23:483–490. [DOI] [PubMed] [Google Scholar]
- 84.Kneppers E, van der Holt B, Kersten MJ, et al. Lenalidomide maintenance after nonmyeloablative allogeneic stem cell transplantation in multiple myeloma is not feasible: results of the HOVON 76 Trial. Blood. 2011; 118:2413–2419. [DOI] [PubMed] [Google Scholar]
- 85.Mathew NR, Baumgartner F, Braun L, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. 2018; 24:282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Bergen CA, van Luxemburg-Heijs SA, de Wreede LC, et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J Clin Invest. 2017; 127:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020; 17:418–434. [DOI] [PubMed] [Google Scholar]
- 88.Yun HD, Waller EK. Finding the sweet spot for donor lymphocyte infusions. Biol Blood Marrow Transplant. 2013; 19:507–508. [DOI] [PubMed] [Google Scholar]
- 89.Straetemans T, Janssen A, Jansen K, et al. TEG001 insert integrity from vector producer cells until medicinal product. Mol Ther. 2020; 28:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vyborova A, Beringer DX, Fasci D, et al. γ9δ2T cell diversity and the receptor interface with tumor cells. J Clin Invest. 2020; 130:4637–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kierkels GJJ, Scheper W, Meringa AD, et al. Identification of a tumor-specific allo-HLA-restricted γδTCR. Blood Adv. 2019; 3:2870–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Straetemans T, Kierkels GJJ, Doorn R, et al. GMP-grade manufacturing of T cells engineered to express a defined γδTCR. Front Immunol. 2018; 9:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith M, Zakrzewski J, James S, et al. Posttransplant chimeric antigen receptor therapy. Blood. 2018; 131:1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]